

Abstract
The role of neuropeptides in synaptic plasticity is less well understood than that of classical transmitters such as glutamate. Here we report the importance of the G-protein-coupled calcitonin gene-related peptide (CGRP1) receptor as a critical link between amygdala plasticity and pain behavior. A key player in emotionality and affective disorders, the amygdala has been implicated in the well documented, but mechanistically unexplained, relationship between pain and affect. Our electrophysiological and pharmacological in vitro (patch-clamp recordings) and in vivo (extracellular single-unit recordings) data show that selective CGRP1 receptor antagonists (CGRP8-37 and BIBN4096BS) in the amygdala reverse arthritis pain-related plasticity through a protein kinase A (PKA)-dependent postsynaptic mechanism that involves NMDA receptors. CGRP1 receptor antagonists inhibited synaptic plasticity in the laterocapsular division of the central nucleus of the amygdala (CeLC) in brain slices from arthritic rats compared with normal controls. The effects were accompanied by decreased neuronal excitability and reduced amplitude, but not frequency, of miniature EPSCs; paired-pulse facilitation was unaffected. The antagonist effects were occluded by a PKA inhibitor. CGRP1 receptor blockade also directly inhibited NMDA-evoked, but not AMPA-evoked, membrane currents. Together, these data suggest a postsynaptic site of action. At the systems level, the antagonists reversed the sensitization of nociceptive CeLC neurons in anesthetized rats in the arthritis pain model. Importantly, CGRP1 receptor blockade in the CeLC inhibited spinal (hindlimb withdrawal reflexes) and supraspinal pain behavior of awake arthritic rats, including affective responses such as ultrasonic vocalizations. This study provides direct evidence for the critical dependence of pain behavior on CGRP1-mediated amygdala plasticity.
Keywords: amygdala, synaptic plasticity, sensitization, pain, neuropeptide, vocalization
Introduction
The amygdala plays an important role in emotional learning and memory and affective disorders such as anxiety and depression (Davis, 1998; Davidson et al., 1999; LeDoux, 2000; Rodrigues et al., 2004) but has only recently been linked to pain processing (Neugebauer et al., 2004). The amygdala includes several anatomically and functionally distinct nuclei. The laterocapsular part of the central nucleus (CeLC) is now defined as the “nociceptive amygdala” because of its high content of neurons that process pain-related information (Bernard and Bandler, 1998; Neugebauer et al., 2004). The CeLC is the target of the spino-parabrachio-amygdaloid pain pathway (Bernard and Bandler, 1998) and also receives affect-related information from the lateral (LA) and basolateral (BLA) amygdala (Neugebauer et al., 2004). Associative learning and plasticity in the LA-BLA circuitry is critical for the emotional evaluation of sensory stimuli and affective states and disorders (LeDoux, 2000; Pare et al., 2004; Rodrigues et al., 2004; Walker and Davis, 2004). Therefore, the CeLC is well positioned to integrate pain-related information with affective content, contribute to the emotional response to pain, and serve as the neuronal interface through which affect modulates pain (Neugebauer et al., 2004).
The mechanisms and behavioral consequences of pain processing in the amygdala are only beginning to emerge. Our previous studies showed synaptic plasticity in the CeLC in the arthritis pain model (Neugebauer and Li, 2003; Neugebauer et al., 2003). This pain-related plasticity depends on presynaptic metabotropic glutamate receptors (Neugebauer et al., 2003; Li and Neugebauer, 2004a) and on postsynaptic NMDA receptor phosphorylation through protein kinase A (PKA) but not PKC (Li and Neugebauer, 2004b; Bird et al., 2005). Using an integrative approach that combines electrophysiology in vitro and in vivo and behavioral analysis, we show here that the calcitonin gene-related peptide 1 (CGRP1) receptor serves as the critical molecule that links these presynaptic and postsynaptic mechanisms and contributes to pain behavior organized at different levels of the pain neuraxis.
CGRP is a 37 amino acid peptide that activates adenylyl cyclase and PKA through G-protein-coupled receptors, including the CGRP1 receptor for which selective antagonists are available (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997; Doods et al., 2000). Previous anatomical data and our present study suggest that CGRP is a molecular marker of the CeLC. The CeLC is delineated by its abundance of CGRP-immunoreactive terminals of fibers from the external lateral parabrachial area (Kruger et al., 1988; Schwaber et al., 1988; Harrigan et al., 1994; de Lacalle and Saper, 2000), which is part of the spino-parabrachio-amygdaloid pain pathway. These terminals innervate CeLC neurons that project to brainstem areas such as the periaqueductal gray (Schwaber et al., 1988; Harrigan et al., 1994), which is important for expression of behavior and descending pain modulation. The central nucleus of the amygdala (CeA) also represents one of the brain areas with the highest levels of CGRP and CGRP receptors (Skofitsch and Jacobowitz, 1985; Van Rossum et al., 1997; Oliver et al., 1998). Whereas the involvement of CGRP in peripheral and spinal pain mechanisms is well established (Galeazza et al., 1995; Neugebauer et al., 1996; Schaible, 1996; Ruda et al., 2000; Sun et al., 2004), less is known about its role in pain-related plasticity in the brain.
Materials and Methods
Arthritis pain model. The mono-arthritis was induced in the left knee joint of adult rats as described in detail previously (Neugebauer and Li, 2003; Neugebauer et al., 2003). A kaolin suspension (4%, 80-100 μl) was injected into the joint cavity through the patellar ligament with a syringe (1 ml, 25 ga, 5/8 inch). After repetitive flexions and extensions of the knee for 15 min, a carrageenan solution (2%, 80-100 μl) was injected into the knee joint cavity, and the leg was flexed and extended for another 5 min. This treatment paradigm reliably leads to inflammation and swelling of the knee within 1-3 h, reaches a maximum plateau at 5-6 h, and persists for days (Neugebauer and Li, 2003; Neugebauer et al., 2003). Electrophysiological and behavior measurements of arthritis pain-related changes were made at the 6 h time point.
In vitro electrophysiology: patch-clamp recording. Amygdala slice preparation. Brain slices containing the CeA were obtained from arthritic rats and normal rats (120-250 g; Sprague Dawley). Rats were decapitated, and the brains quickly were dissected out and blocked in cold (4°C) artificial CSF (ACSF). ACSF contained the following (in mm): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. ACSF was oxygenated and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2. Coronal brain slices (500 μm) were prepared using a Vibroslice (Camden Instruments, London, UK). After incubation in ACSF at room temperature (21°C) for at least 1 h, a single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C), which superfused the slice at ∼2 ml/min.
Whole-cell patch-clamp recording. Whole-cell recordings using the “blind” patch technique were obtained from neurons in the CeLC (see Fig. 7a) as described previously (Neugebauer et al., 2003; Bird et al., 2005). The different nuclei of the amygdala and the subdivisions of the CeA are easily discerned under the microscope (see Fig. 7a). Patch electrodes (4-6 MΩ tip resistance) were made from borosilicate glass capillaries (1.5 and 1.12 mm, outer and inner diameter, respectively; Drummond, Broomall, PA) pulled on a Flaming-Brown micropipette puller (P-80/PC; Sutter Instruments, Novato, CA). The internal solution of the recording electrodes contained the following (in mm): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.4 Na3-GTP, pH adjusted to 7.2-7.3 with KOH (osmolarity adjusted to 280 mOsm/kg with sucrose). After tight (>2GΩ) seals were formed and the whole-cell configuration was obtained, neurons were included in the sample if the resting membrane potential was more negative than -50 mV and action potentials overshooting 0 mV were evoked by direct cathodal stimulation. Voltage and current signals were low-pass filtered at 1 kHz with a dual four-pole Bessel filter (Warner Instruments, Hamden, CT), digitized at 5 kHz (Digidata 1322A interface; Molecular Devices, Union City, CA), and stored on a computer (Dell Pentium 4). Data were also continuously recorded on an ink chart recorder (Gould 3400; Gould Instruments, Valley View, OH). Current- and voltage-clamp [d-SEVC (discontinuous single-electrode voltage clamp)] recordings were made using an Axoclamp-2B amplifier (Molecular Devices) with a switching frequency of 5-6 kHz (30% duty cycle), gain of 3-8 nA/mV, and time constant of 20 ms. Phase shift and anti-alias filter were optimized. The head-stage voltage was monitored continuously on a digital oscilloscope (Gould 400; Gould Instruments) to ensure precise performance of the amplifier. Voltage and current data were analyzed with pClamp9 software (Axon Instruments).
Figure 7.

CGRP8-37 inhibits pain-related behavior (audible and ultrasonic vocalizations and hindlimb withdrawal reflexes) in awake animals with arthritis but not in normal animals. Audible and ultrasonic vocalizations were measured in normal rats (left column) and arthritic rats (right column). Duration of vocalizations was measured as the arithmetic sum of the duration of each individual vocalization event as described previously (Han and Neugebauer, 2005). a, VADs, which continue beyond the actual stimulus and are organized in the limbic forebrain (Borszcz and Leaton, 2003; Han and Neugebauer, 2005), were evoked by noxious (2000 g/30 mm2) stimulation (15 s) of the knee. Application of CGRP8-37 (100 μm, concentration in the microdialysis probe; 15-20 min) into the CeLC of normal rats had no significant effect (left; n = 4; p > 0.05, paired t test). In arthritic animals (6 h after induction), CGRP8-37 (100μm) significantly reduced audible and ultrasonic VADs (right; n = 9; p < 0.05, paired t test). Vocalizations of arthritic animals were expressed as percentage of vocalizations of the same animals before arthritis induction (normal, set to 100%). b, Audible and ultrasonic VDSs, which are organized at the medullary brainstem level (Borszcz and Leaton, 2003; Han and Neugebauer, 2005), were evoked by noxious (2000 g/30 mm2) stimulation (15 s) of the knee. Administration of CGRP8-37 (100μm, concentration in the microdialysis probe; 15-20 min) into the CeLC did not affect the duration of audible and ultrasonic VDSs in normal animals (left; n = 4; p > 0.05, paired t test) but significantly inhibited VDSs of arthritic rats (right; n = 9; p < 0.05, paired t test). c, Hindlimb withdrawal reflexes were evoked by mechanical stimulation (compression) of the knee (15 s) with increasing intensity (steps of 50 g/30 mm2). Withdrawal thresholds, a measure of pain sensitivity, were defined as the minimum stimulus intensity that evoked a withdrawal reflex. Thresholds decreased after arthritis. Application of CGRP8-37 into the CeLC significantly increased the reduced thresholds in arthritic animals (right; n = 6; p < 0.05, paired t test) but had no effect in normal rats (left; n = 4; p > 0.05, paired t test). d, Placement control experiments show that application of CGRP8-37 into the striatum (caudate-putamen dorsolateral to CeLC) did not produce significant changes of audible and ultrasonic vocalizations in arthritic animals (n = 5; p > 0.05, paired t test). Vocalization data in d represent the total duration of VAD plus VDS. Because neither VADs nor VDSs were inhibited by CGRP8-37, the data were pooled for simplification. a-d, CGRP8-37 (100 μm) was administered by microdialysis for 15-20 min. All drug effects were reversible. Bar histograms and error bars represent mean ± SE. *p < 0.05; **p < 0.01.
Synaptic stimulation. A concentric bipolar stimulating electrode (22 kΩ resistance; David Kopf Instruments, Tujunga, CA) was positioned on the afferent fiber tract from the parabrachial area PB (see Introduction) (Bernard and Bandler, 1998; Neugebauer et al., 2004) under microscopic control as described previously (Neugebauer et al., 2003; Bird et al., 2005) and illustrated in Figure 7a. EPSCs were evoked in CeLC neurons by electrical stimulation (150 μs square-wave pulses; Grass S88 stimulator; Grass Instruments, West Warwick, RI) at frequencies below 0.25 Hz. Input-output functions were obtained by increasing the stimulus intensity in 100 μA steps. For evaluation of a drug effect on synaptically evoked responses, the stimulus intensity was adjusted to 75-80% of the intensity required for orthodromic spike generation (Neugebauer et al., 2003; Bird et al., 2005).
Drugs. Calcitonin gene-related peptide fragment 8-37 (CGRP8-37), a selective CGRP1 receptor antagonist (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997), was purchased from Bachem (Torrance, CA). BIBN4096BS, a selective nonpeptide CGRP1 receptor antagonist (Doods et al., 2000), was kindly supplied by Boehringer Ingelheim Pharma (Biberach, Germany). KT5720, a potent and selective PKA inhibitor (Cabell and Audesirk, 1993; Bird et al., 2005), NMDA, and AMPA were all purchased from Tocris Cookson (Ellisville, MO).
Drugs were dissolved in ACSF on the day of the experiment and applied to the brain slice by gravity-driven superfusion in the ACSF. Solution flow into the recording chamber (1 ml volume) was controlled with a three-way stopcock. Drugs were applied for at least 15 min to establish equilibrium in the tissue. Based on initial observations showing that drug effects reached a plateau after 10 min, the 12-15 min time point was selected for the full set of tests (including Input-output functions, current-voltage relationships, paired-pulse facilitation (PPF), and action potential firing properties; see above) and for the comparison of drug effects.
In vivo electrophysiology: extracellular single-unit recording. Animal preparation and anesthesia. Adult rats (250-350 g; Sprague Dawley) were anesthetized with pentobarbital sodium (50 mg/kg, i.p.). A cannula was inserted into the trachea for artificial respiration and to measure end-tidal CO2 levels. A catheter in the jugular vein allowed continuous administration of anesthetic (see below) and fluid support (3-4 ml · kg-1 · h-1 Ringer's lactate solution, i.v.). The carotid artery was catheterized for blood pressure monitoring. Depth of anesthesia was assessed as follows: by regularly testing the corneal blink, hindpaw withdrawal, and tail-pinch reflexes; continuously monitoring the end-tidal CO2 levels (kept at 4.0 ± 0.2%), arterial blood pressure (kept at 135 ± 5 mmHg), heart rate, and electrocardiogram pattern; and checking for abnormal breathing patterns. Core body temperature was maintained at 37°C by means of a homeothermic blanket system. Animals were mounted in a stereotaxic frame (David Kopf Instruments), paralyzed with pancuronium (induction, 0.3-0.5 mg, i.v.; maintenance, 0.3 mg/h, i.v.), and artificially ventilated (3-3.5 ml; 55-65 strokes/min). Constant levels of anesthesia were maintained by continuous intravenous infusion of pentobarbital (15 mg · kg-1 · h-1). A small unilateral craniotomy was performed at the sutura fronto-parietalis level for the recording of CeLC neurons and for drug application by microdialysis (see below) as described previously (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b).
Recording and identification of amygdala neurons. Extracellular recordings were made from single neurons in the CeLC with glass-insulated carbon filament electrodes (3-5 MΩ) as described previously (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b), using the following stereotaxic coordinates (Paxinos and Watson, 1998): 2.0-2.2 mm caudal to bregma; 3.8-4.2 mm lateral to midline; depth of 7-9 mm. The recorded signals were amplified, displayed on analog and digital storage oscilloscopes, fed into a window discriminator, digitized by an interface (CED 1401+; Cambridge Electronics Design, Cambridge, UK), and recorded on a computer (Dell Pentium 4). Spike2 software (version 3; Cambridge Electronics Design) was used for on-line and off-line analysis of single-unit activity. An individual CeLC neuron was identified by the configuration, shape, and height of the recorded action potentials (spikes) in response to innocuous and noxious mechanical stimulation (compression) of the knee as described in detail previously (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b). Mechanical stimuli of innocuous (100 and 500 g/30 mm2) and noxious (1500 and 2000 g/30 mm2) intensity were applied to the knee and other parts of the receptive field (e.g., ankle) by means of a forceps equipped with a force transducer, whose calibrated output was amplified and displayed in grams on an liquid crystal display screen (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b). The output signal was also fed into the Cambridge Electronics Design interface and recorded on the computer for on-line and off-line analysis. Stimulus intensities of 100-500 g/30 mm2 applied to the knee and other deep tissue are considered innocuous because they do not evoke hindlimb withdrawal reflexes in awake rats and are not felt to be painful when tested on the experimenters. Pressure stimuli >1500 g/30 mm2 are noxious because they evoke hindlimb withdrawal reflexes in awake rats and are distinctly painful when applied to the experimenters (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b; Han and Neugebauer, 2005). Stimulus-response functions were obtained with the use of a wide range of innocuous and noxious stimuli (100-2000 g/30 mm2).
In this study, neurons were selected that had a receptive field in the knee and responded more strongly to noxious than innocuous stimuli, because these so-called multireceptive (MR) neurons have been shown in our previous studies to become sensitized consistently in the arthritis pain model (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b). Size and threshold of the total receptive field, background activity, and responses to innocuous and noxious stimuli (see above) were recorded before and for 6-9 h after induction of arthritis in the knee. Innocuous and noxious stimuli (15 s duration each) were applied three times in a control period of at least 2 h before arthritis induction and then every hour after induction of arthritis
Drug application. Known concentrations of CGRP1 receptor antagonists (CGRP8-37 and BIBN4096BS; see above, Drugs) were administered into the CeLC by microdialysis 6 h after induction of arthritis as described in detail previously (Neugebauer and Li, 2003; Li and Neugebauer, 2004a,b). Several hours before the start of the electrophysiological recordings, a microdialysis probe (20 kDa cutoff, membrane length of 2 mm; CMA12; CMA/Microdialysis, North Chelmsford, MA) was lowered vertically into the CeLC and positioned stereotaxically 500 μm anterior to the recording electrode, using the following coordinates (Paxinos and Watson, 1998): 1.8 mm caudal to bregma; 4.0 mm lateral to midline; depth of tip, 9.0 mm. The microdialysis probe was connected with PE50 tubing to an infusion pump (Harvard Apparatus, Holliston, MA) and perfused with ACSF (see above, in vitro experiments) oxygenated and equilibrated to pH 7.4. ACSF was pumped through the fiber throughout the experiment to maintain stable conditions in the tissue. Drugs (see above) were dissolved in ACSF on the day of the experiment and administered into the CeLC at a rate of 5 μl/min for 20 min to establish equilibrium in the tissue.
Background activity and responses to innocuous and noxious stimulation of the knee (see above) were measured every 5 min during drug application. Based on initial observations showing that drug effects reached a plateau after 10 min, the 12-15 min time point was selected for the full set of tests (including stimulus-response functions, see above) and for the comparison of drug effects. The drug concentration in the microdialysis fiber was 100 times that predicted to be needed in the tissue based on data in the literature (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997) and our own in vitro data (this study) because of the concentration gradient across the dialysis membrane (Li and Neugebauer, 2004a,b; Han and Neugebauer, 2005). The numbers given in this article refer to the drug concentrations in the microdialysis fiber.
Behavior: vocalizations and hindlimb withdrawal reflexes
Experimental protocol. On day 1, a guide cannula for drug (and ACSF vehicle) application by microdialysis was stereotaxically inserted into the CeLC. Baseline (pre-arthritis) vocalizations and spinal withdrawal reflexes were measured in normal rats in the afternoon of day 2. The behavioral tests were repeated in the afternoon of day 3 in the same animals 6 h after arthritis induction in one knee (see above). CGRP8-37 was then administered into the CeLC through the microdialysis probe, and behavior was measured at 15 min during the continued drug administration and again at 30 min of washout with ACSF. CGRP8-37 was also tested in a different set of naive animals without arthritis.
Stereotaxic implantation of microdialysis guide cannula and drug administration. As described in detail previously (Han and Neugebauer, 2005), rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.), and a small unilateral craniotomy was performed at the sutura fronto-parietalis level. Using a stereotaxic apparatus (David Kopf Instruments), a guide cannula (CMA/12; CMA/Microdialysis) was inserted through a drill hole into the CeLC or striatum (as a placement control) using the following stereotaxic coordinates (Paxinos and Watson, 1998): CeA, 2.0 mm caudal to bregma; 4.0 mm lateral to midline; depth of 7.0 mm; striatum: 2.2 mm caudal to bregma; 4.5 mm lateral to midline; depth of 5.0 mm. Once inserted, the microdialysis probe protruded beyond the tip of the guide cannula by 2 mm. The cannula was fixed to the skull with dental acrylic (Plastics One, Roanoke, VA). Antibiotic ointment was applied to the exposed tissue to prevent infection. On the day of the experiment, a microdialysis probe (20 kDa cutoff, membrane length of 2 mm; CMA12; CMA/Microdialysis) was inserted through the guide cannula for administration of ACSF (same composition as in the in vitro electrophysiology studies) or drugs into the CeLC (or striatum) at a rate of 5 μl/min using an infusion pump (Harvard Apparatus).
CGRP8-37 and BIBN4096BS (see above) were dissolved in ACSF on the day of the experiment at a concentration 100 times that predicted to be needed based on data in the literature (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997) and our own in vitro data (this study) because of the concentration gradient across the dialysis membrane (Li and Neugebauer, 2004a,b; Han and Neugebauer, 2005). The numbers given in this article refer to the drug concentrations in the microdialysis fiber. ACSF administered alone served as a vehicle control. Behavior was measured at 15 min during continued drug administration and again at 30 min of washout with ACSF.
Audible and ultrasonic vocalizations. Vocalizations were recorded and analyzed as described in detail previously (Han and Neugebauer, 2005). The experimental setup [Han JS, Neugebauer V (2005) U.S. Patent Application 98006/28UTL, pending] included a custom-designed recording chamber, a condenser microphone (audible range, 20 Hz to 16 kHz) connected to a preamplifier and a ultrasound detector (25 ± 4 kHz), filter and amplifier (UltraVox four-channel system; Noldus Information Technology, Leesburg, VA), and data acquisition software (UltraVox 2.0; Noldus Information Technology), which automatically monitored the occurrence of vocalizations within user-defined frequencies (see above) and recorded the number and duration of digitized events (audible and ultrasonic vocalizations) on a computer (Dell Pentium 4) (Han and Neugebauer, 2005; Han et al., 2005). This computerized recording system was set to suppress nonrelevant audible sounds and to ignore ultrasounds outside the defined frequency range (25 ± 4 kHz). Animals were placed in the recording chamber for acclimation 1 h before the vocalization measurements and for habituation (1 h on 2 d). The recording chamber ensured the stable positioning of the animal at a fixed distance from the sound detectors and allowed the reproducible stimulation of the knee joint. The chamber contained openings for the hindlimbs to allow the application of brief (15 s) innocuous (100 g/30 mm2) and noxious (2000 g/30 mm2) mechanical stimuli with a calibrated forceps (same as in the in vivo electrophysiology studies); it also had an opening for the head to permit drug administration into the amygdala through the microdialysis probe inserted into the implanted guide cannula. Rate and duration of vocalizations were recorded during application of the mechanical stimulus [vocalizations during stimulation (VDSs), organized at the medullary level] and in the period immediately after stimulation [vocalization “afterdischarges” (VADs), generated in the limbic forebrain] (Borszcz and Leaton, 2003; Han and Neugebauer, 2005).
Hindlimb withdrawal reflex. The threshold of spinally organized withdrawal reflexes in response to stimulation of the knee was determined subsequently to the vocalization measurements. Mechanical stimuli of increasing intensity (steps of 50 g/30 mm2) were applied to the knee joint by means of a calibrated forceps with a force transducer as in the vocalization experiments. Withdrawal threshold was defined as the minimum stimulus intensity that evoked a withdrawal reflex.
Histology
At the end of each in vivo electrophysiology experiment, the recording site in the CeLC was marked by injecting direct current (250 μA for 3 min) through the carbon filament recording electrode (see Fig. 7b). The position of the microdialysis probe was also confirmed histologically (see Fig. 7c,d). The brain was removed and submerged in 10% Formalin and potassium ferrocyanide. Tissues were stored in 20% sucrose before they were frozen sectioned at 50 μm. Sections were stained with Neutral Red, mounted on gel-coated slides, and cover-slipped. The boundaries of the different amygdala nuclei were easily identified under the microscope. Lesion/recording sites were plotted on standard diagrams (see Fig. 7).
Data analysis and statistics
All averaged values are given as the mean ± SEM. Statistical significance was accepted at the level p < 0.05. GraphPad Prism 3.0 software (GraphPad Software, San Diego, CA) was used for all statistical analysis except when noted.
In vitro electrophysiology. Input-output functions and concentration-response relationships were compared using a two-way ANOVA followed by post hoc tests when appropriate. Concentration-response curves were obtained by nonlinear regression analysis using the formula y = A + (B - A)/[1 + (10C/10X)D], where A is the bottom plateau, B is the top plateau, C is log(IC50), and D is the slope coefficient. The paired t test was used to compare test EPSC amplitudes and PPF evoked by one stimulus intensity before and during drug application. Miniature EPSCs (mEPSCs) were analyzed for frequency and amplitude distributions using the MiniAnalysis program 5.3 (Synaptosoft, Decatur, GA). The Kolmogorov-Smirnov test was used for cumulative distribution analysis of mEPSC amplitude and frequency.
In vivo electrophysiology. Extracellularly recorded single-unit activity (action potentials) was analyzed off-line from peristimulus rate histograms using Spike2 software (version 3; Cambridge Electronics Design). Evoked responses were expressed as spikes (action potentials) per second (Hertz) by subtracting from the total activity during the stimulus (15 s) any background activity (Hertz) in the 15 s preceding the stimulus. Responses before and during drug administration were compared using a paired t test. Concentration-response relationships were obtained and analyzed statistically as described above (see above, In vitro electrophysiology).
Audible and ultrasonic vocalizations. Duration of audible and ultrasonic vocalizations was normalized to pre-arthritis (normal) conditions. The duration is defined as the arithmetic sum (total amount) of the durations of individual vocalization events that occur during (VDS) or after (VAD) a single stimulus. A paired t test was used to compare behavioral changes (vocalizations and withdrawal thresholds) in the same animal before and during drug administration.
Results
Endogenous activation of CGRP1 receptors is required for pain-related synaptic plasticity of CeLC neurons
Whole-cell voltage-clamp recordings of neurons in the CeLC were made in brain slices from untreated normal rats and from rats in which an arthritis pain state had been induced 6 h before (for details, see Neugebauer et al., 2003; Bird et al., 2005). CeLC neurons from arthritic rats showed significantly increased synaptic transmission (Fig. 1a,b), which indicates “synaptic plasticity” because it is preserved in the reduced slice preparation and maintained independently of peripheral or spinal pain mechanisms. Pain-related synaptic plasticity is evident from the increased synaptic strength measured as increased peak amplitudes of monosynaptic EPSCs at the nociceptive PB-CeLC synapse (Neugebauer et al., 2004). Individual current traces show that, compared with a control neuron (Fig. 1a, left), monosynaptic EPSCs were enhanced in a CeLC neuron recorded in a brain slice from an arthritic rat (6 h after induction) (Fig. 1a, right). Monosynaptic EPSCs of progressively larger amplitudes were evoked by electrical synaptic stimulation with increasing intensities, and Input-output relationships were obtained by measuring EPSC peak amplitude (picoamperes) as a function of afferent fiber volley stimulus intensity (microamperes) for each neuron (Fig. 1b). The Input-output function of the PB-CeLC synapse was significantly increased in CeLC neurons from arthritic rats (n = 19) compared with control neurons from normal rats (n = 37), as evidenced by the steeper slope and upward shift at higher stimulus intensities (p < 0.0001; F(1,593) = 60.29, two-way ANOVA followed by Bonferroni's post hoc tests) (Fig. 1b). The sites of synaptic stimulation and patch-clamp recording in the amygdala brain slice are schematically illustrated in Figure 7a.
Figure 1.

Pain-related synaptic plasticity in the amygdala depends in part on the activation of CGRP1 receptors. a, Peak amplitudes of monosynaptic EPSCs, a measure of synaptic strength, were larger in a CeLC neuron recorded in a brain slice from an arthritic rat (right) than in a control neuron from a normal rat (left). Individual traces show monosynaptic EPSCs (average of 8-10 EPSCs) evoked at the PB-CeLC synapse with increasing stimulus intensities. Calibration: 50 ms, 50 pA. b, Input-output functions were measured by increasing the stimulus intensity in 100 μA steps. CeLC neurons from arthritic animals (n = 19) showed significantly enhanced synaptic transmission compared with control neurons (n = 37) (p < 0.0001, F(1,593) = 60.29, two-way ANOVA followed by Bonferroni's post hoc tests). c, A selective CGRP1 receptor antagonist (CGRP8-37, 1 μm) inhibited synaptic plasticity in a CeLC neuron from an arthritic animal (middle trace) but had little effect on basal synaptic transmission in a CeLC neuron from a normal animal (left trace). Likewise, the selective nonpeptide CGRP1 receptor antagonist (BIBN4096BS, 1 μm) inhibited synaptic plasticity in a CeLC neuron from an arthritic animal (right trace). Individual traces show monosynaptic EPSCs (average of 8-10 EPSCs) evoked at the PB-CeLC synapse with the stimulus intensity set to 70-80% of that required for generating maximum EPSC amplitude. Calibration: 50 ms, 50 pA. d, Averaged raw (current) data show that CGRP8-37 (1 μm) inhibited the increased EPSC amplitude in neurons (n=17) from arthritic rats (right; p < 0.05, paired t test) but had no significant effects on the amplitude of EPSCs recorded in control neurons (n = 29) from normal rats (left). e, BIBN4096BS (1 μm) also decreased the EPSC amplitude (averaged current data) significantly (p < 0.05, paired t test; n = 5). f, Concentration-response relationships show that CGRP8-37 was more efficacious in neurons from arthritic rats (n = 17) than in control neurons (n = 29) from normal rats (p < 0.01; F(1,220) = 10.74, two-way ANOVA). Peak amplitudes of monosynaptic EPSCs during each concentration of CGRP8-37 were averaged and expressed as percentage of predrug (baseline) control (100%). CGRP8-37 was applied for at least 15 min, and measurements were made at 12 min. g, Input-output function of the PB-CeA synapse (see b) was significantly reduced by CGRP8-37 (1 μm) in CeLC neurons from arthritic rats (n = 12; p < 0.0001; F(1,242) = 76.32, two-way ANOVA followed by Bonferroni's post hoc tests). Whole-cell voltage-clamp recordings were made from CeLC neurons held at -60 mV. *p < 0.05; **p < 0.01; ***p < 0.001.
Subsequently, we addressed the role of CGRP1 receptor activation in pain-related synaptic plasticity compared with normal synaptic transmission. A selective CGRP1 receptor antagonist (CGRP8-37, 1 μm) inhibited synaptic plasticity in CeLC neurons in slices from arthritic rats but had little effect on basal synaptic transmission in CeLC neurons from normal rats (Fig. 1c,d,f,g). CGRP8-37 inhibited the EPSC peak amplitude in the arthritis pain model significantly and nearly restored the level of synaptic transmission to normal (Fig. 1d; p < 0.05, paired t test; n = 17; Fig. 1c, individual current traces) but had no significant effect on synaptic transmission in control neurons from normal animals (Fig. 1c,d, left) (n = 29). To ensure the selectivity of CGRP1 receptor blockade, we also tested a selective nonpeptide CGRP1 antagonist (BIBN4096BS, 1 μm). BIBN4096BS inhibited synaptic plasticity in CeLC neurons from arthritic rats (Fig. 1e; p < 0.05, paired t test; n = 5; Fig. 1c, right, individual current traces). Synaptic responses were evoked by a stimulus intensity adjusted to 70-80% of that required for generating the maximum EPSC amplitude.
Cumulative concentration-response relationships show that CGRP8-37 inhibited synaptic plasticity in CeLC neurons (n = 29) from arthritic rats more efficaciously than basal synaptic transmission in control neurons (n = 17) from normal rats (Fig. 1f; p < 0.01, F(1,220) = 10.74, two-way ANOVA; Bonferroni's post hoc tests indicate significant differences for individual concentrations). The IC50 did not change significantly in the arthritis pain model compared with normal controls (2.7 and 1.1 nm, respectively; see Materials and Methods). CGRP8-37 also changed the input-output function of the PB-CeLC synapse in the arthritis pain state to the level recorded under normal conditions (Fig. 1e). The inhibitory effects of CGRP8-37 were particularly pronounced at higher stimulus intensities (n = 12; p < 0.0001; F(1,242) = 76.32, two-way ANOVA followed by Bonferroni's post hoc tests). These data suggest that enhanced activation of CGRP1 receptors by the endogenous ligand CGRP represents an important mechanism of pain-related synaptic plasticity in the amygdala.
Postsynaptic rather than presynaptic CGRP1 receptor activation in pain-related synaptic plasticity
The major source of CGRP in the CeLC is the external lateral parabrachial area (Kruger et al., 1988; Schwaber et al., 1988; Harrigan et al., 1994; de Lacalle and Saper, 2000), which is part of the spino-parabrachio-amygdaloid pain pathway (Bernard and Bandler, 1998) and projects heavily to the CeLC. To assess whether CGRP acts on presynaptic or postsynaptic sites in the CeLC, we used a number of well established electrophysiological methods, including the analysis of neuronal excitability (Fig. 2), amplitude and frequency of spontaneous mEPSCs (Fig. 3a-c), and paired-pulse facilitation (Fig. 3d,e). These parameters were measured before and during application of CGRP8-37 in amygdala brain slices from normal and arthritic rats.
Figure 2.

CGRP8-37 inhibits neuronal excitability of CeLC neurons in the arthritis pain model but not under normal conditions. Action potentials were evoked in CeLC neurons by direct (through the patch electrode) intracellular injections of current pulses (250 ms) of increasing magnitude (50 pA steps) before and during CGRP8-37 administration. a, b, CGRP8-37 did not affect the action potential firing rate in CeLC neurons in slices from normal rats. c, d, However, the action potential firing rate was significantly decreased by CGRP8-37 in CeLC neurons from arthritic rats (n = 10; p < 0.0001; F(1,144) = 14.15, two-way ANOVA), suggesting a functional change of CGRP1 receptor activation that has postsynaptic effects in arthritis but not under normal conditions. For the measurement of action potential firing in current clamp, neurons were recorded at -60 mV. Calibration (in a, c): top traces, 100 ms, 25 mV; bottom traces, 100 ms, 150 pA. Symbols and error bars in b and d represent mean ± SE.
Figure 3.

mEPSC analysis and PPF suggest postsynaptic rather than presynaptic effects of CGRP8-37. a, Original current traces of mEPSC recordings in an individual CeLC neuron in the presence of TTX (1μm) showed that CGRP8-37 (1μm) reduced amplitude but not frequency of mEPSCs. Calibration: 1 s, 20 mV. The CeLC neuron was recorded in a slice from an arthritic rat. b, c, Normalized cumulative distribution analysis of mEPSC amplitude and frequency showed that CGRP8-37 caused a significant shift toward smaller amplitudes (b) (p < 0.005; maximal difference in cumulative fraction, 0.175; Kolmogorov-Smirnov test) but had no effect on the interevent interval (frequency) distribution (c). CGRP8-37 selectively decreased mean mEPSC amplitude (p < 0.05, paired t test) but not mEPSC frequency (events per second) in the sample of neurons (n = 4; see bar histograms in d, e). PPF, a measure of presynaptic mechanisms, was not changed by CGRP8-37. PPF was calculated as the ratio of the second and the first of two consecutive EPSCs evoked by two electrical stimuli of equal intensity at increasing interstimulus intervals. Peak EPSC amplitudes were measured as the difference between the current level before the stimulus artifact and the peak of the EPSC. d, Current traces (average of 8-10 EPSCs) recorded in an individual CeLC neuron illustrate that PPF evoked at a 50 ms interval was not affected by CGRP8-37. Calibration: 25 ms, 50 pA. e, CGRP8-37 had no significant effect on PPF at various stimulus intervals in the whole sample of neurons (n = 6; p > 0.05, paired t test), further arguing against a presynaptic action. Symbols and error bars represent mean ± SE. Neurons were recorded in voltage clamp at -60 mV.
Action potentials were evoked in current-clamp mode by direct intracellular current injections of increasing magnitude through the patch electrode. Input-output functions of neuronal excitability were obtained by averaging the frequency of action potentials evoked at each current intensity. CGRP8-37 significantly decreased the Input-output function of CeLC neurons from arthritic rats (Fig. 2c,d)(n = 10; p < 0.0001; F(1,144) = 14.15, two-way ANOVA) but had no significant effect in CeLC neurons from normal rats (Fig. 2a,b) (n = 14; p > 0.05; F(1,206) = 0.44, two-way ANOVA). Accordingly, the analysis of current-voltage relationships in voltage clamp showed that CGRP8-37 decreased the slope conductance in CeLC neurons from arthritic rats significantly (p < 0.01, paired t test; CGRP8-37, 4.88 ± 1.57 nS; predrug control, 5.80 ± 1.57 nS; n = 9). CGRP8-37 had no significant effect on the slope conductance of control neurons from normal animals (p < 0.01, paired t test; CGRP8-37, 4.45 ± 1.39 nS; predrug control, 4.69 ± 1.22 nS; n = 14). In agreement with our previous studies (Neugebauer et al., 2003; Bird et al., 2005), these data also show that the slope conductances of CeLC neurons in the arthritis pain model were increased compared with control neurons. Together, these results suggest that activation of CGRP1 receptors is involved in the pain-related increase of neuronal excitability in the amygdala.
The analysis of spontaneous mEPSCs in the presence of TTX is a well established electrophysiological approach to determine presynaptic versus postsynaptic mechanisms. Presynaptic changes at the transmitter release site affect mEPSC frequency, whereas changes at the postsynaptic membrane would alter mEPSC amplitude (quantal size) (Wyllie et al., 1994; Han et al., 2004). CGRP8-37 decreased the amplitude, but not frequency, of mEPSCs recorded in TTX (1 μm) in slices from arthritic rats (Fig. 3). This postsynaptic effect is illustrated in the current traces recorded in an individual CeLC neuron (Fig. 3a). In the whole sample of neurons (n = 4), CGRP8-37 caused a shift of the normalized cumulative mEPSC amplitude distribution toward smaller amplitudes (p < 0.005, Kolmogorov-Smirnov test) and decreased the mean mEPSC amplitude significantly (p < 0.05, paired t test; n = 4) (Fig. 3b). CGRP8-37 had no effect on the frequency of mEPSCs (see normalized cumulative interevent interval distribution and mean mEPSC frequency in Fig. 3c)(p > 0.05, paired t test; n = 4).
Further arguing against a presynaptic site of action, CGRP8-37 had no significant effect on PPF. PPF refers to the observation that the amplitude of the second of two consecutive evoked EPSCs is larger than the initial EPSC if the interstimulus interval is sufficiently small. Any changes in PPF suggest a presynaptic site of action (McKernan and Shinnick-Gallagher, 1997; Neugebauer et al., 2003; Bird et al., 2005). Thus, if a drug (e.g., CGRP8-37) decreases transmitter release, PPF is enhanced. CGRP8-37 had no significant effect on PPF recorded in CeLC neurons (see individual example in Fig. 3d and summarized data in Fig. 3e). PPF was calculated as the ratio of the second and the first EPSCs evoked by two electrical stimuli of equal intensity. CGRP8-37 application did not significantly change PPF at various interstimulus intervals (n = 6; p > 0.05, paired t test). Together, the results from the analysis of PPF, mEPSCs, and neuronal excitability suggest that endogenous CGRP1 receptor activation occurs at a postsynaptic rather than presynaptic site in CeLC neurons.
CGRP1 receptors act through PKA to modulate NMDA receptor function
Our previous studies showed that postsynaptic NMDA receptor activation though a PKA-dependent mechanism plays a critical role in pain-related synaptic plasticity in the CeLC (Bird et al., 2005). Because CGRP1 receptors are known to couple to the activation of PKA (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997), we tested the hypothesis that endogenous activation of CGRP1 receptors modulates PKA and NMDA receptor function.
Pain-related synaptic plasticity was inhibited in the presence of a PKA inhibitor (KT5720, 1 μm) (Fig. 4), which has been shown before to block the NMDA-mediated component of synaptic transmission in CeLC neurons in the arthritis pain model (Bird et al., 2005). KT5720 was applied by superfusion of the slice (Fig. 4a-c) or directly into the cell through the patch pipette (Fig. 4d-f). In the presence of the PKA inhibitor, CGRP8-37 produced no additional inhibition, suggesting that CGRP1 receptor activation requires PKA because the effects of CGRP8-37 were occluded by the PKA inhibitor. Importantly, the fact that direct intracellular application of KT5720 also prevented the inhibitory effects of CGRP8-37 strongly suggests a postsynaptic site of action. The magnitude of inhibition by KT5720 and CGRP8-37 was comparable with that by an NMDA receptor antagonist (AP-5) reported previously (Bird et al., 2005). Because NMDA receptors contribute to pain-related synaptic plasticity but not normal transmission in the CeLC (Bird et al., 2005), these data suggest the selective involvement of PKA and CGRP1 receptors in NMDA-mediated synaptic plasticity.
Figure 4.
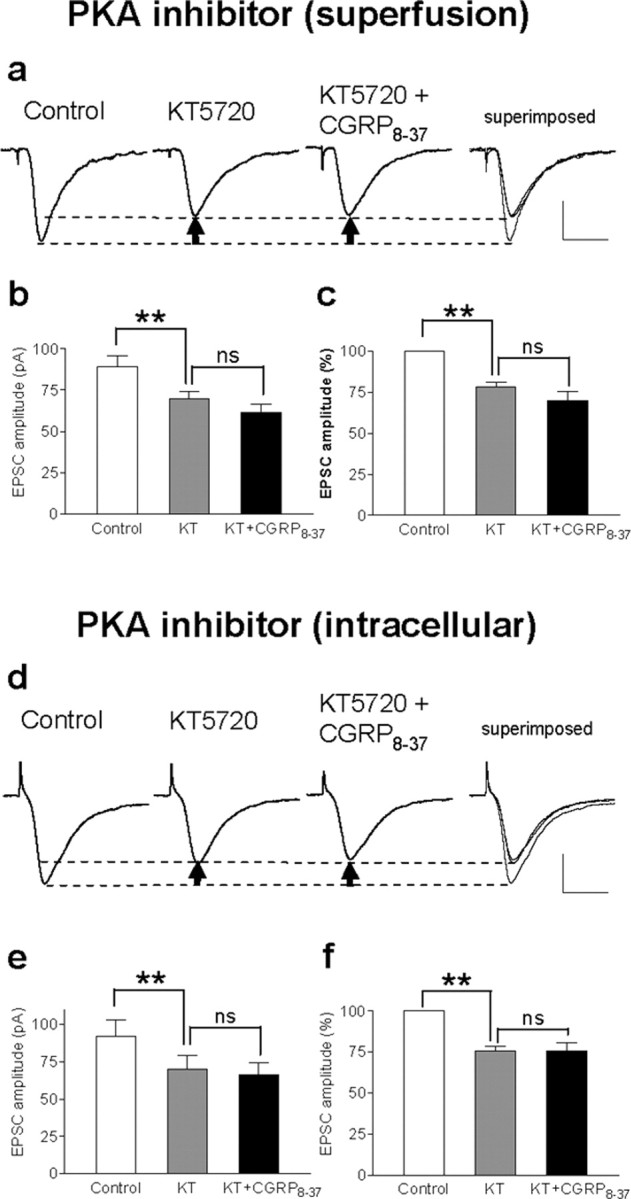
Effects of CGRP8-37 are occluded by a PKA inhibitor. a-c, Superfusion of the slices with a selective membrane-permeable PKA inhibitor (KT5720; 1μm in ACSF) decreased synaptic plasticity and abolished the inhibitory effects of CGRP8-37 (1 μm), suggesting that CGRP1 receptor activation involves PKA activation. a, Individual traces show monosynaptic EPSCs (average of 8-10 EPSCs) in a CeLC neuron in a brain slice from an arthritic rat. Recordings were made before drug application, in the presence of KT5720 alone and during coapplication of CGRP8-37 and KT5720; superimposed traces are shown on the right. Calibration: 20 ms, 40 pA. Arrows indicate difference of peak amplitudes. b, c, Averaged raw (picoamperes; b) and normalized (percentage; c) data show the significant (**p < 0.01, repeated measures ANOVA with Tukey's post hoc test) inhibitory effect of a PKA inhibitor [KT5720 (KT)] but no additional inhibition by CGRP8-37 when coapplied with KT5720. d-f, Direct intracellular application of the PKA inhibitor through the patch pipette filled with internal solution containing KT5720 (1 μm) also occluded the inhibitory effects of CGRP8-37, suggesting a direct postsynaptic mechanism. d, Monosynaptic EPSCs were measured immediately after whole-cell patch configuration was obtained control (left). EPSC amplitude decreased 10 min after the patch formation when the PKA inhibitor had entered the cell. Coapplication of CGRP8-37 (superfusion) did not further reduce EPSC amplitude. e, f, Averaged raw (picoamperes; e) and normalized (percentage; f) data show the significant (**p < 0.01, repeated measures ANOVA with Tukey's post hoc test) inhibitory effect of a PKA inhibitor (KT5720, intracellular application) but no additional inhibition by CGRP8-37. Calibration: 20 ms, 40 pA.
Subsequently, we determined the effects of CGRP8-37 on NMDA- and AMPA-mediated membrane currents, because increased NMDA, but not non-NMDA, receptor function depends on PKA activation in the CeLC in the arthritis pain model (Bird et al., 2005). Membrane currents were evoked by exogenous NMDA (1 mm) (Fig. 5a) and AMPA (Fig. 5b) applied to the recording chamber using the drop application technique (Bird et al., 2005). CGRP8-37 significantly decreased the peak amplitude and area under the curve (total charge) of the NMDA receptor-mediated inward current (p < 0.05, paired t test; n = 5) (Fig. 5a,c). However, membrane currents evoked by exogenous AMPA (30 μm) were not affected by CGRP8-37 in terms of peak amplitude and area under the curve (p > 0.05, paired t test; n = 5) (Fig. 5b,d).
Figure 5.

CGRP8-37 effects involve inhibition of NMDA, but not AMPA, receptor function. a, CGRP8-37 decreased the inward current evoked by application of exogenous NMDA (1 mm in the chamber) in a neuron from an arthritic rat. Calibration: 20 s, 200 pA. The arrrow indicates difference of peak amplitude of NMDA current before and during CGRP8-37 application. b, CGRP8-37 had no effect on the inward current evoked by application of exogenous AMPA (30 μm in the chamber) in a CeLC neuron from an arthritic rat. Calibration: 100s, 200pA. c, Averaged data show the significant inhibitory effect of CGRP8-37 on NMDA-evoked membrane currents in terms of peak current and area under the curve (n=5; paired t test, p < 0.05). d, AMPA-evoked membrane currents were not affected significantly by CGRP8-37 (n = 5; paired t test, p > 0.05). Symbols and error bars represent mean ± SE. Neurons were recorded in voltage clamp at -60 mV. *p < 0.05.
Together with our previous studies (Bird et al., 2005), these data show that CGRP1 receptors contribute to synaptic plasticity through the activation of PKA and PKA-dependent postsynaptic NMDA receptor function.
Endogenous activation of CGRP1 receptors is required for pain-related sensitization of CeLC neurons
Whereas the reduced brain slice preparation allows the definitive analysis of neuronal plasticity maintained in a particular brain area, the processing of pain-related information in identified nociceptive neurons and its behavioral consequence can only be studied at the systems level in the whole animal. To determine the significance of CGRP1 receptor activation, we therefore studied the effects of CGRP1 receptor block on electrophysiological responses in anesthetized animals and on behavioral responses in awake animals.
Extracellular single-unit recordings were made from CeLC neurons in anesthetized rats, and drugs were administered into the CeLC through microdialysis probes as described in detail previously (Neugebauer and Li, 2003; Li and Neugebauer, 2004a). All CeLC neurons included in this study responded more strongly to noxious (painful in the awake subject) than innocuous stimuli and were classified as MR neurons. MR neurons represent the class of amygdala neurons that consistently develop sensitization to afferent inputs in the arthritis pain model (Neugebauer and Li, 2003; Li and Neugebauer, 2004a). Responses to innocuous and noxious stimuli were recorded continuously in the same neuron before and after the induction of a localized mono-arthritis in the knee (Neugebauer et al., 2004). Figure 6a shows an individual example (histograms display the number of action potentials or spikes per second). The CeLC neuron responded more strongly to brief (15 s) noxious than innocuous stimulation (compression) of the knee joint under control conditions. In the arthritis pain state (6 h after induction), the responses of the same neuron increased not only to stimulation of the arthritic knee but also of non-injured tissue in the ankle, paw, and forelimb (data not shown). The increased responsiveness of neurons in the CNS to stimulation of non-injured tissues is generally accepted to indicate a state of “central sensitization” and is now also well documented for CeLC neurons (Neugebauer et al., 2004). Administration of CGRP8-37 (100 μm, concentration in microdialysis fiber; 20 min) into the CeLC next to the recorded neuron strongly reduced the enhanced responses in a reversible manner.
Figure 6.
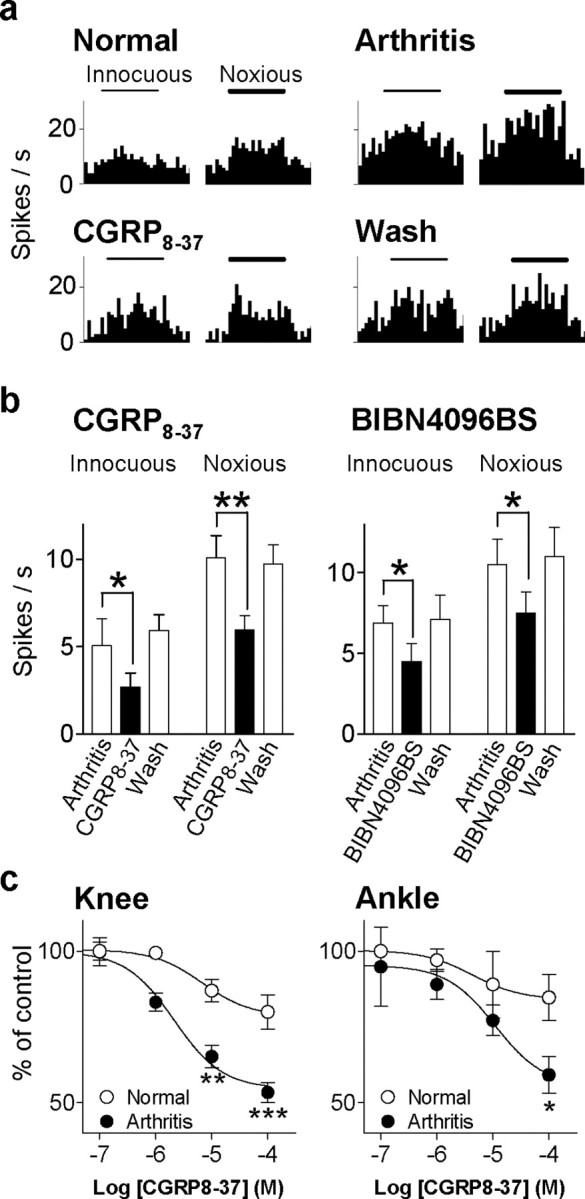
CGRP1 receptor antagonists inhibit nociceptive sensitization of CeLC neurons in anesthetized intact animals. a, Extracellular recordings of the responses of one multireceptive CeLC neuron to innocuous (100 g/30 mm2) and noxious (2000 g/30 mm2) mechanical stimulation of the knee joint before and 6 h after induction of the knee joint arthritis (see Materials and Methods). CGRP8-37 (100μm) was administered into the CeLC by microdialysis for 20 min. Histograms show action potentials (spikes) per second (bin width, 1 s). Horizontal bars indicate duration of stimuli (15 s). b, Averaged raw data (spikes per second) for the sample of neurons tested with CGRP8-37 (100 μm; n = 7) and BIBN4096BS (100 μm; n = 4). Both antagonists significantly reduced the increased responses to normal levels (p < 0.01-0.05, paired t test). The inhibitory effects were reversible after washout for 20-30 min with ACSF in the microdialysis fiber. Bar histograms and error bars represent mean ± SE. *p < 0.05; **p < 0.01. c, Concentration-response relationships show that CGRP8-37 was more efficacious in sensitized neurons 6 h after induction of arthritis (n = 7) than under normal conditions before arthritis (n = 7). The differences of drug effects between arthritis and normal conditions were statistically significant (knee, p < 0.001, F(1,25) = 26.57; ankle, p < 0.05, F(1,25) = 6.36; two-way ANOVA). *p < 0.05; **p < 0.01; ***p < 0.001 (Bonferroni's post hoc tests). Responses to brief (15 s) noxious stimulation of the knee or ankle during each concentration of CGRP8-37 were averaged and expressed as percentage of predrug (baseline) control (100%). CGRP8-37 was administered by microdialysis for 20 min, and measurements were made at 15-20 min. Concentrations shown indicate concentrations in the microdialysis fiber.
The inhibitory effects of CGRP1 receptor antagonists on pain-related sensitization in the amygdala (CeLC) are summarized in Figure 6b. CGRP8-37 (n = 7) and BIBN4096BS (n = 4) significantly (p < 0.01-0.05, paired t test) reduced the enhanced responses of CeLC neurons to stimulation of the arthritic knee with normally innocuous and noxious intensities (see Materials and Methods). Drug effects were reversible after washout for 20-30 min. Antagonists were administered by microdialysis into the CeLC (100 μm, concentration in microdialysis fiber; 20 min). Figure 6c shows the functional change of endogenously activated CGRP1 receptors in the CeLC in the arthritis pain model compared with normal conditions. Concentration-response relationships were obtained for the effects of CGRP8-37 on the responses of CeLC neurons to noxious stimulation of the arthritic knee and the non-injured ankle. CGRP8-37 was more efficacious in sensitized neurons 6 h after induction of arthritis (n = 7) than under normal conditions before arthritis (n = 7). The differences of drug effects between arthritis and normal conditions were statistically significant (knee, p < 0.001, F(1,25) = 26.57; ankle, p < 0.05, F(1,25) = 6.36; two-way ANOVA). The IC50 values were not significantly different (knee, 6.8 and 2.1 μm; ankle 4.3 and 11 μm, normal versus arthritis; concentrations in the microdialysis fiber). CGRP8-37 was administered by microdialysis for 20 min, and measurements were made at 15 min.
Recordings and drug administrations were made into the right CeLC contralateral to the arthritis because of the strong contralateral projection of the spino-parabrachio-amygdaloid pain pathway and our previous studies showing pain-related plasticity in the right (contralateral) CeLC (Neugebauer et al., 2004). All recording sites in the CeLC were confirmed histologically (see Fig. 8b). These data suggest that CGRP1 receptors in the CeLC are activated endogenously in the arthritis pain state in the intact animal and are required for the pain-related sensitization of CeLC neurons.
Figure 8.

Histologic verification of electrophysiological recording sites and drug application sites. a-d, Standard diagrams (adapted from Paxinos and Watson, 1998) show coronal sections through the right brain hemisphere at different levels posterior to bregma (-1.88 to -2.12 mm). a, In vitro patch-clamp recordings were made in the shaded area (CeLC). Stimulating electrode was positioned on the fiber tract of afferents from the parabrachial area (dashed line). The boundaries of the different amygdaloid nuclei as well as the fibers from the brainstem can be easily visualized under the microscope. b, Recording sites of CeLC neurons in the in vivo electrophysiological studies using extracellular single-unit recordings. c, Sites of drug application into the CeLC by microdialysis in the vocalization experiments. d, Sites of drug application into the striatum by microdialysis as placement controls for any effects attributable to drug diffusion. CeM, Medial division of the central nucleus of the amygdala; CeL, lateral division of the central nucleus of the amygdala.
Endogenous activation of CGRP1 receptors in the amygdala is required for pain behavior organized at different levels of the pain neuraxis
To validate the significance of the CGRP1 receptor activation observed in the electrophysiological studies, we analyzed the effect of CGRP8-37 on supraspinally (vocalizations) and spinally (hindlimb withdrawal reflexes) organized behavior in awake animals. Pain-related vocalizations in the audible and ultrasonic range were measured in the same animal before and after induction of arthritis and before and during administration of CGRP8-37 into the CeLC by microdialysis as described previously (Han and Neugebauer, 2005). Audible and ultrasonic vocalizations evoked by noxious stimuli represent nocifensive and affective responses, respectively (Han and Neugebauer, 2005). Vocalizations are further classified as VDSs, which are organized at the medullary brainstem level, and VADs, which outlast the actual stimulus and are organized in the limbic forebrain, particularly the amygdala (Borszcz and Leaton, 2003; Han and Neugebauer, 2005).
The duration of audible and ultrasonic vocalizations of the VAD and VDS types increased in the arthritis pain model (6 h after induction) (Fig. 7a,b). Administration of CGRP8-37 (100 μm, concentration in the microdialysis probe; 15-20 min) into the CeLC inhibited the duration of audible and ultrasonic VADs (Fig. 7a, right) and VDSs (Fig. 7b, right) evoked by noxious (2000 g/30 mm2) stimulation (15 s) of the arthritic knee (n = 9). The inhibitory effects of CGRP8-37 were significant (p < 0.01-0.05; paired t test) and reversible. In contrast, CGRP8-37 had no significant effect on the vocalizations of naive (non-arthritic) animals (n = 5) (Fig. 7a,b, left). Predrug vocalizations were measured during administration of ACSF through the microdialysis probe, thus serving as vehicle controls. Drugs were administered into the right CeLC contralateral to the arthritis because of the strong contralateral projection of the spino-parabrachio-amygdaloid pain pathway and our previous studies showing pain-related plasticity in the right CeLC (Neugebauer et al., 2004). All animals had guide cannulas for the microdialysis probes implanted on the day before the behavioral tests. The positions of the microdialysis probes in the CeLC were verified histologically (Fig. 8c).
The vocalization data suggest that chemical inactivation of the CeLC by CGRP8-37 inhibits pain responses organized in the brainstem (VDS) and limbic forebrain (VAD). Next we determined the contribution of CGRP1 receptors in the CeLC to pain responses organized at the level of the spinal cord. Hindlimb withdrawal reflexes in response to stimulation (compression) of the knee were measured before and after induction of arthritis and before and during drug application (Fig. 7c). Mechanical stimuli of increasing intensity (steps of 50 g/30 mm2) were applied to the knee joint. Withdrawal threshold was defined as the minimum stimulus intensity that evoked a withdrawal reflex (Han and Neugebauer, 2005). Administration of CGRP8-37 (100 μm; 15 min) into the CeLC had no significant effect on the withdrawal thresholds measured in naive (non-arthritic) animals (n = 4) (Fig. 7c, left). After arthritis induction, hindlimb withdrawal thresholds decreased, suggesting increased pain sensitivity (Fig. 7c, right). Administration of CGRP8-37 (100 μm; 15 min) into the CeLC partially reversed the effects of arthritis by increasing the hindlimb withdrawal thresholds significantly (n = 6; p < 0.05, paired t test), suggesting an important role of CGRP1 receptor activation in the amygdala in the descending modulation of spinal nociceptive processing.
As a control for any drug effects attributable to diffusion from the microdialysis probe to other brain areas, microdialysis probes were stereotaxically inserted into the striatum (caudate-putamen) for drug application in a separate set of animals as placement controls (Fig. 7d). The striatum was selected because it is located adjacent (dorsolateral) to the CeLC but does not form direct projections to the CeLC (Neugebauer et al., 2004; Han and Neugebauer, 2005). Thus, drug application into this area should not have any effect on CeLC-mediated functions. This site, however, is sufficiently close to the CeLC to be useful as a control for drug diffusion. Administration of CGRP8-37 (100 μm, 15 min; n = 5) (Fig. 7d) into the striatum had no significant effects on the audible and ultrasonic vocalizations and withdrawal reflexes (data not shown) in arthritic animals evoked by stimulation of the knee (p > 0.05, paired t test). The positions of the microdialysis probes in the CeLC were verified histologically (Fig. 8d).
Discussion
The present study is the first to show that the endogenous activation of CGRP1 receptors in the nociceptive amygdala (CeLC) contributes critically to pain-related synaptic plasticity in the CeLC and consequently to pain behavior. Our integrative approach of in vitro and in vivo electrophysiology and behavioral analysis allowed us to determine the cellular mechanisms of CGRP1 receptor function and their significance at the systems level. The major findings are as follows. (1) Selective CGRP1 receptor antagonists (CGRP8-37 and BIBN4096BS) inhibited synaptic plasticity and neuronal excitability in CeLC neurons in vitro in the arthritis pain model induced in vivo. (2) Analysis of spontaneous miniature EPSCs, PPF, and membrane effects indicated a postsynaptic rather than presynaptic mechanism. (3) The occlusion of CGRP8-37 effects by a PKA inhibitor and the direct inhibition of NMDA, but not AMPA, receptor activation by CGRP8-37 suggested that CGRP1 receptors couple to PKA activation and NMDA receptor function. (4) CGRP8-37 and BIBN4096BS reversed the pain-related sensitization of nociceptive CeLC neurons recorded in vivo. (5) Chemical inactivation of the CeLC by CGRP8-37 inhibited spinally (withdrawal reflexes) and supraspinally (vocalizations) organized pain behavior in awake animals.
The amygdala is well positioned to play an important role in the clinically important reciprocal relationship between pain and emotional-affective states (Rhudy and Meagher, 2001; Gallagher and Verma, 2004). The amygdala has long been known to be critically involved in mechanisms of fear, anxiety, and depression (Davis, 1998; Davidson et al., 1999; LeDoux, 2000; Rodrigues et al., 2004). More recent research has linked the amygdala also to the pain system (Neugebauer et al., 2004). The present study makes a major contribution toward a better understanding of the pain-affect relationship by providing valuable novel information about mechanisms of plasticity in the nociceptive amygdala and their behavioral consequences. Assuming that affective states and disorders can mimic pain-related plasticity in the amygdala, they would be able to gain access to the modulation of pain and pain behavior through the nociceptive amygdala. Although this hypothesis awaits proof, the present study sets the stage for mechanistic research into the pain-affect interaction.
Our previous studies showed plastic changes in the nociceptive amygdala in an arthritis pain model (Neugebauer and Li, 2003; Neugebauer et al., 2003; Li and Neugebauer, 2004a,b; Bird et al., 2005). Plasticity was measured as increased synaptic transmission in the nociceptive parabrachio-amygdaloid pathway, enhanced processing of nociceptive signals (sensitization), and increased neuronal excitability of CeLC neurons, which would result in increased output functions of the amygdala. Mechanisms and consequences of such plastic changes are only beginning to emerge (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Pain-related plasticity in the nociceptive amygdala (CeLC) depends on the activation and upregulation of presynaptic metabotropic glutamate receptors of the mGluR1 subtype (Neugebauer et al., 2003; Li and Neugebauer, 2004a), which can regulate the release of glutamate and other transmitters and modulators, including neuropeptides (Cartmell and Schoepp, 2000). Postsynaptically, normally “silent” NMDA receptors become functional through phosphorylation, but not upregulation, that depends on PKA but not PKC (Li and Neugebauer, 2004b; Bird et al., 2005). Which molecule, however, can account for PKA activation and thus serve as the missing link between presynaptic and postsynaptic sites?
CGRP is a prime candidate for several reasons. The exclusive source of CGRP in the amygdala is the lateral parabrachial area (Kruger et al., 1988; Schwaber et al., 1988; Harrigan et al., 1994; de Lacalle and Saper, 2000), which is part of the spino-parabrachio-amygdaloid pain pathway (Bernard and Bandler, 1998). CGRP-immunoreactive terminals target specifically the CeLC and innervate CeLC neurons that project to brainstem areas such as the periaqueductal gray (Schwaber et al., 1988; Harrigan et al., 1994; Xu et al., 2003). The central nucleus of the amygdala also contains particularly high levels of CGRP binding sites (Van Rossum et al., 1997; Oliver et al., 1998) but no CGRP mRNA-expressing or CGRP-immunoreactive neurons (Van Rossum et al., 1997). This mismatch suggests that the endogenous activation of CGRP receptors observed in the present study is attributable to release of CGRP from the spino-parabrachio-amygdaloid pain pathway but not from intrinsic circuits.
CGRP binds to G-protein-coupled receptors, which activate adenylyl cyclase and cAMP-dependent PKA (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997). Pharmacologically, two classes of CGRP receptors have been proposed (CGRP1 and CGRP2 receptors), which have no significant affinity for calcitonin-like peptides (Poyner, 1996; Wimalawansa, 1996; Oliver et al., 1998; Hasbak et al., 2003). CGRP1 but not CGRP2 receptors have been cloned (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997) and are expressed at particularly high levels in the central nucleus of the amygdala (Oliver et al., 1998). Specific antagonists such as the C-terminal fragment CGRP8-37 and the nonpeptide compound BIBN4096BS are available for CGRP1 but not CGRP2 receptors (Poyner, 1996; Wimalawansa, 1996; Van Rossum et al., 1997; Doods et al., 2000). CGRP1 receptors consist of three different proteins: the calcitonin receptor-like receptor (CRLR), receptor activity-modifying protein (RAMP1), and the receptor component protein (RCP) (Hasbak et al., 2003). RAMP1 defines the ligand-binding site, whereas RCP couples the receptor to signal transduction pathways (Hasbak et al., 2003). The low nanomolar affinities for CGRP8-37 measured in the present study are consistent with the binding to the CRLR component of the CGRP1 receptor (Oliver et al., 1998; Hasbak et al., 2003). The change in efficacy of CGRP8-37 can be the result of increased CGRP release as well as RCP-mediated enhanced coupling of the CGRP1 receptor to second messengers, including PKA activation.
The present study shows that CGRP1 serves as the critical molecule to link presynaptic and postsynaptic mechanisms of pain-related plasticity in the CeLC and contributes to pain behavior organized at different levels of the pain neuraxis. Patch-clamp experiments using CGRP8-37, PKA inhibition, and NMDA receptor activation suggest that CGRP1 receptor activation can modulate NMDA receptor function through PKA (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The synaptic component inhibited by block of PKA and/or CGRP1 receptors was comparable with that mediated by NMDA receptors in CeLC plasticity shown previously (Bird et al., 2005), suggesting the selective involvement of PKA and CGRP1 receptors in NMDA-mediated synaptic plasticity. This conclusion is also supported by the direct inhibitory effect of CGRP8-37 on NMDA-, but not AMPA-, evoked membrane currents (Fig. 5). Our data further suggest that CGRP1 receptor activation occurs at the postsynaptic rather than presynaptic site (Figs. 2, 3). CGRP8-37 inhibited action potential firing evoked by direct depolarizing current injections into the neuron (postsynaptic site). PPF, a measure of presynaptic changes, was not affected. Amplitude distribution (quantal size), but not frequency, of spontaneous miniature EPSCs was decreased by CGRP8-37. CGRP8-37 inhibited the membrane current evoked by direct application of NMDA.
Whereas the involvement of CGRP receptors in peripheral and spinal pain mechanisms is well documented (Galeazza et al., 1995; Neugebauer et al., 1996; Schaible, 1996; Ruda et al., 2000; Sun et al., 2004), less is known about the role of CGRP receptors in the brain in models of prolonged or persistent pain states. One laboratory reported that administration of exogenous CGRP into periaqueductal gray (Yu et al., 2003), nucleus accumbens (Li et al., 2001), or amygdala (Xu et al., 2003) had antinociceptive behavioral effects in naive animals. Our electrophysiological data show that CGRP1 antagonists inhibit the processing of nociceptive signals in CeLC neurons in the arthritis model of persistent pain. Our behavioral studies further suggest that block of CGRP1 receptors in the CeLC inhibits spinally and supraspinally organized pain behavior, which is consistent with the inhibition of amygdala-mediated descending pain facilitation (Gebhart, 2004; Neugebauer et al., 2004; Vanegas and Schaible, 2004). Vocalizations have been used successfully previously to determine higher integrated pain behavior (Borszcz and Leaton, 2003; Han and Neugebauer, 2005; Han et al., 2005; Ko et al., 2005). Whereas our antagonist study suggests that endogenous activation of CGRP1 receptors in the amygdala produces pain behavior through descending facilitation, one behavioral study reported antinociceptive effects of exogenous CGRP administration into the central nucleus of the amygdala (Xu et al., 2003) (see above). However, these experiments were done in normal rats (not in a pain model). Additionally, drugs were administered into the left amygdala. In the present study, we targeted the right amygdala because our previous electrophysiological in vivo and in vitro studies showed pain-related plasticity in the right amygdala (Neugebauer and Li, 2003; Neugebauer et al., 2003) and our behavioral data indicated that the right amygdala is coupled to pain facilitation in the arthritis pain model (Han and Neugebauer, 2005). This is consistent with a strong contralateral projection of the spino-parabrachio-amygdaloid pain pathway (Bernard and Bandler, 1998; Neugebauer et al., 2004) (arthritis was induced in the left knee in this and our previous studies). It remains to be determined whether lateralization or differences between normal conditions and persistent pain can account for this difference.
In conclusion, the present study shows for the first time that the endogenous activation of CGRP1 receptors in the CeLC is critically involved in pain-related plasticity (in vitro and in vivo electrophysiology) and contributes to nociceptive and affective pain responses (audible and ultrasonic vocalizations, respectively). Therefore, CGRP1 receptors in the amygdala may be a novel therapeutic target for pain relief. Furthermore, they can play an important role in the pain-affect interaction if affective states and disorders indeed mimic pain-related plasticity in the amygdala and thus gain access to pain modulation. Our proposed model (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) will allow testing of this hypothesis.
Footnotes
This work was supported by National Institutes of Health Grants NS38261 and NS11255 and by John Sealy Memorial Endowment Fund Grant 2521-04. We thank Dr. William D. Willis for his helpful comments on this manuscript. We also thank Dr. Henri Doods (Boehringer Ingelheim Pharma, Biberach, Germany) for the generous gift of BIBN4096BS.
Correspondence should be addressed to Dr. Volker Neugebauer, Department of Neuroscience and Cell Biology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555-1069. E-mail: voneugeb@utmb.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510717-12$15.00/0
References
- Bernard J-F, Bandler R (1998) Parallel circuits for emotional coping behaviour: new pieces in the puzzle. J Comp Neurol 401: 429-436. [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V (2005) Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol (Lond) 564: 907-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borszcz GS, Leaton RN (2003) The effect of amygdala lesions on conditional and unconditional vocalizations in rats. Neurobiol Learn Mem 79: 212-225. [DOI] [PubMed] [Google Scholar]
- Cabell L, Audesirk G (1993) Effects of selective inhibition of protein kinase C, cyclic AMP-dependent protein kinase, and Ca2+-calmodulin-dependent protein kinase on neurite development in cultured rat hippocampal neurons. Int J Dev Neurosci 11: 357-368. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75: 889-907. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Abercrombie H, Nitschke JB, Putnam K (1999) Regional brain function, emotion and disorders of emotion. Curr Opin Neurobiol 9: 228-234. [DOI] [PubMed] [Google Scholar]
- Davis M (1998) Anatomic and physiologic substrates of emotion in an animal model. J Clin Neurophysiol 15: 378-387. [DOI] [PubMed] [Google Scholar]
- de Lacalle S, Saper CB (2000) Calcitonin gene-related peptide-like immunoreactivity marks putative visceral sensory pathways in human brain. Neuroscience 100: 115-130. [DOI] [PubMed] [Google Scholar]
- Doods H, Hallermayer G, Wu D, Entzeroth M, Rudolf K, Engel W, Eberlein W (2000) Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol 129: 420-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeazza MT, Garry MG, Yost HJ, Strait KA, Hargreaves KM, Seybold VS (1995) Plasticity in the synthesis and storage of substance P and calcitonin gene-related peptide in primary afferent neurons during peripheral inflammation. Neuroscience 66: 443-458. [DOI] [PubMed] [Google Scholar]
- Gallagher RM, Verma S (2004) Mood and anxiety disorders in chronic pain. In: Progress in pain research and management, Vol 27, Psychosocial aspects of pain: a handbook for health care providers, Pt II, Evaluating pain patients (Dworkin RH, Breitbard WS, eds), pp 139-178. Seattle: IASP.
- Gebhart GF (2004) Descending modulation of pain. Neurosci Biobehav Rev 27: 729-737. [DOI] [PubMed] [Google Scholar]
- Han JS, Neugebauer V (2005) mGluR1 and mGluR5 antagonists in the amygdala inhibit different components of audible and ultrasonic vocalizations in a model of arthritic pain. Pain 113: 211-222. [DOI] [PubMed] [Google Scholar]
- Han JS, Bird GC, Neugebauer V (2004) Enhanced group III mGluR-mediated inhibition of pain-related synaptic plasticity in the amygdala. Neuropharmacology 46: 918-926. [DOI] [PubMed] [Google Scholar]
- Han JS, Bird GC, Li W, Neugebauer V (2005) Computerized analysis of audible and ultrasonic vocalizations of rats as a standarized measure of pain-related behavior. J Neurosci Methods 141: 261-269. [DOI] [PubMed] [Google Scholar]
- Harrigan EA, Magnuson DJ, Thunstedt GM, Gray TS (1994) Corticotropin releasing factor neurons are innervated by calcitonin gene-related peptide terminals in the rat central amygdaloid nucleus. Brain Res Bull 33: 529-534. [DOI] [PubMed] [Google Scholar]
- Hasbak P, Opgaard OS, Eskesen K, Schifter S, Arendrup H, Longmore J, Edvinsson L (2003) Investigation of CGRP receptors and peptide pharmacology in human coronary arteries. Characterization with a nonpeptide antagonist. J Pharmacol Exp Ther 304: 326-333. [DOI] [PubMed] [Google Scholar]
- Ko SW, Chatila T, Zhuo M (2005) Contribution of CaMKIV to injury and fear- induced ultrasonic vocalizations in adult mice. Mol Pain 1: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger L, Sternini C, Brecha NC, Mantyh PW (1988) Distribution of calcitonin gene-related peptide immunoreactivity in relation to the rat central somatosensory projection. J Comp Neurol 273: 149-162. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155-184. [DOI] [PubMed] [Google Scholar]
- Li N, Lundeberg T, Yu LC (2001) Involvement of CGRP and CGRP1 receptor in nociception in the nucleus accumbens of rats. Brain Res 901: 161-166. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V (2004a) Differential roles of mGluR1 and mGluR5 in brief and prolonged nociceptive processing in central amygdala neurons. J Neurophysiol 91: 13-24. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V (2004b) Block of NMDA and non-NMDA receptor activation results in reduced background and evoked activity of central amygdala neurons in a model of arthritic pain. Pain 110: 112-122. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607-611. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W (2003) Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol 89: 716-727. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Rumenapp P, Schaible H-G (1996) Calcitonin gene-related peptide is involved in the spinal processing of mechanosensory input from the rat's knee joint and in the generation and maintenance of hyperexcitability of dorsal horn neurons during development of acute inflammation. Neuroscience 71: 1095-1109. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW (2003) Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci 23: 52-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS (2004) The amygdala and persistent pain. The Neuroscientist 10: 221-234. [DOI] [PubMed] [Google Scholar]
- Oliver KR, Wainwright A, Heavens RP, Hill RG, Sirinathsinghji DJS (1998) Distribution of novel CGRP1 receptor and adrenomedullin receptor mRNAs in the rat central nervous system. Brain Res Mol Brain Res 57: 149-154. [DOI] [PubMed] [Google Scholar]
- Pare D, Quirk GJ, Ledoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1-9. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. New York: Academic.
- Poyner D (1996) Pharmacology of receptors for calcitonin gene-related peptide and amylin. Trends Pharmacol Sci 16: 424-429. [DOI] [PubMed] [Google Scholar]
- Rhudy JL, Meagher MW (2001) The role of emotion in pain modulation. Curr Opin Psychiatry 14: 241-245. [Google Scholar]
- Rodrigues SM, Schafe GE, Ledoux JE (2004) Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron 44: 75-91. [DOI] [PubMed] [Google Scholar]
- Ruda MA, Ling QD, Hohmann AG, Peng YB, Tachibana T (2000) Altered nociceptive neuronal circuits after neonatal peripheral inflammation. Science 289: 628-631. [DOI] [PubMed] [Google Scholar]
- Schaible H-G (1996) On the role of tachykinins and calcitonin gene-related peptide in the spinal mechanisms of nociception and in the induction and maintenance of inflammation-evoked hyperexcitability in spinal cord neurons (with special reference to nociception in joints). Prog Brain Res 113: 423-441. [DOI] [PubMed] [Google Scholar]
- Schwaber JS, Sternini C, Brecha NC, Rogers WT, Card JP (1988) Neurons containing calcitonin gene-related peptide in the parabrachial nucleus project to the central nucleus of the amygdala. J Comp Neurol 270: 416-426. [DOI] [PubMed] [Google Scholar]
- Skofitsch G, Jacobowitz DM (1985) Calcitonin gene-related peptide: detailed immunohistochemical distribution in the central nervous system. Peptides 6: 721-745. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Lawand NB, Lin Q, Willis WD (2004) Role of calcitonin gene-related peptide in the sensitization of dorsal horn neurons to mechanical stimulation after intradermal injection of capsaicin. J Neurophysiol 92: 320-326. [DOI] [PubMed] [Google Scholar]
- Van Rossum D, Hanish U-K, Quirion R (1997) Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev 21: 649-678. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG (2004) Descending control of persistent pain: inhibitory or facilitatory? Brain Res Rev 46: 295-309. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M (2004) Are fear memories made and maintained by the same NMDA receptor-dependent mechanisms? Neuron 41: 680-682. [DOI] [PubMed] [Google Scholar]
- Wimalawansa SJ (1996) Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocr Rev 17: 533-585. [DOI] [PubMed] [Google Scholar]
- Wyllie DJ, Manabe T, Nicoll RA (1994) A rise in postsynaptic Ca2+ potentiates miniature excitatory postsynaptic currents and AMPA responses in hippocampal neurons. Neuron 12: 127-138. [DOI] [PubMed] [Google Scholar]
- Xu W, Lundeberg T, Wang YT, Li Y, Yu L-C (2003) Antinociceptive effect of calcitonin gene-related peptide in the central nucleus of amygdala: activating opioid receptors through amygdala-periaqueductal gray pathway. Neuroscience 118: 1015-1022. [DOI] [PubMed] [Google Scholar]
- Yu LC, Weng XH, Wang JW, Lundeberg T (2003) Involvement of calcitonin gene-related peptide and its receptor in anti-nociception in the periaqueductal grey of rats. Neurosci Lett 349: 1-4. [DOI] [PubMed] [Google Scholar]