

Abstract
Here, we describe the generation of a novel transgenic mouse model of human tauopathy. The rTg(tauP301L)4510 mouse expresses the P301L mutation in tau (4R0N) associated with frontotemporal dementia and parkinsonism linked to chromosome 17. Transgene expression was driven by a forebrain-specific Ca2+ calmodulin kinase II promoter system resulting in high levels of expression in the hippocampus and neocortex. Importantly, transgene expression in this model is induced via the tetracycline-operon responsive element and is suppressed after treatment with doxycycline. Continued transgene expression in rTg(tauP301L)4510 mice results in age-dependent development of many salient characteristics of hereditary human dementia. From an early age, immunohistochemical studies demonstrated abnormal biochemical processing of tau and the presence of pathological conformation- and phosphorylation-dependent epitopes. Neurofibrillary tangle (NFT) pathology was first observed in the neocortex and progressed into the hippocampus and limbic structures with increasing age. Consistent with the formation of NFTs, immunoblots indicated an age-dependent transition of accumulating tau species from Sarkosyl soluble 55 kDa to insoluble hyperphosphorylated 64 kDa. Ultrastructural analysis revealed the presence of straight tau filaments. Furthermore, the effects of tauP301L expression on spatial reference memory were longitudinally tested using the Morris water maze. Compared with nontransgenic age-matched control littermates, rTg(tauP301L)4510 mice developed significant cognitive impairments from 4 months of age. Memory deficits were accompanied by gross forebrain atrophy and a prominent loss of neurons, most strikingly in hippocampal subdivision CA1. Collectively, these data describe a novel transgenic mouse that closely mimics human tauopathy and may represent an important model for the future study of tau-related neurodegenerative disease.
Keywords: tau, transgenic, neurodegeneration, hippocampus, Alzheimer, FTDP-17
Introduction
Neurodegenerative diseases characterized by the pathological accumulation of tau include Alzheimer's disease (AD), frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), Pick's disease, progressive supranuclear palsy, and corticobasal degeneration (Iqbal et al., 2005). All of these conditions are characterized by the progressive formation of neurofibrillary tangles (NFTs), intracellular neuronal lesions composed of insoluble, conformationally abnormal, hyperphosphorylated accumulations of tau. Although it is known that abnormal processing of tau is central to the formation of NFTs and that NFTs are intimately linked with neurodegeneration during dementia, elucidation of the underlying mechanisms remains enigmatic. In addition to NFT, postmortem analysis of AD brain reveals two pathological hallmarks: β-amyloid (Aβ) plaques and neuronal loss. Thus, what also remains to be determined is the relationship between tau and Aβ and the true significance of tau in AD.
Physiologically a highly soluble protein, tau is functionally known to bind microtubules and is critical in the processes of neuronal outgrowth and axonal integrity (Weingarten et al., 1975). The adult human brain expresses six different tau isoforms that result from alternative splicing (Goedert et al., 1989). The efficacy of tau in binding to microtubules is greatly influenced by the exclusion/inclusion of exon 10, yielding tau species with three or four microtubule binding repeats, respectively. Supporting a central role for tau in dementia, genetic studies show that missense mutations in the tau gene (Mtapt) induce the development of neurodegenerative diseases such as FTDP-17 (Hutton et al., 1998). Unknown is how mutations in tau induce pathological consequences. Because mutations in tau have been shown to decrease microtubule binding, a loss of functionality has been proposed (Hasegawa et al., 1998; Hong et al., 1998). However, enhanced microtubule binding by four-repeat tau, as a result of mutations in intron 10 and exon 10 (N279K, S305N), suggests a gain of function may also promote FTDP-17 (Clark et al., 1998; Hutton et al., 1998; Spillantini et al., 1998b).
Transgenic mice expressing mutations in amyloid precursor protein (APP) have been an essential tool in AD research, with recent studies identifying the most significant Aβ species (S. Lesné, J. Cleary, L. Kotilinek, R. Kayed, C. C. Glabe, and K. Ashe, unpublished observations). However, APP transgenic mice are limited as models of AD by the absence of NFT and little or no neuronal loss (Irizarry et al., 1997; Hardy and Selkoe, 2002; Urbanc et al., 2002). Here, we attempted to generate a transgenic model that would closely mimic the clinical features of human tauopathy. The P301L mutation in exon 10 is the most common mutation linked to FTDP-17 and is pathologically characterized by the presence of NFT and neuronal loss in the forebrain (Hutton et al., 1998). Previously, tau pathology was described in a P301L mouse model (JNPL3), but transgene expression was driven under MoPrP, and NFTs were mainly present in hindbrain and spinal cord (Lewis et al., 2000). To study the effects of tauP301L in the brain regions most severely affected in AD and FTDP-17, we expressed transgenic tauP301L driven by a Ca2+/calmodulin kinase II (CaMKII) promoter system designed for specific transgene expression in the forebrain. The high levels of transgenic tauP301L expression achieved induced age-dependent development of the three major pathological hallmarks of human tauopathy: 1) memory impairment, 2) NFT, and 3) neuron loss. Here, we present an extensive age-dependent immunocytochemical, pathological, behavioral, and biochemical description of the regulatable Tg(tauP301L)4510 [rTg(tauP301L)4510] mice that includes comparison to human FTDP-17 tissue and investigation of spinal pathology. We believe this model may provide an important tool for investigating the mechanism(s) underlying neurodegeneration and cognitive impairment in age-related neurodegenerative disease.
Materials and Methods
Animals. Briefly, rTg(tauP301L)4510 mice were generated using a system of responder and activator transgenes. Mice expressing the activator transgenes were derived following a generous gift from Dr. E. Kandel (Columbia University, New York, NY) (Mayford et al., 1996) and successively backcrossed a minimum of five times onto a 129S6 background strain. Responder mice were maintained in the FVB/N strain. Mice were screened by PCR using the primer pairs 5′-GATTAACAGCGCATTAGAGCTG-3′ and 5′-GCATATGATCAATTCAAGGCCGATAAG-3′ for activator transgenes and 5′-TGAACCAGGATGGCTGAGCC-3′ and 5′-TTGTCATCGCTTCCAGTCCCCG-3′ for responder transgenes. All experiments described in this study were conducted in full accordance with American Association for the Accreditation of Laboratory Animal Care and Institutional Animal Care and Use Committee guidelines, with every effort made to minimize the number of animals used.
Transgene constructs. The tau responder transgene tauP301L construct cDNA, which encodes human four-repeat tau lacking the N-terminal sequences (4R0N), was modified such that the tauP301L transgene (containing exons 1, 4-5, 7, and 9-13, intron 13, and exon 14) driven by tetracycline-operon responsive element (TRE) was placed in the context of the mouse prion protein gene (prnp) transcribed but untranslated sequences, which were derived from the MoPrP.Xho expression vector (Borchelt et al., 1996). First, the SalI fragment of a previously created tauP301L transgene (starting 29 bases upstream of the start methionine and extending 1228 bases downstream of the stop codon) (Lewis et al., 2000), including the entire coding sequence of tau plus the P301L mutation, was inserted into the unique XhoI site of MoPrP.Xho to generate prnp.tauP301L. Next, the XbaI fragment of prnp.tauP301L, including partial sequences of prnp introns 1 and 2, along with exons 2-3 and the tauP301L open reading frame, was cloned into the unique XbaI site in the inducible expression vector pTRE (Clontech, Palo Alto, CA), resulting in the plasmid pTRE.prnp.tauP301L. The resultant DNA was digested with XhoI and NgoM IV enzymes, fractionated, and purified by electroelution, followed by organic extraction. Purified fragments containing a modified tau transgene were introduced by microinjection into the pronuclei of donor FVB/N embryos, using standard techniques.
In situ hybridization. Sagittal cryostat sections (15 μm) were fixed in 4% paraformaldehyde, dehydrated, and hybridized with a human tau-specific oligomer (5′-CTTTCAGGCCAGCGTCCGTGTCACCCTCTTGGTC-3′) 3′-end labeled with α 35S dATP. Sections were hybridized at 37°C overnight in buffer containing 4× SSC, 1× Denhardt's solution, 50% v/v deionized formamide, 10% w/v dextran sulfate, 200 mg/μl herring sperm DNA, and 0.03% β-mercaptoethanol. Control sections were hybridized in the presence of a 50- to 100-fold molar excess of unlabeled oligonucleotide. After hybridization, the sections were stringently washed three times with 1× SSC at 50°C, dehydrated, and exposed to βmax hyperfilm (Amersham Biosciences, Piscataway, NJ) for 2-4 d. After exposure to hyperfilm, slides were dipped in hypercoat emulsion, LM-1 (Amersham Biosciences), and exposed at 4°C for 8 d. Slides were developed using Kodak (Rochester, NY) D-19, fixed using Hypam fixative (Ilford Imaging, Paramus, NJ), and counterstained with toluidine blue.
Immunohistochemistry. Hemibrains were immersion fixed in 10% formalin for 24-48 h and embedded in paraffin. Serial sections were cut at 5 μm using a microtome, mounted onto CapGap slides, and rehydrated according to standard protocols. Mounted slides were pretreated with a citrate buffer, 6.0 pH, in a Black & Decker (Hampstead, MD) steamer for 30 min, with a 10 min cool down. Standard 2 d immunostaining procedures using peroxidase-labeled streptavidin and DAB chromagen on an automated TechMate 500 capillary gap immunostainer (Ventana Medical Systems, Tucson, AZ) were used. Hematoxylin counterstaining was used to provide cytological detail. Hematoxylin and eosin, Congo red, thioflavin-S, Gallyas, and modified Bielschowsky silver staining were performed using standard histological techniques. All primary antibody concentrations were titered to provide optimal staining (Table 1). No positive labeling was observed for pathological tau epitopes in nontransgenic mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Table 1.
Antibodies used
|
Antibody |
Species |
Specificity |
Dilution |
Source |
|---|---|---|---|---|
| Tau-5 | Mouse | Tau; human/mouse specific | 1:5000 | Biosource |
| T-14 | Mouse | Tau; human specific, 141-178 aa | 1:300 | Zymed |
| MC-1 | Mouse | Tau; conformational epitope, 7-9 and 326-330 aa | 1:8000 | P. Davies |
| Alz-50 | Mouse | Tau; conformational epitope, 3-15 and 312-322 aa | 1:50 | P. Davies |
| CP-13 | Mouse | Tau; pSer202 | 1:2000 | P. Davies |
| TG-3 | Mouse | Tau; conformational, pThr231/pSer235 | 1:200 | P. Davies |
| PG-5 | Mouse | Tau; pSer409 | 1:200 | P. Davies |
| AT-8 | Mouse | Tau; pSer202/pThr205 | 1:4000 | Innogenetics |
| PHF-1 | Mouse | Tau; pSer396/pSer404 | 1:3000 | P. Davies |
| NeuN | Mouse | NeuN | 1:4000 | Chemicon |
| E-1 | Rabbit | Tau, human specific, 19-33 aa | 1:2000 | S.-H. Yen |
| GAPDH | Mouse | GAPDH | 1:3000 | Biodesign International |
| GFAP | Rabbit | GFAP | 1:20,000 | Daku (Carpinteria, CA) |
| SMI-31 |
Mouse |
Phosphorylated neurofilament |
1:8000 |
Sternberger (Jarrettsville, MD) |
Biochemistry. Mouse brain tissue for biochemical studies was rapidly dissected and quickly frozen in isopentane for storage at -80°C. To generate forebrain lysates, olfactory bulbs, corticolimbic and subcortical brain stem structures, and the cerebellum were all removed. Frozen hemi-forebrains were thawed and mixed gently in a mortar with a few strokes of a pestle in 10 wet weight volumes of ice-cold Tris-buffered saline (TBS) containing protease inhibitor mixture, phenylmethylsulfonyl fluoride (PMSF), phenenthroline monohydrate, and Phosphatase Inhibitor Cocktails I and II (all from Sigma, St. Louis, MO), at a final dilution of 1:100. To estimate total levels of mouse and human tau, protein corrected (Bradford assay) brain extracts were diluted in reducing sample buffer, electrophoresed on 10% Tris-HCl gels (Bio-Rad, Hercules, CA), and transferred onto 0.45 μm polyvinylidene difluoride membranes (Millipore, Bedford, MA). Briefly, blots were processed with primary antibodies Tau-5 (Biosource, Camarillo, CA), T14 (Zymed, San Francisco, CA), and α-tubulin (Sigma) and visualized using enhanced chemiluminescence reagents (Pierce, Rockford, IL), followed by exposure onto hyperfilm (Kodak).
Sarkosyl extractions. Brain homogenate (90 μl) was ultracentrifuged at 150,000 × g for 15 min at 4°C. Supernatant (S1; soluble 1 fraction) was removed for analysis, and the pellet was rehomogenized in 10 mm Tris, pH 7.4, 0.8 m NaCl, 10% sucrose, 1 mm EGTA, and 1 mm PMSF and ultracentrifuged as before. Supernatant was incubated with a final volume of 1% Sarkosyl for 1 h at 37°C before ultracentrifugation at 150,000 × g for 30 min at 4°C. The pellet was resuspended in 20 μl of Tris-EDTA (10 mm Tris, pH 8.0, and 1 mm EDTA) and labeled P3 (Sarkosyl-insoluble tau). Equal loading for the S1 fraction was determined by protein assay with bicinchoninic acid (B9643; Sigma) and Copper (II) Sulfate (C2284; Sigma) against BSA standards. Equal loading for the P3 fraction was based on original brain weight (protein from 1.35 mg of tissue was loaded for each sample). Standard Western protocol was used with samples run on 10% Tris-glycine gels (EC6078Box; Invitrogen, San Diego, CA). Blots were blocked in 5% nonfat milk in TBS with 0.05% Tween 20 and hybridized with primary antibody [E1 human tau polyclonal (1:10,000; gift from S.-H. Yen, Mayo Clinic, Jacksonville, FL) overnight; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal (1:3000; Biodesign International, Saco, ME) for 1 h], followed by appropriate secondary antibody and visualization with Western Chemiluminescent ECL reagent (Pierce). Soluble and Sarkosyl-insoluble tau species were electrophoresed beside 10 ng of recombinant 4R0N human tau run on the same gel for quantification purposes. Soluble tau levels were adjusted for GAPDH levels to reflect equal loading. Band density from film exposed within the linear range was measured using Image-Quant 5.0 software (Molecular Dynamics, Sunnyvale, CA).
Behavioral analysis. Spatial reference memory was measured using the Morris water maze tailored to more rapid learning in the 129FVBF1 background strain (Westerman et al., 2002). Mice were handled 60 s per day for 10 d during the 2 weeks before the initiation of testing. Prehandling was designed to condition the mice to manipulations that would be experienced during introduction and removal from the testing pool and included a 20 s exposure to water at a depth of 1 cm. Mice were tested longitudinally from 2.5 to 9.5 months at ∼6 week intervals. An additional group of mice was tested at 1.3 months of age, independent of the longitudinal study. At each age tested, mice received visible platform training for 3 d (six trials per day) and hidden platform training for 6 d (four trials per day). At 2.5 months, visible platform training preceded hidden platform training, whereas hidden platform training preceded visible platform training at subsequent time points. For 1.3-month-old mice, visible platform training preceded hidden platform training. The spatial cues and hidden platform location were changed at each age tested. Four probe trials of 30 s duration were performed 20 h after 8, 12, 16, and 24 hidden training trials. The mean target quadrant occupancy of all four probes was calculated. All trials were monitored using a computerized tracking system (Noldus EthoVision 3.0; Noldus Information Technology, Wageningen, The Netherlands), and performance measures were extracted using Wintrack (Wolfer et al., 2001).
Statistical analyses. Statistical analysis consisted of ANOVA and repeated-measures ANOVA (RMANOVA). Post hoc comparisons were performed using unpaired t tests, Fisher's PLSD, or paired t tests with p values of <0.05 considered significant.
Results
Generation of transgenic mice expressing mutant human tauP301L
Here, we describe the creation of a transgenic mouse model expressing a mutation in human tau that is linked to hereditary tauopathy (Hutton et al., 1998). A responder transgene was generated consisting of a TRE placed upstream of a cDNA encoding human four-repeat tau with the P301L mutation, lacking both N-terminal inserts (4R0N tauP301L). An activator transgene in a second mouse line consisted of the tet-off open reading frame (Gossen and Bujard, 1992) placed downstream of CaMKII promoter elements. This system has been shown to result in expression from the TRE that is restricted to forebrain structures (Mayford et al., 1996). Mice harboring responder or activator transgenes were bred to generate bigenic progeny containing both transgenes (+/+, referred to as tau pos) (Fig. 1A). Remarkably, we found the highest levels of brain P301L in mice harboring the lowest number of responder transgenes (Fig. 1B). As expected, transgenic tau mRNA expression in bigenic mice was primarily restricted to structures in the forebrain (Fig. 1C,D) and predominantly restricted to neuronal cell types. The highest expression was seen in the hippocampus and cortex (Fig. 1D) and was detectable throughout all fields of the hippocampal formation and in all layers of the cortex, except for lamina I, in which there are no cell bodies. In support of in situ hybridization findings, immunoblots indicated high expression of tauP301L in the hippocampus and cortex (Fig. 1E), with no detectable transgene expression in the spinal cord. Previous biochemical analyses revealed expression levels of tauP301 in bigenic rTg(tauP301L)4510 mice to represent ∼13 U of tau (1 U is equivalent to the level of endogenous murine tau) (SantaCruz et al., 2005). Our data confirm previous findings describing the postnatal expression of CaMKII-driven transgenes (Mayford et al., 1996). We detected expression of transgenic tau species that labeled positively with human-specific tau antibody T14 (Zymed) beginning at age postnatal day 7 (P7) (Fig. 1F).
Figure 1.

Generation and characterization of rTg(tauP301L)4510 mice. A, Diagrammatic representation of activator and responder transgenes in bigenic mice. B, Highest levels of tauP301L expression were achieved in mice harboring the lowest number of responder transgene copies. C, Specific transgene expression in the forebrain was confirmed by in situ hybridization in 2- and 4-month-old rTg(tauP301L)4510 mice. Emulsion-dipped slides revealed silver grains clustered over individual neurons, of tau-positive mice only, indicating expression was restricted to neuronal cells types and that the majority of neuronal cells in the hippocampus and cortex expressed transgenic transcripts. D, Composite images (16 images at ×20) of emulsion-dipped slides from 4-month-old tau-positive and tau-negative mice. E, Biochemical studies confirmed high expression of tauP301L in the hippocampus and cortex and no detectable expression in the spinal cord of 2.5-month-old mice. F, Western blot analysis of postnatal forebrain indicated the earliest expression of transgenic tauP301L in P7 mice. Mouse and human tau were detected with tau-5 antibody (Biosource), and human-specific tau was probed for with T14 (Zymed). To demonstrate the high levels of transgene expression, tau-5 immunoblots were loaded with 3 μg of tau-positive forebrain extract compared with 30 μg of tau-negative lysate. T14 and α-tubulin blots were run in parallel after equal loading of 3 μg of protein across all lanes. neg, Negative; pos, positive; Tg, transgenic;α-Tub,α-tubulin; Olf, olfactory bulb; Ctx, cortex; Hpp, hippocampus; Cbm, cerebellum; BnSt, brainstem; Spn, Spinal cord.
Pathological tau conformation and phosphorylation
Postmortem analyses of brain tissue from FTDP-17 and AD patients revealed the deposition of biochemically altered tau species. Normally a highly soluble physiological protein, pathological phosphorylation and conformational changes result in translocation of insoluble tau species from the axon and accumulation in the cell body. We observed positive labeling with antibodies specifically directed at pathological species of tau (Table 2). Abnormal conformations of tau were detected from an early age (Fig. 2). Using MC-1 antibody, pathological conformations of tau were present in the hippocampus and neocortex of 2.5-month-old rTg(tauP301L)4510 mice (Fig. 2A,B). Early changes in tau phosphorylation were observed after processing with monoclonal antibody CP-13. Interestingly, the pattern of CP-13 staining was high in the dendrites and cell bodies of young mice but more prominent in the cell bodies of older mice (Fig. 2C,D). The number of neurons exhibiting somatodendritic accumulation of tau increased in an age-dependent manner, was similar between gender, and highly consistent when mice of the same age were compared. We observed substantial staining at older ages with antibodies that recognize epitopes consistent with the formation of pre-tangles (accumulations of nonargyrophilic hyperphosphorylated tau in the neuronal cell body) in human tauopathies. Pre-tangles were first observed in the cortex and appeared at later ages in limbic structures after immunohistochemical processing with TG-3, AT-8, and PG-5 antibodies (Fig. 3E-J). The number of late-stage tangles and morphologically abnormal neurons also increased with age, with copious PHF-1-positive cells present from 5 months of age (Fig. 3K,L; Table 2). At ages ≥5 months, prominent positive labeling was also observed for pathological epitopes detected by AT-100 (Innogenetics, Temse, Belgium), 12E8 (Elan, San Francisco, CA), and Alz-50 (Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY) antibodies (data not shown).
Table 2.
Semiquantification of positive tau labeling hierarchy in rTg(tauP301L)4510
|
|
n |
MC-1 |
CP-13 |
TG-3 |
PG-5 |
AT-8 |
PHF-1 |
Biel. |
|---|---|---|---|---|---|---|---|---|
| CA1/subiculum | ||||||||
| 1.3months | 7-9 | ++ | ++ | + | − | − | − | − |
| 2.5 months | 7-9 | ++ | +++ | + | + | + | + | − |
| 4-5.5 months | 4-9 | ++ | +++ | +++ | ++ | ++ | ++ | + |
| 7-8.5 months | 2-8 | +++ | +++ | +++ | +++ | +++ | +++ | ++ |
| Cortex | ||||||||
| 1.3 months | 9 | + | ++ | − | − | + | + | − |
| 2.5 months | 7-9 | + | ++ | + | + | + | + | + |
| 4-5.5 months | 4-10 | ++ | +++ | +++ | ++ | ++ | + | ++ |
| 7-8.5 months |
2-8 |
+++ |
+++ |
+++ |
+++ |
+++ |
++ |
++ |
Biel., Bielschowsky silver staining; −, no positive labeling; +, occasional; ++, moderate; +++, prominent.
Figure 2.

Accumulation of pathological tau species in rTg(tauP301L)4510 brain. Immunohistochemical studies revealed the progression of abnormal tau conformation and phosphorylation. A, B, Conformational alterations in tau detected with MC-1 are shown in 2.5-month-old rTg(tauP301L)4510 hippocampus. C, D, At 2.5 months, CP-13-positive labeling was prominent in axons and cell bodies of CA1 hippocampal neurons. Increased numbers of neurons positive for phosphorylation sites associated with the formation of pre-tangles were observed in the hippocampus (E, F, I, J) and cortex (G, H, K, L) of older mice. Original magnifications: A, C, E, G, I, K, 40×; B, D, F, H, J, L, 100×. No positive labeling was observed after parallel processing of control tissue (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 3.

Progression of NFT pathology in rTg(tauP301L)4510 brain. Formation of Bielschowsky silver-positive NFTs in the rTg(tauP301L)4510 cortex (Ctx) and hippocampus (Hpp) was age dependent and consistent with forebrain atrophy. A-D, The number of cortical NFTs increased with age, and cortical atrophy was obvious in mice >5 months of age. The arrows in A indicate rare NFTs in the cortex of 2.5-month-old rTg(tauP301L)4510 mice. Low-power photomicrographs reveal severe hippocampal degeneration with increasing age (E-H), an effect that was most striking in CA1 (I-L). Original magnifications: A-D, 20×; E-H, 4×; I-L, 100×.
Parallel studies using human FTDP brain tissue supported the relevance of the phosphorylation- and conformation-specific antibodies used (Fig. 4). Although we observed very few Bielschowsky silver positive NFTs, many neurons were positively labeled with the antibodies used in rTg(tauP301L)4510 mouse tissue. In human tissue, we observed a diffuse pattern of tau labeling with Alz-50, (Fig. 4A), AT-8, and CP-13 (data not shown), consistent with glial accumulation of pathological tau species in FTDP brain. Collectively, immunohistochemical studies revealed an age-dependent progression of tau processing in rTg(tauP301L)4510 that results in pathophysiological deposition of tau as mature tangles in brain regions that are severely affected in human tauopathies and AD.
Figure 4.

Positive tau labeling in human FTDP brain tissue mirrors rTg(tauP301L)4510. Parallel immunohistochemical processing of human FTDP-17 (P301L) tissue showed similar staining patterns in the rTg(tauP301L)4510 brain shown in Figure 2 A-F. G-J, In addition to Bielschowsky staining, standard histological stains Gallyas silver and thioflavin-S confirmed the presence of mature tangles in the 10-month-old rTg(tauP301L)4510 cortex. Original magnifications: A-H, 100×; I, J, 20×. neg, Negative; pos, positive.
Age-dependent progression of NFT pathology
We studied the development and progression of neuropathology in rTg(tauP301L)4510 by detection of NFTs using traditional Bielschowsky silver stains. Low-power magnification photomicrographs illustrate the temporal and topographical progression of NFT formation (Fig. 3). Occasional argyrophilic tangles were first observed in the cortex of rTg(tauP301L)4510 mice at 2.5 months of age (Fig. 3A). The density of argyrophilic tangle-like inclusions in the cortex increased with age (Fig. 3A-D). Similar to FTDP, where cortical pathology precedes the appearance of hippocampal lesions, NFTs were not consistently observed in limbic structures until ∼5 months of age (Fig. 3F). NFT pathology progressed within the hippocampal formation after a distinct subregional pattern of distribution. Mature tangles occurred initially in CA1 pyramidal neurons, spread to CA2, and by 8.5 months of age included pyramidal neurons in CA3 and granular neurons of the dentate gyrus (Fig. 3E-L). The presence of mature NFTs in rTg(tauP301L)4510 mouse forebrain was confirmed by Gallyas and thioflavin-S staining (Fig. 4H,J).
Age-dependent neurodegeneration in rTg(tauP301L)4510
To evaluate the neurodegenerative consequences of transgenic tauP301L expression and NFT formation, we measured whole-brain weight in mice from P1 to P16 (Fig. 5A). At the youngest age examined, we observed no significant difference between brain weight in tau-positive mice versus control littermates. However, when compared with control values, a small but consistent reduction in brain weight (4-7%) was observed in tau-Tg mice up to 4 months of age. This finding was consistent with biochemical data showing the earliest detectable expression of transgene at P7 (Fig. 1E). At ≥5 months of age, gross brain weights were significantly lower in rTg(tauP301L)4510 mice. This effect was attributable to specific atrophy of the forebrain (Fig. 5B), the target of our CaMKII promoter-driven transgene. Histological analyses of formalin-fixed brain tissue in 10-month-old mice indicated a striking degree of degeneration in the hippocampus and neocortex (Fig. 5C-J). High-power magnification revealed massive neuronal loss, which was most apparent in hippocampal subdivision CA1 (Fig. 5C-F). These observations were supported by identical findings after immunohistochemical processing with neuron-specific marker NeuN (Fig. 5G-J). In addition, consistent with the presence of degenerating neurons, forebrains of 10-month-old rTg(tauP301L)4510 mice showed prominent GFAP-positive labeling of reactive astrocytes (Fig. 5K-N).
Figure 5.

Age-dependent forebrain degeneration in rTg(tauP301L)4510 mice. A, Compared with nontransgenic, age-matched control littermates, rTg(tauP301L)4510 mice exhibited a significant decrease in whole-brain weight from an early age. Because of the small numbers examined at several ages, mice were binned into P1-P14, P28 to 4 months, and 5.5-16 months for statistical analysis. ANOVA revealed that rTg (tauP301L)4510 mice exhibited a significant decrease in brain weight starting at ∼ 1 month. Data for transgene ANOVA are as follows: P28 to 4 months: F(1,80) = 37.47, p < 0.0001; 5.5-16 months: F(1,45) = 130.7, p < 0.0001. Data for tau-negative and tau-positive mice, respectively, are as follows: P1, n = 8, n = 2; P7, n = 8, n = 2; P14, n = 12, n = 4; P28, n = 6, n = 4; 1.3 months, n = 35, n = 9; 2.5 months, n = 17, n = 7; 4 months, n = 3, n = 1; 5.5 months, n = 9, n = 7; 7 months, n = 2, n = 3; 10 months, n = 13, n = 9; 16 months, n = 1, n = 3. Tau-negative mice, 7 months of age, shown here were part of a concurrent study and received doxycycline (200 mg/kg) in their chow starting at 5.5 months of age. Previous studies have shown no effect of doxycycline supplementation on brain weight in nontransgenic mice (SantaCruz et al., 2005). Error bars represent SEM. **p < 0.01. B, Macroscopic photographs show forebrain atrophy in a representative 10-month-old tau-Tg mouse compared with control. Histological staining with hematoxylin and eosin (H&E) revealed massive neuronal loss in the frontal cortex and hippocampus in 10-month-old rTg(tauP301L)4510 mice (D, F) compared with control (C, E). Identical observations were made after immunohistochemical labeling with neuron-specific marker NeuN [tau negative (G, I), tau positive (H, J)]. Neuronal loss in 10-month-old rTg(tauP301L)4510 mice was accompanied by GFAP-positive reactive astrocytes (L, N), which were rarely observed in age-matched controls (K, M). Positive staining in immersion-fixed control tissue was mainly vascular. Original magnifications: C, D, G, H, 4×; K, L, 20×; E, F, I, J, M, N, 100×. neg, Negative; pos, positive.
Despite substantial atrophy, rTg(tauP301L)4510 mice age in a relatively normal manner. Consistent with previous P301L models, we noted the development of a specific external phenotype as the mice aged. From ∼9.5 months of age, the most severely affected rTg(tauP301L)4510 mice exhibited decreased ambulation and body weight and developed a hunched posture with hind-limb dysfunction and tail rigor. Surprisingly, rTg(tauP301L)4510 mice continued to age, with the oldest living mice currently >20 months of age.
Biochemical analysis of tau solubility
We studied the biochemical properties of tau in rTg(tauP301L)4510 mice at ages ranging from 2.5 to 10 months. Physiologically a highly soluble protein, we detected high levels of ∼55 kDa tau in the soluble fraction across all ages studied (Fig. 6). Tau accumulations in NFTs consist of insoluble twisted filaments and can be isolated from AD brain based on Sarkosyl insolubility (Greenberg and Davies, 1990). At 2.5 months of age, Sarkosyl-insoluble tau species were 55 kDa (Fig. 6). Consistent with hyperphosphorylation, we observed a shift from 55 to 64 kDa with increasing age. At ≥5.5 months, there was a prominent 64 kDa band and little or no 55 kDa Sarkosyl-insoluble tau species. The 64 kDa tau species is a hyperphosphorylated form of 4R0N tau, because the only tau species present after dephosphorylation comigrated with 4R0N recombinant tau (data not shown). Significantly, the insoluble 64 kDa tau species observed in this mouse model comigrates with a major hyperphosphorylated tau species observed in human NFTs in AD and other tauopathies (Buee et al., 2000). We observed a decrease in the expression of Sarkosyl-insoluble 64 kDa tau at 8.5 months, which may reflect degeneration of tangle-bearing neurons. To study the ultrastructural nature of tau accumulation in rTg(tauP301L)4510, we conducted electron-microscopic analyses. The neuronal inclusions in this line were composed of a mass of straight tau filaments that occasionally formed a herring bone pattern (Fig. 7).
Figure 6.

NFT pathology is accompanied by parallel changes in tau biochemistry. Representative immunoblots show soluble and Sarkosyl-insoluble tau species detected in forebrain homogenates of rTg(tauP301L)4510 mice aged 2.5-10 months. Western blots revealed an age-dependent transition from soluble 55 kDa tau to Sarkosyl-insoluble hyperphosphorylated 64 kDa tau. Human-specific tau species were detected with E1, and equal loading was ensured after probing for GADPH. Positive tau bands were referenced to 64 kDa tau-positive control (P3) and 60 kDa molecular weight markers (arrow).
Figure 7.
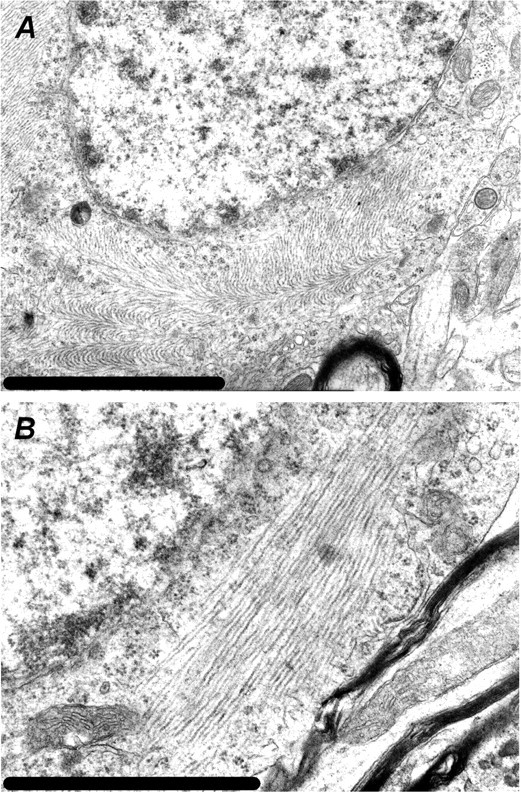
Ultrastructure analysis reveals the presence of straight filaments. Representative photomicrographs by electron microscopy on glutaldehyde-fixed brain tissue from a 10-month-old rTg(tauP301L)4510 mouse are shown. Tau pathology consisted of straight tau filaments that occasionally formed a herring bone pattern. Scale bars: A, 1 μm; B, 500 nm.
Age-dependent development of cognitive impairments
The effect of tauP301L expression on spatial reference memory in rTg(tauP301L)4510 mice was examined using the Morris water maze (Westerman et al., 2002). To investigate the development and progression of cognitive impairment in rTg(tauP301L)4510 mice, we longitudinally studied performance at 2.5, 4.5, 7, and 9.5 months of age. After training sessions with visible and hidden platforms, we probed for retention of spatial reference memory by analyzing the mean time spent swimming in the target quadrant (Fig. 8A) and search bias (Fig. 8B). We observed the first indications of impaired spatial reference memory at 2.5 months of age, as represented by a slightly reduced search bias for the target quadrant compared with control mice (Fig. 8B). However, the retention of spatial memory became more dramatically impaired as the mice aged. When compared with nontransgenic age matched control littermates, spatial reference memory was significantly impaired in rTg(tauP301L)4510 mice at all ages tested after 4 months of age. In fact, at 7 and 9.5 months of age, rTg(tauP301L)4510 mice were severely impaired with target quadrant occupancies indicating random swimming (25%). Independently, we tested spatial reference memory at 1.3 months of age, the youngest age compatible with this behavioral protocol. At 1.3 months, we observed no significant abnormalities during the probe trials, indicating no major deficits in the retention of spatial memory in very young mice. Thus, the retention of spatial reference memory declined dramatically in rTg(tauP301L)4510 mice in an age-dependent manner.
Figure 8.

rTg(tauP301L)4510 mice develop age-dependent memory deficits. Longitudinal studies of rTg(tauP301L)4510 mice [2.5-9.5 months; n = 26 control and n = 9 rTg(tauP301L)4510] in the Morris water maze revealed cognitive impairments in retention of spatial reference memory. An independent study examined behavioral performance in 1.3-month-old mice [n = 31 control and n = 10 rTg(tauP301L)4510]. A, Compared with control mice, the mean time spent swimming in the target quadrant during probe trials decreased in rTg(tauP301L)4510 mice with increasing age. RMANOVA data are as follows: transgene: F(1,33) = 80.1, p < 0.0001; age versus transgene: F(3,99) = 7.46, p < 0.001; age, rTg(tauP301L)4510 mice: F(3,24) = 5.82, p < 0.01. Individual quadrant occupancy shows search bias as the percentage of time spent in each of the four quadrants (mean off our probe trials). B, Differences between the percentage of time spent in the target and opposite quadrants compared with all other quadrants was determined using an ANOVA with Fisher's PLSD post hoc analysis. Parallel analyses indicate increasing impairment in spatial navigation in hidden (C) but not cued (D) platform phases of the water-maze protocol. C, RMANOVA data for hidden platform mean path length are as follows: transgene: F(1,33) = 96.17, p < 0.0001; age versus transgene: F(3,99) = 12.02, p < 0.0001; age, rTg(tauP301L)4510 mice: F(3,24) = 4.64, p = 0.01. During the first 2 d of cued training in the longitudinal study, both nontransgenic and rTg(tauP301L)4510 mice developed shorter path lengths with repeated testing (age versus transgene RMANOVA: F(3,99) = 2.58, p = 0.06). D, Although nontransgenic mice also improved with repeated testing during the final day of cued training (age RMANOVA: F(3,75) = 26.47, p < 0.0001), rTg(tauP301L)4510 mice exhibited stable performance (age RMANOVA: F(3,24) = 2.08, p = 0.13). Transgenic control mice aged 7 and 9.5 months shown here were part of a concurrent study and received doxycycline (200 mg/kg) in their chow starting at 5.5 months of age. Previous studies have shown no effect of doxycycline supplementation on spatial reference memory performance in nontransgenic mice (SantaCruz et al., 2005). #p < 0.05, ⋄p < 0.01, *p < 0.001, **p < 0.0001. neg, Negative; pos, positive; Opp, opposite.
We also observed cognitive impairments in the acquisition phases of behavioral testing. The mean distance to locate the hidden platform was consistently longer for rTg(tauP301L)4510 mice versus age-matched controls (Fig. 8C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We observed stable performance in control mice but detected a significant transgene effect in rTg(tauP301L)4510 mice that worsened with age. In younger mice with comparable target quadrant occupancies during probe trials, this may reflect increased time spent swimming in close proximity to the platform before mounting it and may represent a mild age-independent impairment in rTg(tauP301L)4510 mice. However, progression of acquisition impairment during hidden platform training was not attributable to an equivalent decline during cued training. This is supported by the observation that both bigenic and control mice display shorter path lengths with increasing age during the first 2 d of cued training and that transgenic mice have stable performance during the final day of cued training, which also suggests normal visual ability (Fig. 8D; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Although bigenic mice did not reach the efficiency achieved by nontransgenic controls (Fig. 8D; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Assessment of motor function in rTg(tauP301L)4510 mice
Previous transgenic mouse models expressing human tau driven by nonspecific neuronal promoters have described the development of spinal pathology and motor impairment in aged animals (Spittaels et al., 1999; Lewis et al., 2000; Terwel et al., 2005). To investigate these possibilities in our model, we dissected spinal cords from 10-month-old rTg(tauP301L)4510 mice and performed histological and immunohistochemical analyses. Supporting the specificity of the CaMKII forebrain-targeted promoter system used, we did not observe any evident reduction in the density of motor neurons in the anterior horn of the spinal cord. Although spinal cords of bigenic mice appeared thinner than littermate controls, we did not observe a decrease in motor neuron density in cervical, lumbar, or sacral sections of the spinal cord when processed with NeuN (Fig. 9A-D). However, we did observe prominent atrophy of the dorsal corticospinal tracts that was accompanied by a marked loss of neurofilament (Fig. 9E,F). Consistent with the preservation of muscle fibers in upper motor lesions, skeletal muscle integrity appeared normal and comparable to transgenic controls (Fig. 9G,H).
Figure 9.

Loss of motor input induces degeneration of dorsal corticospinal tracts. The spinal cord of 10-month-old mice showed no evident loss of motor neurons in the anterior horn of rTg(tauP301L)4510 mice (B, D) versus control mice (A, C). However, examination of corticospinal tract tissue indicated a marked loss of neurofilament (E, F). We observed no differences in skeletal muscle integrity when hematoxylin and eosin-processed rTg(tauP301L)4510 tissue was compared with control (G, H). Most severely affected 10-month-old rTg(tauP301L)4510 mice displayed a phenotypic grasping reflex in response to tail hang (I, J). Original magnifications: A, B, 4×; C-F, 40×; G, H, 100×. neg, Negative; pos, positive.
To test the hypothesis that rTg(tauP301L)4510 mice develop functional motor deficits, we measured swim speed across all ages (Fig. 10). Data obtained from visible, hidden, and probe trials indicate comparable swim speeds in mice aged ≤2.5 months (Fig. 10A-C). To ensure that decreased time spent swimming in the target quadrant (Fig. 8A,B) was not as a result of a motor disability, we examined swim speed at ≥4 months. Data from hidden platform and probe trials revealed faster swim speeds in tau-positive versus tau-negative mice at 4-7 months of age, possibly reflecting a difference in search strategy (Janus, 2004). Mean swim speeds were lower in 9.5-month-old rTg(tauP301L)4510 mice compared with controls. However, closer analysis of swim speed during the entire 30 s probe indicated tau-positive mice are capable of swimming at speeds comparable to those measured in control mice (Fig. 10D,E). These data were strongly influenced by an age-dependent increase in latency to start swimming in rTg(tauP301L)4510 mice (Fig. 10F), which may reflect the impaired cognitive abilities of these mice. This effect was supported by longer latencies to traverse a balance beam, reduced exploration in open-field tests (data not shown), and the development of a dystonic posture with tail rigor at 9.5 months of age. As described previously for other transgenic mouse models expressing the P301L tau mutation (Lewis et al., 2000; Terwel et al., 2005), rTg(tauP301L)4510 mice displayed clasping and limb retraction when lifted by the tail (Fig. 9I,J). Collectively, these data reveal a motor impairment in aged rTg(tauP301L)4510 mice that is age dependent and appears secondary to loss of motor cortex input.
Figure 10.

Assessment of motor function in rTg(tauP301L)4510 mice. Direct assessment of motor ability was measured by analysis of swim speed during each phase of behavioral testing. A-C, Mean swim speeds for mice aged 1.3 months and for mice tested longitudinally from 2.5 to 9.5 months during the visible, hidden, and probe trials, respectively. Age versus transgene RMANOVA data are as follows: A, F(3,99) = 31.33, p <0.0001; B, F(3,99) = 14.03, p <0.0001; C, F(3,99) = 16.30, p <0.0001. Older rTg(tauP301L)4510 mice at 9. 5 months of age didnot achieve the mean swim speed recorded in tau-negative controls until the final 10 s of probe trials (D, E; transgene RMANOVA, 9.5 months: F(1,33) = 19.74, p < 0.0001), an effect that was associated with a longer latency to begin swimming (F; age versus transgene RMANOVA: F(3,99) = 47.55, p < 0.0001; tau-positive age RMANOVA: F(3,24) = 18.87, p < 0.0001); age versus transgene RMANOVA: F(3,99) = 47.55, p < 0.0001). Data for 1.3-month-old mice [n = 31 control and n = 10 rTg(tauP301L)4510] and 2.5- to 9.5-month-old mice [n = 26 control and n = 9 rTg(tauP301L)4510] are shown. #p < 0.05, ⋄p < 0.01, *p < 0.001, **p < 0.0001. neg, Negative; pos, positive.
Discussion
Our rTg(tauP301L)4510 mice develop age-dependent progression of tau pathology that is highly comparable to human tauopathy. Expression of tauP301L results in significant impairments in spatial reference memory, NFT pathology, and severe forebrain atrophy. Rapid progression of tau pathology in rTg(tauP301L)4510 mice was dependent on the relatively high levels of transgene expression achieved (∼13 U). This was achieved after the observation that fewer responder transgene copies improved the expression of tauP301L. These data suggest that competition for transactivator proteins by multiple transgene copies lowered the efficiency of transcription. Importantly, transgene is strongly expressed in forebrain structures such as the neocortex and hippocampus, supporting this mouse as a relevant model of AD and FTDP-17 neurodegeneration. Another advantage of this promoter system is the avoidance of early motor deficits associated with direct transgene expression in the spinal cord and peripheral nervous system (Spittaels et al., 1999; Lewis et al., 2000; Terwel et al., 2005). Moreover, expression of tauP301L in this model is regulatable and can be suppressed after administration of doxycycline. This sophisticated system supports experiments designed to test hypotheses focusing on the effects of transgene withdrawal (SantaCruz et al., 2005).
In contrast to a recent model expressing the R406W tau mutation (Ikeda et al., 2005) (∼20% penetrance), we observed a highly consistent phenotype in bigenic mice that occurred with 100% penetrance. We believe the rapid onset of tau pathology (∼2.5 months) and the aggressive progression of cognitive decline and neurodegeneration avail an efficient and economical experimental model. Supporting the importance of transgene level, an additional line of mice expressing ∼7 U of tauP301L did not develop pre-tangles until 14.5 months of age or argyrophilic mature tangles until 20 months of age. At 2.5 months, the earliest pathological tau epitopes detected in rTg(tauP301L)4510 mice were those dependent on abnormal conformational changes in tau. Prominent positive labeling was observed in the hippocampus and cortex of 2.5-month-old rTg(tauP301L)4510 mice using MC-1 and Alz-50 conformation-dependent antibodies. These findings suggest conformational alterations in tau may be an early and critical factor in subsequent pathological cascades. This hypothesis is supported by a comprehensive study by Terwel et al. (2005) that compared progression of tau pathology in mice expressing human tauP301L versus wild-type human tau (tau-4R2N). The authors concluded that the conformation state of tau was the critical difference in the efficacy of tau aggregation. Although wild-type tau-4R2N was relatively more hyperphosphorylated, only tauP301L was prone to aggregation. Because structural modification appears to precede many markers of phosphorylation in rTg(tauP301L)4510, altered conformation may facilitate or promote hyperphosphorylation of tau, an established hallmark of tauopathy.
In older mice, positive labeling with antibodies raised against pre-tangle epitopes increased with age. Importantly, pre-tangles detected in rTg(tauP301L)4510 mice were morphologically and tinctorially identical to those observed in parallel studies using human tissue. The pattern of tau phosphorylation appeared to be hierarchal with MC-1 and CP-13 eptiopes preceding TG-3, AT-8, and PG-5. By 10 months of age, immunohistochemical processing of rTg(tauP301L)4510 revealed strong labeling with all phospho-antibodies used and dominant staining with PHF-1, a marker of late-stage tangles. Consistent with the formation of NFTs, biochemical studies revealed an age-dependent transition from soluble 55 kDa to hyperphosphorylated Sarkosyl-insoluble 64 kDa tau. Concurrent analyses of tau ultrastructure exposed the presence of both straight and herring bone-shaped filaments. In human tauopathies, tau inclusions encompass a wide range of filament morphologies (i.e., PHF, straight, twisted ribbon, rope like), but in all cases, the formation of these lesions is associated with neurodegeneration (Spillantini et al., 1998a). Interestingly, a similar pattern of tau filaments was observed in the JNPL3 mice, despite the fact that the NFT in this model occurred in a completely different population of neurons and on a different mouse strain background (Lewis et al., 2000, 2001). These data imply that the morphology of the tau filaments in these two models reflect an inherent property of the 4R0N P301L human tau species expressed in both mouse lines.
The presence of mature argyrophilic tangles was confirmed using traditional histopathological techniques. NFTs were observed after staining with Bielschowsky silver, Gallyas, and thioflavin-S protocols. Again, the age-dependent formation of NFTs described in rTg(tauP301L)4510 mirrors human disease and the progression of NFT pathology in FTDP-17, the condition linked to the P301L mutation (Hutton et al., 1998). Unlike AD in which NFT pathology is observed in the limbic structures before the cortex, both rTg(tauP301L)4510 mice and patients with FTDP-17 experience early cortical pathology that precedes that in the hippocampus. However, it is possible that in rTg(tauP301L)4510 mice, this effect may be a nonspecific consequence of promoter efficacy rather than a differential indication of brain region vulnerability to tau pathology.
Supporting a direct neurodegenerative effect of tauP301L expression, NFT pathology was accompanied by severe forebrain atrophy and massive neuronal loss in the neocortex and hippocampus. Remarkably, gross brain weights were reduced to ∼60% of control values by 9.5 months of age, a statistic that underestimates specific forebrain atrophy. Although the rate of atrophy appeared to slow after 10 months of age, the remaining forebrain tissue in 16-month-old rTg(tauP301L)4510 mice was measured at ∼33% of littermate controls. Most striking, neuronal loss occurred in hippocampal CA1, the same subdivision that showed high levels of biochemically abnormal tau (conformation and phosphorylation). However, the number of granule neurons in the dentate gyrus was equally affected. Our previous study estimated the total number of neurons in CA1 of 5.5-month-old rTg(tauP301L) mice to represent ∼30% of those counted in nontransgenic age-matched controls (SantaCruz et al., 2005). This is an important consequence of tauP301L expression, because most previous transgenic models expressing mutant tau do not exhibit or report neuron loss in the brain, a key feature of human disease. On the other hand, mice expressing non-mutant human tau, in the absence of mouse tau, developed extensive cell death between the ages of 8 and 17 months (Andorfer et al., 2003). Most recent studies of this model suggest the mechanism of tau-mediated neuronal death may involve the re-expression of cell-cycle proteins, indicating incomplete re-entry of the cell cycle (Andorfer et al., 2005). However, we did not observe aberrant cell-cycle events in rTg(tauP301L)4510 brain (M. Ramsden, C. Forster, J. Lewis, and K. Ashe, unpublished data). Alternatively, aberrant phosphorylation of tau has been shown to be central to the process of neurodegeneration following in a recent study that showed neuronal loss in the brains of inducible transgenic mice overexpressing the Cdk5 regulator p25 (Cruz et al., 2003). Elucidating the mechanism of neuronal death in rTg(tauP301L)4510 mice is a major goal of future studies.
In addition to NFT pathology and neuronal degeneration, rTg(tauP301L)4510 mice developed age-dependent impairments in cognitive ability. When we tested spatial reference memory in the Morris water maze, we detected memory deficits that were significant from 4.5 months of age. This aspect of cognitive function was assessed because of the highly sensitive nature of the Morris water maze and the dependence of spatial reference memory on the hippocampus (Morris et al., 1982). As the mice aged, cognitive impairments worsened, and at 9.5 months, the mean probe performance was equal to random swimming, indicating little or no retention of spatial reference memory. Importantly, we did not observe an impairment in spatial reference memory at 1.3 months of age, the youngest mice we were able to behaviorally characterize. These data suggest that the significant age-dependent decline in retention of spatial reference memory was not a direct effect of transgene expression on postnatal brain development. Our previous study dissociated the progression of NFT pathology as the underlying cause of memory loss in this transgenic model (SantaCruz et al., 2005). Critically, what remains to be determined is the specific tau species responsible for inducing cognitive impairment in rTg(tauP301L)4510. We hypothesize aberrant changes in tau phosphorylation, conformation, or oligomerization may be central to this process. Impairments in rodent spatial reference memory can be induced after increased kinase activation or phosphatase inhibition (Sun et al., 2003, 2005). Moreover, memory impairments induced after hyperphosphorylation of tau can be reversed in mice after application of kinase inhibitors (Liu et al., 2003).
A common phenotype in tau transgenic models is the appearance of motor impairment. Previous studies expressing transgenic tau under the control of neuronal promoters have described motor deficits that result after spinal pathology. This phenomenon has been described in models expressing P301L, P301S, and non-mutant human tau (Lewis et al., 2000; Allen et al., 2002; Terwel et al., 2005). Consistent with spinal degeneration and peripheral neuropathy, these studies reveal loss of motor neurons in the anterior horn and atrophy of skeletal muscle. Motor deficits in 10-month-old rTg(tauP301L)4510 mice appear to be independent of anterior horn motor neuron degeneration, and the integrity of skeletal muscle fibers is comparable to control mice. In contrast to previous reports, motor impairment in rTg(tauP301L)4510 is consistent with loss of upper motor input after cortical degeneration, which is further supported by evident loss of neurofilament and demyelination of the dorsal corticospinal tracts. As a possible consequence of cognitive impairment, rTg(tauP301L)4510 mice develop an age-dependent increase in the time taken to start swimming. However, these deficits in older mice did not negatively impact ability to perform in spatial memory tests given their ability to achieve comparable mean swim speeds during the probe trials.
In our previous study, we manipulated transgene expression in rTg(tauP301L)4510 to investigate the role of NFT in the development of cognitive deficit (SantaCruz et al., 2005). Surprisingly, we discovered a recovery of spatial reference memory after transgene suppression, suggesting potential for cognitive improvement in patients with early-stage dementia. Moreover, progression of NFT pathology in the absence of memory dysfunction dissociated NFT formation and cognitive decline. These data support the hypothesis that deleterious effects on neuronal function precede overt structural changes, such as formation of NFT and neuron loss. Future studies with rTg(tauP301L)4510 aim to elucidate the mechanism(s) of neuronal death and memory loss in this model. Revelation of the underlying biochemical cascades may present novel therapeutic targets for the treatment of age-related tauopathies, such as AD and FTDP-17.
Footnotes
This work was supported by National Institutes of Health Grants P01-AG15343 and R01-026252 (K.H.A.) and R01-NS46355 (J.L.). We thank N. Nash, M. Sherman, and S. Nelson for technical expertise, P. Sharpe for secretarial assistance, Drs. P. Davies and S. H. Yen for the generous gifts of antibodies, and Dr. E. Kandel for the generous donation of activator mice.
Correspondence should be addressed to Dr. Karen H. Ashe, Department of Neurology, University of Minnesota Medical School, Minneapolis, MN 55455. E-mail: hsiao005@umn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510637-11$15.00/0
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and non-apoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22: 9340-9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86: 582-590. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25: 5446-5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal 13: 159-163. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33: 95-130. [DOI] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC (1998) Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 95: 13103-13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40: 471-483. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3: 519-526. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89: 5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437: 207-210. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282: 1914-1917. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: 702-705. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, Ishiguro K, Hasegawa M, Yen SH, Chishti MA, Harigaya Y, Abe K, Okamoto K, St George-Hyslop P, Westaway D (2005) Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol 166: 521-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: 198-210. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT (1997) Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C (2004) Search strategies used by APP transgenic mice during navigation in the Morris water maze. Learn Mem 11: 337-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402-405. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487-1491. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, Xu H, Wang JZ (2003) Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem 87: 1333-1344. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678-1683. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297: 681-683. [DOI] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: 476-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B (1998a) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8: 387-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998b) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 155: 2153-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ (2003) Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118: 1175-1182. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang X, Liu S, Wang Q, Wang J, Bennecib M, Gong CX, Sengupta A, Grundke-Iqbal I, Iqbal K (2005) Bilateral injection of isoproterenol into hippocampus induces Alzheimer-like hyperphosphorylation of tau and spatial memory deficit in rat. FEBS Lett 579: 251-258. [DOI] [PubMed] [Google Scholar]
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F (2005) Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 280: 3963-3973. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT (2002) Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc Natl Acad Sci USA 99: 13990-13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72: 1858-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer DP, Madani R, Valenti P, Lipp HP (2001) Extended analysis of path data from mutant mice using the public domain software Wintrack. Physiol Behav 73: 745-753. [DOI] [PubMed] [Google Scholar]