

Abstract
DYT1 is the most common inherited dystonia. Currently, there are no preventive or curative therapies for this dominantly inherited disease. DYT1 dystonia is caused by a common three-nucleotide deletion in the TOR1A gene that eliminates a glutamic acid residue from the protein torsinA. Recent studies suggest that torsinA carrying the disease-linked mutation, torsinA(ΔE) acts through a dominant-negative effect by recruiting wild-type torsinA [torsinA(wt)] into oligomeric structures in the nuclear envelope. Therefore, suppressing torsinA(ΔE) expression through RNA interference (RNAi) could restore the normal function of torsinA(wt), representing a potentially effective therapy regardless of the biological role of torsinA. Here, we have generated short hairpin RNAs (shRNAs) that mediate allele-specific suppression of torsinA(ΔE) and rescue cells from its dominant-negative effect, restoring the normal distribution of torsinA(wt). In addition, delivery of this shRNA by a recombinant feline immunodeficiency virus effectively silenced torsinA(ΔE) in a neural model of the disease. We further establish the feasibility of this viral-mediated RNAi approach by demonstrating significant suppression of endogenous torsinA in mammalian neurons. Finally, this silencing of torsinA is achieved without triggering an interferon response. These results support the potential use of viral-mediated RNAi as a therapy for DYT1 dystonia and establish the basis for preclinical testing in animal models of the disease.
Keywords: dystonia, RNA interference, torsina, dyt1, gene therapy, FIV
Introduction
DYT1 dystonia is the most common form of hereditary dystonia. This dominantly inherited disorder usually presents during childhood as localized involuntary muscle contractions that subsequently generalize, resulting in a disabling motor phenotype (Fahn et al., 1998). To date, there is no effective treatment for DYT1 dystonia.
DYT1 dystonia is caused by a 3 bp deletion in TOR1A, resulting in the loss of a glutamic acid residue in torsinA (Ozelius et al., 1997). TorsinA is an ATPase associated with diverse cellular activities (AAA) protein (Hanson and Whiteheart, 2005) that resides primarily in the endoplasmic reticulum (ER) (Hewett et al., 2000; Kustedjo et al., 2000). Interestingly, the mutant form of torsinA [torsinA(ΔE)] accumulates in the nuclear envelope (NE), leading to the formation of NE-derived membranous inclusions known as spheroid bodies that accumulate in the cytoplasm (Gonzalez-Alegre and Paulson, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004). Despite this recent discovery, the normal function of torsinA and its role in disease pathogenesis remain unknown.
Based on the known properties of other AAA proteins, torsinA is predicted to function as an oligomeric structure and torsinA(ΔE) to act through a dominant-negative effect (Breakefield et al., 2001; Torres et al., 2004; Hanson and Whiteheart, 2005). This hypothesis is experimentally supported by recent reports (Goodchild and Dauer, 2004; Naismith et al., 2004; Torres et al., 2004). Therefore, silencing expression of torsinA(ΔE) represents a potentially useful therapy. However, because the function of wild-type torsinA [torsinA(wt)] remains unknown, specific suppression of torsinA(ΔE) while preserving levels of torsinA(wt) might be required. The importance of allele specificity is underscored by the perinatal lethality of torsinA-null mice (Dauer and Goodchild, 2004).
RNA interference (RNAi) is a cellular pathway for posttranscriptional gene silencing (Mello and Conte, 2004). The ability to manipulate RNAi in the laboratory is being exploited to develop novel therapies for human disease (Davidson and Paulson, 2004; Stevenson, 2004). There are several reasons why DYT1 dystonia is an ideal disease in which to explore therapeutic RNAi-mediated gene silencing. First, because DYT1 likely results from a dominant-negative effect, a reduction in torsinA(ΔE) would restore the normal function of torsinA(wt). Second, using in vitro-synthesized short interfering RNA (siRNA), we have previously demonstrated that the 3 nt difference between wt and mutant alleles is sufficient to permit specific silencing of the mutant gene (Gonzalez-Alegre et al., 2003). Third, because DYT1 dystonia is caused by a single common mutation, this therapeutic approach would be applicable to all patients. Fourth, as a disorder of neuronal function rather than neuronal loss, the DYT1 dystonia phenotype may be reversible. Finally, current therapies available for DYT1 dystonia are of limited efficacy.
Here, we have generated short hairpin RNAs (shRNAs) that mediate allele-specific silencing of torsinA(ΔE) and rescue cells from its dominant-negative effect, and established the feasibility of this approach by suppressing expression of endogenous torsinA in mammalian neurons without triggering an interferon response. These results support the potential of viral-mediated RNAi as a therapeutic strategy for DYT1 dystonia.
Materials and Methods
Plasmid and virus construction. The various torsinA expression plasmids used for this study have been described previously (Gonzalez-Alegre et al., 2003). To generate the U6shRNA constructs, we used a PCR-based approach described previously (Harper et al., 2005), with primers designed to generate the different hairpins shown in Figure 1: five hairpins targeting torsinA(ΔE) (U6shTAmut1-5) and three control hairpins, one targeting torsinA(wt) (U6shTAwt), one targeting a sequence common for both for human and murine torsinA (U6shTAcom), and a mismatched sequence not targeting either allele as shown previously (U6shTAmis) (Gonzalez-Alegre et al., 2003). The PCR product was ligated into the pCR2.1 vector using the TOPO-TA Cloning kit (Invitrogen, San Diego, CA) and the product verified by sequencing. GFP-guide and GFP-passenger expression vectors were generated by PCR amplification of enhanced green fluorescent protein (eGFP), fused to the sequences described in Figure 7. The PCR product was then inserted into the multiple cloning site of pcDNA3.1 (Invitrogen). Additional plasmids included GFP-Q19 (GFP fused to a stretch of 19 glutamines), torsinA(wt)-GFP (a kind gift from Dr. P. Hanson, Washington University, St. Louis, MO) (Naismith et al., 2004), and HA-torsinA(ΔE) (Gonzalez-Alegre et al., 2003).
Figure 1.

Allele-specific silencing of transiently expressed torsinA(ΔE) in Cos7 cells. A, Target cDNA region of torsinA(ΔE) and different short hairpin RNAs used in this study. B, Western blot results showing the effect of different shRNAs on torsinA(wt)-GFP or HA-torsinA(ΔE) expression. α-Tubulin is shown as a loading control. C, Similar experiment performed with shmis, shTAcom, and shTAmut2-5. D, Quantification of results from three experiments as in B and C. Error bars indicate SE. mis, U6shTAmis; mut, U6shTAmut5; com, U6shTAcom; torA, torsinA; tub, α-tubulin.
Figure 7.

Preferential shRNA strand participation in the RNAi pathway. A, Shown is the sequence of the targeted torsinA(ΔE) cDNA region used to generate the GFP-guide and GFP-passenger reporter constructs, underlining the specific sequence used for each, with a schematic representation of the resulting coding region. B, Western blot analysis of transiently transfected Cos7 cells showing the effect of different shRNAs on GFP-guide and GFP-passenger. α-Tubulin is shown as a loading control. C, Quantification of results from three experiments as in B. Error bars indicate SE. com, U6shTAcom; mut, U6shTAmut; tub, α-tubulin; WB, Western blot.
Feline immunodeficiency virus (FIV) pseudotyped with VSV-G (vesicular stomatitis virus G) were generated by the Gene Transfer Vector Core of the University of Iowa (www.uiowa.edu/~gene/) as described previously (Brooks et al., 2002). In summary, HEK293 cells were transfected with vector, envelope, and packaging plasmids, followed by collection of supernatants and particle concentration. The vector plasmid was based on the pVETL backbone containing a GFP reporter gene driven by a cytomegalovirus (CMV) promoter as shown in Figure 2 A. The hairpin was digested out of pCR2.1 using EcoRI, and ligated into the PmeI site of pVETL(CMV)GFP.
Figure 2.

Allele-specific silencing of torsinA(ΔE) in a simulated heterozygous state. Cos7 cells were transiently transfected with torsinA(wt)-GFP, HA-torsinA(ΔE), and different pVETL-(GFP)shRNAs. A, Schematic diagram of pVETL constructs used for expression of shRNAs and the GFP reporter. B, Western blot analysis showing allele-specific suppression of HA-torsinA(ΔE) by shTAmut5, and potent suppression of both forms of torsinA by shTAcom. C, Quantification of three experiments as in B. Error bars indicate SE. mis, U6shTAmis; com, U6shTAcom; mut5, U6shTAmut5; torA, torsinA; tub, α-tubulin; WB, Western blot.
Cell culture, transfection, and infection. Culture, maintenance, differentiation, and inducibility of PC6-3 torsinA(wt) and torsinA(ΔE) clonal cell lines have been reported previously (Gonzalez-Alegre and Paulson, 2004). Cos7 cells were cultured as described previously (Gonzalez-Alegre et al., 2003). Primary neuronal cultures were obtained from the cerebral cortex of embryonic day 16 wild-type B6C3F1/J mice (The Jackson Laboratory, Bar Harbor, ME) as described previously (Meberg and Miller, 2003) with minor modifications.
Transient transfections were performed using Lipofectamine Plus (Invitrogen) according to the manufacturer's instructions in 6- or 12-well plates with cells at 70-90% confluence. For transient cotransfection of torsinA- and shRNA-expressing constructs, we used a shRNA:target molar ratio of 2-3:1. For infection with recombinant FIV, the virus was diluted in serum-free OptiMEM (Invitrogen) and cells were infected at a multiplicity of infection of 10 or 25 for 3 h, replacing it then with serumcontaining media. To activate the interferon response, the corresponding cells in 12- or 24-well dishes were incubated with 50,000 or 100,000 U of interferon α2a (Santa Cruz Biotechnology, Santa Cruz, CA), respectively, for 12 h before harvesting.
Western blot analysis. Denatured total cell lysates were obtained at the indicated times, run in SDS-PAGE, transferred to nitrocellulose membranes, and blotted using various antibodies as described previously (Gonzalez-Alegre et al., 2003). For quantification experiments, the resulting blots were scanned, and the pixel count and intensity of immunoreactive bands were quantified using the Scion Image software (Scion, Frederick, MD) (Gonzalez-Alegre et al., 2003). We normalized the torsinA signals based on the amount of protein loaded (using the α-tubulin signal as an internal reference) and expressed the result as a percentage of the signal obtained in the control lane.
The following antibodies were used to detect torsinA in immunoblots: for overexpressed torsinA, a polyclonal rabbit antiserum generated against a torsinA-maltose-binding protein fusion protein or mouse monoclonal DMG10 (1:1000); and for endogenous protein, mouse monoclonal DM2A8 (1:250) (all a kind gift from Dr. V. Ramesh, Massachusetts General Hospital, Boston, MA) (Sharma et al., 2005). Other antibodies used were mouse monoclonal anti-RNA-regulated protein kinase (PKR), rabbit polyclonal anti-signal transducer and activator of transcription 1 (STAT1) and goat polyclonal anti-phosphorylated STAT1 (all from Santa Cruz), mouse monoclonal anti-α-tubulin (Sigma), mouse monoclonal anti-GFP (MBL, Woburn, MA), and mouse monoclonal 12CA5 [anti-hemagglutinin (HA)] (Roche, Basel, Switzerland).
Immunofluorescence. Cells were fixed in 4% paraformaldehyde in PBS, processed, and mounted in slides as described previously (Gonzalez-Alegre and Paulson, 2004). Fluorescence visualization of cells was performed with a Zeiss Axioplan fluorescence microscope (Zeiss, Thornwood, NY), and digital images were collected on red, green, and blue fluorescence channels using an Axiocam HRm (Zeiss) digital camera. Digital images were assembled using Adobe Photoshop 6.0.
To model the dominant-negative effect of torsinA(ΔE), PC6-3 torsinA(ΔE)-inducible cell lines were transiently transfected with torsinA(wt)-GFP. We randomly selected 100 GFP-expressing cells per experiment and determined the percentage in which the GFP signal was recruited into cytoplasmic inclusions. This was performed with or without inducing torsinA(ΔE) transcription by the addition of doxycycline. To quantify the number of inclusions per cell, we randomly selected 50 cells with GFP-cytoplasmic inclusions and counted the number of inclusions per cell, grouping them as follows: <3, 3-5, 6-10, and >10 inclusions/cell. All quantification results are expressed as mean ± SD.
Results
Allele-specific suppression of torsinA(ΔE)
Based on the target sequence identified in our previous study using in vitro-synthesized siRNA (Gonzalez-Alegre et al., 2003), we designed and constructed a hairpin transcribed from a U6 promoter to target torsinA(ΔE) specifically (U6shTAmut) (Fig. 1A). Coexpression of U6shTAmut with HA-torsinA(ΔE) in Cos7 cells showed potent suppression of torsinA(ΔE) levels measured by Western blot analysis (Fig. 1B). However, when cotransfected with torsinA(wt)-GFP, this shRNA partially silenced torsinA(wt) (Fig. 1B,D) and therefore lacked silencing specificity. To optimize allele specificity, we screened four additional U6shRNAs for specific targeting of torsinA(ΔE) (shTAmut2-5) (Fig. 1A). Although U6shTAmut2 preferentially silenced torsinA(ΔE), it lacked robust specificity much like U6shTAmut. In contrast, U6shTAmut3, -4, and -5 induced potent silencing of torsinA(ΔE) without interfering with torsinA(wt) expression (Fig. 1C,D). U6shTAmut3 and -4 have an identical targeting sequence and differ only in the loop region (Fig. 1A). We did not find a significant difference in silencing efficacy between these two, suggesting that the use of either loop does not influence the efficacy or specificity of the construct. Because of constraints derived from the primary sequence of the targeted gene, U6shTAmut3 and -4 encode a potential premature U6 termination of transcription signal (TTTTT) in their sense strand. However, their silencing efficacy was similar to U6shTAmut5, suggesting the entire hairpin was transcribed. Although it is possible that the proximity of this sequence to the initiation of transcription complex precluded transcriptional termination, we elected to use U6shTAmut5 to avoid the potential problem of generating truncated transcripts. We also designed a hairpin targeting a sequence shared by mouse and human torsinA upstream of the area of the deletion (U6shTAcom). As shown in Figure 1, C and D, U6shTAcom potently suppressed expression of both alleles.
To further demonstrate allele specificity, we designed experiments to coexpress U6shTAmut5 with wt and torsinA(ΔE) and determine the preferential target when exposed to both alleles. We first cloned U6shTAmut5 into pEVTL(CMV)GFP, the shuttle vector subsequently used to generate recombinant FIV (Fig. 2A), and then cotransfected this construct into Cos7 cells concurrently with torsinA(wt)-GFP and HA-torsinA(ΔE). The different tags allow us to distinguish the two forms of torsinA by their different molecular weights and use of specific antibodies. As shown in Figure 2, B and C, U6shTAmut5 showed exquisite preference for the mutated allele even in the presence of torsinA(wt), demonstrating that the 3 nt difference allows the shRNA to specifically silence the targeted message. In contrast, U6shTAcom robustly silenced both alleles.
RNA-mediated rescue of torsinA(wt) recruitment into spheroid bodies
Although much remains to be learned about the function of torsinA, it is clear that the glutamic acid deletion causes torsinA(ΔE) to be accumulated in the NE, where it recruits torsinA(wt), likely through a dominant-negative effect (Gonzalez-Alegre and Paulson, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004). We previously generated inducible neural cell lines to study torsinA biology. In these clonal cell lines, the addition of doxycycline to the culture media induces the synthesis of either torsinA(wt) (which diffusely distributes throughout the ER) or torsinA(ΔE) (which relocalizes to the NE, leading to the formation of NE-derived cytoplasmic inclusions known as spheroid bodies) (Gonzalez-Alegre and Paulson, 2004). To model and quantify the potential dominant-negative effect of torsinA(ΔE), we transiently transfected these cell lines with torsinA(wt)-GFP. TorsinA(wt)-GFP is normally detected by fluorescence microscopy as a diffuse cytoplasmic signal consistent with ER localization, although some cells develop a small number of cytoplasmic inclusions (Fig. 3A). However, when torsinA(ΔE) expression was induced, the GFP signal was recruited to multiple spheroid bodies in most cells (Fig. 3A,D), providing an easily quantifiable phenotype of the recruitment of torsinA(wt) by torsinA(ΔE).
Figure 3.
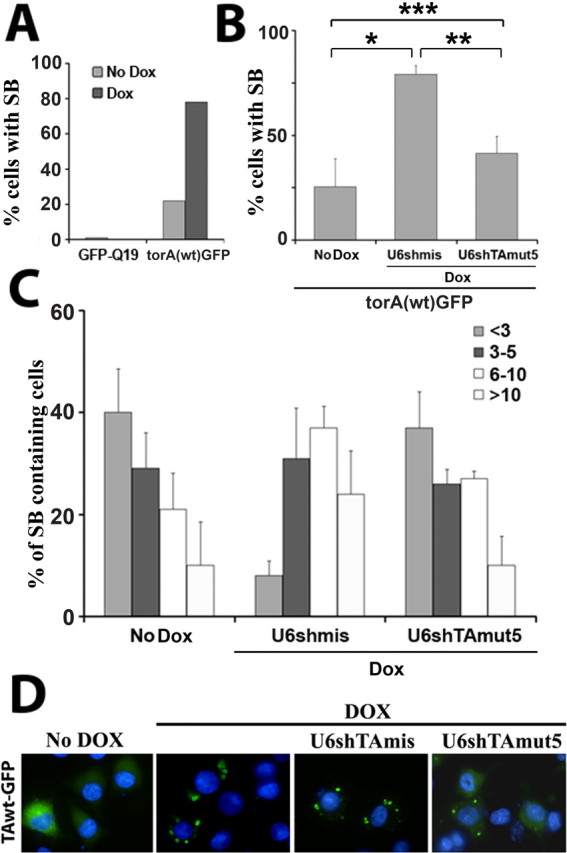
RNA-mediated rescue of torsinA(wt) recruitment into spheroid bodies. A, TorsinA(ΔE)-inducible neural cell lines, which, after the addition of doxycycline, synthesize untagged torsinA(ΔE) and generate spheroid bodies, were transfected with GFP-Q19 or torsinA(wt)-GFP, with or without doxycycline, showing recruitment of torsinA(wt)-GFP, but not GFP-Q19, into spheroid bodies. Some noninduced cells, only expressing torsinA(wt)-GFP, also developed some cytoplasmic inclusions. B, shTAmut5 reversed the torsinA(ΔE)-mediated recruitment of torsinA(wt)-GFP into cytoplasmic inclusions to levels similar to noninduced cells. Results represent the percentage of GFP-positive cells with inclusions. Statistical differences, determined by paired t test, are indicated by asterisks (*p < 0.05; **p = 0.06; ***p = NS). C, Quantification of inclusions in 50 randomly selected inclusion-containing cells per experiment (<3, 3-5, 6-10, >10 inclusions/cell). Expression of torsinA(ΔE) dramatically increased the number of inclusions per cell, and this was reversed by shTAmut5 to the levels of noninduced cells. Results represent mean (±SD) of three independent experiments. D, Representative fluorescence microscopy images of torsinA(ΔE)-mediated recruitment of torsinA(wt)-GFP into cytoplasmic inclusions, and of rescue by shTAmut5. Blue, DAPI nuclear staining. SB, Spheroid bodies; Dox, doxycycline; torA, torsinA.
Based on the findings described in Figure 2, we hypothesized that RNAi suppression of torsinA(ΔE) by U6shTAmut5 would rescue the recruitment of torsinA(wt)-GFP into spheroid bodies. As shown in Figure 3, B and D, U6shTAmut5 partially reversed the recruitment of GFP into cytoplasmic inclusions. Furthermore, in cells still containing GFP inclusions, the number was restored to control levels (Fig. 3C). We conclude that U6shTAmut5 selectively silences torsinA(ΔE) and rescues the recruitment of torsinA(wt) into spheroid bodies, restoring its normal subcellular localization and presumably allowing it to perform its normal function.
Silencing torsinA using recombinant FIV
We inserted the therapeutic shRNA into a vehicle capable of delivery to CNS neurons. We used FIV because of its proven ability to transduce neurons in vivo, its capacity to mediate nuclear access of the encoded transgene, its capability to integrate in the host genome leading to long-term gene expression, and the fact that it is not a human pathogen (Davidson and Breakefield, 2003). If successful, this virus could be considered for testing in animal and human trials. FIV vectors were engineered to coexpress eGFP (FIVeGFP), which enabled simultaneous monitoring of delivery and efficacy of silencing in transduced cells. We generated FIVeGFP encoding U6shTAmut5 or U6shTAcom. The inducible torsinA(ΔE) cell lines were infected with these viral vectors, with vector expressing only GFP serving as a control. Transduction efficiency was >90% as determined by the presence of GFP signal detected by immunofluorescence (not shown). As shown in Figure 4, A and B, U6shTAmut5 strongly suppressed torsinA(ΔE) expression as measured and quantified by Western blot analysis. We previously showed the presence of a higher molecular weight torsinA-immunoreactive band in the nuclear fraction of cells overexpressing torsinA(ΔE). As suggested before (Gonzalez-Alegre and Paulson, 2004), this band could represent aberrant torsinA oligomers in the NE, although this remains to be proven. As shown in Figure 4A, silencing torsinA(ΔE) reduced the levels of this band (Fig. 4A, arrowhead), likely reflecting decreased accumulation of torsinA(ΔE) in the NE. If torsinA(ΔE) is not accumulating in the NE, spheroid bodies should not be generated. We quantified the number of cells containing spheroid bodies by immunofluorescence, confirming a dramatic reduction in cells treated with FIVeGFP.shTAmut5 (Fig. 4C,D). Interestingly, we detected increased diffuse torsinA signal seen in cells treated by FIVeGFP.shTAmut5, which may represent rescue of endogenous torsinA from recruitment into spheroid bodies (Fig. 4C). These findings suggest that FIVeGFP.shTAmut5 mediates potent silencing of torsinA(ΔE), preventing both its accumulation in the NE and the formation of spheroid bodies. FIVeGFP.U6shTAcom was also highly effective in reducing torsinA(ΔE) levels (Fig. 4A,B), demonstrating that this recombinant virus could prove useful as a research tool to suppress endogenous torsinA.
Figure 4.

FIVeGFP.shRNA-mediated silencing of torsinA(ΔE) prevents formation of spheroid bodies. A, Western blot analysis of PC6-3 torsinA(ΔE)-expressing cell lines, showing potent suppression of torsinA(ΔE) expression by FIVeGFP.shTAcom and shTAmut5 but not a FIVeGFP control. The formation of a higher molecular weight, torsinA-immunoreactive band of ∼85 kDa (arrow), was also suppressed. GFP, FIVeGFP; com, FIVeGFP.shTAcom; mut5, FIVeGFP.shTAmut5. B, Quantification of results from three experiments as in A. C, Immunofluorescence image, with GFP as a reporter for transduced cells, showing prevention of spheroid body formation by FIVeGFP.shTAmut5 but not by FIVeGFP. Blue, DAPI nuclear staining. D, Quantification of the presence of spheroid bodies in three independent experiments in nontransduced control cells or cells transduced by different viral vectors. Error bars indicate SE. DOX, Doxycycline; com, U6shTAcom; mut5, U6shTAmut5; tub, α-tubulin; WB, Western blot; torA, torsinA.
The above results demonstrate silencing of the targeted transcript in overexpression systems. Next, we sought to determine whether endogenous torsinA can be effectively suppressed in mammalian neurons by RNAi, the situation we would encounter when treating human disease. We chose to silence torsinA in primary neuronal cultures derived from wild-type mice using U6shTAcom. Five days after infection with the different FIV vectors, primary neurons were harvested for protein analysis. The results demonstrate significant silencing of endogenous torsinA by FIVeGFP.U6shTAcom, whereas viruses encoding U6shTAmut5 or only GFP did not reduce torsinA levels (Fig. 5A,B). Neurons transduced with FIVeGFP.U6shTAcom, and therefore GFP-positive, also displayed reduced torsinA immunostaining (Fig. 5C). Thus, endogenous torsinA can be efficiently suppressed by FIV-delivered shRNAs in mammalian neurons.
Figure 5.

Silencing endogenous torsinA in wild-type primary neuronal cultures. Murine primary neuronal cultures were transduced with different FIVeGFP constructs and assayed for silencing of torsinA. A, Western blot analysis showing endogenous torsinA suppression by FIVeGFP.shTAcom. The arrowhead indicates torsinA at the expected molecular weight of 37.8kDa. Three additional nonspecific bands are recognized by the DM2A8 antibody. Tubulin is shown as a loading control. B, Quantification of Western blot signal as in A. Error bars indicate SE. C, Indirect immunofluorescence showing decreased torsinA signal in neurons transduced by FIVeGFP.shTAcom (arrowheads), but not FIVeGFP or FIVeGFP.shTAmut5, when compared with surrounding nontransduced cells. tub, α-Tubulin; WB, Western blot.
Assessing potential adverse effects of FIV-delivered RNAi
One advantage of manipulating posttranscriptional gene silencing with short, double-stranded RNAs is the theoretical lack of activation of the interferon response, a problem that has hindered the use of previous antisense oligonucleotides (Scherer and Rossi, 2003). Although siRNAs shorter than 30 nt should not activate this pathway, experimental evidence in this area has been conflicting (Bridge et al., 2003; Sledz et al., 2003; Fish and Kruithof, 2004; Heidel et al., 2004; Pebernard and Iggo, 2004; Scacheri et al., 2004; Hornung et al., 2005; Judge et al., 2005). The divergent results reported could reflect the different sequences targeted, the methodology used, the design of the hairpin, or the ability of the targeted tissue to develop an interferon response. Therefore, we directly assessed the state of the interferon response in cell types targeted with the different FIV vectors. As a positive control, nontransduced cells were treated with interferon-α. PC6-3 cells treated with interferon-α displayed an upregulation of interferon-responsive genes, including PKR and STAT-1, and increased phosphorylation (activation) of STAT-1, demonstrating the ability of this cell line to activate this pathway (Fig. 6A). However, transduction with the various FIV vectors did not measurably increase expression of those proteins, despite achieving effective target silencing (Fig. 6A). Nevertheless, lack of RNAi-induced activation of the interferon response in this immortalized cell line does not ensure that we would not encounter that problem in the CNS, where a glial-based immune surveillance system could be more sensitive to such intervention. However, when the same recombinant viruses were used to transduce primary neuronal cultures, which also contain glial cells, expression of the interferon-responsive genes remained unchanged (Fig. 6B). In summary, these findings suggest that FIV-delivered shRNA can mediate silencing of torsinA in brain tissue without triggering an interferon response.
Figure 6.

Assessment of the interferon response in neuronal cells transduced by FIVeGFP.shRNA. A, Western blot analysis of PC6-3 torsinA(ΔE)-inducible cell lines showing increased PKR and STAT1 signal and STAT1 phosphorylation when treated with interferon-α, but not with the different FIVeGFP constructs, despite effective torsinA silencing. B, Western blot analysis of the same experiment performed in primary neuronal cultures, showing similar results. GFP, FIVeGFP; com, FIVeGFP.shTAcom; mut5, FIVeGFP.shTAmut5; DOX, doxycycline; IFN, interferon; torA, torsinA; tub, α-tubulin.
Additional potential problems with this therapeutic approach are off-target effects independent of an interferon response and the risk of overly occupying the endogenous RNAi machinery with exogenous shRNA. Although siRNA are double stranded, only one strand (antisense or guide) mediates degradation of the targeted message; the complementary (passenger) strand should be degraded rather than incorporated into the silencing complex. Preferential incorporation of the antisense strand into the RNA-induced silencing complex (RISC) should improve silencing efficacy and minimize possible adverse effects caused by incorporation of the passenger strand into RISC. Evidence suggests that the thermodynamic stability of base-pairing at the 5′ end of each siRNA strand determines which one participates in the RNAi pathway (Khvorova et al., 2003; Schwarz et al., 2003). The thermodynamic profile of our allelespecific hairpins [assessed with the Sfold software (http://sfold.wadsworth.org)] (Ding et al., 2004) favors incorporation of the antisense strand into the RISC complex (data not shown). To test this experimentally, we designed a system to indirectly measure each strand's participation in the RNAi pathway. To generate a reporter construct that assesses antisense strand-mediated silencing, we inserted the targeted sequence of torsinA at the 3′ end of GFP cDNA (Fig. 7A). A second construct to test passenger strand-mediated silencing was generated by placing the complementary (template) strand of the targeted region of torsinA at the 3′ end of GFP (Fig. 7A). The first construct (GFP-guide) represents a target for the antisense strand of our torsinA shRNA, whereas the second construct (GFP-passenger) would be target for the passenger strand. Ideally, torsinA shRNA would silence the GFP-guide and not the GFP-passenger construct. As a negative control, we used U6shTAcom, because the sequence targeted by this hairpin is not included in either reporter construct. Both reporter constructs were expressed at similar levels in Cos7 cells and displayed a diffuse cellular distribution by fluorescence microscopy (data not shown). As shown in Figure 7, B and C, and as predicted from the thermodynamic stability analysis, the different U6shTAmut constructs preferentially silenced GFP-guide, with minor to no effect on GFP-passenger levels. These results document preferential participation of the antisense strand in the RNAi pathway, therefore potentially minimizing shRNA-derived adverse effects.
Discussion
In this study, we generated a lentivirus vector that potently and specifically silences torsinA(ΔE), and established its therapeutic potential for DYT1 dystonia. This conclusion is supported by the demonstration of robust, allele-specific silencing of human torsinA(ΔE), which prevented its accumulation in the NE and the formation of spheroid bodies, and rescued cells from a dominant-negative effect of torsinA(ΔE) over torsinA(wt). In addition, viral-mediated delivery of this hairpin in neuronal cells was highly efficient and suppressed endogenous torsinA expression in mammalian neurons without triggering an interferon response. With these results, we have created the tools required to test in vivo the therapeutic value of RNAi in animal models of DYT1 dystonia.
Recent studies have shown successful silencing of neurodegenerative disease genes in vivo, with improvement in behavioral and pathological phenotypes in animal models (Xia et al., 2004; Harper et al., 2005; Ralph et al., 2005; Raoul et al., 2005). However, DYT1 dystonia is a disorder of neuronal dysfunction rather than of neuronal loss, suggesting potential reversibility of the disease phenotype. Therefore, we predict that, using the vector generated in this study, we could achieve significant suppression of torsinA(ΔE) expression in vivo, reversing the molecular abnormalities that lead to neuronal dysfunction in DYT1 dystonia.
Allele-specific silencing of torsinA(ΔE)
DYT1 dystonia is caused by a single common mutation in torsinA that probably exerts a dominant-negative effect on torsinA(wt), recruiting it to the NE. Based on our the current understanding of how AAA proteins function, the most likely model of torsinA action is that it forms oligomeric rings bound to ATP, interacts with substrate proteins, and mediates a conformational change in the substrate protein on ATP hydrolysis, resulting in release of the substrate and disassembly of the complex (Hanson and Whiteheart, 2005). The presence of torsinA(ΔE) in oligomers likely prevents them from completing the cycle, rendering them in a “locked state.” This is supported by the finding that a dominant-negative form of torsinA, able to bind but not hydrolyze ATP, also accumulates in the NE (Goodchild and Dauer, 2004; Naismith et al., 2004). Preferential suppression of torsinA(ΔE), as shown here, would restore functional torsinA(wt) oligomers, allowing them to carry out their as-yet-unknown biological function. In demonstrating that specific silencing of torsinA(ΔE) allowed torsinA(wt) to regain its normal distribution, we have provided strong experimental support for this view. Thus, the therapeutic approach explored in this study is potentially applicable to DYT1 dystonia, although the function of torsinA remains unknown.
Allele-specific silencing of neurological disease-causing genes has not been accomplished in vivo yet. To achieve that goal in DYT1 dystonia, we will need to employ genetically engineered animals. Although transgenic mice expressing human torsinA(ΔE) are clearly very important for torsinA research, the sequence differences between human and mouse torsinA preclude an in vivo analysis of allele specificity. Furthermore, they would not be able to demonstrate effective silencing of endogenously expressed torsinA, because the target would be an overexpressed transgene. To verify allele-specific silencing of endogenous torsinA(ΔE), we will need to wait for the characterization of knock-in models of DYT1 dystonia. Nevertheless, we have successfully silenced endogenous torsinA in mouse neurons. Together with the demonstration of allele specificity, we have provided enough evidence to predict that U6shTAmut5 will selectively silence endogenously expressed torsinA(ΔE) in vivo. Furthermore, our nonspecific hairpin could be used to study the normal function of torsinA in vivo.
Generating a recombinant lentivirus to silence torsinA in vivo
For successful therapeutic silencing, we will need to selectively suppress torsinA(ΔE) and restore the normal distribution of torsinA(wt) in the neurons responsible for the development of the disease. Functional studies performed in DYT1 patients suggest that the striatum is the most likely area of dysfunction (Walker and Shashidharan, 2003) and therefore the anatomical target. FIV effectively transduces neurons in the basal ganglia (Davidson and Breakefield, 2003), as demonstrated by reversing pathology in an animal model of lysosomal storage disease after gene replacement in the striatum (Brooks et al., 2002). If knock-in models of DYT1 dystonia display a measurable phenotype, silencing the mutant allele with these tools will serve to define the brain region responsible for DYT1 dystonia.
Assessing potential adverse effects of RNAi-mediated silencing in neuronal cells
The recent studies demonstrating effective silencing of disease-causing genes in the CNS have not directly addressed the potential adverse effects derived from this therapy (Xia et al., 2004; Harper et al., 2005; Ralph et al., 2005; Raoul et al., 2005). One concern is that the activation of an interferon response could cause nonspecific transcriptional and translational alterations. FIV does not trigger a significant immune response in the brain (Davidson and Breakefield, 2003). Furthermore, FIV preferentially transduces neurons, avoiding potential interferon response activation by transduced glial cells. In this study, we demonstrated that neither the viral vector nor the shRNAs used trigger this protective pathway. Moreover, we designed a novel system that demonstrated preferential antisense strand participation in the silencing pathway, thus minimizing the potential off-target effects that could be mediated by the passenger strand. However, this result does not rule out other possible off-target effects by, for example, co-opting the RNAi machinery. These factors will need to be further addressed in future studies in vivo, to assess the safety of this approach before planning human trials.
In conclusion, we have generated a therapeutic lentiviral vector for DYT1 dystonia and demonstrated its efficacy in neuronal cells, thus establishing the basis for future preclinical studies in animal models of DYT1 dystonia.
Footnotes
This work was supported by a Menkes/Markham Fellowship from the Dystonia Medical Research Foundation (P.G.-A.) and National Institutes of Health Grant NS050210 (P.G.-A., B.L.D., H.L.P.). We are grateful to Dr. Edgardo Rodriguez for his helpful comments and suggestions.
Correspondence should be addressed to Dr. Pedro Gonzalez-Alegre, Department of Neurology, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, Iowa City, IA 52242-1101. E-mail: pedro-gonzalez-alegre@uiowa.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510502-08$15.00/0
References
- Breakefield XO, Kamm C, Hanson PI (2001) TorsinA: movement at many levels. Neuron 31: 9-12. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R (2003) Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 34: 263-264. [DOI] [PubMed] [Google Scholar]
- Brooks AI, Stein CS, Hughes SM, Heth J, McCray Jr PM, Sauter SL, Johnston JC, Cory-Slechta DA, Federoff HJ, Davidson BL (2002) Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc Natl Acad Sci USA 99: 6216-6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Goodchild R (2004) Mouse models of torsinA dysfunction. Adv Neurol 94: 67-72. [PubMed] [Google Scholar]
- Davidson BL, Breakefield XO (2003) Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci 4: 353-364. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Paulson HL (2004) Molecular medicine for the brain: silencing of disease genes with RNA interference. Lancet Neurol 3: 145-149. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chan CY, Lawrence CE (2004) Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res 32: W135-W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, Bressman SB, Marsden CD (1998) Classification of dystonia. Adv Neurol 78: 1-10. [PubMed] [Google Scholar]
- Fish RJ, Kruithof EK (2004) Short-term cytotoxic effects and long-term instability of RNAi delivered using lentiviral vectors. BMC Mol Biol 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Paulson HL (2004) Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci 24: 2593-2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Miller VM, Davidson BL, Paulson HL (2003) Toward therapy for DYT1 dystonia: allele-specific silencing of mutant TorsinA. Ann Neurol 53: 781-787. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT (2004) Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA 101: 847-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engone, will work. Nat Rev Mol Cell Biol 6: 519-529. [DOI] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL (2005) RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci USA 102: 5820-5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel JD, Hu S, Liu XF, Triche TJ, Davis ME (2004) Lack of interferon response in animals to naked siRNAs. Nat Biotechnol 22: 1579-1582. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO (2000) Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet 9: 1403-1413. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11: 263-270. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I (2005) Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 23: 457-462. [DOI] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209-216. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MH, Cravatt BF (2000) Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem 275: 27933-27939. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW (2003) Culturing hippocampal and cortical neurons. Methods Cell Biol 71: 111-127. [DOI] [PubMed] [Google Scholar]
- Mello CC, Conte Jr D (2004) Revealing the world of RNA interference. Nature 431: 338-342. [DOI] [PubMed] [Google Scholar]
- Naismith TV, Heuser JE, Breakefield XO, Hanson PI (2004) TorsinA in the nuclear envelope. Proc Natl Acad Sci USA 101: 7612-7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, GusellaJF, Breakefield XO (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17: 40-48. [DOI] [PubMed] [Google Scholar]
- Pebernard S, Iggo RD (2004) Determinants of interferon-stimulated gene induction by RNAi vectors. Differentiation 72: 103-111. [DOI] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M (2005) Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med 11: 429-433. [DOI] [PubMed] [Google Scholar]
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P (2005) Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med 11: 423-428. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, Collins FS (2004) Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 101: 1892-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer LJ, Rossi JJ (2003) Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol 21: 1457-1465. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199-208. [DOI] [PubMed] [Google Scholar]
- Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO (2005) Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci 25: 5351-5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR (2003) Activation of the interferon system by short-interfering RNAs. Nat Cell Biol 5: 834-839. [DOI] [PubMed] [Google Scholar]
- Stevenson M (2004) Therapeutic potential of RNA interference. N Engl J Med 351: 1772-1777. [DOI] [PubMed] [Google Scholar]
- Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG (2004) Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated ΔE-torsinA mutant. Proc Natl Acad Sci USA 101: 15650-15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RH, Shashidharan P (2003) Developments in the molecular biology of DYT1 dystonia. Mov Disord 18: 1102-1107. [DOI] [PubMed] [Google Scholar]
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, Paulson HL, Yang L, Kotin RM, Davidson BL (2004) RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 10: 816-820. [DOI] [PubMed] [Google Scholar]