

Abstract
The intracellular mechanisms that bias mammalian neural precursors to generate neurons versus glial cells are not well understood. We demonstrated previously that the growth factor-regulated mitogen-activated protein kinase kinase (MEK) and its downstream target, the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors, are essential for neurogenesis in cultured cortical precursor cells (Ménard et al., 2002). Here, we examined a role for this pathway during cortical cell fate determination in vivo using in utero electroporation of the embryonic cortex. These studies demonstrate that inhibition of the activity of either MEK or the C/EBPs inhibits the genesis of neurons in vivo. Moreover, the MEK pathway mediates phosphorylation of C/EBPβ in cortical precursors, and expression of a C/EBPβ construct in which the MEK pathway phosphorylation sites are mutated inhibits neurogenesis. Conversely, expression of a C/EBPβ construct, in which the same sites are mutated to glutamate and therefore are “constitutively” phosphorylated, enhances neurogenesis in the early embryonic cortex. A subpopulation of precursors in which C/EBP activity is inhibited are maintained as cycling precursors in the ventricular/subventricular zone of the cortex until early in postnatal life, when they have an enhanced propensity to generate astrocytes, presumably in response to gliogenic signals in the neonatal environment. Thus, activation of an MEK-C/EBP pathway in cortical precursors in vivo biases them to become neurons and against becoming astrocytes, thereby acting as a growth factor-regulated switch.
Keywords: neural stem cells, MEK, in utero electroporation, neurogenesis, gliogenesis, cortical development, ERK, transcription factors
Introduction
Development of the mammalian nervous system is determined by a complex interplay between intrinsic genetic mechanisms and extrinsic cues such as growth factors, an interplay perhaps best studied in the developing cortex (Lillien, 1998; Shen et al., 1998; Ferguson and Slack, 2003). In vivo, cortical neurogenesis occurs early, whereas gliogenesis occurs later, peaking postnatally. The same sequential genesis of neurons and then glia also occurs in embryonic cortical precursor cultures (Qian et al., 2000; Barnabé-Heider et al., 2005), and recent work indicates that this timed sequence is regulated by growth factors in the neural environment. For example, the gliogenic cytokine cardiotrophin-1 is expressed by newly born neurons and acts as a feedback timer to regulate the onset of cortical astrocyte formation (Barnabé-Heider et al., 2005). Conversely, ligands that signal via receptor tyrosine kinases regulate cortical neurogenesis; fibroblast growth factor 2 (FGF2) is necessary for neurogenesis (Raballo et al., 2000) and platelet-derived growth factor (PDGF) and the neurotrophins enhance the genesis of neurons in the presence of FGF2 (Ghosh and Greenberg, 1995; Williams et al., 1997; Park et al., 1999; Barnabé-Heider and Miller, 2003). However, the intracellular mechanisms that allow precursors to integrate these diverse signals to ultimately generate a neuron versus a glial cell are still not well understood.
What are the intracellular growth factor-mediated signals that regulate neurogenesis? We recently demonstrated that mitogen-activated protein (MAP) kinase kinase (MEK), which is downstream of many growth factor receptors, promotes neurogenesis from cultured cortical precursors without apparently regulating either survival or proliferation (Ménard et al., 2002; Barnabé-Heider and Miller, 2003). In addition, we showed that one of the targets of MEK, the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors, is essential for cortical precursors to generate neurons in culture (Ménard et al., 2002), where it directly promotes transcription of at least one early pan-neuronal gene, Tα1 α-tubulin (Miller et al., 1987; Gloster et al., 1994, 1999). The C/EBP family is composed of basic leucine zipper DNA-binding proteins (C/EBPs α, β, γ, δ, ϵ, and ζ) that recognize a common DNA-binding sequence (Johnson and Williams, 1994) and that are expressed in virtually all tissues, including the developing and adult brain (Sterneck and Johnson, 1998; Sterneck et al., 1998; Nadeau et al., 2005).
Because previous work indicates that the neurogenic basic helix-loop-helix (bHLH) proteins are also essential for cortical neurogenesis (Nieto et al., 2001; Sun et al., 2001), we hypothesized that cortical precursors are biased to generate neurons by their expression of neurogenic bHLHs and that growth factors in their immediate environment activate MEK, which in turn instructs those precursors to generate neurons via phosphorylation and activation of the C/EBP family. Here, we directly tested this hypothesis in vivo, using in utero electroporation to genetically manipulate cortical precursors in the embryonic telencephalon. Our data indicate that MEK-mediated phosphorylation of the C/EBPs is essential for cortical precursors to generate neurons and that at the same time activated C/EBP promotes neurogenesis, it inhibits astrocyte formation, thereby acting as a growth factor-regulated fate switch in vivo.
Materials and Methods
Culture of cortical precursor cells. Cortical precursors were cultured as described previously (Toma et al., 2000; Ménard et al., 2002; Barnabé-Heider and Miller, 2003). Briefly, cortices were dissected from embryonic day 12 (E12) to E13 CD1 mouse embryos in ice-cold HBSS (Invitrogen, Gaithersburg, MD) and transferred to Neurobasal medium (Invitrogen) containing 500 μm l-glutamine (Cambrex Biosciences, Hopkinton, MA), 2% B27 supplement (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 40 ng/ml FGF2 (Collaborative Biomedical Products, Bedford, MA). The tissue was mechanically triturated with a plastic pipette and plated onto two-well chamber slides (Nunc, Naperville, IL) precoated with 2% laminin and 1% poly-d-lysine (Collaborative Biomedical Products). Cell density was 250,000 cells/well for two-well chamber slides.
Transfection and CNTF stimulation of precursor cultures and HEK 293 cells. Two to 4 h after plating of precursor cells, 2 μg of DNA and 3 μl of Fugene 6 (Roche, Welwyn Garden City, UK) with 100 μl of Opti-MEM (Invitrogen) for each well was mixed, incubated for 30-45 min at room temperature, and added to the cultures. One-half of the medium was changed after 24 h, and 50 ng/ml ciliary neurotrophic factor (CNTF) (Peprotech, Rocky Hill, NJ) was added in some experiments. For transfection of human embryonic kidney 293 (HEK 293) cells, 4 μg of DNA and 10 μl of Lipofectamine 2000 (Invitrogen) with 250 μl of Opti-MEM was added to 70% confluent cells in six-well plates.
In utero electroporation. A plasmid expressing a nuclear-localized green fluorescent protein (GFP) driven from the elongation factor 1 promoter was used with either an empty CMV500 vector, or plasmids encoding acidic C/EBP (A-C/EBP) (Ménard et al., 2002), hemagglutinin (HA)-tagged dominant-negative (DN)-MEK (Mansour et al., 1994; Shalin et al., 2004), or C/EBPβ mutants in which threonine-217 was mutated to glutamate (the T217E phosphorylation mimic or CA-C/EBPβ) or in which threonine-217 and threonine-188 were mutated to alanine (C/EBPβ T/A phosphorylation mutant) (both were a kind gift from Dr. Peter Johnson, National Cancer Institute, Frederick, MD). The introduction of DNA by in utero electroporation was performed essentially as described previously (Ohtsuka et al., 1999; Barnabé-Heider et al., 2005). Briefly, CD1 pregnant mice were anesthetized with isoflurane and nitrous oxide, a midline incision was performed to access the embryos, and ∼1 μl of DNA solution was injected into the E14-E15 lateral ventricle. The solution contained the GFP plasmid (1.3 μg total) and the plasmid of interest or the empty vector (3.9 μg total) and 0.05% trypan blue as a tracer. After injection, electroporation was performed using a square electroporator CUY21 EDIT (TR-Tech, Tokyo, Japan) to deliver five 50 ms pulses of 50 V with 950 ms intervals per embryo. Embryos were reimplanted in utero, and 3-7 d later, the embryonic or neonatal brains were harvested, fixed in 4% paraformaldehyde (PFA) (Sigma, St. Louis, MO) at 4°C overnight, cryoprotected in 30% sucrose at 4°C overnight, and embedded in OCT compound. The brains were kept at -80°C until cryosectioned (16 μm) and analyzed.
Immunocytochemistry. For immunocytochemistry of cultured cells, cells were washed with HEPES-buffered saline (HBS) and fixed with 4% PFA for 15 min, permeabilized with 0.2% NP-40 (Roche) in HBS, and blocked with buffer containing 6% normal goat serum (NGS) (Jackson ImmunoResearch, West Grove, PA) and 0.5% bovine serum albumin (BSA) (Jackson ImmunoResearch) for 1-2 h at room temperature. Cells were then incubated with primary antibodies in HBS containing 3% NGS and 0.25% BSA at 4°C overnight. After washing with HBS, cells were incubated with secondary antibodies prepared in HBS containing 3% NGS and 0.25% BSA at room temperature for 1 h. Samples were then washed with HBS, counterstained with Hoechst 33258 (1:2000; Sigma) for 2 min, and mounted with GelTol (Fisher Scientific, Houston, TX). For immunocytochemistry of tissue sections, sections were warmed and washed, postfixed with 4% PFA for 10-15 min, blocked, and permeabilized with 10% BSA and 0.3% Triton X-100. The M.O.M. blocking kit (Vector Laboratories, Burlingame, CA) was then used according to the manufacturer's protocol. Sections were incubated with primary antibodies at 4°C overnight, washed with PBS, and incubated with secondary antibodies at room temperature for 1 h. They were then counterstained with Hoechst 33258 for 2 min and mounted with GelTol. The primary antibodies used were mouse anti-GFP (1:1000; Invitrogen), rabbit anti-GFP (1:500; Chemicon, Temecula, CA), mouse anti-HA (1:400; Boehringer Mannheim, Mannheim, Germany), mouse anti-HuD (1:200; Invitrogen), mouse anti-Ki67 (1:200; PharMingen, Heidelberg, Germany), mouse anti-NeuN (1:200; Chemicon), rabbit anti-C/EBPβ (C-19) (1: 800; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-βIII-tubulin (1:500; Covance, Princeton, NJ), rabbit anti-GFAP (1:800; Chemicon), mouse anti-MAP2 (1:400; Sigma), rabbit anti-phospho-C/EBPβ (pC/EBPβ) (1:500; Cell Signaling Technology, Beverly, MA), mouse anti-nestin (1:400; Chemicon), mouse anti-S100β (1:1000; Sigma), and biotinylated anti-musashi (1:500; a kind gift from Dr. Hideyuki Okano, Keio University, Tokyo, Japan). The secondary antibodies used were indocarbocyanine (Cy3)-conjugated goat anti-mouse and anti-rabbit IgG (1:400; Jackson ImmunoResearch), FITC-conjugated anti-mouse and anti-rabbit IgG (1:200; Jackson ImmunoResearch), dichlorotriazinyl amino fluorescein-conjugated streptavidin (1: 2000; Jackson ImmunoResearch), and Cy3-conjugated streptavidin (1: 2000; Jackson ImmunoResearch).
Western blots and immunoprecipitations. For biochemical analysis, cells were washed with ice-cold HBSS and lysed directly in the dish with radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, pH 7.2, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1% Na deoxycholate, and 0.1% v/v SDS) supplemented with protease inhibitor mixture (Boehringer Mannheim) and 1.5 mm sodium vanadate. Lysates were scraped into Eppendorf tubes, rocked for 10 min at 4°C, and cleared by centrifugation. Protein concentration was determined using the bicinchoninic acid assay (Pierce, Rockford, IL) and BSA as a standard. Equal amounts of protein (40-50 μg) were boiled in sample buffer, separated by 10-15% SDS-PAGE gels, and transferred to 0.2 μm nitrocellulose membranes for 3 h at 0.75 A at 4°C. Membranes were blocked in 5% skim milk powder in TBS and 0.5% Tween 20 (TBS-T) for 2 h at room temperature and incubated overnight at 4°C with primary antibodies. The primary antibodies used were mouse anti-HA (1:1000; Boehringer Mannheim), rabbit anti-GFP (1:1000; Chemicon), rabbit anti-phosphorylated extracellular signal-regulated kinase (pErk) (1:5000; Promega, Madison, WI), rabbit anti-Erk (K-23) (1:5000; Santa Cruz Biotechnology), and rabbit anti-C/EBPβ (C-19) (1:500; Santa Cruz Biotechnology). After washing with TBS-T, membranes were incubated with secondary antibodies, HRP-conjugated goat anti-mouse or anti-rabbit (1:10,000; Boehringer Mannheim), in blocking solution for 2 h at room temperature. Detection was performed using the ECL chemiluminescence reagent (Amersham Biosciences, Arlington Heights, IL) and XAR x-ray films (Eastman Kodak, Rochester, NY).
For immunoprecipitation, cortical precursors [0, 2, and 4 d in vitro (DIV)] from CD1 mice were lysed as described above. Equal amounts of protein were incubated with 10 μl of the pan-C/EBP (Δ198) antibody (Santa Cruz Biotechnology) overnight at 4°C and incubated for 2 h with 30 μl of protein A-Sepharose (Sigma). The precipitated proteins were collected by centrifugation, washed three times with RIPA buffer, boiled with sample buffer, loaded on a 15% SDS-PAGE gel, and transferred to a nitrocellulose membrane. The membrane was blocked with 3% BSA in TBS-T and processed as detailed above using rabbit anti-pC/EBPβ (1: 500; Cell Signaling Technology) as the primary antibody.
Microscopy and confocal analysis. For quantitation of immunocytochemistry, >300 cells per condition were counted and analyzed. Digital image acquisition was performed with Northern Eclipse software (Empix, Mississauga, Ontario, Canada) using a Sony (Tokyo, Japan) XC-75CE CCD video camera. For quantitation of immunocytochemistry on tissue sections, brains were chosen for comparison that showed a similar anatomical distribution and level of GFP expression with regard to the total cortex. After sectioning, three to four brain sections at the same anatomical level per embryo were analyzed using a Zeiss (Oberkochen, Germany) Pascal confocal microscope and the manufacturer's software. A mean of four scans taken with a 40× objective were computed for each image. In all graphs, error bars indicate SEM, and the statistics were performed using either one-way ANOVA with Student-Newman-Keuls post hoc test or the Student's t test, as indicated in the figures.
Results
MEK activity is important for cortical neurogenesis in vivo
We demonstrated previously that the growth factor-regulated kinase MEK is not required for the survival or proliferation of cultured cortical precursors but is instead essential for those precursors to generate neurons (Ménard et al., 2002; Barnabé-Heider and Miller, 2003). To ask whether MEK activation was also required for cortical neurogenesis in vivo, we performed in utero electroporation with an HA-tagged DN-MEK (Mansour et al., 1994; Shalin et al., 2004). Plasmids encoding a nuclear-targeted GFP and DN-MEK or empty vector were coelectroporated after injection into the E15 lateral ventricles, and embryos were reimplanted and analyzed 1-3 d later. In some cases, a plasmid encoding a nuclear-targeted β-galactosidase (LacZ) was also used as a control. One day after electroporation, only cells in the ventricular zone/subventricular zone (VZ/SVZ) of the telencephalon were transfected (data not shown), as reported previously (Ohtsuka et al., 1999; Barnabé-Heider et al., 2005). Of these transfected cells, ∼85% expressed the proliferation marker Ki67, indicating that they were dividing precursors. These electroporated cells maintained expression of the transfected construct until at least postnatal day 7 (P7) (data not shown). Importantly, immunocytochemistry revealed that, at all time points analyzed, the vast majority of cells were positive for both GFP and the HA-tagged DN-MEK in cortices coelectroporated with both of these plasmids (Fig. 1a) (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). Biochemical confirmation that these plasmids were appropriately expressed was obtained by performing Western blot analysis on electroporated cortices, where either a GFP-expressing plasmid or GFP-plus-HA-tagged DN-MEK-expressing plasmids were injected into one lateral ventricle (Fig. 1b).
Figure 1.
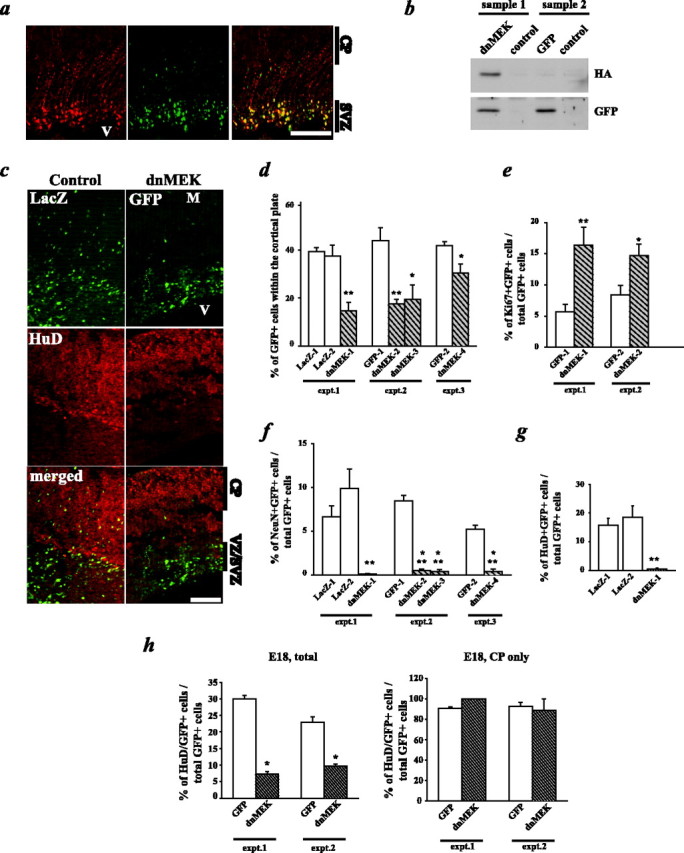
DN-MEK inhibits cortical neurogenesis in vivo. In utero electroporations of precursor cells in the E14-E15 telencephalic VZ/SVZ are shown, with plasmids encoding GFP and HA-tagged DN-MEK, or GFP and the empty vector or a LacZ plasmid. Three days after electroporation, coronal sections of the embryonic telencephalon were analyzed by immunocytochemistry for GFP or LacZ and several markers. a, Photomicrographs of a section from an E18 brain that was cotransfected with plasmids encoding GFP and HA-tagged DN-MEK and immunostained for GFP (green) and HA (red). The right panel shows the merged image. V, Ventricle; CP, cortical plate. Note that the majority of the GFP-positive cells also express HA-tagged DN-MEK. b, Western blot analysis of cortices electroporated with GFP and HA-tagged DN-MEK (sample 1) or with GFP and the empty vector (sample 2). The nonelectroporated hemisphere from the same animal was run as a control for each sample. The membrane was probed for the HA epitope and GFP. c, Photomicrographs taken on a light microscope of sections spanning the ventricle (V) to the meninges (M) through E18 cortices of embryos transfected with the LacZ vector (left 3 panels) or with the DN-MEK plasmid (right 3 panels). In each set of three panels, the top shows the GFP or LacZ immunostaining, the middle shows the neuron-specific HuD immunostaining, and the bottom shows the merged image. d, Quantitation of sections similar to those shown in c for the percentage of GFP- or LacZ-positive cells present within the cortical plate. Transfected cells within the cortex of three littermate pairs, three to four sections per embryo, were counted to obtain these numbers. *p < 0.05; **p < 0.01, relative to control-transfected sections (ANOVA or Student's t test). e, Quantitation of confocal microscopic analysis of GFP and the proliferation marker Ki67. Transfected cells within the cortex of three littermate pairs, four sections per embryo, were counted to obtain these numbers. *p < 0.05; **p < 0.01, relative to control-transfected sections (ANOVA or Student's t test). f, Quantitation of confocal microscopic analysis of GFP and the mature neuron-specific marker NeuN, as described in e. **p < 0.01, ***p < 0.001, relative to control-transfected sections (ANOVA or Student's t test). g, Quantitation of confocal microscopic analysis of GFP and the early neuron-specific marker HuD expression, as described in e. **p < 0.01, relative to control-transfected sections (ANOVA or Student's t test). h, Quantitation of confocal microscopic analysis of GFP and HuD expression as in g, showing the percentage of HuD-positive transfected cells through the entire cortex (left; E18, total) versus the percentage in the cortical plate (right; E18, CP only) in the same sections. Scale bars: a, 200 μm; c, 100 μm. Error bars indicate SEM.
Having established the efficacy of this procedure, we then asked whether DN-MEK inhibited neurogenesis. Analysis of control electroporated brains revealed that at E18, 3 d after electroporation, many GFP- or LacZ-expressing control cells were still in the VZ/SVZ, but many had also migrated into the cortical plate, which contains newly born neurons (Fig. 1c, left). Quantitation demonstrated that ∼40-50% of the total control transfected cells had migrated into the cortical plate region (Fig. 1d). In contrast, many more DN-MEK-expressing cells remained in the VZ/SVZ, with only 15-30% migrating into the cortical plate region (Fig. 1c,d). To ask whether this lack of migration indicated that DN-MEK had inhibited the genesis of neurons, we performed immunocytochemistry for two neuronal markers, HuD, an early marker (Fig. 1c) (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material), and NeuN, a somewhat later marker. Confocal microscopy and quantitation revealed that ∼20-30% of the GFP-expressing control cells expressed HuD, and ∼10% NeuN (Fig. 1f-h). In contrast, 1-10% of the cells expressing DN-MEK were positive for HuD, and <1% for NeuN (Fig. 1f-h). More detailed spatial analysis revealed that, of the transfected cells that were in the cortical plate, >80% were positive for HuD, and this percentage was similar for both control and DN-MEK-transfected populations (Fig. 1h), indicating that some DN-MEK-expressing cells do become postmitotic neurons and that it is these cells that migrate to the cortical plate. In contrast, no HuD-positive cells were observed in the VZ/SVZ, nor were any GFAP-positive astrocytes observed (data not shown). Instead, many of the DN-MEK-expressing cells in the VZ/SVZ continued to proliferate, as indicated by immunocytochemical analysis for Ki67; ∼15% of the total DN-MEK-expressing cells were Ki67-positive, compared with 5-8% of control cells (Fig. 1e). Thus, inhibition of MEK inhibits neurogenesis and inappropriately maintains many cortical precursors as dividing cells within the VZ/SVZ.
C/EBPβ is phosphorylated by the MEK-ERK pathway during neurogenesis
One of the targets of the MEK pathway is C/EBPβ, which is phosphorylated at threonine-188 by ERK and threonine-217 by ribosomal S6 kinase (Rsk), both of which are kinases downstream of MEK (Davis, 1995). To ask whether C/EBPβ phosphorylation by the MEK pathway occurred coincident with neurogenesis, we used an antibody specific for C/EBPβ phosphorylated at the ERK site, threonine-188. Initially, we examined cultured cortical precursors isolated from E12.5 mouse cortex and plated in the presence of FGF2. We showed previously that, at the time of plating, these cells are dividing, nestin-positive precursors that, for the first 5 d, generate only neurons but then also generate astrocytes and oligodendrocytes (Slack et al., 1998; Toma et al., 2000; Ménard et al., 2002; Barnabé-Heider and Miller, 2003; Barnabé-Heider et al., 2005). Western blot analysis of pan-C/EBP immunoprecipitations showed that phospho-C/EBPβ could be detected in freshly isolated cortical precursors, as well as in later cortical cultures that contained both precursors and neurons (Fig. 2d). Double-label immunocytochemical analysis of precursors cultured for 5 d revealed that a subpopulation of nestin-positive cells were immunoreactive for phospho-C/EBPβ, as were almost all MAP2-positive newly born neurons (Fig. 2a). In contrast, phospho-C/EBPβ was not detectably expressed in cells that were positive for S100β, a marker of non-neuronal precursors and young astrocytes. To confirm that phospho-C/EBPβ was downstream of MEK in these cells, we transfected precursor cultures with GFP alone or GFP plus DN-MEK and, 5 d later, performed immunocytochemistry. This analysis revealed that DN-MEK significantly reduced the percentage of transfected cells expressing detectable levels of phospho-C/EBPβ (Fig. 2e). Thus, phospho-C/EBPβ levels are highest in a subset of neuronal precursors and in newly born neurons, and this C/EBP phosphorylation is downstream of MEK, consistent with a potential role in neurogenesis. To ask whether a similar pattern of C/EBPβ phosphorylation was seen in vivo, we cryosectioned E18 brains and performed double-label immunocytochemistry for phospho-C/EBPβ and the neuronal marker HuD, or the precursor markers nestin (all precursors) or NG2 (oligodendrocyte-biased precursors). This analysis revealed that a subpopulation of nestin-positive precursors in the VZ/SVZ were positive for phospho-C/EBPβ (Fig. 2c), as were many HuD-positive neurons in the cortical plate (Fig. 2b), whereas the large majority of NG2-positive cells did not express phospho-C/EBPβ (data not shown).
Figure 2.

C/EBP is phosphorylated via the MEK-Erk pathway in newly born neurons in culture and in vivo. a, Double-label immunocytochemical analysis of cortical precursor cells after 3 DIV for pC/EBPβ (red) and MAP2, nestin, or S100β (green). Arrows represent cells that coexpress both markers and arrowheads represent cells that are positive for pC/EBPβ only. b, c, Double-label immunocytochemistry on sections of the E18 cortex for pC/EBPβ (red) and HuD (b) or nestin (c) (both in green). In b and c, the right panel is a higher-magnification micrograph of sections similar to those in the left panel. Arrows represent cells that coexpress both markers and arrowheads represent cells that stained for pC/EBPβ only. d, Western blot analysis for pC/EBPβ in cortical precursor cell lysates that were immunoprecipitated with an antibody specific for all C/EBP family members. Cortical precursors were analyzed immediately after plating (0 DIV) and after 2 and 4 d of culture (2 DIV, 4 DIV). e, Quantitation of the percentage of transfected, phospho-C/EBPβ-positive cells in cortical precursor cultures transfected with GFP alone or with GFP plus DN-MEK. *p < 0.05; Student's t test. Scale bar, 100 μm. Error bars indicate SEM.
C/EBPβ phosphorylation is essential for neurogenesis in culture
We showed previously that the C/EBP family is essential for cortical neurogenesis in culture using a dominant-negative A-C/EBP in which the basic DNA binding domain is mutated to acidic residues so that it, and any wild-type C/EBP with which it dimerizes, can no longer bind to DNA (Ménard et al., 2002). Moreover, we showed that C/EBPβ mutated to glutamate at threonine-217 (thereby acting as a mimic of C/EBPβ phosphorylated at the Rsk site) was sufficient to enhance neurogenesis in culture (Ménard et al., 2002) (Fig. 3d). To ask whether C/EBP phosphorylation was also necessary for C/EBP neurogenic actions, we performed the converse experiment, using a C/EBPβ mutant in which both threonine-188 and threonine-217 were mutated to alanine and could thus no longer be phosphorylated in response to MEK activation (C/EBP T/A mutant) (Fig. 3a,b). E12.5 cortical precursors were cotransfected in culture with GFP and the C/EBP T/A mutant on the day of plating and, 4 d later, were analyzed immunocytochemically for neuron-specific βIII-tubulin (Fig. 3c). Quantitation demonstrated that the C/EBPβ T/A mutant decreased the number of neurons that were generated from cortical precursors by approximately twofold (Fig. 3d). Thus, C/EBP phosphorylation via the MEK pathway is both necessary and sufficient for neurogenesis in cultured cortical precursors.
Figure 3.

Expression of a C/EBP phosphorylation mutant at the Erk and Rsk sites inhibits neurogenesis in culture. a, Schematic of C/EBPβ wild-type protein. Two activation domains (ADI and ADII) are situated to the N terminus (NH2), whereas the DNA-binding domain (DBD) and the leucine zipper dimerization domain (LZ) are located at the C terminus (COOH). The two phosphorylation sites investigated in this study, threonine-188 (T188; a substrate for ERK) and threonine-217 (T217; a substrate for Rsk), are also shown. b, Western blot analysis for the different C/EBPβ mutants expressed in HEK 293 cells showing expression of proteins of the appropriate sizes. c, Double-label immunocytochemical analysis for GFP (green) and βIII-tubulin (red) at 4 DIV on precursor cells that were transfected with either GFP plus the empty vector or GFP and the C/EBPβ T/A phosphorylation mutant. Arrows represent cells that coexpress both markers and arrowheads represent cells that were transfected but that do not express βIII-tubulin. Cells were counterstained with Hoechst (blue) to show all of the nuclei in the field. d, Quantitation of data similar to that shown in c for precursor cells transfected with GFP plus empty vector, GFP and the C/EBPβ T/A mutant, or GFP and the CA-C/EBPβ phosphorylation mimic, cultured for 4 DIV and immunostained for βIII-tubulin. *p < 0.01, relative to control-transfected cultures (ANOVA or Student's t test). e, Quantitation of data similar to that shown in c for precursor cells stimulated with CNTF after transfection with plasmids encoding GFP plus empty vector, GFP plus the C/EBP T/A mutant, or GFP plus A-C/EBP. Cells were immunostained for GFAP after 4 DIV. *p < 0.01, relative to control-transfected cultures (ANOVA or Student's t test). Scale bar, 100 μm. Error bars indicate SEM.
In addition to regulating neurogenesis, when the C/EBP family is completely inhibited using A-C/EBP, precocious cytokine-induced gliogenesis is enhanced (Ménard et al., 2002) (Fig. 3e). To ask whether phosphorylation of C/EBPs plays any role in this effect on gliogenesis, we transfected the C/EBPβ T/A mutant into cortical precursors and exposed them to CNTF. In contrast to A-C/EBP, this phosphorylation mutant had no effect on astrocyte formation, as monitored by immunocytochemistry for the astrocyte protein GFAP (Fig. 3e). These data argue that phosphorylation by the MEK pathway is not important for C/EBP-mediated inhibition of astrocyte formation, consistent with our previous work showing that inhibition of MEK itself also does not affect cytokine-induced gliogenesis (Ménard et al., 2002; Barnabé-Heider and Miller, 2003; Barnabé-Heider et al., 2005). Thus, although C/EBPs promote neurogenesis and inhibit gliogenesis, the mechanisms by which they regulate these two events are dissociable.
C/EBP phosphorylation by the MEK pathway is essential for neurogenesis in vivo
To ask whether C/EBPs were essential for cortical neurogenesis in vivo, as suggested by our culture studies, we performed in utero electroporation experiments. The lateral ventricles of E15 embryos were injected with plasmids encoding A-C/EBP and GFP, electroporation was performed and the brains were harvested 3 d later. Analysis of sections through the cortex of these brains demonstrated that the vast majority of the cells transfected with A-C/EBP remained in the VZ/SVZ and did not migrate to the cortical plate, results similar to those seen with DN-MEK (Fig. 1d). Specifically, in control brains, 40-50% of the GFP-positive cells had migrated into the cortical plate over the 3 d period, but only ∼10% of the GFP-positive cells coexpressing A-C/EBP were in the cortical plate region (Fig. 4d). Immunocytochemistry for the neuronal markers NeuN and HuD, and quantitation by confocal microscopy (Fig. 4c,e,f) revealed that this deficit in migration was attributable to the fact that A-C/EBP inhibited cortical neurogenesis; 20-30% and 5-15% of the control cells expressed HuD and NeuN, respectively, whereas only ∼5% and 1-2% of the A-C/EBP-expressing cells expressed these two markers. Moreover, as seen for DN-MEK, none of the A-C/EBP-expressing cells in the VZ/SVZ expressed NeuN or HuD, whereas transfected cells in the cortical plate region were almost all positive for HuD, whether they expressed GFP alone or GFP plus A-C/EBP. Thus, C/EBP activity is essential for cortical neurogenesis in vivo, and when it is inhibited, cells remain in the VZ/SVZ and do not express neuronal genes.
Figure 4.
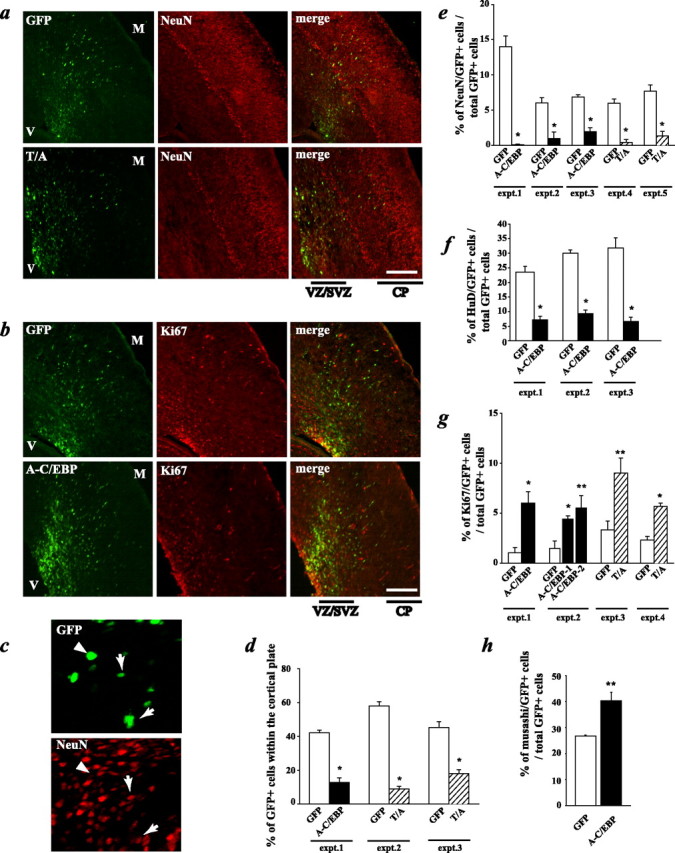
Inhibition of C/EBPs decreases neurogenesis and maintains cells as proliferating precursors in vivo. In utero electroporation of precursor cells in the E14-E15 telencephalic VZ/SVZ is shown, with plasmids encoding GFP and A-C/EBP, GFP and the C/EBPβ T/A phosphorylation mutant (T/A), or with GFP and the empty vector. Three days after electroporation, coronal sections of the embryonic telencephalon were analyzed by immunocytochemistry for GFP and several markers. a, Photomicrographs taken on a light microscope of sections spanning the ventricle (V) to the meninges (M) through E18 cortices of embryos cotransfected with GFP and the empty vector (top 3 panels) or with GFP and the C/EBPβ T/A mutant (bottom 3 panels). In each set of three panels, the left shows the GFP immunostaining, the middle shows the neuron-specific NeuN immunostaining, and the right shows the merged image. b, Photomicrographs of E18 cortex sections from brains that were cotransfected with GFP and the empty vector (top) or GFP and A-C/EBP (bottom) and immunostained for the proliferation marker Ki67. c, Confocal image of colocalization for NeuN (in red) and GFP (in green) in a section through the cortical plate region. Arrows represent cells that coexpress both markers, and arrowheads represent cells that stain for GFP only. d, Quantitation of sections similar to those shown in a for the percentage of GFP-positive cells present in the cortical plate region. Transfected cells within the cortex of three littermate pairs, three to four sections per embryo, were counted to obtain these numbers. *p < 0.05, relative to control-transfected sections (ANOVA or Student's t test). e-h, Quantitation of confocal microscopic analysis of GFP and the mature neuronal marker NeuN (e), GFP and the early neuronal marker HuD (f), GFP and the proliferation marker Ki67 (g), and GFP and the proliferation marker musashi (h) in cortical sections similar to those shown in a-c. Transfected cells within the cortex of littermate pairs, four sections per embryo, were counted to obtain these numbers. *p < 0.05, **p < 0.01, relative to control-transfected sections (ANOVA or Student's t test). Scale bars, 100 μm. Error bars indicate SEM.
To ask whether, as seen for DN-MEK, the A-C/EBP-expressing cells that did not become neurons were maintained as proliferating precursors, we performed immunocytochemistry for the proliferation marker Ki67 and for the neural precursor marker musashi (Kaneko et al., 2000). Quantitation by confocal microscopy revealed that a significantly greater percentage of A-C/EBP-expressing cells versus control cells were Ki67 positive (Fig. 4b,g) (supplemental Fig. 1d, available at www.jneurosci.org as supplemental material). Moreover, ∼40% of the A-C/EBP-expressing cells were positive for musashi, whereas only ∼25-30% of control cells expressed this precursor marker (Fig. 4h). Analysis of adjacent sections for GFAP revealed that, as seen in culture (Ménard et al., 2002), none of the A-C/EBP-expressing cells became astrocytes (data not shown), presumably because of a lack of gliogenic signals in the neural environment at this time point (Barnabé-Heider et al., 2005). Thus, inhibition of the C/EBP family inhibits neurogenesis and maintains cells in the VZ/SVZ as cycling precursor cells.
To ask whether MEK pathway-mediated phosphorylation of C/EBPs was important for the neurogenic actions of these transcription factors, we performed similar in utero electroporation experiments using the C/EBPβ T/A phosphorylation mutant. E15 cortices were electroporated either with plasmids encoding the C/EBPβ T/A mutant plus GFP or with empty vector plus GFP, and embryos were analyzed 3 d later. These experiments revealed that the C/EBPβ T/A mutant gave results similar to those seen with A-C/EBP, with regard to all of the parameters measured. Specifically, the vast majority of the C/EBPβ T/A mutant-expressing cells remained in the VZ/SVZ (Fig. 4d), where many of them were proliferating, as monitored by expression of Ki67 (Fig. 4g). In addition, significantly fewer of the C/EBPβ T/A mutant-expressing cells were positive for NeuN, arguing that this mutant prevented precursors from adopting a neuronal phenotype (Fig. 4a,e) (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material). Finally, as seen for A-C/EBP, none of the C/EBP T/A mutant-expressing cells was positive for GFAP at this time point (data not shown). Thus, MEK-mediated phosphorylation of C/EBPs is essential to promote cortical neurogenesis in vivo as it is in culture.
Phosphorylation of C/EBPβ is sufficient to promote neurogenesis at early embryonic time points in vivo
Because our data indicated that C/EBP phosphorylation was essential for neurogenesis in vivo as it was in culture, we asked whether enhanced C/EBP phosphorylation was sufficient to promote neurogenesis in vivo. Initially, we performed in utero electroporations of E15 mouse embryos, cotransfecting GFP plus the phosphorylation mimic CA-C/EBP or GFP plus the empty vector control plasmid. Embryos were analyzed 3 d later, and cortical sections were examined by immunocytochemistry. Initially, we noted that there was no difference in the migration of control versus CA-C/EBP-expressing cells; in both cases, ∼50% of the GFP-positive cells were in the cortical plate region (Fig. 5e). Confocal microscopic quantitation of sections immunostained for NeuN and Ki67 also revealed no differences between control and CA-C/EBP-expressing cells (Fig. 5a-d) (supplemental Fig. 1c,d, available at www.jneurosci.org as supplemental material), arguing that, at this developmental time point, enhanced C/EBP phosphorylation did not promote neurogenesis.
Figure 5.
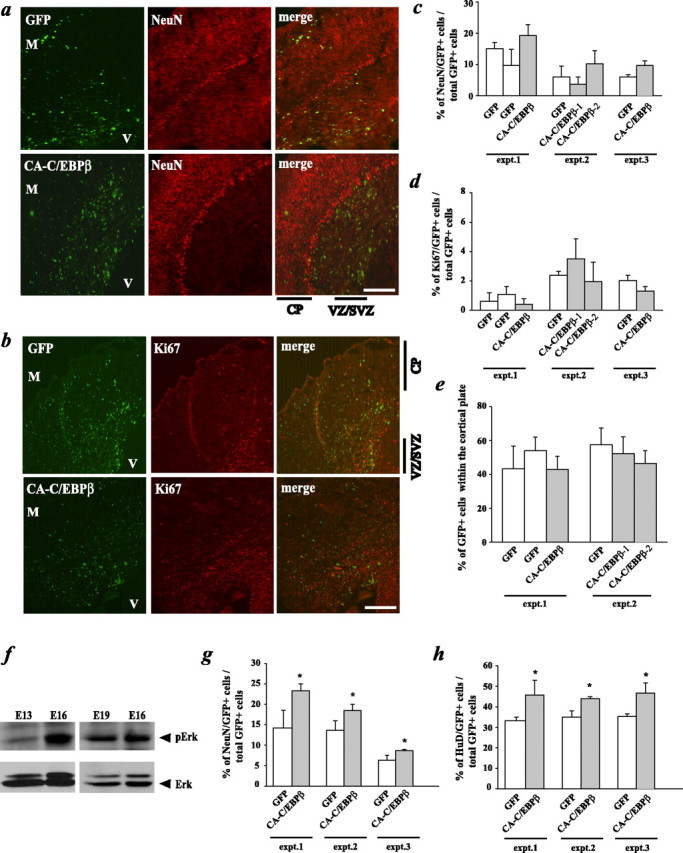
A C/EBPβ phosphorylation mimic promotes cortical neurogenesis in vivo at E13-E16 but not at E15-E18 when endogenous ERK activation is already maximal. a-e, In utero electroporation of precursor cells in the E15 cortex with plasmids encoding either GFP and the C/EBPβ phosphorylation mimic (CA-C/EBPβ) or GFP and the empty vector. Three days after electroporation, coronal sections of the embryonic telencephalon were analyzed by immunocytochemistry for GFP and several markers. a, b, Photomicrographs taken on a light microscope of sections spanning the ventricle (V) to the meninges (M) through E18 cortices of embryos cotransfected with GFP and the empty vector (top 3 panels) or with GFP and CA-C/EBPβ (bottom 3 panels). Sections were immunostained for GFP and NeuN (a) or GFP and Ki67 (b). c, d, Quantitation of confocal microscopic analysis of GFP and NeuN (c) or GFP and Ki67 (d) in sections similar to those shown in a and b. Transfected cells within the cortex of littermate pairs, four sections per embryo, were counted to obtain these numbers. e, Quantitation of sections similar to those shown in a for the percentage of GFP-positive cells present within the cortical plate region. Transfected cells within the SVZ of two littermate pairs, three to four sections per embryo, were counted to obtain these numbers. f, Western blot analysis for pERKs in equal amounts of protein isolated from the cortex at embryonic days 13-19. The blots were reprobed for total ERKs as a loading control. g, h, In utero electroporation of precursor cells in the E13 cortex with plasmids as described for a-e. Sections were analyzed at E16 by immunostaining and quantitated by confocal microscopy for GFP and NeuN (g) or GFP and HuD (h). Transfected cells within the cortex of littermate pairs, four sections per embryo, were counted to obtain these numbers. *p < 0.05, relative to control-transfected sections (ANOVA or Student's t test). Scale bars, 100 μm. CP, Cortical plate. Error bars indicate SEM.
One potential explanation for these results is that during midgestation the environment is maximally proneurogenic, the MEK-C/EBP pathway is already fully activated, and thus, enhanced phosphorylation of C/EBP cannot promote neurogenesis further. To ask whether this was the case, we performed Western blot analysis for activated, phosphorylated ERKs, which are downstream of MEK and up-stream of the C/EBPs. This analysis (Fig. 5f) revealed that phospho-ERK levels were robustly increased between E13 and E16 and were maintained until E19. We therefore predicted that enhanced C/EBP phosphorylation might promote neurogenesis between E13 and E16, before maximal activation of the MEK pathway occurred. To test this prediction, we electroporated the same constructs into the cortices of mouse embryos at E13 and analyzed them 3 d later. Analysis by immunocytochemistry and confocal microscopy revealed a modest but significant increase in the number of CA-C/EBP-expressing cells that were positive for the neuronal markers NeuN and HuD relative to controls (Fig. 5g,h). Thus, phosphorylation of C/EBPs is sufficient to enhance neurogenesis at early but not later embryonic time points, likely because activation of the MEK-C/EBP pathway is already maximal at these later time points.
C/EBPs inhibit gliogenesis in vivo as they do in culture
These in vivo data demonstrated that inhibition of the MEK-C/EBP pathway maintained increased numbers of undifferentiated precursors within the VZ/SVZ from E15 to E18. To ask about the ultimate fate of these cells, we performed similar in utero electroporation experiments with A-C/EBP at E15 and analyzed the cortex at P3. At this stage, ∼75% of the transfected cells were present in the cortical plate region, and this number was similar whether cells were transfected with A-C/EBP plus GFP or with GFP alone. Of these, >95% were neurons, as indicated by expression of HuD (Fig. 6a). These data therefore suggest that A-C/EBP delayed but apparently did not permanently inhibit precursors from becoming neurons, although the transient nature of the transfections also makes it possible that A-C/EBP levels had decreased sufficiently by these later time points to allow neurogenic C/EBP-mediated transcription to occur. If this interpretation is correct, then one prediction is that the delayed, later-differentiating neurons would be more superficially located in the neonatal cortex. To test this prediction, we analyzed the localization of GFP-positive neurons that had differentiated from precursors transfected with or without A-C/EBP at E15. In control transfected brains, by P3, all of the GFP-positive cells in the cortical plate were found within cortical layer II/III (Fig. 6b, left), with ∼65% of the transfected neurons located in the upper one-half of this layer (Fig. 6c). In contrast, in A-C/EBP-transfected brains, almost all of the GFP-positive neurons were found within the upper one-half of layer II/III (Fig. 6b, right; c), indicating that they were, as predicted, more superficially located.
Figure 6.

Inhibition of C/EBPs increases gliogenesis after birth. In utero electroporation of precursor cells in the E15 cortex is shown, with plasmids encoding GFP and A-C/EBP, GFP and the C/EBPβ T/A mutant, or GFP and the empty vector. Animals were analyzed at postnatal day 3, and coronal sections through the cortex were examined by immunocytochemistry for GFP and the neuronal marker HuD (a) or the glial marker GFAP (d, e). a, Quantitation of confocal microscopic analysis of GFP and HuD expression in the cortical plate region. Transfected cells within the cortex of nonlittermates, four sections per embryo, were counted to obtain these numbers. The two groups were not significantly different. b, Photomicrographs of coronal sections through the P3 neocortex of brains that were transfected with GFP (green) plus control plasmid (left) or A-C/EBP plasmid (right) and counterstained with Hoechst (blue) to show cell nuclei. The cortical layers are denoted to the side of each panel. Layer II/III is demarcated with hatched lines, and the solid line represents the boundary between the upper and lower halves of this layer. c, Percentage of GFP-positive cells in the upper one-half of neocortical layer II/III in P3 brains that were transfected at E15 with GFP plus control plasmid or A-C/EBP plasmid, quantitated from sections similar to those shown in b. n = 3 control and 3 A-C/EBP-transfected brains. *p < 0.05. d, Confocal image of colocalization for GFP (green) and GFAP (red). Arrows represent cells that coexpress both markers and the arrowhead represents a cell that stained for GFP only. e, Quantitation of confocal microscopic analysis of GFP and GFAP expression in the VZ/SVZ. Transfected cells within the cortex of nonlittermates, four sections per embryo, were counted to obtain these numbers. *p < 0.05, relative to control-transfected sections (ANOVA or Student's t test). Scale bars: b, 100 μm; d, 10 μm. Error bars indicate SEM.
This analysis also revealed that, at P3, none of the transfected cells in the VZ/SVZ expressed HuD or NeuN. We therefore asked whether any of these cells were astrocytes, because (1) our previous culture data indicated that inhibition of C/EBPs enhanced astrocyte formation in response to a gliogenic, cytokine environment (Ménard et al., 2002), and (2) the early neonatal cortex is a cytokine-rich, gliogenic environment (Morrow et al., 2001; Barnabé-Heider et al., 2005). Immunocytochemical analysis for GFAP revealed that a significantly higher percentage of A-C/EBP-transfected cells expressed GFAP relative to their control transfected counterparts (Fig. 6d,e), an average of 25 and 43% of control versus A-C/EBP-transfected cells, respectively. Interestingly, transfection of the T/A C/EBPβ mutant had no effect on the percentage of VZ/SVZ cells that became astrocytes (Fig. 6e), findings similar to what we had observed in culture (Fig. 3e). These data therefore support the hypothesis that C/EBP activation promotes neurogenesis at the same time that it inhibits astrocyte formation in response to a gliogenic environment, and that the neurogenic and gliogenic mechanisms are dissociable.
Discussion
The data presented here support three major conclusions. First, activation of the growth factor-regulated MEK pathway is essential for cortical precursors to generate neurons in vivo, and when this activation is inhibited, many precursors remain in the VZ/SVZ in an undifferentiated state. Second, MEK pathway activation leads to C/EBP phosphorylation in a subset of precursors, and this phosphorylation is necessary and sufficient to promote neurogenesis. Finally, in the absence of C/EBP family activation, precursors initially remain in the VZ/SVZ as cycling precursors but then have an enhanced propensity to generate astrocytes in the late embryonic/early neonatal environment, presumably when they are exposed to extrinsic, gliogenic cues. Thus, growth factor-mediated activation of the MEK-C/EBP pathway promotes the genesis of neurons from cortical precursors, and, at the same time, C/EBP activation apparently inhibits the genesis of astrocytes when the same precursors are exposed to a gliogenic environment. The C/EBPs, therefore, act as growth factor-regulated switches that directly couple alterations in the neural environment to cell-fate choices during embryogenesis.
Our experiments demonstrate that, even at E18, some precursors “escape” the effects of inhibiting MEK or C/EBPs, commit to being neurons, and migrate to the cortical plate, and that this escape is much more pronounced by P3. One explanation for this finding is that, at E15, we are transfecting multiple populations of VZ/SVZ precursors, including biased neuronal progenitors that are undergoing terminal mitosis at the time of transfection and thus no longer require MEK-C/EBP pathway activation. A second explanation is a technical one; these transfections are transient, and levels of expression and inhibition differ from cell to cell. Thus, it is likely that MEK-C/EBP pathway activity is still sufficient in some transfected cells to promote neurogenesis. This technical limitation would become more important at later time points after transfection. Finally, other pathways may be able to promote neurogenesis, albeit perhaps less efficiently, in the absence of MEK-C/EBP pathway activation. Interestingly, regardless of the explanation, our data demonstrate that these delayed precursors then generate neurons that are appropriately more superficially localized. These data are reminiscent of similar findings with cortical precursors that were transiently inhibited from differentiating by Notch pathway activation (Mizutani and Saito, 2005), and suggest that these delayed precursors can still ultimately respond appropriately to developmental cues.
We therefore propose a model in which growth factors in the extrinsic environment play a key role in dictating the timing of neurogenesis versus gliogenesis in the embryonic cortex. Specifically, we propose that early embryonic cortical precursors are biased to become neurons by expression of positively acting bHLHs (Nieto et al., 2001; Sun et al., 2001) but that these biased precursors do not generate neurons until tyrosine kinase receptor ligands in the neural environment signal via the MEK-C/EBP pathway. At the same time, the C/EBPs inhibit cortical precursors from generating astrocytes in response to low levels of embryonic neural cytokines such as neuropoietin and cardiotrophin-like cytokine (Uemura et al., 2002; Derouet et al., 2004). Such an inhibitory mechanism is necessary, because early cortical precursors are competent to generate astrocytes if cytokine levels in their environment are sufficiently high (Barnabé-Heider et al., 2005). Later in embryogenesis, the cortical environment alters to become increasingly gliogenic (Morrow et al., 2001), in large part because of synthesis of the cytokine cardiotrophin-1 by newly born cortical neurons (Barnabé-Heider et al., 2005). Over this same time period, cortical precursors become sensitized to cytokines via a variety of mechanisms, including increased expression of epidermal growth factor (EGF) receptor (Burrows et al., 1997; Sun et al., 2005) and/or components of the gliogenic Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (He et al., 2005), and demethylation of astrocyte-specific genes (Takizawa et al., 2001). Ultimately, when neuron-derived cytokine levels become sufficiently high, these sensitized precursors respond by generating astrocytes instead of neurons. Interestingly, gliogenic signaling via the gp130 cytokine receptor and STAT3 not only promotes the genesis of astrocytes but also inhibits the genesis of neurons (Bonni et al., 1997; Barnabé-Heider et al., 2005), thereby ensuring the efficiency of the neurogenic to gliogenic switch. Thus, although intrinsic alterations in precursor cell bias and responsiveness are clearly important, we propose that it is growth factors in the developing environment that ultimately dictate when and how many of these different cell types are generated.
What are the growth factors that activate the MEK-C/EBP pathway and thereby promote neurogenesis? Previous work has identified a number of candidate receptor tyrosine kinase ligands, including FGF2, neurotrophins, heparin-binding (HB)-EGF, PDGF, and IGF1. Specifically, FGF2 is mitogenic for cortical precursors (Lukaszewicz et al., 2002) and is essential for normal neurogenesis (Raballo et al., 2000). Cortical precursors express the neurotrophin receptors tyrosine receptor kinase B (TrkB) and TrkC, and ablation of TrkB or the neurotrophins BDNF and/or neurotrophin-3 (NT-3) perturb the development of these precursors (Barnabé-Heider and Miller, 2003; Medina et al., 2004). HB-EGF is expressed in the embryonic VZ/SVZ (Nakagawa et al., 1998) and promotes neurogenesis in the adult CNS (Jin et al., 2003). Cultured cortical precursors express PDGF receptor and respond to PDGF with enhanced neurogenesis (Johe et al., 1996; Williams et al., 1997; Park et al., 1999). Finally, overexpression of IGF1 from the nestin promoter causes increased proliferation and neuron number in the embryonic cortex (Popken et al., 2004). What are the sources of these growth factors during neurogenesis? Cortical precursors of the VZ/SVZ themselves express BDNF, NT-3 (Maisonpierre et al., 1990; Fukumitsu et al., 1998), FGF2 (Raballo et al., 2000), and HB-EGF (Nakagawa et al., 1998). Intriguingly, endothelial cells also express BDNF (Kim et al., 2004), FGF2 (Albuquerque et al., 1998), PDGF, and HB-EGF (Arkonac et al., 1998), and embryonic endothelial cells have recently been shown to promote self-renewal and neurogenesis in neural stem cells (Shen et al., 2004). Thus, it is likely that cortical precursors are exposed to diverse ligands from multiple sources, and signaling via these growth factors could converge on the MEK-C/EBP pathway to ultimately regulate the genesis of neurons. It is also likely that these same growth factors regulate survival and proliferation of cortical precursors via other pathways such as the phosphatidylinositol 3-kinase-Akt pathway (Barnabé-Heider and Miller, 2003; Sinor and Lillien, 2004), and thus, the precise repertoire of responses is likely defined by the relative levels and/or time course of activation of these different pathways.
How do the C/EBPs promote neurogenesis? Precedent for the C/EBP family of transcription factors acting to regulate cellular differentiation in response to growth factors derives from studies on a number of developing systems, most notably during hematopoiesis (Yamanaka et al., 1998) and adipogenesis (Lane et al., 1999). In these systems, various members of the C/EBP family act in concert with cell type-specific determination signals to promote or repress transcription of genes essential for terminal differentiation. We propose a similar mechanism for neurogenesis in which the C/EBPs collaborate with proneurogenic bHLHs to ultimately promote neurogenesis by transcription of neuron-specific genes such as Tα1 α-tubulin (Ménard et al., 2002). Interactions between these two families of transcription factors might be mediated by the transcriptional coactivators p300/CREB (cAMP response element-binding protein) binding protein (CBP) (Goodman and Smolik, 2000), which bind to both neurogenic bHLHs such as neurogenin1 (Sun et al., 2001) and NeuroD/BETA2 (Mutoh et al., 1998), as well as to C/EBPβ (Mink et al., 1997). C/EBPs and neurogenic bHLHs might also cooperate to transcriptionally activate a neuron-specific gene expression program by binding to separate consensus sites within the same genes. Alternatively, the C/EBPs might promote neurogenesis by sequestering activating transcription factor 5 (ATF5), another transcription factor of the leucine zipper family that maintains neural precursors in an undifferentiated state (Angelastro et al., 2003, 2005). Previous work has shown that C/EBPs and ATFs can heterodimerize (Shuman et al., 1997) and that the heterodimers bind to different DNA binding sites than do either ATF or C/EBP homodimers (Haas et al., 1995). Thus, the relative levels and activation state of ATF5 versus C/EBPs may well determine whether or when a precursor differentiates into a neuron.
One of the interesting conclusions deriving from our work is that activation of the C/EBPs not only promotes neurogenesis but also inhibits gliogenesis. Neurogenic bHLHs have a similar ability to bias precursors to become neurons while at the same time inhibiting them from becoming glial cells (Tomita et al., 2000; Nieto et al., 2001; Sun et al., 2001). For bHLHs such as Neurogenin1, these two effects are apparently mediated via different mechanisms, with the proneurogenic effects being transcriptionally mediated and the antigliogenic effects resulting from Neurogenin1-mediated sequestration of p300/CBP (Sun et al., 2001). For the C/EBPs, these two activities also appear to be dissociable, because inhibition of total C/EBP family activity promoted neurogenesis and inhibited gliogenesis, but inhibition of C/EBP phosphorylation by the MEK family only affected neurogenesis. However, although these two functions are dissociable, both the proneurogenic and antigliogenic effects may be transcriptionally mediated, because C/EBPs both enhance and repress transcription (Lekstrom-Himes and Xanthopoulos, 1998), and phosphorylation differentially regulates these transcriptional actions.
In conclusion, our data support the hypothesis that activation of a MEK-C/EBP pathway is essential for cortical precursors to generate neurons in vivo, and that the C/EBPs act as switches to promote neurogenesis and inhibit gliogenesis. Such a growth factor-regulated intracellular switch provides a mechanism for integrating intrinsic cellular programs with the external neural environment, thereby ensuring the genesis of appropriate numbers of neurons at the appropriate time in embryogenesis.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research. F.D.M. holds the Canada Research Chair in Developmental Neurobiology. We thank Peter Johnson for the C/EBP plasmids and for his advice and guidance, Goichi Miyoshi and Masami Sakamoto for their help with the in utero electroporation technique, Hideyuki Okano for the musashi antibody, Andrée Gauthier for her generosity with experimental samples, and David Kaplan and the members of the Miller/Kaplan laboratories for frequent discussions.
Correspondence should be addressed to Dr. Freda D. Miller, Room 3203, Black Wing, Developmental Biology, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. E-mail: fredam@sickkids.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510747-12$15.00/0
References
- Albuquerque ML, Akiyama SK, Schnaper HW (1998) Basic fibroblast growth factor release by human coronary artery endothelial cells is enhanced by matrix proteins, 17beta-estradiol, and a PKC signaling pathway. Exp Cell Res 245: 163-169. [DOI] [PubMed] [Google Scholar]
- Angelastro JM, Ignatova TN, Kukekov VG, Steindler DA, Stengren GB, Mendelsohn C, Greene LA (2003) Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J Neurosci 23: 4590-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelastro JM, Mason JL, Ignatova TN, Kukekov VG, Stengren GB, Goldman JE, Greene LA (2005) Downregulation of activating transcription factor 5 is required for differentiation of neural progenitor cells into astrocytes. J Neurosci 25: 3889-3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkonac BM, Foster LC, Sibinga NE, Patterson C, Lai K, Tsai JC, Lee ME, Perrella MA, Haber E (1998) Vascular endothelial growth factor induces heparin-binding epidermal growth factor-like growth factor in vascular endothelial cells. J Biol Chem 273: 4400-4405. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Miller FD (2003) Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci 23: 5149-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJL, Porsche C, Sendtner M, Kaplan DR, Miller FD (2005) Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron 48: 253-265. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME (1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278: 477-483. [DOI] [PubMed] [Google Scholar]
- Burrows RC, Wancio D, Levitt P, Lillien L (1997) Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 19: 251-267. [DOI] [PubMed] [Google Scholar]
- Davis RJ (1995) Transcriptional regulation by MAP kinases. Mol Reprod Dev 42: 459-467. [DOI] [PubMed] [Google Scholar]
- Derouet D, Rousseau F, Alfonsi F, Froget J, Hermann J, Barbier F, Perret D, Diveu C, Guillet C, Preisser L, Dumont A, Barbado M, Morel A, deLap-eyriere O, Gascan H, Chevalier S (2004) Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci USA 101: 4827-4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KL, Slack RS (2003) Growth factors: can they promote neurogenesis? Trends Neurosci 26: 283-285. [DOI] [PubMed] [Google Scholar]
- Fukumitsu H, Furukawa Y, Tsusaka M, Kinukawa H, Nitta A, Nomoto H, Mima T, Furukawa S (1998) Simultaneous expression of brain-derived neurotrophic factor and neurotrophin-3 in Cajal-Retzius, subplate and ventricular progenitor cells during early development stages of the rat cerebral cortex. Neuroscience 84: 115-127. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89-103. [DOI] [PubMed] [Google Scholar]
- Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD (1994) The Tα1 α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci 14: 7319-7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD (1999) Early induction of Tα1 α-tubulin transcription in neurons of the developing nervous system. J Comp Neurol 405: 45-60. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553-1577. [PubMed] [Google Scholar]
- Haas NB, Cantwell CA, Johnson PF, Burch JB (1995) DNA-binding specificity of the PAR basic leucine zipper protein VBP partially overlaps those of the C/EBP and CREB/ATF families and is influenced by domains that flank the core basic region. Mol Cell Biol 15: 1923-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE (2005) A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci 8: 616-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, Greenberg DA (2003) Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell 2: 175-183. [DOI] [PubMed] [Google Scholar]
- Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD (1996) Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev 10: 3129-3140. [DOI] [PubMed] [Google Scholar]
- Johnson PF, Williams SC (1994) CCAAT/enhancer binding proteins (C/EBPs) In: Liver gene expression (Yaniv M, Tronche F, eds), pp 231-258. Austin, TX: Landes.
- Kaneko Y, Sakakibara S, Imai T, Suzuki A, Nakamura Y, Sawamoto K, Ogawa Y, Toyama Y, Miyata Y, Okano H (2000) Musashi1: an evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev Neurosci 22: 139-153. [DOI] [PubMed] [Google Scholar]
- Kim H, Li Q, Hempstead BL, Madri JA (2004) Paracrine and autocrine functions of brain-derived neurotrophin factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem 279: 33538-33546. [DOI] [PubMed] [Google Scholar]
- Lane MD, Tang QQ, Jiang MS (1999) Role of the CCAAT enhancer binding proteins (C/EBPs) in adipocyte differentiation. Biochem Biophys Res Commun 266: 677-683. [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes J, Xanthopoulos KG (1998) Biological role of the CCAAT/enhancer binding protein family of transcription factors. J Biol Chem 273: 28545-28548. [DOI] [PubMed] [Google Scholar]
- Lillien L (1998) Progenitor cells: what do they know and when do they know it? Curr Biol 8: R872-R874. [DOI] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Kennedy H, Dehay C (2002) Contrasting effects of basic fibroblast growth factor and neurotrophin 3 on cell cycle kinetics of mouse cortical stem cells. J Neurosci 22: 6610-6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD (1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5: 501-509. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265: 966-970. [DOI] [PubMed] [Google Scholar]
- Medina DL, Sciarretta C, Calella AM, Von Bohlen Und Halbach O, Unsicker K, Minichiello L (2004) TrkB regulates neocortex formation through the Shc/PLCgamma-mediated control of neuronal migration. EMBO J 23: 3803-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Salvelson A, Yang XM, Lederfein D, Barnabé-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD (2002) An essential role for a MEK-C/EBP pathway during growth factor-mediated cortical neurogenesis. Neuron 36: 597-610. [DOI] [PubMed] [Google Scholar]
- Miller FD, Naus CC, Bloom FE, Milner RJ (1987) Differential regulation of α-tubulin isotypes during neuronal maturation. J Cell Biol 105: 3065-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink S, Haenig B, Klempnauer KH (1997) Interaction and functional collaboration of p300 and C/EBPbeta. Mol Cell Biol 17: 6609-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Saito T (2005) Progenitors resume generating neurons after temporary inhibition of neurogenesis by Notch activation in the mammalian cerebral cortex. Development 132: 1295-1304. [DOI] [PubMed] [Google Scholar]
- Morrow T, Song MR, Ghosh A (2001) Sequential specification of neurons and glia by developmentally regulated extracellular factors. Development 128: 3585-3594. [DOI] [PubMed] [Google Scholar]
- Mutoh H, Naya FJ, Tsai MJ, Leiter AB (1998) The basic-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes Dev 12: 820-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Hein P, Fernandes KJL, Peterson AC, Miller FD (2005) A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol Cell Neurosci 29: 525-535. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Sasahara M, Hayase Y, Haneda M, Yasuda H, Kikkawa R, Higashiyama S, Hazama F (1998) Neuronal and glial expression of heparin-binding EGF-like growth factor in central nervous system of prenatal and early-postnatal rat. Brain Res Dev Brain Res 108: 263-272. [DOI] [PubMed] [Google Scholar]
- Nieto M, Schuurmans C, Britz O, Guillemot F (2001) Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron 29: 401-413. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R (1999) Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J 18: 2196-2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JK, Williams BP, Alberta JA, Stiles CD (1999) Bipotent cortical progenitor cells process conflicting cues for neurons and glia in a hierarchical manner. J Neurosci 19: 10383-10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O'Kusky JR, D'Ercole AJ (2004) In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci 19: 2056-2068. [DOI] [PubMed] [Google Scholar]
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S (2000) Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28: 69-80. [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM (2000) Basic fibroblast growth factor (FGF2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 20: 5012-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Zirrgiebel U, Honsa KJ, Julien JP, Miller FD, Kaplan DR, Sweatt JD (2004) Neuronal MEK is important for normal fear conditioning in mice. J Neurosci Res 75: 760-770. [DOI] [PubMed] [Google Scholar]
- Shen Q, Qian X, Capela A, Temple S (1998) Stem cells in the embryonic cerebral cortex: their role in histogenesis and patterning. J Neurobiol 36: 162-174. [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S (2004) Endothelial cells stimulates self-renewal and expand neurogenesis of neural stem cells. Science 304: 1338-1340. [DOI] [PubMed] [Google Scholar]
- Shuman JD, Cheong J, Coligan JE (1997) ATF-2 and C/EBPalpha can form a heterodimeric DNA binding complex in vitro Functional implications for transcriptional regulation. J Biol Chem 272: 12793-12800. [DOI] [PubMed] [Google Scholar]
- Sinor AD, Lillien L (2004) Akt-1 expression level regulates CNS precursors. J Neurosci 24: 8531-8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD (1998) A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol 140: 1497-1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneck E, Johnson PF (1998) CCAAT/enhancer binding protein beta is a neuronal transcriptional regulator activated by nerve growth factor receptor signaling. J Neurochem 70: 2424-2433. [DOI] [PubMed] [Google Scholar]
- Sterneck E, Paylor R, Jackson-Lewis V, Libbey M, Przedborski S, Tessarollo L, Crawley JN, Johnson PF (1998) Selectively enhanced contextual fear conditioning in mice lacking the transcriptional regulator CCAAT/enhancer binding protein delta. Proc Natl Acad Sci USA 95: 10908-10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Nadal-Viciens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME (2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104: 365-376. [DOI] [PubMed] [Google Scholar]
- Sun Y, Goderie SK, Temple S (2005) Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron 45: 873-886. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T (2001) DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell 1: 749-758. [DOI] [PubMed] [Google Scholar]
- Toma JG, El-Bizri H, Barnabé-Heider F, Aloyz R, Miller FD (2000) Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci 20: 7648-7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Moriyoshi K, Nakanishi S, Guillemot F, Kageyama R (2000) Mammalian achaete-scute and atonal homologs regulate neuronal versus glial fate determination in the central nervous system. EMBO J 19: 5460-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura A, Takizawa T, Ochiai W, Yanagisawa M, Nakashima K, Taga T (2002) Cardiotrophin-like cytokine induces astrocyte differentiation of fetal neuroepithelial cells via activation of STAT3. Cytokine 18: 1-7. [DOI] [PubMed] [Google Scholar]
- Williams BP, Park JK, Alberta JA, Muhlebach SG, Hwang GY, Roberts TM, Stiles CD (1997) A PDGF-regulated immediate early gene response initiates neuronal differentiation in ventricular zone progenitor cells. Neuron 18: 553-562. [DOI] [PubMed] [Google Scholar]
- Yamanaka R, Lekstrom-Himes J, Barlow C, Wynshaw-Boris A, Xanthopoulos KG (1998) CCAAT/enhancer binding proteins are critical components of the transcriptional regulation of hematopoiesis. Int J Mol Med 1: 213-221. [DOI] [PubMed] [Google Scholar]