

Adaptation to the environment is one of the fundamental regulatory processes in biology and is found among both simple and complex organisms. In a changing environment, simple organisms enhance species survival by high rates of spontaneous mutation achieved by several means: short maturation rates, rapid rates of reproduction, recombination through sexual reproduction, and large numbers of offspring. Then, by a process of natural selection, organisms that are adapted to their environment will survive and multiply. The process of natural selection also affects complex multicellular organisms and promotes adaptive changes. However, this simple strategy for survival becomes less effective in multicellular organisms as the ecological niche becomes more complex and the rates of maturation and fertility decrease. As a result, changes in the environment outpace the rate of genetic evolutionary change, which is limited by generation time. How do multicellular organisms produce adaptive change without genetic mutation?
One solution to this problem is the development of complex physiological and behavioral systems coordinated by a CNS. The nervous system permits rapid adaptation to changing environmental conditions without genetic mutation (Kandel, 1984) by coordinating inputs from the internal and external environment via receptors and directing a complicated physiological response through various effector systems to maintain homeostasis. In many instances, homeostasis is maintained by reflexes and fixed action patterns in response to a stimulus. A problem arises when a stimulus or event in the environment does not determinately predict a condition or appropriate response to that condition.
To cope with environmental complexity and ambiguity, an organism requires mechanisms that allow experience to affect relatively long-lasting changes in behavior. With a nervous system, the organism can accomplish this through a mechanism that has been called learning (Scott, 1965; Hilgard and Bower, 1975). According to the cellular connectionist hypothesis of Tanzi (1894) and Ramon y Cajal (1894), behavior modification is achieved by the strengthening of preexisting connections and by recombining potential combinations of dormant pathways between neural pathways that mediate innate response laid down during development or by the growth of new connections. Thus, recombination of connections between structural pathways increases the information storage capacity of the nervous system.
So, how does the nervous system generate the diversity of cell types and connections and form long-term changes in synaptic connections as a result of experience (memory) with only 20,000-30,000 genes in the vertebrate genome? Although somatic mutation in neuronal precursors through retrotransposon hopping has been proposed (Muotri et al., 2005) to generate diversity in the nervous system, as it does in the immune system, it is more likely that permanent changes in gene expression patterns are achieved through permanent changes in chromatin remodeling without changes in DNA sequence. Such epigenetic mechanisms are likely used to maintain both cell memory (how the cell maintains its differentiated characteristic) as well as maintain and strengthen synaptic connections that maintain long-term changes in behavior. Thus, the concept of chromatin remodeling addresses a key challenge in neurobiology of how stable changes in gene expression are induced in neurons and glia to produce long-lasting changes in behavior.
In this symposium, Goodman et al. discuss use of a new approach called serial analysis of chromatin occupancy (SACO) to interrogate the entire genome for the occupancy of all of the binding sites of a given transcription factor. This new approach allows us to begin to identify networks of genes in an unbiased manner that are affected by the condition being studied. Dr. Champagne's work highlights the impact of the environment on DNA methylation of gene promoters affecting production of the corresponding proteins. Her work is a dramatic example of gene-environment interactions, demonstrating how nurturing in early development affects behavior later in life. The work also shows a strong correlation between DNA methylation and behavior. Mayford and Korzus describe how normal consolidation of short-term to long-term memory requires histone acetylation by the histone acetyltransferase (HAT) activity of cAMP response element-binding protein (CREB) binding protein (CBP) and how this process is affected when the HAT activity of CBP is knocked out in an animal model. Their work may point to a new target for therapeutics for a variety of neurodegenerative diseases. Kumar et al. demonstrate how acute and chronic cocaine administration induces distinct changes in chromatin structure in striatum, a brain region important for cocaine action. They also provide data that implicate the transcription factor ΔFosB in mediating some of these effects and directly relate these changes in chromatin structure to the behavioral actions of cocaine.
Genome-wide analysis of transcription factor binding sites (by R.H.G., S.I., J.D., and G.M.)
Why study gene networks?
Basically, cellular identity is determined by the constellation of transcription factors expressed at critical times during development. Additionally, cellular homeostasis and adaptation are regulated by the interactions of these factors with specific networks of genomic targets. These target genes are not induced (or repressed) in isolation but rather are regulated in a coordinated manner. Determining which transcription factors are present in a cell is relatively easy. Identifying transcription factor targets is more difficult.
Traditional methods for analyzing transcription factor activity
In the drug abuse field, analyses of transcription factor binding most often have relied on in vitro assays. One such approach is the electrophoretic mobility shift assay (EMSA), which measures the binding of a transcription factor to a DNA element, usually an oligonucleotide, by monitoring its migration through a nondenaturing polyacrylamide gel. Bound DNA fragments migrate more slowly than free ones, resulting in a mobility shift, and competition experiments using wild-type and mutant oligonucleotides can be used to determine the specificity of binding. From a crude cellular extract, binding of a specific transcription factor can be detected using immunological agents (so-called super-shift assays). Related measurements (fluorescence anisotropy assays) monitor the ability of a transcription factor to retard the rotation of a DNA fragment in solution. Although fluorescence measurements are more quantitative than EMSA, they require highly purified components and cannot be used to assay crude extracts. Surface plasmon resonance can be used to determine on and off rates of protein-DNA interactions and similarly involve purified or recombinant components. Cell-based assays use reporter genes (luciferase and β-galactosidase) fused to DNA regulatory elements. By introducing these genes into cultured cells, one can monitor the ability of a cotransfected or endogenous transcription factor to activate (or repress) transcription.
Traditional methods do not reflect in vivo transcription factor binding events
What do these assays reveal about transcription of genes in their native context? Unfortunately, rather little. EMSA, fluorescence anisotropy, and surface plasmon resonance measure the binding of factors to naked DNA, which normally does not exist in eukaryotic cells. DNA is packaged in chromatin, which limits transcription factor access. Moreover, the magnitude of this chromatin block is now known to be a critical component of transcriptional regulation. Transfected reporter genes do not recapitulate this chromatin regulation, as demonstrated by Cha-Molstad et al. (2004), who showed that the binding of one of the best-characterized transcription factors, CREB, to cAMP-regulated enhancer (CRE) sequences in native genes varied in a cell-specific manner. CREB binds to the endogenous somatostatin CRE in cortical neurons, for example, but not to the same element in PC12 cells. This illustrates the difficulty in predicting whether a transcription factor binding site is occupied in vivo without actually measuring binding directly.
Methods for identifying in vivo transcription factor-DNA interactions
In vivo genomic footprinting. Fortunately, methods exist that allow transcription factor binding to be analyzed in native chromatin. In vivo genomic footprinting detects the ability of a bound transcription factor to protect specific DNA sequences from chemical cleavage. Although this approach cannot distinguish the binding of one transcription factor from another, it can determine the location of the binding site fairly precisely. The nucleotide sequence of the protected DNA element can often be used to predict the identity of the interacting factor, although this can be misleading because it is not uncommon for multiple factors to bind to the same DNA element.
Chromatin immunoprecipitation assay. A more powerful approach is the chromatin immunoprecipitation (ChIP) assay. Initially developed to monitor histone modifications, ChIP can detect binding of specific transcription factors to native chromatin (Wells and Farnham, 2002). In this method, transcription factors are reversibly cross-linked to their binding sites using formaldehyde, the DNA is sonicated to generate fragments of 500-1000 bp in length, and specific transcription factor-DNA complexes are immunoprecipitated using specific antibodies. Quantification of binding is achieved by PCR amplifying the DNA fragment surrounding the suspected binding site. Because of the size of the sonicated DNA fragments, ChIP does not have the spatial resolution of in vivo footprints. The use of immunological reagents makes it possible to determine which specific factors are binding, however. Also, unlike in vivo genomic footprinting, ChIP can detect factors, such as coactivators and corepressors, that interact with DNA indirectly.
ChIP-on-chip assay. Although ChIP is extremely useful for asking whether an individual binding site is occupied, it was not designed to assess transcription factor-DNA interactions on a global level. Theoretically, one could clone and sequence ChIP fragments to identify the family of gene targets for any particular transcription factor. In practice, however, this procedure has not yielded much information. A variant of the ChIP assay, termed ChIP-on-chip, extends the power of this analysis. In this procedure, ChIP DNA obtained using antibodies to a particular transcription factor is used to probe a microarray representing suspected promoter sequences. ChIP-on-chip assays have been extremely useful in analyzing yeast transcriptional networks, in which the promoters are well characterized (Lee et al., 2002). Initial attempts to apply this technique to metazoan genomes used microarrays containing CpG islands, which tend to be enriched in promoter sequences (Weinmann et al., 2002).
Limitations of promoter-based ChIP-on-chip analysis. More recently, microarrays containing specific gene promoters have been used (Odom et al., 2004). Using this approach, one can interrogate thousands of putative promoter regions relatively inexpensively. (The cost of a promoter microarray is less than $1000.) Although informative, this type of analysis has two limitations. First, mammalian genomic databases remain somewhat poorly annotated. It is straightforward to identify protein-coding regions, but the transcriptional initiation sites are often unknown or mischaracterized. Second, transcription factors do not necessarily bind near initiation sites. Thus, promoter-based ChIP-on-chip analysis may miss the majority of transcriptional regulatory elements. This concern was illustrated by Cawley et al. (2004), who used a tiled microarray representing all nonrepetitive sequences on chromosomes 21 and 22 to interrogate binding of three transcription factors, SP1, p53, and c-myc. (This approach was possible because chromosomes 21 and 22, the two smallest chromosomes, represent only 1-2% of the genome.) Surprisingly, only ∼20% of the transcription factor binding sites localized to known promoter regions. The remainder were dispersed throughout the genome and drove the expression of noncoding transcripts, some of which overlapped in a sense or antisense orientation with known protein-coding genes. These regulatory elements would not be detected by microarrays that interrogate only known promoters. Thus, if noncoding RNAs are biologically important (a matter that is still under debate), promoter-based ChIP-on-chip assays provide only a limited picture of the “transcriptome.”
A solution to this limitation is to use microarrays that interrogate the entire genome. Problems with such so-called “tiled” microarrays are cost, reproducibility, and statistical analysis. A single experiment that includes sufficient replicates to ensure confidence in the analysis currently costs more than $200,000, which is well beyond most investigators' budgets. ChIP-on-chip assays are also limited by the same problems that plague conventional microarray experiments, such as hybridization efficiency and differential incorporation of dyes, sensitivity of the technique to sample preparation and labeling protocols, choice of appropriate reference, etc. Furthermore, the unique distribution of ChIP-on-chip data, which is skewed in the direction of enrichment, raises statistical issues that are just now being recognized. For example, it is not possible to determine a priori what percentage of the distribution will be enriched. Much of the statistical methodology developed for normalization of conventional microarrays exploits the fact that the data are symmetrical and assumes that the bulk of the data are invariant. Because this is not the case for ChIP-on-chip data (using either promoter-based or tiled arrays), new approaches must be developed to address the systematic sources of variation inherent to this technique.
SACO identifies transcription factor binding events in an unbiased manner. An alternative method was developed recently that overcomes these limitations and allows interrogation of entire mammalian genomes in an unbiased manner. SACO combines chromatin immunoprecipitation with a modification of a technique termed Long SAGE. [SAGE (Serial Analysis of Gene Expression) is a method that was developed to analyze mixtures of RNA.] The underpinning of SACO is that a 21 nucleotide sequence generated from immunoprecipitated chromatin [termed genomic signature tags (GSTs)] can be localized to a unique position within a mammalian genome 75% of the time (Fig. 1). Thus, by collecting, sequencing, and localizing GSTs, one can identify (and potentially quantify) transcription factor binding events in an unbiased manner. Data presenting the power of SACO for analysis of CREB binding sites were described recently by Impey et al. (2004). Since the publication of the SACO method, several papers have appeared that used similar approaches, attesting to its utility (Chen and Sadowski, 2005; Kim et al., 2005; Labhart et al., 2005; Roh et al., 2005).
Figure 1.
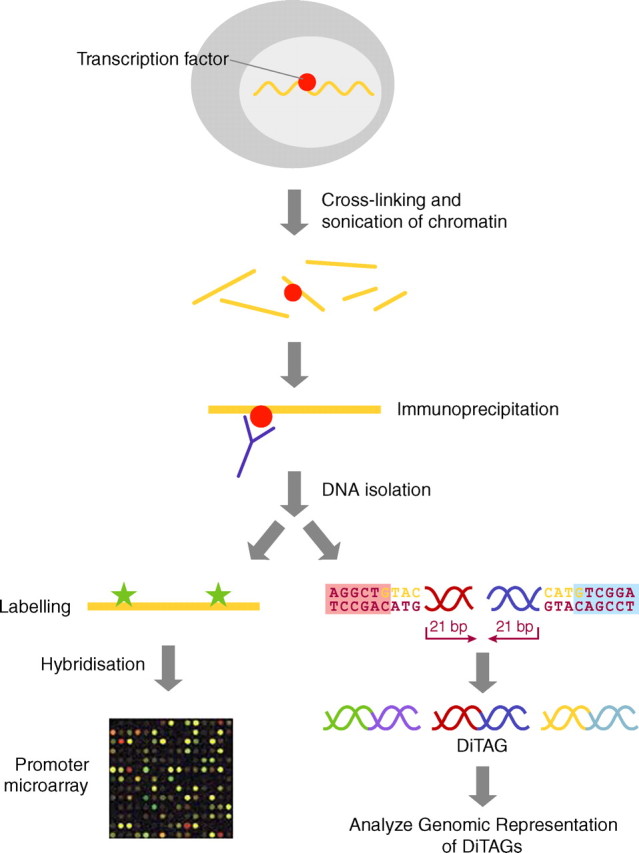
Schematic of hybridization (ChIP-on-chip) and sequence (SACO)-based genomic screens for transcription factor binding sites. For the details of the methods, see Odom et al. (2004) and Impey et al. (2004). Figure adapted from Servitja and Ferrer (2004)
Like any immunoprecipitation-based method, some of the SACO targets will represent nonspecific background. One can overcome this problem simply by sequencing more genomic tags. Presumably, nonspecific interactions occur randomly; specific interactions are reproducible. Thus, by analyzing a large number of tags, one detects multiple hits only for authentic transcription factor binding regions. Although it might seem that identifying clustered GSTs would occur rather infrequently, this has not been the case. Clustering occurs because the design of the method tends to localize tags adjacent to a restriction enzyme recognition site that occurs on average every 250 base pairs. Although more expensive than promoter-based ChIP-on-chip assays, SACO (and related sequence-based techniques) are considerably more affordable (approximately $5000-$10,000 per experiment) than tiled genomic arrays. Although tiled arrays will undoubtedly become less expensive, this cost differential is likely to continue for the foreseeable future because of advances in DNA sequencing (pyrophosphate sequencing, for example). Nonetheless, construction of libraries for sequence-based approaches is more laborious and cannot be farmed out to commercial vendors.
SACO analysis reveals the complement of bound transcription factors
What information can you get from analysis of entire genomes? Clearly, considerable insight can be gained from promoter-based ChIP-on-chip assays (Odom et al., 2004; Zhang et al., 2005). Their main limitation is that they only analyze genes that are already known. Many transcriptional regulatory elements are found inside genes or in intergenic regions. Some of these regulatory elements drive the expression of antisense transcripts; indeed, 20% of human transcripts are predicted to form senseantisense pairs (Chen et al., 2005). (Antisense regulation might lower transcript levels more rapidly than simply ceasing transcription and conceivably could direct stable changes in chromatin structure.) How this mode of regulation might contribute to drug abuse behavior has not been examined.
A surprising result of the CREB SACO study was that CREB binding sites were commonly located in bidirectional promoters. Thus, the CRE that controls c-fos expression, for example, also regulates expression of a noncoding RNA transcribed in the opposite direction (Impey et al., 2004). Many of these novel promoters would not be detected without genome-wide screens. Another class of regulatory elements revealed by the more comprehensive screens controls the expression of micro RNAs. These 19-25 nucleotide molecules are generated from transcripts typically localized within introns, untranslated regions, and noncoding genes (for review, see Kim, 2005). Indeed, some micro-RNAs are produced from introns of genes whose exons not only have no coding potential but also have an exceedingly short half-life. In these cases, the exons serve simply to demarcate the introns, which provide the full functional activity of the gene. Some small nucleolar RNAs are organized similarly.
Several transcription factors (CREB, c-fos, fra1, and ΔFosB) have emerged as possible mediators of drug abuse behavior. Determining how alterations in these factors cause specific neuronal changes requires elucidation of their genomic targets. Clearly, many of these targets will be known protein-coding genes, and the number of these potential targets alone is daunting. The prevalence of noncoding transcription and the likelihood that noncoding RNAs provide important aspects of biological regulation justify the expense of genome-wide analyses of transcription factor binding sites, however. Whether noncoding RNAs will be found that control specific aspects of neuronal function or drug abuse behavior remains unknown. Nevertheless, it is worth noting that a large fraction of micro RNAs are expressed specifically within the CNS. The explosion of evidence that noncoding RNAs contribute to cellular adaptation makes it a good bet that genome-wide screens will greatly expand the repertoire of molecular mechanisms that regulate behavioral responses to drugs of abuse.
Environmental regulation of epigenetic modification (by F.A.C.)
The early environment, consisting of both the prenatal and postnatal periods, has a profound effect on gene expression and adult patterns of behavior. This has been demonstrated experimentally in mammals through manipulation of mother-infant interactions involving prenatal stress, maternal deprivation, and postnatal “handling” of offspring. In these established paradigms, the question remaining involves mechanism: how do events occurring early in development exert long-term effects on gene expression, resulting in stable changes in phenotype of the organism? The answer may involve the same mechanisms, which, at a cellular level, allow for the differentiation of genetically identical cells. In the process of development, cellular differentiation is achieved through gene silencing, involving the attachment of methyl groups to DNA (Jones and Taylor, 1980). Once DNA is methylated, transcriptional factors are blocked from gaining access to the gene, and thus expression is effectively silenced (Razin, 1998). Although DNA methylation is only one of many epigenetic modifications that can alter gene expression, this modification is particularly stable, and patterns of methylation are maintained after cell division (Razin, 1998). Thus, epigenetic modification, involving DNA methylation is an ideal candidate mechanism for examining the long-term effects of early environment.
Early rearing experiences and individual differences in stress responsivity
In mammals, quality of the early environment is conferred by the mother, both through nutritional and behavioral investment. In rodents, observations of mother-infant interactions during the first week postpartum indicate that there are stable natural variations in maternal behavior, particularly licking/grooming (LG), which are critical for shaping stress responsivity of adult offspring (Meaney, 2001; Champagne et al., 2003). Offspring born to mothers who exhibit high levels of LG are less anxious in a novel environment and have an attenuated corticosterone response to stress compared with offspring of low-LG mothers (Liu et al., 1997; Caldji et al., 1998). These behavioral and physiological characteristics are associated with expression of hippocampal glucocorticoid receptors (GRs), such that adult offspring of low-LG mothers have reduced levels of GRs compared with off-spring of high-LG mothers (Liu et al., 1997). Cross-fostering studies confirm that these phenotypes are mediated by variations in maternal care received during the postpartum period (Francis et al., 1999).
Epigenetic regulation of glucocorticoid receptor expression
The regulatory regions that reside within the GR promoter are critical for determining the transcriptional activity of this gene. Analysis of the methylation status of the GR 17 promoter in offspring of high- and low-LG mothers indicates that, indeed, epigenetic modifications mediate the long-term effects of LG on offspring phenotype (Weaver et al., 2004). Mapping of the methyl groups attached to the GR promoter indicate increased methylation at several sites within the promoter, and in particular, at a binding site for the transcription factor nerve growth factor-inducible factor A (NGFI-A). Differential methylation of this site is not present in day 20 embryos or at postnatal day 1, when LG is equally high among both high- and low-LG mothers (Champagne et al., 2003), but differences emerge during the first week postpartum and remain after weaning and into adulthood. Chromatin immunoprecipitation analysis using hippocampal tissue from offspring of high- and low-LG mothers confirms that, in this differentially methylated tissue, NGFI-A binding is reduced in low-LG offspring, resulting in differences in GR expression.
Plasticity of epigenetic modification
The establishment of adult patterns of DNA methylation during the postnatal period suggests that critical periods exist after which levels of gene expression are relatively stable. However, pharmacological manipulations in adulthood are capable of targeting the epigenome and altering gene expression and hence phenotype. In the case of the GR 17 promoter, this can be achieved in a reversible manner by central administration of trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor. TSA, by increasing histone acetylation, activates chromatin, leading to DNA demethylation. After 1 week of daily central infusions of TSA, adult offspring of low-LG mothers exhibit a remarkable change in phenotype (Weaver et al., 2004). On measures of corticosterone response to stress, these TSA-treated offspring are indistinguishable from adult offspring of high-LG mothers. These alterations in phenotype correspond to increased GR receptors and decreased GR 17 promoter methylation.
Epigenetic regulation may also be achieved though S-adenosylmethionine (SAM), a methyl donor critical to DNA methylation (Detich et al., 2003). In vivo levels of SAM are dependent on dietary choline, folates, vitamin B12, and methionine (Cooney, 1993). In adult wild-type mice, 2 weeks of daily subcutaneous injections of methionine produce a downregulation of both GAD67, an enzyme necessary for GABA production, and reelin, a protein involved in neuronal migration in the developing brain (Tremolizzo et al., 2002). This treatment regimen increases SAM, increases methylation of the reelin promoter region, alters prepulse inhibition, and reduces social recognition (Tremolizzo et al., 2002, 2005). Conversely, subcutaneous injections of valproate, a histone deacetylase inhibitor, increases expression of both GAD67 and reelin, suggesting that plasticity in these epigenetic modifications can occur in adulthood and can result in both upregulation and downregulation of gene expression.
Transmission of epigenetic modification across generations
Although the transmission of traits over generations has long been the domain of genetics, there is increasing evidence in support of nongenomic and epigenetic inheritance in mammals. Natural variations in maternal care in the rat are transmitted from mother to female offspring, such that the offspring of high-LG mothers are themselves high in LG and offspring of low-LG mothers exhibit low levels of LG toward their own offspring (Francis et al., 1999; Champagne et al., 2003). Thus, generations of females inherit the maternal style exhibited by their mothers, and, hence, differential GR 17 promoter methylation is passed from one generation to the next via maternal behavior (Fig. 2). This transmission is dependent on the quality of postpartum care, such that cross-fostered offspring will exhibit maternal LG that corresponds to that of their adopted mother rather than their biological mother.
Figure 2.

Illustration of the behavioral transmission of epigenetic modifications across generations via maternal care. Variations in maternal LG are transmitted from mother (F0) to female offspring (F1), and differential methylation of the 17 promoter region of the GR is likewise induced by F0 LG. Variation in LG of female offspring (F1) is then transmitted to the F2 generation, as are patterns of GR methylation.
In contrast to this example of behavioral transmission, there is also evidence for the inheritance of DNA methylation patterns that involves the incomplete removal of epigenetic modifications in the germ line. This has been demonstrated experimentally in mice with the “viable yellow” Avy agouti allele (Morgan et al., 1999; Waterland and Jirtle, 2003). The expression of this allele produces a yellow coat color, obesity, diabetes, and susceptibility to cancer (Miltenberger et al., 1997), and extensive variation in phenotype is produced by differential methylation of the Avy promoter. The degree of Avy promoter methylation is transmitted from mother to offspring such that pseudoagouti female mice, who have the same genotype as yellow agouti females but are characterized by normal coat color and body weight attributable to epigenetic silencing of the Avy promoter, give birth to a higher percentage of pseudoagouti offspring compared with yellow agouti females (Wolff, 1978). Furthermore, embryo transfer of fertilized oocytes from yellow agouti to non-agouti females produces no pseudoagouti offspring, suggesting that maternal environment does not mediate this epigenetically determined phenotype (Morgan et al., 1999). However, modifications in the expression of Avy agouti allele can be produced through dietary intake of methionine, such that, among offspring born to mothers placed on high methionine diets, there is a shift toward a pseudoagouti phenotype (Wolff et al., 1998; Waterland and Jirtle, 2003). Thus, the degree of Avy promoter methylation and hence agouti phenotype can be passed from one generation to the next via maternal epigenetic inheritance but can also be modified by maternal environment.
The importance of the mother in transmitting epigenetic modifications across generations is clear, but there is also evidence for paternal inheritance. In mice, expression of the axin-fused (AxinFu) allele results in a phenotype characterized by kinks in the tail (Rakyan et al., 2003). Offspring born to males expressing AxinFu have a higher frequency of this phenotype than do offspring born to males in which the AxinFu allele has been epigenetically silenced through DNA methylation. Analysis of the AxinFu allele in mature sperm of these males suggests that methylation patterns persist in the gametes, allowing for the epigenetic inheritance to offspring. Paternal epigenetic inheritance has also been observed in offspring born to mothers exposed to toxins. In utero exposure to estrogenic and antiandrogenic endocrine disruptors reduces fertility in male offspring, and these effects are transmitted to subsequent generations (Anway et al., 2005). Analysis of DNA methylation levels in F2 and F3 generation offspring confirm that the transmission of these effects involves epigenetic modifications. Thus, paternal epigenetic inheritance can be modified by environment and shape the development of subsequent offspring. These studies suggest that the persistent effects of experiences occurring early in development are mediated by epigenetic modifications that can be transmitted to multiple generations of offspring. DNA methylation, by silencing gene expression, is critical to this process and can be targeted in later life to alter patterns of gene expression and behavior. The capacity for epigenetic changes to create a cellular memory of these environmental events suggests that a Lamarkian mode of inheritance may indeed be possible and provide a mechanism through which adaptive phenotypes emerge. However, many questions remain. What type of environmental events are capable of exerting these changes? How do these events exert changes in DNA methylation? Are these epigenetic changes tissue specific? Exploration of the degree of complexity that epigenetic modifications confer on the genome will allow us to address these questions and further our understanding of gene-environment interactions.
Histone acetylation is required for long-term memory consolidation (by M.M. and E.K.)
One of the central questions in neuroscience is, what are the cellular and molecular changes in the brain that are responsible for the establishment of long-lasting memories? It has been known for almost 50 years that inhibitors of protein synthesis given during initial learning, either systemically or to specific brain regions, allow animals to learn and remember a given task for up to a few hours but lead to a severe impairment in memory lasting 24 h or longer (Andrew, 1980; Davis and Squire, 1984). This phenomenon has been observed in many species from invertebrates, such as Drosophila and Aplysia, to vertebrates, such as fish, rats, and mice. This finding has led to the view that the establishment of short-term memory involves the modification of preexisting proteins, whereas the stabilization of these changes into a long-lasting memory trace requires the induction of the new gene expression. This presumed conversion of short-term memory into long-term memory, defined by the blockade with protein synthesis inhibitors, is often referred to as memory consolidation.
CREB is a critical element in memory consolidation
Studies in invertebrates of the transcriptional signaling events that may underlie this memory consolidation have identified the transcription factor CREB as a critical component. Memory is generally thought to involve changes in synaptic connectivity. This has been most thoroughly studied in Aplysia in which a form of long-term synaptic plasticity induced by cAMP underlies a simple form of learning. These neurons show both a short-lasting and a long-lasting form of synaptic plasticity, and inhibitors of CREB-dependent transcription block selectively the long-lasting plasticity (Dash et al., 1990; Abel and Kandel, 1998). In Drosophila, genetic studies using CREB-blocking transgenes suggest that CREB is also critical for the consolidation of short-term to longterm memory in a simple pavlovian conditioning task (Yin and Tully, 1996). In mice, genetic studies using partial knock-outs of CREB and CREB dominant-negative transgenes have also demonstrated a requirement for CREB-dependent signaling in longterm memory consolidation (Bourtchuladze et al., 1994; Kida et al., 2002; Pittenger et al., 2002).
Possible function of CREB binding protein
These results across multiple species suggest that CREB-dependent gene induction is a critical and conserved step in the consolidation of long-term memory. However, transcriptional activation requires recruitment of a large number of proteins in addition to transcription factors such as CREB. Of particular interest with regard to learning and memory is the CREB binding protein, or CBP (Chrivia et al., 1993). CBP is a transcriptional coactivator that interacts with multiple transcriptional regulators and facilitates the assembly of the basic transcriptional machinery (Janknecht, 2002). In addition to its function as a platform for the recruitment of the transcriptional machinery, CBP also acts as an HAT (Ogryzko et al., 1996). Histone acetylation is one mechanism for the local and global control of chromatin structure and is essential for achieving high-level gene expression (Janknecht, 2002). A role for CBP in higher cognitive function is suggested by the finding that Rubinstein-Taybi syndrome (RTS), a disorder in humans characterized by growth and psychomotor delay, abnormal anatomy, and severe mental retardation (Rubinstein and Taybi, 1963; Hennekam et al., 1990), is caused by heterozygous mutations at the CBP locus (Petrij et al., 1995).
CBP-dependent histone acetylation is required for memory consolidation
To test the function of CBP in the adult brain and specifically the role of CBP-mediated HAT activity, we generated transgenic mice carrying a dominant-negative CBP transgene that specifically blocks HAT activity (Korzus et al., 2004). The mice were generated using the inducible tet system to allow for regulated expression of the transgene. We found that the mutant mice exhibit a long-term memory deficit, whereas the encoding of new information and short-term memory are normal. This was consistent with the idea that the HAT function of CBP is required for the consolidation of short-term to long-term memory. Similar deficits in long-term memory were also observed in heterozygous null mice (Bourtchouladze et al., 2003; Alarcon et al., 2004). However, one of the great difficulties in using genetically modified mice to study adult brain function is that it is usually difficult to determine whether the observed phenotype results from an acute effect on gene function or is attributable to some developmental abnormality in neuronal circuitry. This is particularly relevant in terms of CBP function because it is clear that CBP is important for transcriptional activation by a broad range of transcription factors in addition to CREB and the fact that, in the human disorder RTS, the absence of a single allele of CBP leads to clear abnormalities in development. We were able to distinguish between these possibilities by suppressing expression of the dominant-negative CBP HAT transgene in adult animals, which showed that the long-term memory deficits were suppressed when the transgene was suppressed. These results demonstrate that the observed behavioral phenotypes are not attributable to irreversible alterations in brain development but rather attributable to an acute requirement for CBP HAT function.
The acetylation state of histones is a general regulator of chromatin structure and transcriptional viability at specific loci (Fischle et al., 2003). Transcriptional activation involves not only the induction of transcription factors but also the removal of promoter-specific repressors as well as histone deacetylases (Rosenfeld and Glass, 2001). This can be achieved by recruiting specific coactivators of transcription, such as CBP, that facilitate HAT-dependent nucleosomal remodeling and promote transition from silent to transcriptionally permissive chromatin (Rosenfeld and Glass, 2001). Subsequent nucleosome acetylation alters chromatin structure and mediates gene-specific removal of epigenetically controlled repression (Rosenfeld and Glass, 2001; Fischle et al., 2003; Jaenisch and Bird, 2003). This represents an attractive mechanism for regulating long-term transcriptional changes associated with long-lasting forms of neuronal and behavioral plasticity.
Inhibitors of histone deacetylase may ameliorate memory deficits
In addition to proteins with acetyltransferase function such as CBP, there is also a class of proteins that function to remove the acetyl groups from histones and convert chromatin to an inactive state. There are a number of pharmacological agents at various stages of clinical development, primarily in the area of cancer treatment, that inhibit these histone deacetylases and lead to a generalized increase in histone acetylation. We tested the ability of these deacetylase inhibitors to reverse the long-term memory deficits caused by expression of the CBP HAT dominant-negative transgene. We found that the inhibitors had no detectable effect on the baseline behavior of mice when administered systemically, but they reversed the long-term memory impairment in the CBP HAT transgenic animals. In a separate study (Alarcon et al., 2004), histone deacetylase inhibitors were also effective in reversing the memory phenotype in the CBP heterozygous null mouse. These results confirm the role for acute HAT activity in the adult brain for the establishment of long-term memory. In addition, they suggest a potential therapeutic intervention in RTS and potentially other diseases in the nervous system that may involve impairments in CBP function.
Significance of specific histone modification in memory consolidation
What might be the significance of a requirement for specific histone modification in the consolidation of long-term memory? The induction of a memory trace is thought to occur via activity-dependent changes in synaptic efficacy such as long-term potentiation. These forms of synaptic plasticity require activation of NMDA receptors and are mediated by the subsequent NMDA receptor-induced Ca2+ signal (Davis et al., 1992; Bear and Malenka, 1994). It is known that both CREB and CBP are required for Ca2+-induced transcription in cultured hippocampal and cortical neurons (Hardingham et al., 1999; Hu et al., 1999; Impey et al., 2002). One possible effect of this dual-pathway system for transcriptional activation is outlined in Figure 3. By inducing the covalent modification of histones and subsequent opening of repressed chromatin structure, CBP could open up a time window whereby, after the initial chromatin modification, the requirement for histone acetyltransferase function at given target locus would no longer exist. After the initial CBP-mediated response to a short-lasting Ca2+ signal leading to acetylation-dependent alteration of local chromatin structure, CBP HAT or even CBP platform function would be dispensable for subsequent steps. In fact, activation of CREB [or CREM (cAMP response element modulator), a CREB family member] by other coactivators has been described (Fimia et al., 1999; Conkright et al., 2003). This could allow for prolonged elevation of transcription in response to an initial learning event by maintaining transcription even after signals to CREB and CBP were no longer present.
Figure 3.

Acetylation is a critical requirement for long-term memory consolidation. The suggested model takes into account five observations: (1) NMDA receptor-dependent neuronal activation is required for memory formation (Davis et al., 1992), (2) neuronal activity-dependent transcription is required for memory consolidation (Andrew, 1980; Davis and Squire, 1984), (3) NMDA-dependent transcription requires the concurrent activation of transcription factors such as CREB and coactivators such as CBP (Chawla et al., 1998; Impey et al., 2002), (4) behaviorally induced CREB phosphorylation is transient and does not correlate with peak gene induction (Stanciu et al., 2001), and (5) CBP acetyltransferase activity is required for long-term memory consolidation but not for short-term memory (Korzus et al., 2004). This model suggests that CBP-mediated histone acetylation is a limiting step in the transcriptional activation controlling memory consolidation. Initial steps include induction of transient CREB phosphorylation, CBP activation, and CBP-mediated histone acetylation at a specific transcriptional unit in response to the initial learning event. Subsequently, the prolonged elevated transcription required for memory consolidation could be maintained by CBP and CREB phosphorylation-independent nuclear mechanisms, even after signals to CREB and CBP are no longer present. This ongoing transcription would remain open until the competing deacetylase-dependent mechanisms suppress transcription (modified from Korzus et al., 2004). Ac, Acetyl groups; Pol II, RNA polymerase II; TBP, TATA-binding protein.
We found that CBP histone acetyltransferase deficiency in the adult brain results in cognitive dysfunction. These studies demonstrated not only the suitability of these animal models for gaining new insights in the molecular etiology of RTS but also in the assessment of new therapeutic approaches. Besides RTS, CBP acetyltransferase-targeting drugs might be considered in neurodegenerative disorders such as Huntington's disease, in which CBP can be sequestrated by direct interaction with the huntingtin protein (Steffan et al., 2000, 2001). CBP is just one of many histone acetylases present in neurons, and acetylation is just one of several types of covalent modification that can impact chromatin structure and thereby transcriptional viability. It is likely that these mechanisms play a significant role in a variety of long-term behavioral and neuronal plasticity.
Chromatin remodeling and drug addiction (by A.K., W.R., D.E.H.T., and E.J.N.)
The fact that drugs of abuse induce neuroadaptations in brain through regulation of gene expression raises the possibility that chromatin remodeling at specific gene promoters may be a key mechanism involved. Here, we briefly summarize recent evidence that drugs such as cocaine cause histone acetylation at specific promoters and that direct regulation of histone acetylation modifies biochemical and behavioral responses to cocaine. Together, these early data support the hypothesis that chromatin remodeling is an important regulatory mechanism underlying the neural and behavioral plasticity associated with drug addiction.
Epigenetic mechanisms and neuropsychiatric disorders
Modifications in chromatin, a process called chromatin remodeling, are important determinants of gene expression. Chromatin remodeling involves numerous types of posttranslational modifications on core histone tails of the nucleosome. These modifications include acetylation, methylation, phosphorylation, ubiquitinylation, and ADP-ribosylation, which in turn alter the structural interactions between DNA and histones (Strahl and Allis, 2000; Hake et al., 2004). DNA (cytosine residues) can also be directly modified by methylation. HATs and HDACs mediate acetylation and deacetylation, respectively, one of the most critical posttranslational modifications. Chromatin remodeling also involves enzymes that alter the structure of chromatin by disrupting the nucleosome in an ATP-dependent manner and thereby repositioning the nucleosomes (Fig. 4). The best-studied example is the SWI-SNF (mating switching and sucrose nonfermenting) complex. Together, these regulatory proteins act on a target promoter, and likely throughout a gene, to increase or decrease its condensation and thereby the accessibility of nearby DNA to the basal transcriptional machinery (Cairns, 2001; Narlikar et al., 2002). As just one example, acetylation of histones H3 and H4, or phosphoacetylation of H3, at gene promoters, via decondensation of the nearby chromatin, is linked to increased gene activity, whereas deacetylation with repression/silencing of the gene.
Figure 4.

Scheme of proposed chromatin remodeling events at a cocaine-activated gene. A shows the repressed state of chromatin, in which a site-specific repressor (Rep) recruits an HDAC complex, which removes acetyl groups (Ac) from histone N-terminal tails. Gene inactivation may also involve other modifications, such as methylation (Me) of histone tails. B shows the active state of chromatin around a cocaine-activated gene (e.g., Cdk5), in which a cocaine-induced transcriptional activator (e.g., ΔFosB-JunD) recruits a HAT and chromatin remodeling complex (SWI-SNF), which induces acetylation (and perhaps demethylation) of histone tails and repositioning of nucleosomes. These actions facilitate the binding of general transcription factors and the basal transcriptional apparatus [e.g., transcription factor IID (TFIID) and RNA polymerase II (Pol II)] to the promoter.
Although this knowledge of chromatin remodeling mechanisms has come primarily from studies of developmental and cancer biology, recent findings from humans and animal models suggest a role for such mechanisms in neuropsychiatric phenomena as well. Certain disorders, such as Rubinstein-Taybi syndrome, Coffin-Lowry syndrome, X-linked α-thalessemia, and Rett syndrome, arise from mutations in genes encoding proteins involved in the chromatin remodeling machinery (Cho et al., 2004). Other disorders, which develop later in life, have genetic predilections but also require environmental exposures that appear to act, at least in part, through epigenetic modifications, i.e., changes in proteins associated with DNA that affect gene expression. Indeed, recent investigations have underscored a critical role for chromatin remodeling in neuronal function, including synaptic plasticity and learning and memory, as well as in the treatments for neuropsychiatric disorders (Guan et al., 2002; Huang et al., 2002; Crosio et al., 2003; Alarcon et al., 2004; Korzus et al., 2004; Li et al., 2004; Tsankova et al., 2004; Levensen and Sweatt, 2005). This symposium focuses on accumulating evidence in the addiction field.
Regulation of chromatin remodeling by drugs of abuse
The striatum, in particular the ventral striatum or nucleus accumbens, is a major neural substrate for the addicting actions of cocaine and other drugs of abuse. Numerous studies have documented that acute or chronic exposure to a drug of abuse alters the expression of particular genes in striatum, and, increasingly, regulation of individual genes is being linked to the behavioral abnormalities that characterize addiction (McClung and Nestler, 2003; Kalivas, 2004, Yao et al., 2004; Carlezon et al., 2005). A dramatic feature of addiction is the striking longevity of these behavioral abnormalities, which indicates that drugs of abuse cause extremely long-lived changes in the brain. One possible mechanism for such stable plasticity is chromatin remodeling.
To investigate this possibility, we are using ChIP assays to study changes in chromatin structure associated with drug action at particular gene promoters known to be regulated in addiction models. In this assay, striatum from control and drug-treated animals is fixed in Formalin to cross-link DNA-protein complexes. DNA is then sheared into small fragments, and the DNA-protein complexes are immunoprecipitated using an antibody directed against the DNA-binding protein of interest. After immunoprecipitation, cross-linking is reversed, proteins are removed by proteinase K treatment, and the DNA is analyzed by real-time PCR to quantify the degree to which a gene of interest was associated with that DNA-binding protein in the intact tissue. In our initial study, we examined genes that represent distinct drug mechanisms: c-fos, which is activated in striatum by acute cocaine but desensitized by chronic cocaine, FosB, activated by both acute as well as chronic cocaine, and BDNF and cyclin-dependent kinase 5 (Cdk5), activated by cocaine chronically but not acutely (Bibb et al., 2001; Nestler, 2001; Grimm et al., 2003; McClung and Nestler, 2003).
We found that cocaine induces H4 acetylation and H3 phosphoacetylation at the c-fos gene promoter in striatum after acute administration (Kumar et al., 2005a). Time course analysis shows that these histone modifications can be seen as early as 30 min after drug exposure and return to normal within 3 h. Acute cocaine administration induces a similar transient increase in H4 acetylation at the FosB gene. A major distinction between the two genes, however, concerns their regulation by chronic cocaine. The ability of cocaine to induce histone modifications at the c-fos promoter completely desensitizes after repeated cocaine exposure, whereas cocaine continues to induce histone modifications at the FosB promoter, with H3, rather than H4, acetylation observed under these conditions. These ChIP data are consistent with the knowledge that induction of c-fos by cocaine desensitizes with repeated cocaine, whereas the FosB gene only shows partial desensitization (McClung et al., 2004).
Consistent with previous observations that both the BDNF and Cdk5 genes are induced in striatum by chronic but not acute cocaine exposure, we found that changes in histones associated with these genes occur after chronic cocaine only (Kumar et al., 2005a). Thus, repeated cocaine administration, including chronic self-administration, increases H3 acetylation at both genes, an effect seen at least 24 h after the last cocaine exposure.
We are also using ChIP assays to study the association of the transcription factor ΔFosB with cocaine-regulated genes. ΔFosB, a product of the FosB gene, accumulates uniquely during chronic drug exposure attributable to its long half-life (McClung et al., 2004). We found increased association of ΔFosB with the Cdk5 gene in striatum after chronic cocaine exposure (Kumar et al., 2005a). Furthermore, inducible overexpression of ΔFosB in striatum of adult bitransgenic mice, which is sufficient for induction of Cdk5 expression in vivo (Bibb et al., 2001), causes increased binding of ΔFosB with the Cdk5 gene. Chronic cocaine administration, or inducible overexpression of ΔFosB, causes increased binding of Brg1 (a component of the SWI-SNF complex) with the Cdk5 gene (Kumar et al., 2005a). Together, these results support a scheme whereby the gradual accumulation of ΔFosB, in response to chronic cocaine administration, recruits chromatin remodeling factors, such as specific HATs to induce H3 acetylation as well as Brg1-containing chromatin remodeling complexes, to its target genes such as Cdk5. In contrast, we found no association between ΔFosB and the BDNF gene, consistent with evidence that BDNF is not a target of this transcription factor (McClung and Nestler, 2003). In addition, cocaine activation of the BDNF gene is not associated with increased binding of Brg1. These observations indicate that different chromatin remodeling complexes are involved in the various genomic effects of cocaine even within the same brain region. One possibility is that the induction of BDNF by cocaine occurs via another transcription factor, such as CREB (McClung and Nestler, 2003).
One consistent observation in our studies to date is that the acute effects of cocaine are associated predominantly with H4 acetylation, whereas the chronic effects of the drug are restricted to H3 acetylation. This switch from H4 acetylation acutely to H3 acetylation chronically holds even for individual genes (e.g., FosB) that are induced under both conditions. A similar switch from H4 to H3 acetylation was also observed recently in hippocampus in response to acute and chronic seizures (Tsankova et al., 2004). The molecular mechanism involved in this switch is not known, but it is conceivable that different HATs or HDACs associate with particular genes after acute versus repeated perturbation of the brain. Additional work is needed to study this and alternative explanations.
Histone acetylation modulates biochemical and behavioral responses to cocaine
We are using HDAC inhibitors to explore whether cocaine-induced changes in histone acetylation are relevant for the biochemical and behavioral effects of the drug. Acute cocaine administration induces global levels of histone acetylation in striatum, and this effect is potentiated by systemic coadministration of sodium butyrate or trichostatin A, two structurally distinct HDAC inhibitors (Kumar et al., 2005a). A very similar pattern was observed for cocaine induction of c-fos mRNA and for increased association of phosphoacetylated H3 with the c-fos gene promoter in striatum, both of which showed synergistic responses after the coadministration of HDAC inhibitors and cocaine. These data establish a clear correlation between alterations in histone acetylation and regulation of gene expression in striatum.
A similar correlation was evident for the behavioral effects of cocaine: administration of sodium butyrate, at a dose that does not alter locomotor activity, almost doubled the locomotor response to cocaine (Kumar et al., 2005a; Theobald et al., 2005). Likewise, trichostatin A augments the rewarding effects of cocaine as measured in the place conditioning paradigm. Conversely, increased HDAC activity in striatum specifically achieved via the viral-mediated overexpression of HDAC4 in this region, had the opposite effect, and dramatically reduced the place conditioning effects of cocaine (Kumar et al., 2005a; Renthal et al., 2005). Our findings thereby establish connections among the biochemical and behavioral effects of cocaine with respect to regulation of histone acetylation in striatum.
HDAC inhibitors are being explored as treatments for several diseases, such as certain cancers, Huntington's disease, and spinal muscular atrophy. Our findings raise the possibility that drugs aimed at HDACs, or perhaps other chromatin remodeling factors, could potentially be explored as novel treatments for drug addiction.
Future directions
ChIP assays have enabled a major technical advance in neuroscience. Before the advent of this technology, studies of gene regulation in brain in vivo were severely limited and relied on in vitro studies to gain insight into in vivo mechanisms. Now it is possible for the first time to study such transcriptional mechanisms in vivo and to understand, with increasing complexity, how chronic cocaine administration leads to the long-term regulation of its target genes.
ChIP also makes it possible to investigate, in an open-ended way, the range of genes that are regulated within a given brain region in vivo. Thus, by performing ChIP, followed by analysis of the immunoprecipitated DNA on intergenic microarrays, it is possible to gain a genome-wide appreciation of genes regulated in striatum by cocaine and identify which of these genes are influenced by ΔFosB (Kumar et al., 2005b). Such ChIP-on-chip assays promise to provide unprecedented insight into the molecular mechanisms underlying addiction.
The experiments described above clearly represent the very early stages of understanding drug-induced changes in chromatin structure that may be involved in the pathophysiology of drug addiction. Future research must be aimed at delineating how cocaine, presumably via regulation of dopamine receptors and intracellular signaling cascades, leads to the regulation of specific transcription factors such as ΔFosB and CREB and how regulation of these factors causes changes in histone acetylation, as well as presumably many other histone, and even DNA, modifications at particular target genes (Fig. 4). We also need to learn how other drugs of abuse affect chromatin remodeling in striatum and other drug-sensitive brain regions. This work will identify the precise steps by which different transcriptional complexes are formed at each of the genes regulated after acute or chronic drug exposure. This new knowledge has the potential of dramatically expanding our appreciation of the factors that control individual susceptibility to cocaine addiction as well as providing fundamentally new pathways at which to intervene to one day treat or prevent the development of an addictive disorder.
Footnotes
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R37 DK45423 (R.H.G., S.I., J.D., G.M.), by a fellowship from the Canadian Institutes of Health Research (F.A.C.), by National Institute of Mental Health Grant R01 MH57368-05 (M.M., E.K.), and grants from the National Institute on Drug Abuse (A.K., W.R., D.E.H.T., E.J.N.). The introductory remarks are based on the introduction to the doctoral dissertation by J.D.P. J.D.P. thanks Joseph S. Camardo and Eric R. Kandel for helpful discussions.
The order of authors is based on the order of symposium sections.
Correspondence should be addressed to Christine M. Colvis, Division of Basic Neurosciences and Behavioral Research, National Institute on Drug Abuse, 6001 Executive Boulevard, Bethesda, MD 20892-9555. E-mail: ccolvis@nida.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510379-11$15.00/0
References
- Abel T, Kandel E (1998) Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev 26: 360-378. [DOI] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A (2004) Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947-959. [DOI] [PubMed] [Google Scholar]
- Andrew RJ (1980) The functional organization of phases of memory consolidation. In: Advances in the study of behaviour (Hinde RA, Beer C, Bunsel M, eds), pp 337-367. New York: Academic.
- Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308: 1466-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC (1994) Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4: 389-399. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Chen JS, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P (2001) Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature 410: 376-380. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T (2003) A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA 100: 10518-10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59-68. [DOI] [PubMed] [Google Scholar]
- Cairns BR (2001) Emerging roles for chromatin remodeling in cancer biology. Trends Cell Biol 11: S15-S21. [DOI] [PubMed] [Google Scholar]
- Caldji C, Tannenbaum B, Sharma S, Francis D, Plotsky PM, Meaney MJ (1998) Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proc Natl Acad Sci USA 95: 5335-5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon Jr WA, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28: 436-445. [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116: 499-509. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S, Goodman RH (2004) Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci USA 101: 13572-13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne FA, Francis DD, Mar A, Meaney MJ (2003) Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol Behav 79: 359-371. [DOI] [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H (1998) CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science 281: 1505-1509. [DOI] [PubMed] [Google Scholar]
- Chen J, Sadowski I (2005) Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc Natl Acad Sci USA 102: 4813-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sun M, Hurst LD, Carmichael GG, Rowley JD (2005) Genome-wide analysis of coordinate expression and evolution of human cis-encoded sense-antisense transcripts. Trends Genet 21: 326-329. [DOI] [PubMed] [Google Scholar]
- Cho KS, Elizondo LI, Boerkoel CF (2004) Advances in chromatin remodeling and human diseases. Curr Opin Genet Dev 14: 308-315. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365: 855-859. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12: 413-423. [DOI] [PubMed] [Google Scholar]
- Cooney CA (1993) Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging 57: 261-273. [PubMed] [Google Scholar]
- Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P (2003) Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 116: 4905-4914. [DOI] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER (1990) Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature 345: 718-721. [DOI] [PubMed] [Google Scholar]
- Davis H, Squire L (1984) Protein synthesis and memory: a review. Psychol Bull 96: 518-559. [PubMed] [Google Scholar]
- Davis S, Butcher SP, Morris RG (1992) The NMDA receptor antagonist d-2-amino-5-phosphonopentanoate (d-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro J Neurosci 12: 21-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detich N, Hamm S, Just G, Knox JD, Szyf M (2003) The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem 278: 20812-20820. [DOI] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, Sassone-Corsi P (1999) CBP-independent activation of CREM and CREB by the LIM-only protein ACT. Nature 398: 165-169. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172-183. [DOI] [PubMed] [Google Scholar]
- Francis D, Diorio J, Liu D, Meaney MJ (1999) Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 286: 1155-1158. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y (2003) Time dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci 23: 742-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER (2002) Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111: 483-493. [DOI] [PubMed] [Google Scholar]
- Hake SB, Xiao A, Allis CD (2004) Linking the epigenetic language of covalent modifications to cancer. Br J Cancer 90: 761-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H (1999) Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium. Neuron 22: 789-798. [DOI] [PubMed] [Google Scholar]
- Hennekam RC, Stevens CA, Van de Kamp JJ (1990) Etiology and recurrence risk in Rubinstein-Taybi syndrome. Am J Med Genet Suppl 6: 56-64. [DOI] [PubMed] [Google Scholar]
- Hilgard ER, Bower GH (1975) Theories of learning, p 17. Englewood Cliffs, NJ: Prentice-Hall.
- Hu SC, Chrivia J, Ghosh A (1999) Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron 22: 799-808. [DOI] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R (2002) Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22: 8422-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH (2002) Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34: 235-244. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer J, Yochum GS, Boss JM, McWeeney SK, Dunn JJ, Mandel G, Goodman RH (2004) Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 119: 1041-1054. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet [Suppl] 33: 245-254. [DOI] [PubMed] [Google Scholar]
- Janknecht R (2002) The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol Histopathol 17: 657-668. [DOI] [PubMed] [Google Scholar]
- Jones PA, Taylor SM (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20: 85-93. [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2004) Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4: 23-29. [DOI] [PubMed] [Google Scholar]
- Kandel ER (1984) Steps toward a molecular grammar for learning: Explorations into the nature of memory. In: Medicine, Science, and Society (Isselbacker KJ, ed), Symposium celebrating the Harvard Medical School Bicentennial. New York: Wiley.
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ (2002) CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5: 348-355. [DOI] [PubMed] [Google Scholar]
- Kim A, Bhinge AA, Morgan XC, Iyer VR (2005) Mapping DNA protein interactions in large genomes by sequence tag analysis of genomic enrichment. Nat Methods 2: 47-53. [DOI] [PubMed] [Google Scholar]
- Kim VN (2005) MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 6: 376-385. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ (2005a) Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48: 303-314. [DOI] [PubMed] [Google Scholar]
- Kumar A, Sikder D, Renthal W, Choi KH, Truong H, Russo SJ, Theobald DEH, Self DW, Kodadek TJ, Nestler EJ (2005b) Global maps of histone acetylation and gene regulatory networks in the nucleus accumbens after chronic cocaine using ChIP on chip. Soc Neurosci Abstr 31: 451.4. [Google Scholar]
- Labhart P, Karmakar S, Salicru EM, Egan BS, Alexiadis V, O'Malley BW, Smith CL (2005) Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc Natl Acad Sci USA 102: 1339-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae Science 298: 799-804. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD (2005) Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6: 108-118. [DOI] [PubMed] [Google Scholar]
- Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, Konradi C, Akbarian S (2004) Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem 90: 1117-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ (1997) Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 277: 1659-1662. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ (2003) Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat Neurosci 11: 1208-1215. [DOI] [PubMed] [Google Scholar]
- McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ (2004) ΔFosB: a molecular switch for long-term adaptation. Mol Brain Res 132: 146-154. [DOI] [PubMed] [Google Scholar]
- Meaney MJ (2001) Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 24: 1161-1192. [DOI] [PubMed] [Google Scholar]
- Miltenberger RJ, Mynatt RL, Wilkinson JE, Woychik RP (1997) The role of the agouti gene in the yellow obese syndrome. J Nutr 127: 1902S-1907S. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E (1999) Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23: 314-318. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retro-transposition. Nature 16: 435:903-910. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan H, Kingston RE (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475-487. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303: 1378-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953-959. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH (1995) Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376: 348-351. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER (2002) Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34: 447-462. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, Whitelaw E (2003) Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci USA 100: 2538-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón y Cajal S (1894) La fine structure des centros nerveux: the Croonian Lecture. Proc R Soc Lond B Biol Sci B55: 443-468. [Google Scholar]
- Razin A (1998) CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J 17: 4905-4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Russo SJ, Tsankova N, Neve RL, Olson EN, Nestler EJ (2005) Epigenetic control of cocaine reward by histone deacetylases in the nucleus accumbens. Soc Neurosci Abstr 31: 451.3. [Google Scholar]
- Roh T-Y, Cuddapah S, Zhao K (2005) Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 19: 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Glass CK (2001) Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem 276: 36865-36868. [DOI] [PubMed] [Google Scholar]
- Rubinstein JH, Taybi H (1963) Broad thumbs and toes and facial abnormalities. Am J Dis Child 105: 588-608. [DOI] [PubMed] [Google Scholar]
- Scott JP (1965) Animal behavior, p 93. Chicago: University of Chicago.
- Servitja JM, Ferrer J (2004) Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia 47: 597-613. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Radulovic J, Spiess J (2001) Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res Mol Brain Res 94: 15-24. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97: 6763-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila Nature 413: 739-743. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45. [DOI] [PubMed] [Google Scholar]
- Tanzi E (1893) I fatti e le induzioni dell'odierna istologia del sistema nervoso. Riv Sper Fren Med Leg 19: 419-472. [Google Scholar]
- Theobald DEH, Kumar A, Renthal W, Nestler EJ (2005) An investigation into the behavioral and biochemical sequalae of histone deacetylase (HDAC) inhibitors. Soc Neurosci Abstr 31: 451.2. [Google Scholar]
- Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A (2002) An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci USA 99: 17095-17100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, Tueting P, Rodriguez-Menendez V, Costa E, Guidotti A (2005) Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry 57: 500-509. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ (2004) Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci 24: 5603-5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23: 5293-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7: 847-854. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ (2002) Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev 16: 235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J, Farnham PJ (2002) Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods 26: 48-56. [DOI] [PubMed] [Google Scholar]
- Wolff GL (1978) Influence of maternal phenotype on metabolic differentiation of agouti locus mutants in the mouse. Genetics 88: 529-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff GL, Kodell RL, Moore SR, Cooney CA (1998) Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J 12: 949-957. [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG (2004) Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41: 625-638. [DOI] [PubMed] [Google Scholar]
- Yin JC, Tully T (1996) CREB and the formation of long-term memory. Curr Opin Neurobiol 6: 264-268. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA 102: 4459-4464. [DOI] [PMC free article] [PubMed] [Google Scholar]