

Abstract
During the development of in vivo amyloid imaging agents, an effort was made to use micro-positron emission tomography (PET) imaging in the presenilin-1 (PS1)/amyloid precursor protein (APP) transgenic mouse model of CNS amyloid deposition to screen new compounds and further study Pittsburgh Compound-B (PIB), a PET tracer that has been shown to be retained well in amyloid-containing areas of Alzheimer's disease (AD) brain. Unexpectedly, we saw no significant retention of PIB in this model even at 12 months of age when amyloid deposition in the PS1/APP mouse typically exceeds that seen in AD. This study describes a series of ex vivo and postmortem in vitro studies designed to explain this low retention. Ex vivo brain pharmacokinetic studies confirmed the low in vivo PIB retention observed in micro-PET experiments. In vitro binding studies showed that PS1/APP brain tissue contained less than one high-affinity (Kd = 1-2 nm) PIB binding site per 1000 molecules of amyloid-β (Aβ), whereas AD brain contained >500 PIB binding sites per 1000 molecules of Aβ. Synthetic Aβ closely resembled PS1/APP brain in having less than one high-affinity PIB binding site per 1000 molecules of Aβ, although the characteristics of the few high-affinity PIB binding sites found on synthetic Aβ were very similar to those found in AD brain. We hypothesize that differences in the time course of deposition or tissue factors present during deposition lead to differences in secondary structure between Aβ deposited in AD brain and either synthetic Aβ or Aβ deposited in PS1/APP brain.
Keywords: transgenic mouse, amyloid-β, plaques, neuroimaging, positron emission tomography, benzothiazole
Introduction
Modification of the amyloid-binding dye thioflavin-T showed that neutral benzothiazole-anilines could bind to amyloid with low nanomolar affinity, enter the brain in amounts sufficient for imaging with positron emission tomography (PET), and clear rapidly from normal brain tissue (Klunk et al., 2001; Mathis et al., 2002). A structure-activity study of benzothiazole compounds suggested that a hydroxylated derivative, termed Pittsburgh Compound-B (PIB; [11C]6-OH-BTA-1), had brain clearance properties typical of many useful PET radiotracers (Mathis et al., 2003). Using in vivo multiphoton microscopy, PIB has been shown to label individual amyloid plaques in transgenic (Tg) mouse models of Alzheimer's disease (AD) within 3 min after intravenous injection and clear rapidly from normal brain parenchyma (Bacskai et al., 2003).
In the first human study using PIB in 16 mild AD patients and 9 healthy controls, there was a robust group difference in the PIB retention pattern (Klunk et al., 2004b), and this has been replicated in subsequent studies (Price et al., 2005). Compared with controls, AD patients typically showed twofold to threefold greater retention of PIB in areas of association cortex known to contain large amounts of amyloid deposits in AD. In areas known to be relatively unaffected by amyloid deposition, such as the cerebellum, PIB retention was equivalent in AD patients and controls.
During the development of PIB for in vivo imaging of AD plaque pathology, we sought to make use of micro-PET imaging technology (Cherry, 2001) and Tg mouse models of amyloid deposition. Micro-PET imaging in Tg mice could be an ideal complement to the other screening tests used to identify optimal lead imaging agents (Mathis et al., 2003). For this purpose, we chose the presenilin-1 (PS1)/amyloid precursor protein (APP) Tg mouse model because of the rapid and extensive nature of fibrillar amyloid deposition in these mice (McGowan et al., 1999). The PS1/APP mouse develops amyloid-β (Aβ) deposits beginning at 3 months of age, and these are quite extensive by 8-9 months of age (McGowan et al., 1999).
Surprisingly, initial PIB micro-PET studies in PS1/APP mice up to 12 months of age showed no significant retention compared with PS1 controls (Klunk et al., 2004a). Toyama et al. (2005) have reported similar micro-PET findings in Tg mice using PIB (also known as [11C]6-OH-BTA-1). To understand why the binding of PIB appears so different in human and Tg mouse brain, we undertook a series of in vivo, ex vivo, and in vitro studies examining the interaction of PIB with brain tissue in PS1/APP mice that were characterized immunohistochemically and biochemically for the extent of Aβ deposition. Here, we report that despite the presence of large amounts of insoluble Aβ deposits, PIB binds very poorly to PS1/APP mouse brain tissue. The binding of PIB to PS1/APP mouse brain closely resembles the binding of PIB to synthetic Aβ fibrils and differs considerably from the binding of PIB to homogenates of postmortem AD brain because of the low number of high-affinity PIB binding sites per mole of Aβ.
Materials and Methods
Chemicals
All reagents were purchased from Sigma (St. Louis, MO), unless noted otherwise.
Mice
Double Tg PS1/APP mice were generated by mating male APP Tg2576 (K670N/M671L) mice (Taconic Farms, Germantown, NY) (Hsiao et al., 1996) with female M146L PS1 mice (mutant PS1, line 6.2; University of South Florida, Tampa, FL) (Duff et al., 1996)]. The Tg 2576 mice were derived from a C57B6/SJL × C57B6 background, whereas the M146L PS1 animals were derived from a Swiss-Webster/B6D2F1× B6D2F1 background (Holcomb et al., 1998). Food and water were available ad libitum, and a 12 h light/dark cycle was maintained. Mice were studied at 6, 9, and 12 months of age with micro-PET and at 15 months of age for postmortem biochemical and histological studies. Swiss-Webster mice (25-35 g; Harlan Sprague Dawley, Indianapolis, IN) were studied at 3 months of age. All studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Brain tissue from two 28-month-old PS1/APP mice and age-matched PS1 controls was provided by Dr. Karen Duff (Nathan Kline Research Institute, New York University, New York, NY).
Postmortem human brain tissue
Autopsy tissue was obtained from the University of Pittsburgh Alzheimer's Disease Research Center Brain Bank. Clinical and postmortem diagnoses were made by standardized procedures as described in detail previously (Lopez et al., 2000). All five AD brains represented fairly advanced stages of pathology, as is typical of postmortem tissue. Three control brains were selected for having no significant evidence of neuritic plaques or cerebrovascular amyloid. In addition, there were no neurofibrillary tangles in the frontal cortex of the control brains. Samples of mediofrontal cortex gray matter were dissected at autopsy from 1-cm-thick coronal slices and frozen at -80°C.
In vivo micro-PET imaging
PET imaging of PS1/APP and PS1 littermate control mice was performed using a micro-PET P4 scanner (Concorde Microsystems, Knoxville, TN) at 6, 9, and 12 months of age. At each age, one pair of mice consisting of a PS1/APP and a PS1 littermate was imaged in the scanner simultaneously. Before PET imaging, mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and immobilized with Velcro and elastic restraints. Animals were positioned in the scanner field of view (FOV) such that the cranium occupied the central 2 cm of the axial FOV, at which volume resolution and sensitivity of the micro-PET system are maximal. Animals were given injections in the lateral tail vein with 100-150 μCi of high-specific activity [11C]PIB (>1000 Ci/mmol) dissolved in <200 μl of sterile isotonic saline. PET emission data were collected in list mode for 60 min, commencing with the injection of [11C]PIB. A region of interest was drawn on three to five contiguous image planes encompassing the entire cerebrum and used to sample each frame of the dynamic micro-PET image to determine the function representing the time-varying radioactivity concentration averaged over the entire cerebrum. Arbitrary micro-PET counts were converted to an absolute measure of radioactivity concentration (microcurie/cubic centimeter or microcurie/gram assuming 1 g/cc) using a phantom-derived calibration factor before being normalized to the injected dose (ID) of [11C]PIB and the body mass of the animal (%ID-kilogram/gram). This normalization enables the comparison of brain radioactivity concentrations assessed by other methods as well as across animals of different weights and even across species.
Ex vivo brain tissue radioassay
Ex vivo regional brain radioactivity concentrations were assessed in PS1/APP mice (n = 4; age, 15 months) and control littermates (n = 4) after the injection of 100-150 μCi of [11C]PIB. In addition, nine Swiss-Webster mice were given injections to compare the radioactivity concentration measured dynamically using the micro-PET system with the concentration determined using a well-type gamma counter. Mice were killed by intraperitoneal injection of 100 mg/kg pentobarbital, followed by thoracotomy, cardiac puncture to obtain a terminal blood sample (for carbon-11 counting and weighing), and cardiac excision. PS1/APP and PS1 mice were killed 15 min after injection of [11C]PIB. The Swiss-Webster mice were killed at 2, 30, and 60 min after injection (n = 3 at each time point).
The brains were be removed and dissected into forebrain and cerebellum, which were weighed and counted using a gamma counter (model 5003; Packard Bioscience, Meridian, CT) with an overall sensitivity for detecting 511 keV annihilation photons of ∼35%, which is far in excess of the 1-2% overall system sensitivity of the micro-PET P4 scanner. Raw counts were decay-corrected and converted to an absolute measure of radioactivity concentration (microcurie/gram of brain) using a constant efficiency factor and normalized to both ID and body mass (%ID-kilogram/gram). After the carbon-11 radioactivity was quantified, the PS1/APP and PS1 brains were hemisectioned. One half was frozen at -80°C (for ELISA and postmortem [3H]PIB binding), and the other half was placed in 4% paraformaldehyde (for histological studies). The carbon-11 radioactivity was allowed to decay to background for at least 10 half-lives (>200 min) before the brain samples were further processed for histology, ELISA, or postmortem [3H]PIB binding.
Histological and immunohistochemical techniques
Brain hemispheres for histological/immunohistochemical (IHC) evaluation were immersion-fixed in cold 4% paraformaldehyde for 24 h and cryoprotected by sequential immersion into 15 and 30% sucrose in 0.1 m phosphate buffer, pH 7.4. Forty-micrometer-thick coronal brain sections were collected serially and stored at -20°C in cryoprotectant (0.3 g/ml sucrose, 0.01 g/ml polyvinylpyrrolidone-40, and 30% v/v ethylene glycol in 0.05 m sodium phosphate buffer, pH 7.4), as described previously (Watson et al., 1986), until processed further. Throughout the anteroposterior length of the hippocampus, three sets of five adjacent tissue sections were processed for IHC evaluation using antibodies against total Aβ (6E10), Aβ1-40 and Aβ1-42, or histological staining with 1,4-bis(3-carboxy-4-hydroxyphenylethenyl)-benzene (X-34) (Styren et al., 2000) and 2-(4′-methylaminophenyl)-6-cyanobenzothiazole (6-CN-BTA-1), a highly fluorescent analog of PIB (Mathis et al., 2003). Before histological/IHC evaluation, sections were washed in PBS. For IHC evaluation, endogenous peroxidase activity was inhibited by incubating the tissue in 1.5% H2O2 in 0.1 m PBS for 30 min. Sections were rinsed in 0.1 m PBS, followed by permeabilization and blockade with 0.2% Triton-X and 5% normal goat serum in 0.1 m PBS, and incubated overnight in primary antisera at 4°C. After washing in 0.1 m PBS, sections were incubated in the appropriate biotinylated IgG (Vector Laboratories, Burlingame, CA) in 5% normal serum and 0.1 m PBS for 1 h, washed in 0.1 m PBS, and incubated with ABC (Vector Laboratories) in 0.1 m PBS for 1 h at room temperature. The sections were washed repeatedly, and the color reaction was developed using a 0.05% solution of 3′,3-diaminobenzidine tetra-hydrochloride and 0.03% H2O2. Sections were washed in 0.1 m PBS, mounted on gelatinized slides, dehydrated in alcohols, cleared in xylene, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA).
X-34 and 6-CN-BTA-1 histological staining procedures were performed on tissue sections mounted on gelatin-coated slides. X-34 staining was performed as described previously (Styren et al., 2000). Briefly, tissue sections were washed in PBS and incubated in 100 μm X-34 solution in 40% ethanol and 60% distilled H2O (dH2O), pH 10, for 10 min. After washing in water, there was a 2 min differentiation in 0.2% NaOH in 80% ethanol, followed by a 10 min wash in water. For 6-CN-BTA-1 staining, slides were placed in PBS for 2 min, followed by application of 100 nm 6-CN-BTA-1 in PBS for 10 min in the dark. Slides were subsequently dipped three times in dH2O, differentiated for 2 min in a solution of 0.2% NaOH in 80% ethanol, and dipped again in dH2O. Both X-34- and 6-CN-BTA-1-stained slides were coverslipped using Fluoromount (Southern Biotechnology, Birmingham, AL) and examined on an Olympus (Melville, NY) microscope equipped with a fluorescent attachment.
ELISA analysis of Aβ levels
Extraction of Aβ from brain tissue. Frozen mouse hemibrains or fresh-frozen human brain samples were homogenized (150 mg/ml) in a tissue homogenization buffer (250 mm sucrose, 20 mm Tris base, 1 mm EDTA, 1 mm EGTA, plus protease inhibitors: 10 μg/ml each of leupeptin hemisulfate salt, antipain HCl, pepstatin-A, and 4-(2-aminoethyl)benzenesulfonylfluoride) using a glass vessel and piston-type Teflon pestle, and Aβ was extracted essentially as described previously (Refolo et al., 2000, 2001). Briefly, soluble Aβ was extracted using cold 0.4% diethylamine in 100 mm NaCl, spun at 135,000 × g for 1 h at 4°C, and neutralized by adding 0.5 m Tris-HCl, pH 6.8. The pellet was further extracted using 70% cold formic acid, sonicated for 1 min, spun as above, and neutralized (1 m Tris base, 0.5 m Na2HPO4, and 0.05% NaN3).
ELISA for soluble and insoluble Aβ. The soluble and insoluble Aβ extracts were additionally diluted in ELISA buffer [20 mm sodium phosphate, 2 mm EDTA, 400 mm NaCl, 0.2% BSA, 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate choline acetyltransferase, 0.4% Block Ace (Serotec, Raleigh, NC), and 0.05% NaN3, pH 7.0], and Aβ1-40 and Aβ1-42 concentration was measured by sandwich ELISA as described previously (Koldamova et al., 2005). Briefly, 6E10 monoclonal antibody (Signet, Dedham, MA) was used as a capture antibody. Horseradish peroxidase-conjugated anti-Aβ40 and anti-Aβ42 (Genetics Company, Schlieren, Switzerland), which are specific for Aβ ending at residues 40 and 42, respectively, were used as detection antibodies. Aβ values, based on standard curves using synthetic Aβ1-40 and Aβ1-42 peptide standards (Bachem Biosciences, King of Prussia, PA), were expressed as picomoles per gram of wet brain.
Quantitative postmortem binding of [3H]PIB
Binding to brain homogenates was performed with slight modifications of a procedure described in detail previously (Klunk et al., 2003). Briefly, 150 mg/ml tissue homogenates prepared in tissue homogenization buffer for ELISA analysis were diluted 150-fold in PBS to 1 mg/ml. The Kd and Bmax values for [3H]PIB binding to brain homogenates were determined using increasing concentrations of [3H]PIB between 0.2 and 1 nm and, for total PIB concentrations from 1 to 1000 nm, a constant 1 nm concentration of [3H]PIB (specific activity, 61.4 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO) plus additional unlabeled PIB. The labeled and unlabeled PIB were prepared in 900 μl of PBS, binding was initiated by the addition of 100 μl of the 1 mg/ml brain homogenate (in triplicate), and the samples were incubated at 22°C for 60 min. For comparison of [3H]PIB binding to brain homogenates in human control and AD brain and PS1 and PS1/APP mouse brain, 1 nm [3H]PIB was incubated with 100 μg of tissue in 1 ml of PBS. Nonspecific binding was defined as the number of counts remaining in the presence of 1 μm unlabeled PIB. The binding mixtures were filtered through a Whatman (Maidstone, UK) GF/B glass filter via a Brandel (Gaithersburg, MD) M-24R cell harvester and washed rapidly five times with 3 ml of PBS. The filters are counted in Cytoscint-ES after thorough vortexing. Results were corrected for nonspecific, nondisplaceable binding in the presence of 1 μm PIB and expressed as picomoles of [3H]PIB bound per gram of wet tissue weight in the homogenate. Linearity of [3H]PIB binding was confirmed using 1 nm [3H]PIB and 20-500 μg/ml of the AD brain homogenate with the highest level of [3H]PIB binding. Likewise, linearity with respect to [3H]PIB was confirmed using 100 μg of tissue per milliliter and 1-6 nm [3H]PIB. Binding results between groups were compared using an unpaired sample, unequal variance t test.
To facilitate straightforward comparison of the in vitro [3H]PIB binding data from human AD brain to in vivo data obtainable from PET tracer studies, the data also were expressed in terms of binding potential (BP), where BPtotal = Bmax/Kd (Mintun et al., 1984). To calculate this unitless measure, Bmax was first converted from units of picomoles of [3H]PIB bound per gram of brain to nanomolar units by assuming that 1 g of brain = 1 ml (picomoles/gram = picomoles/milliliter = nanomoles/liter). Division by Kd in nanomolar units directly results in BP.
Results
Micro-PET studies
Preliminary micro-PET studies were performed in pentobarbital-anesthetized PS1/APP mice and PS1 littermate controls at 6, 9, and 12 months of age (one PS1/APP-PS1 pair imaged simultaneously at each age). After intravenous injection of 100-150 μCi of [11C]PIB, dynamic imaging data were collected for 60 min and the time-radioactivity course was established (Fig. 1). As expected, the time-activity curve from the PS1 control mice (Fig. 1, open triangles) showed rapid brain entry and clearance. These pharmacokinetics were quantitatively very similar to those observed in previously reported ex vivo studies in Swiss-Webster mice (Mathis et al., 2003) (Fig. 1, filled circles). Although PS1 mice have elevated levels of soluble Aβ1-42 (Duff et al., 1996), neither PS1 nor Swiss-Webster mice have brain amyloid deposits. Surprisingly, the time-activity curves of the PS1/APP mice were nearly identical to that of the PS1 littermate controls, even at 12 months of age (Fig. 1, open squares). The normalized [11C]PIB concentration (%ID-kilogram/gram) in the brain of PS1/APP mice equilibrated at 100-120% of that observed in the PS1 mice between 15 and 60 min (i.e., nonsignificantly higher in PS1/APP brain). This was unexpected given the previously reported extensive amyloid plaque deposition in the PS1/APP mouse, which begins by 3 months of age and is well established by 8 months of age (McGowan et al., 1999; Takeuchi et al., 2000; Wengenack et al., 2000). Subsequently, a very similar finding of low [11C]PIB retention was reported in Tg2576 mice (Toyama et al., 2005). Because of this unanticipated low retention of [11C]PIB, we suspended the micro-PET studies until we could explore the nature of PIB binding to PS1/APP brain in the more detailed ex vivo and in vitro studies described below. When possible, we compared PIB binding to PS1/APP brain with PIB binding to postmortem AD brain tissue.
Figure 1.

Micro-PET time-activity curves show no difference in the retention of [11C]PIB in PS1/APP and PS1 mice. Brain radioactivity was determined with micro-PET in a 12-month-old PS1/APP mouse (□) and a PS1 littermate (▵) imaged simultaneously. [11C]PIB retention is expressed as %ID per gram of brain normalized to the body weight (in kilograms). The [11C]PIB retention from an ex vivo study with Swiss-Webster (Sw-Web) mice (n = 3 at each time point; •) is also shown. Inset, Axial (left) and sagittal (right) image of the PS1/APP mouse.
Ex vivo [11C]PIB binding in PS1/APP mice
Compared with in vivo micro-PET (∼2% detection efficiency), the higher counting efficiency of the gamma-well counter (∼35%) increases the sensitivity to detect differences in the in vivo retention of [11C]PIB between PS1/APP and PS1 mice. In addition, we performed the ex vivo studies on older (15 months of age) mice that would be expected to have even higher levels of amyloid deposition. [11C]PIB injections similar to those administered in the micro-PET study were given to four 15-month-old PS1/APP mice and four littermate controls 15 min before injection of pentobarbital and cardiac exanguination, followed by removal of the brain and ex vivo gamma counting to detect retained [11C]PIB. The 15 min time point was based on a trade-off between maximizing the time for clearance of free and nonspecifically bound [11C]PIB and minimizing the time for radioactive decay to retain good counting statistics (because of the ∼20 min t1/2 of carbon-11). At 15 min after injection, the amount of [11C]PIB retained in PS1 forebrain was 0.041 ± 0.010%ID-kg/g, and that in PS1/APP forebrain was 0.064 ± 0.024%ID-kg/g. This 56% increase was not significant (p = 0.13) and was mainly attributable to a single high outlier in the PS1/APP group (Fig. 2). This outlier did not prove to have significantly higher Aβ levels in the ELISA studies described below (data not shown). The importance of correcting brain uptake for body weight can be seen in Figure 2. At 15 months of age, the PS1 mice had 22% higher body weight than the PS1/APP mice, and therefore %ID/gram values are artifactually increased in the PS1/APP mice relative to the heavier PS1 mice, although the difference was still not significant (p = 0.08).
Figure 2.
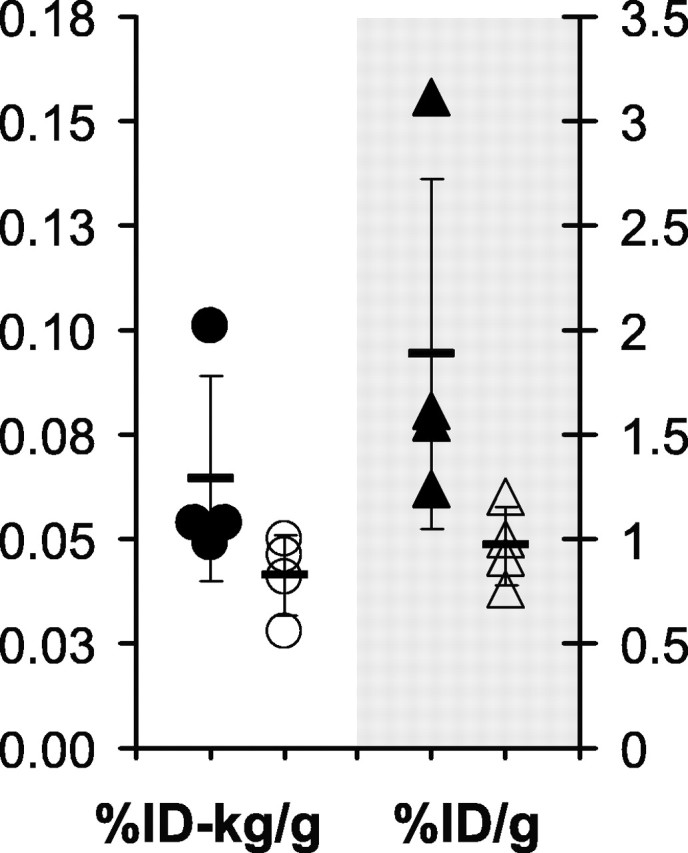
Ex vivo assessment of radioactivity shows no significant difference in the retention of [11C]PIB in PS1/APP and PS1 mice. Fifteen-month-old PS1/APP mice (filled symbols) and PS1 littermate controls (open symbols) were given intravenous injections of [11C]PIB in the tail vein and killed 15 min later. The brains were removed rapidly, and retained radioactivity was assessed in a gamma counter. The same data are expressed as %ID per gram on the right and as the weight-normalized %ID-kilogram/gram on the left. Neither show a significant difference.
Histochemical and biochemical analysis of the PS1/APP mice
To verify that the particular mice used for the ex vivo studies had the expected degree of brain amyloid deposition, the brains of these PS1/APP and PS1 mice were submitted to histochemical and biochemical analysis. After gamma counting as described above, the forebrains (cerebellum and brainstem removed) were bisected along the midline; half was immersion-fixed in 4% paraformaldehyde for histology, and the other half was frozen for biochemical analysis. IHC analysis showed the expected degree and type of amyloid deposition in all four PS1/APP mice and the lack of amyloid deposition in all four PS1 mice (Fig. 3). It has been previously reported that PS1/APP mice have a heavy plaque burden with levels approaching that seen in the human AD brain by ∼6 months of age (Takeuchi et al., 2000; Wengenack et al., 2000). The 15-month-old PS1/APP mouse brains stained heavily with 6E10 antibody (total Aβ) (Fig. 3A) and C-terminal-specific antibodies to Aβ1-40 and Aβ1-42 (Fig. 3C,E). As is typical of PS1/APP mice, staining to Aβ1-40 predominated. Plaques in the PS1/APP mice stained heavily with the highly fluorescent, fibril-specific Congo red derivative X-34 (Fig. 3G) (Styren et al., 2000). PIB itself is only weakly fluorescent and difficult to detect in tissue sections, but a derivative of PIB in which the 6-hydroxy group is replaced by a 6-cyano group (6-CN-BTA-1) is more fluorescent (Mathis et al., 2003). When stained with 100 nm 6-CN-BTA-1, numerous plaques are detected in PS1/APP brain (Fig. 3I). PS1 brain tissue did not stain with any of the amyloid stains (Fig. 3B,D,F,H,J).
Figure 3.
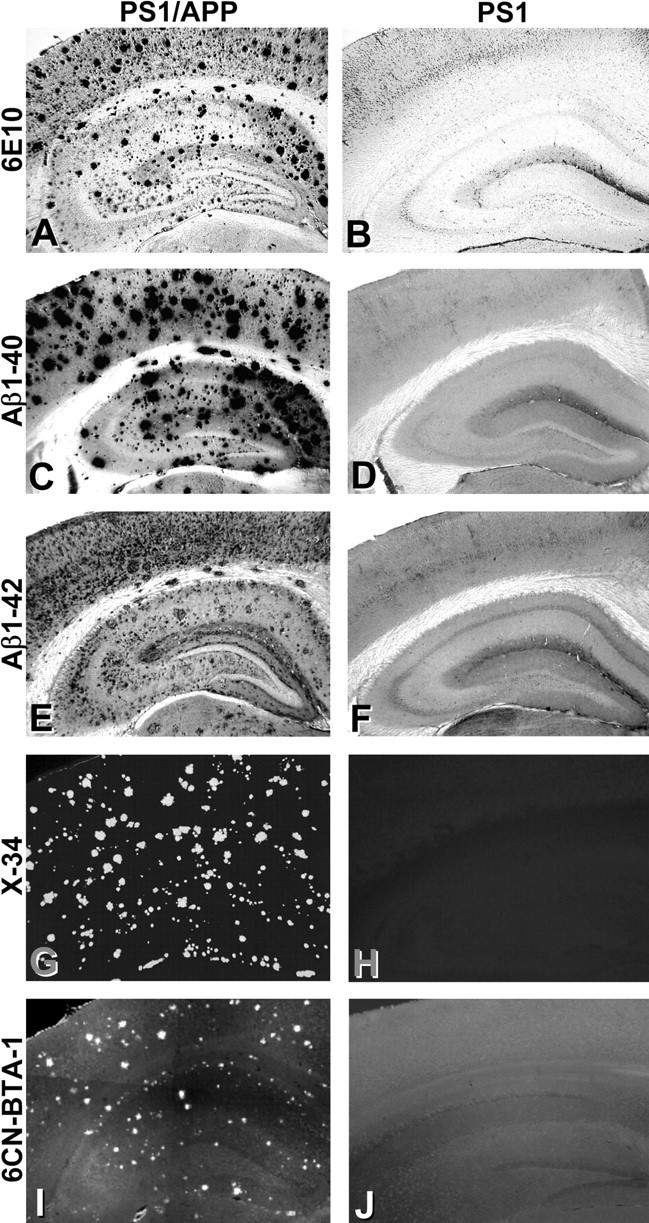
Histological analysis shows extensive amyloid deposition in the PS1/APP mice but not the PS1 mice. Tissue sections from PS1/APP (A, C, E, G, I) and PS1 (B, D, F, H, J) mice were immunostained with antibodies to total Aβ (A, B; 6E10), Aβ1-40 (C, D), and Aβ1-42 (E, F). Sections also were stained with the fluorescent Congo red derivative X-34 (G, H), which stains fibrillar amyloid, and the fluorescent PIB analog 6-CN-BTA-1 (I, J).
The Aβ content also was determined in the frozen half of the forebrains used in the ex vivo studies. The PS1 mice had undetectable amounts of human Aβ, as expected. The PS1/APP mice had very large amounts of insoluble Aβ1-40 and Aβ1-42 that exceeded that observed in typical human AD brain by 7- to 40-fold (Table 1). Soluble Aβ in PS1/APP brain exceeded that in human AD brain by 180- to 200-fold but accounted for only 4-5% of total Aβ. These histological and biochemical analyses confirmed the presence of large amounts of typical Aβ deposits in the mice studied and rule out the lack of Aβ as a cause for poor [3H]PIB binding.
Table 1.
Biochemical analysis of PS1/APP and human AD brain tissue
|
|
Aβ1-40 (pmol/g wet weight) |
Aβ1-42 (pmol/g wet weight) |
Total Aβ (pmol/g wet weight) |
|---|---|---|---|
| Insoluble | |||
| PS1/APP (n = 4) | 35,002 ± 21,266 | 12,838 ± 8453 | 47,840 ± 05,288 |
| Human AD (n = 5) | 764 ± 455 | 1715 ± 565 | 2479 ± 904 |
| Soluble | |||
| PS1/APP (n = 4) | 1056 ± 602 | 573 ± 36 | 1629 ± 980 |
| Human AD (n = 5) | 5.4 ± 4.9 | 3.2 ± 2.73 | 8.6 ± 6.1 |
| % Soluble | |||
| PS1/APP (n = 4) | 3.9 ± 9.7% | 4.9 ± 9.5% | 4.2 ± 2.6% |
| Human AD (n = 5) |
0.73 ± 3.36% |
0.19 ± 9.06% |
0.36 ± 6.14% |
In vitro [3H]PIB binding analysis of human AD brain and PS1/APP brain
Human brain
Binding to postmortem human brain homogenates was used extensively in the development of amyloid imaging agents, including PIB (Klunk et al., 1995, 2003; Mathis et al., 2003). This in vitro method of binding analysis appears to be the most sensitive technique available to detect the binding of compounds to Aβ and to demonstrate differences between amyloid-containing brain and amyloid-free control brain under conditions that mimic the low nanomolar concentrations attainable in PET studies (Klunk et al., 2003). In addition, in vitro binding to brain homogenates negates the effects of variable brain entry and clearance that could affect binding in vivo.
Scatchard analysis demonstrates a high-affinity [3H]PIB binding site in AD brains (n = 5) having a Kd of 2.5 ± 0.2 nm and a Bmax for [3H]PIB binding sites of 1407 ± 328 pmol/g wet weight (BP = 563 ± 131). In two of the five AD brain homogenates, a lower-affinity [3H]PIB binding site with Kd values of ∼250 nm and Bmax values of ∼13,000 pmol/g (BP ∼50) was observed (Fig. 4). Human control brain (n = 3) showed only this low-affinity component (Fig. 4). In the presence of the high-affinity binding component of AD brain, these low-affinity sites would not contribute significantly to in vivo binding at [11C]PIB concentrations around 1 nm (see Discussion). In AD brain, the ratio of the [3H]PIB Bmax to total Aβ was 0.57 ± 0.072 mol of [3H]PIB bound per mole of total insoluble Aβ. In human brain, there was a very good correlation between the Bmax for [3H]PIB binding and total Aβ levels (r = 0.91; p = 0.03) and Aβ1-42 levels (r = 0.92, p = 0.03) (Fig. 5A). There was a trend toward a correlation between the Bmax for [3H]PIB binding and Aβ1-40 levels (r = 0.75), but this did not reach statistical significance with this small number of samples (p = 0.13).
Figure 4.

Scatchard analyses of [3H]PIB binding shows a significant high-affinity binding component in human AD brain but not in PS1/APP brain. Left, This human AD brain (▪) showed a large high-affinity component (open arrow) with a Kd value of 2.8 nm and a Bmax value of 1780 pmol/g (BP = 636) and a low-affinity component with a Kd value of 264 nm and a Bmax value of 11,000 pmol/g (BP = 42). The human control brain (□) showed only the low-affinity component with a Kd value of 242 nm and a Bmax value of 15,000 pmol/g (BP = 62). Right, This PS1/APP brain (•) showed a very small high-affinity component (filled arrow) with a Kd value of 0.10 nm and a Bmax value of 7.78 pmol/g (BP = 78), along with a much larger low-affinity component with a Kd value of 32 nm and a Bmax value of 4300 pmol/g (BP = 134). The PS1 brain (○) showed the low-affinity component with a Kd value of 79 nm and a Bmax value of 4200 pmol/g (BP = 53).
Figure 5.

The binding of [3H]PIB reflects the total amount of Aβ in AD brain but not in PS1/APP brain. Correlation of the Bmax for [3H]PIB binding (A) or correlation of the amount of [3H]PIB bound (B) (using a concentration of 1 nm [3H]PIB) to levels of Aβ1-40 (♦), Aβ1-42 (▪), and total insoluble Aβ (▴) in AD brain (n = 5) is shown. C, Correlation analogous to that in B performed in PS1/APP (PSAPP) brain (n = 4).
In addition to determining Bmax (which can be heavily influenced by binding at saturating concentrations of [3H]PIB in the high nanomolar and low micromolar range), it is important to determine the amount of [3H]PIB bound at concentrations in the low nanomolar range that are attainable in PET studies. The amount of [3H]PIB bound to human AD brain homogenates in the presence of 1 nm [3H]PIB is tightly correlated with the Bmax and BP for [3H]PIB binding to the same brain samples (r = 0.99; p < 0.0001) and thus to the amount of Aβ in those brain samples (Fig. 5B). The amount of 1 nm [3H]PIB bound to human AD brain homogenates is significantly correlated to total Aβ (r = 0.92; p = 0.03) and Aβ1-42 (r = 0.92; p = 0.03). As was the case with Bmax, there was a trend toward a correlation between the amount of 1 nm [3H]PIB bound to human AD brain homogenates and Aβ1-40 (r = 0.79), but again this did not reach statistical significance (p = 0.11). This indicates that the amount of 1 nm [3H]PIB bound to human AD brain homogenates is a good indication of both the Bmax and BP for [3H]PIB binding and for the amount of total insoluble Aβ in that AD brain homogenate. [3H]PIB binding did not correlate significantly with soluble Aβ, and it should be noted that, on a molar basis, the amount of [3H]PIB bound was 40-fold higher than the amount of soluble Aβ in the AD brain homogenates.
Tg mouse brain
Scatchard analysis showed a very small high-affinity [3H]PIB binding component in PS1/APP mouse brain (Fig. 4, filled arrow) having a calculated Kd value of 0.1 nm and Bmax value of 7.8 pmol/g wet weight. Because of the very small nature of this high-affinity component relative to the low-affinity background, these Kd and Bmax values are of doubtful accuracy, as is the resultant value of BP (BP = 78). The low-affinity site had a Kd value of 32 nm and a Bmax value of 4300 pmol/g (BP = 134), suggesting that most of [3H]PIB binding to PS1/APP brain occurs at this low-affinity component (see Discussion). A very similar low-affinity component was present in PS1 brain (Fig. 4). [3H]PIB binding to homogenates of 15-month-old PS1/APP mouse brain (n = 4) did not correlate with Aβ content in the manner observed in AD brain (Fig. 5). Essentially, no relationship was observed between the amount of 1 nm [3H]PIB bound to PS1/APP brain homogenates and the amount of Aβ1-40 (r = 0.29; p > 0.9), Aβ1-42 (r = 0.19; p > 0.9), or total insoluble Aβ (r = 0.21; p > 0.9) present in the homogenates (Fig. 5C).
The amount of 1 nm [3H]PIB bound to various brain homogenates is shown in Table 2. Despite the very large difference in amounts of Aβ, the amount of 1 nm [3H]PIB bound to PS1/APP brain exceeded that bound to PS1 brain by only 3.5-fold. Brain homogenates from two 28-month-old PS1/APP mice also were studied and showed [3H]PIB binding of 3.2- and 2.8-fold higher than age-matched PS1 controls (i.e., no greater than 15-month-old mice). In contrast, the amount of 1 nm [3H]PIB bound to AD brain exceeded human control brain by >15-fold. When compared in terms of [3H]PIB binding per mole of total insoluble Aβ, the binding of [3H]PIB in PS1/APP brain homogenates was 40-fold less than that in AD brain (0.004 vs 0.167 mol [3H]PIB/mol Aβ; p = 0.0001).
Table 2.
PIB binding to homogenates from 15-month-old PS1/APP and PS1 mice and human AD and control brain in the presence of 1 nm [3H]PIB
|
|
PIB bound (pmol/g wet weight) |
Fold difference |
|---|---|---|
| PS1/APP (n = 4) | 166 ± 61 | 3.5 |
| PS1 (n = 4) | 47.5 ± 51 | |
| Human AD (n = 4) | 415 ± 51 | 16.3 |
| Human control (n = 3) |
25.4 ± 46 |
|
In vitro [3H]PIB binding to aggregated, synthetic Aβ1-40 and Aβ1-42
The high-affinity Kd for [3H]PIB binding to synthetic Aβ is very similar to that determined in binding studies with AD brain homogenates (Table 3). There was no significant difference in the affinity of [3H]PIB for Aβ1-40 and Aβ1-42 (Fig. 6). However, when normalized to the amount of Aβ present, the high-affinity Bmax determined for [3H]PIB binding to aggregated, synthetic Aβ was ∼1000-fold lower for both Aβ1-40 and Aβ1-42 than that observed in AD brain but was very similar to the Bmax observed for the high-affinity binding site in PS1/APP brain (Table 3).
Table 3.
Scatchard analysis of [3H]PIB binding to aggregated, synthetic Aβ1-40 and 1-42 fibrils and PS1/APP and AD brain homogenates
|
|
Kd (nm) (High Aff) |
Bmaxa (High Aff) |
BPb (High Aff) |
Kd (nm) (Low Aff) |
Bmaxa (Low Aff) |
BPb (Low Aff) |
BPb (total) |
% High Aff 1 nm PIBd |
|---|---|---|---|---|---|---|---|---|
| Aβ1-40 | 1.02 | 0.61 | c | 50.1 | 8.1 | c | c | 79 |
| Aβ1-42 | 0.91 | 0.45 | c | 51.5 | 9.6 | c | c | 73 |
| PS1/APP | 0.10 | 0.21 | 78 | 32.5 | 119 | 134 | 212 | 37 |
| AD |
2.5 |
565 |
563 |
264e
|
3888e
|
50 |
613 |
92 |
High Aff, High affinity; Low Aff, low affinity.
Picomoles of [3H]PIB bound per nanomole of Aβ.
BP = Bmax/Kd after conversion of Bmax values to nanomolar units.
BP data are not meaningful for synthetic Aβ because the concentration of the Aβ is arbitrarily set and not based on in vivo concentrations.
Expected percentage of PIB bound to the high-affinity binding site in the presence of 1 nm PIB. This was determined from the BP values because binding at tracer concentrations is proportional to BP.
The low-affinity component is detectable in less than half of AD brain samples.
Figure 6.

The binding of [3H]PIB to synthetic Aβ1-40 and Aβ1-42 is nearly identical, and the Bmax is very small as in PS1/APP brain (see Table 3).
To determine whether there was a soluble factor in AD brain necessary for optimal [3H]PIB binding that was missing in and could be transferred to PS1/APP brain or synthetic preparations of Aβ fibrils, a mixing experiment was performed. The addition of AD brain homogenates to PS1/APP and PS1 homogenates or preparations of synthetic Aβ1-40 or Aβ1-42 fibrils showed no synergistic increase in binding (Fig. 7). Binding to the mixture was very close to the sums of the two preparations. Another similar experiment was performed in which serum amyloid protein was added to the mouse brain homogenate, and again no augmentation of [3H]PIB binding was observed (data not shown).
Figure 7.

Mixing experiments do not indicate the presence of an easily transferable factor responsible for high-affinity [3H]PIB binding in AD brain. Homogenates of AD, PS1/APP, and PS1 brain (containing 100μg of tissue) and suspensions of Aβ1-40 and Aβ42-1 fibrils (containing 200 pmol of Aβ) or the mixtures indicated were incubated with 1 nm [3H]PIB in a total volume of 1 ml. The picomoles of [3H]PIB bound are indicated inside or above the bars. The amount of [3H]PIB bound to the mixtures is approximately equal to the sum of the binding to the two individual components, indicating no synergy.
Discussion
Aβ plaque-depositing Tg mice are an obvious model system for the development of PET tracers aimed at imaging amyloid. In the PS1/APP mouse, the time course of amyloid deposition is among the fastest, and the density of fibrillar Aβ deposits is among the highest, of any Tg mouse model (McGowan et al., 1999), making it a likely candidate to show in vivo retention of a fibril-selective radiotracer. Therefore, the failure to detect a significant difference in [11C]PIB retention in the brains of PS1/APP mice compared with PS1 littermate controls as reported above was unexpected.
By histological and biochemical analysis, the 15-month-old PS1/APP mice studied here showed the typical, extensive amyloid deposition described previously in this mouse model. Total, insoluble Aβ levels were 30-fold higher than those observed in AD brain, yet [3H]PIB binding was much lower. Based on the amount of Aβ present in the PS1/APP brain homogenate, the Bmax for the binding of [3H]PIB to PS1/APP brain more closely resembled the Bmax for the binding of [3H]PIB to synthetic Aβ in that both were ∼1000-fold lower than the Bmax for the binding of [3H]PIB to AD brain. This suggests that there is a very low efficiency for forming high-affinity PIB binding sites during in vitro aggregation of synthetic Aβ and a similarly low efficiency during plaque deposition in vivo in the PS1/APP mouse. In AD brain homogenates, PIB bound with high-affinity and total binding to AD brain was >15-fold higher than in control human brain. There was a tight correlation between PIB binding to AD brain and levels of insoluble Aβ. There was very little high-affinity binding of PIB to PS1/APP brain homogenates and total binding was increased only threefold compared with PS1 brain. No correlation was observed between PIB binding and levels of Aβ in PS1/APP brain. This suggests that, at the low nanomolar concentrations typically used in PET studies, the majority of PIB binding to PS1/APP brain is not mediated by Aβ. Because binding at tracer concentrations is proportional to the BP of each binding site, the BP data presented in Table 3 can be used to estimate that only ∼37% of the binding to PS1/APP brain would be to the high-affinity component (compared with ∼92% high-affinity binding in AD brain).
In contrast to AD brain, the PS1/APP mouse shows higher levels of Aβ1-40 than Aβ1-42. However, this is unlikely to be the explanation for poor PIB binding because we have seen similarly low PIB binding in other APP Tg mouse models in which Aβ1-42 predominates (data not shown), and the current study shows that PIB binds similarly to synthetic Aβ1-40 and Aβ1-42 (Fig. 6). Another possible explanation for the low PIB binding in PS1/APP brain could be that PIB does not bind directly to Aβ but binds to a component of human Aβ plaques that is missing in mouse brain. If this component is loosely associated with human plaques, then the addition of human amyloid deposits to PS1/APP brain or synthetic, aggregated Aβ would be expected to synergistically increase the binding of PIB, but this was not observed. Thus, if there is a plaque-associated factor other than Aβ important for the binding of PIB to human plaques, it must be very tightly associated with the plaque during the aggregation process. It is not likely that any missing component is tau related, because at the nanomolar concentrations used in PET studies and this study, PIB binding to tau deposits (i.e., neurofibrillary tangles) cannot be detected in AD brain (Klunk et al., 2003).
Evidence that the high-affinity PIB binding site measured in AD brain could be on the Aβ molecule itself or on a molecule that associates with Aβ with a 1:1 stoichiometry comes from the tight correlation between PIB binding and levels of insoluble Aβ in AD brain. In addition, the fact that the high-affinity Kd component in AD brain is very similar to that found on synthetic Aβ suggests that the high-affinity PIB binding site in AD brain may be present on Aβ itself. Additional evidence that the PIB-binding component of AD brain is on Aβ comes from binding studies with PIB analogs. Despite the low Bmax for high-affinity PIB binding, PIB binding to pure Aβ is not obscured by other sources of nonspecific binding as it is in the PS1/APP mice. This has allowed a comparison of rank-order potencies for the binding of PIB analogs to synthetic Aβ fibrils and AD brain across a range of Ki values from 2 to 3000 nm (Klunk et al., 2003). Correlation of potencies between two pharmacological systems is classic evidence for a structurally common binding site in those two systems (Guidotti et al., 1979). The strong correlation between the potencies of PIB analogs in the Aβ and AD brain binding assays (r = 0.9, with a slope near unity) suggests that a very similar binding site is being measured in both assays (Klunk et al., 2003). This has two implications. First, the high-affinity PIB binding site in AD brain is likely to be on the Aβ molecule itself. Second, this implies that when a high-affinity PIB binding site is formed on synthetic Aβ in vitro, this site is very similar to those found in AD brain, although the frequency of formation is only 1/1000th that in AD brain. Thus, it appears that Aβ itself may contain the entire PIB binding site and other molecules are not absolutely required.
The pathology of AD develops over one or more decades, whereas mouse pathology develops over months. The long time frame of deposition in human brain allows for many post-translational and post-deposition modifications including oxidation, cross-linking, racemization, N- and C-terminal truncation, and others. In addition, even if Aβ alone (in the proper conformation) is sufficient for high-affinity PIB binding, this does not exclude the likely possibility that other molecules could be involved in a scaffolding function during the Aβ aggregation process in AD brain. This scaffolding could increase the frequency with which Aβ assembles to form high-affinity PIB binding sites. A alternative possibility is that high- and low-affinity PIB binding sites are formed at similar frequencies in vivo and in vitro but that Aβ deposits formed in vivo with low-affinity PIB binding sites happen to be cleared more efficiently over the decades-long process that occurs in the human disease, leaving a large percentage of Aβ deposits with high-affinity PIB binding sites.
The phenomenon of low PIB binding would not be expected to be unique to the PS1/APP strain, and indeed preliminary experiments in our laboratory with Tg2576 and several proprietary strains show the same phenomenon. This is consistent with a recent report by Toyama et al. (2005) showing little retention of [11C]PIB (called [11C]6-OH-BTA-1 in that report) in 22-month-old Tg2576 mice. To our knowledge, none of the laboratories working to develop amyloid imaging agents has reported the successful application of micro-PET imaging with their agents in Tg mouse models of AD.
Using micromolar concentrations of PIB that are 1000-fold greater than levels achievable in PET studies that use high, specific-activity ligands, plaques in PS1/APP mice can be detected in vitro and ex vivo by histological staining and in vivo using multiphoton microscopy (Bacskai et al., 2003). Use of low, specific-activity PIB for PET studies to attain these micromolar concentrations would be expected to decrease the ratio of specific to nonspecific binding, so other factors must explain the success of ex vivo and in vivo microscopy. These microscopic techniques take advantage of a relatively small FOV that can exclude some of the potential sources of nonspecific background binding that would be detected using micro-PET or ex vivo radioassays. In addition, similar to the β-sheet detecting characteristics of thioflavin-T (the structural prototype for PIB) (LeVine, 1993), the actual binding of PIB to Aβ can lead to increased fluorescence compared with PIB bound to background structures making plaque-bound PIB preferentially detectable.
It is not clear whether the findings presented here have significance limited only to in vivo model systems used for the development of PIB and structurally related amyloid-binding compounds, or whether the implications extend beyond that. It may be that PIB can act as a probe for the structure of amyloid proteins at the molecular level. The differences in structure between synthetic Aβ, Aβ in Tg mouse models, and Aβ in AD brain suggested by the differences in PIB binding may have no significance beyond this, or they could be a cause of the differences seen in the tissue reaction to these deposits in AD and Tg mouse brain. If other macromolecules are important as scaffolding for the AD-like aggregation of Aβ or if macromolecules become important components of the Aβ fibril structure, then the differences in PIB binding may draw our attention to molecular targets that are important for the development of improved Tg mouse models or, more significantly, are important for the development of new therapeutic agents.
Footnotes
This work was supported in part by grants from the Alzheimer's Association (TLL-01-3381) and the National Institutes of Health (K02 AG001039, R01 AG020226, R01 AG018402, and AG005133). We thank Dr. Karen Duff for provision of the 28-month-old PS1/APP and PS1 brain tissue and for guidance on maintaining our breeding colony. We thank Barb Isanski for her role on the histological studies and the families that made brain tissue available to the University of Pittsburgh Alzheimer's Disease Research Center Brain Bank.
Correspondence should be addressed to Dr. William E. Klunk, Western Psychiatric Institute and Clinic, Room 1422 Thomas Detre Hall, 3811 O'Hara Street, Pittsburgh, PA 15213-2593. E-mail: klunkwe@upmc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510598-09$15.00/0
References
- Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT (2003) Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci USA 100: 12462-12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry SR (2001) Fundamentals of positron emission tomography and applications in preclinical drug development. J Clin Pharmacol 41: 482-491. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 96: 710-713. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Baraldi M, Schwartz JP, Costa E (1979) Molecular mechanisms regulating the interactions between the benzodazepines and GABA receptors in the central nervous system. Pharmacol Biochem Behav 10: 803-807. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Debnath ML, Pettegrew JW (1995) Chrysamine-G binding to Alzheimer and control brain: autopsy study of a new amyloid probe. Neurobiol Aging 16: 541-548. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA (2001) Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci 69: 1471-1484. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA (2003) The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23: 2086-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Debnath ML, Holt DP, Wang Y, Huang G-f, Shao L, Lefterov I, Koldamova R, Ikonomovic M, DeKosky ST, Mathis CA (2004a) Amyloid deposits in transgenic PS1/APP mice do not bind the Amyloid PET Tracer, PIB, in the same manner as human brain amyloid. Neurobiol Aging 25 [Suppl 2]: 232-233. [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B (2004b) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 55: 306-319. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS (2005) The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem 280: 4079-4088. [DOI] [PubMed] [Google Scholar]
- LeVine H (1993) Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci 2: 404-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Klunk W, Saxton J, Hamilton RL, Kaufer DI, Sweet RA, Cidis Meltzer C, Wisniewski S, Kamboh MI, DeKosky ST (2000) Res evaluation and diagnosis of probable Alzheimer's disease over the last two decades: I. Neurology 55: 1854-1862. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT, Holt DP, Wang Y, Huang GF, Debnath ML, Klunk WE (2002) A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett 12: 295-298. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE (2003) Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem 46: 2740-2754. [DOI] [PubMed] [Google Scholar]
- McGowan E, Sanders S, Iwatsubo T, Takeuchi A, Saido T, Zehr C, Yu X, Uljon S, Wang R, Mann D, Dickson D, Duff K (1999) Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis 6: 231-244. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Raichle ME, Kilbourn MR, Wooten GF, Welch MJ (1984) A quantitative model for the in vivo assessment of drug binding sites with positron emission tomography. Ann Neurol 15: 217-227. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA (2005) Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab, in press. [DOI] [PubMed]
- Refolo LM, Pappolla MA, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K (2000) Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: 321-331. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE (2001) A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 8: 890-899. [DOI] [PubMed] [Google Scholar]
- Styren SD, Hamilton RL, Styren GC, Klunk WE (2000) X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J Histochem Cytochem 48: 1223-1232. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao AK, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T (2000) Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 157: 331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama H, Ye D, Ichise M, Liow JS, Cai L, Jacobowitz D, Musachio JL, Hong J, Crescenzo M, Tipre D, Lu JQ, Zoghbi S, Vines DC, Seidel J, Katada K, Green MV, Pike VW, Cohen RM, Innis RB (2005) PET imaging of brain with the beta-amyloid probe, [11C]6-OH-BTA-1, in a transgenic mouse model of Alzheimer's disease. Eur J Nucl Med Mol Imaging 32: 593-600. [DOI] [PubMed] [Google Scholar]
- Watson Jr RE, Wiegand SJ, Clough RW, Hoffman GE (1986) Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides 7: 155-159. [DOI] [PubMed] [Google Scholar]
- Wengenack TM, Whelan S, Curran GL, Duff KE, Poduslo JF (2000) Quantitative histological analysis of amyloid deposition in Alzheimer's double transgenic mouse brain. Neuroscience 101: 939-944. [DOI] [PubMed] [Google Scholar]