

Abstract
Early events in Alzheimer's disease (AD) pathogenesis implicate the accumulation of β-amyloid (Aβ) peptide inside neurons in vulnerable brain regions. However, little is known about the consequences of intraneuronal Aβ on signaling mechanisms. Here, we demonstrate, using an inducible viral vector system to drive intracellular expression of Aβ42 peptide in primary neuronal cultures, that this accumulation results in the inhibition of the Akt survival signaling pathway. Induction of intraneuronal Aβ42 expression leads to a sequential decrease in levels of phospho-Akt, increase in activation of glycogen synthase kinase-3β, and apoptosis. Downregulation of Akt also paralleled intracellular Aβ accumulation in vivo in the Tg2576 AD mouse model. Overexpression of constitutively active Akt reversed the toxic effects of Aβ through a mechanism involving the induction of heat shock proteins (Hsps). We used a small-interfering RNA approach to explore the possibility of a link between Akt activity and Hsp70 expression and concluded that neuroprotection by Akt could be mediated through downstream induction of Hsp70 expression. These results suggest that the early dysfunction associated with intraneuronal Aβ accumulation in AD involve the associated impairments of Akt signaling and suppression of the stress response.
Keywords: Alzheimer, intracellular, amyloid, Akt, Hsp, stress response
Introduction
Scientific efforts are increasingly directed to understand the earliest events of neuronal dysfunction, synaptic and neuronal loss, and dementia in Alzheimer's disease (AD) (Wirths et al., 2004). Although extracellular fibrillar β-amyloid (Aβ) is the major constituent of amyloid plaques and can cause neurotoxicity both in culture and in vivo (Yankner et al., 1990; Kowall et al., 1992; Pike et al., 1993), intraneuronal Aβ accumulation precedes the deposition of amyloid plaques and the appearance of neurofibrillary tangles and correlates with the first indications of cognitive deficits (Gouras et al., 2000; D'Andrea et al., 2001; Oddo et al., 2003; Billings et al., 2005). During early stages of the disease, intracellular accumulation of Aβ appears to compromise neuronal function, synaptic strength, and survival (Yang et al., 1998; Chui et al., 2001; Oddo et al., 2003; Takahashi et al., 2004; Billings et al., 2005). Consistent with clinical observations on the importance of intracellular accumulation, we and others have reported on the toxic effect of intraneuronal Aβ accumulation in model systems (LaFerla et al., 1995; Zhang et al., 2002; Magrane et al., 2004).
The effect of intraneuronal Aβ expression on signaling pathways has not been thoroughly explored. Previous work using a transgenic mouse model in which Aβ42 was expressed intraneuronally showed that cell loss occurred through a p53-dependent mechanism (LaFerla et al., 1996). This observation was confirmed when synthetic Aβ peptides microinjected into neurons were shown to be cytotoxic through a p53-Bax pathway (Zhang et al., 2002). Using an inducible adenoviral-based system, we studied the effects of intracellular Aβ42 expression and reported that the cellular stress response was an important modulator of intraneuronal Aβ toxicity (Magrane et al., 2004). Changes in expression of αB-crystallin and heat shock protein 16 (Hsp16), among others, in response to intracellular Aβ42 expression have also been studied (Link et al., 1999, 2003).
The serine/threonine protein kinase B, also known as Akt, is highly expressed in the brain and has an important role in neuronal survival (Brunet et al., 2001; Brazil et al., 2002). The Akt signaling pathway is altered in diverse neurological conditions, including spinocerebellar ataxia type 1 (Chen et al., 2003; Emamian et al., 2003), amyotrophic lateral sclerosis (Wagey et al., 1998), Huntington's disease (Humbert et al., 2002; Gines et al., 2003), and schizophrenia (Emamian et al., 2004). Insulin resistance in diabetes and the genetic variability in the insulin signal transduction pathway critically impair Akt signaling and are associated with increased risk for AD (Liolitsa et al., 2002; Ho et al., 2004). Several studies have shown that familial AD mutations caused a downregulation of the phosphatidyl inositol 3-kinase (PI3K)-Akt pathway; however, the role of intracellular Aβ was not examined (Kashour et al., 2003; Ryder et al., 2004). In addition, presenilin mutations also sensitize neurons to undergo apoptosis, a process that has been shown to include downregulation of Akt (Weihl et al., 1999; Baki et al., 2004).
The elucidation of details in the signaling pathways affected by intracellular Aβ accumulation is important not only to understand the pathogenesis of AD but also instrumental to design effective treatment and prevention. Here, we examine the effects of Aβ42 on signal transduction pathways involved in neuron survival. Our data suggest a role for the Akt pathway in intracellular Aβ toxicity. In addition, we show that Akt activation is required to induce the cellular stress response that protects neurons against Aβ42 toxicity.
Materials and Methods
Antibodies. The following antibodies were used: anti-Aβ 6E10 (1:300; Signet Laboratories, Dedham, MA) and anti-Aβ42 (1:150; Chemicon, Temecula, CA); JHC Specific anti-phospho-Akt Ser473 (1:200), monoclonal anti-phospho-Akt Ser473 (587F11; 1:100), polyclonal anti-phospho-Akt Ser473, anti-phospho-glycogen synthase kinase-3β (GSK-3β) Ser9, anti-phospho-extracellular signal-regulated kinase 1/2 (ERK1/2), and anti-phospho-c-Jun N-terminal protein kinase (JNK) (1:1000) (all from Cell Signaling Technology, Beverly, MA); anti-Akt, anti-GSK-3β, anti-ERK1/2, anti-JNK, anti-p53, and anti-Hsp90 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA); anti-phospho-GSK-3α (Tyr279)/β (Tyr216) (1:1000; Biosource International, Camarillo, CA); anti-Hsp70 (1:100-5000), anti-Hsp40 (1:10,000), anti-Hsp27 (1:1000), and anti-Hsc70 (1:5000; Stressgen Biotechnologies, San Diego, CA); anti-α isoform of tubulin (1:1000; Sigma, St. Louis, MO); anti-HA tag (1:6000; Abcam, Cambridge, MA), anti-β-galactosidase (1:500; Promega, Madison, WI). Secondary antibodies were used as follows: fluorescent cyanine 2 (Cy2)- and Cy3-conjugated (Jackson ImmunoResearch, West Grove, PA) and peroxidase-conjugated (Dako, High Wycombe, UK).
Adenoviral vector construction. Recombinant, E1-deleted replication-defective adenoviral constructs were produced by standard techniques (Graham and Prevec, 1995). The AdTet-On, AdTRE-LacZ, AdTRE-Aβ42, Ad-Hsp70, and Ad-APP751 viruses have been described previously (Magrane et al., 2004). Ad-myrAkt and Ad-dnAkt expressed an active and inactive form of Akt, respectively, as described previously (Fujio and Walsh, 1999). Ad-HSP-Luc vector expressed the transgene luciferase under the control of the Hsp70B promoter (Smith et al., 2002). All viruses were grown to high titer in human embryonic kidney 293 cells and purified by cesium chloride density gradient ultracentrifugation. Viral titer was determined by plaque assay.
Neuronal cultures. For rat primary cultures, cortices were dissected from neonatal day 1 Sprague Dawley rat brains (Charles River Laboratories, Wilmington, MA), and neurons were prepared as described previously (Magrane et al., 2004). Primary cortical cultures were infected 7 d after plating at multiplicities of infection (m.o.i.) ranging from 50 to 100. AdTet-On and AdTRE-LacZ or AdTRE-Aβ42 were added into culture for 18 h at a 1:5 ratio, and transgene expression was induced by 1 μg/ml doxycycline (Dox; Sigma) for 24 h. SH-SY5Y human neuroblastoma cells were grown in DMEM supplemented with 10% FBS, 2 mm l-glutamine, and antibiotics.
Tg2576 mouse brains. Tg2576 mice were described previously (Hsiao et al., 1996). Littermate animals at 2, 6, 12, and 16 months of age (n = 3 each) were anesthetized and perfused with PBS. Brains were obtained and cut in eight pieces (first longitudinally and then in four pieces each) and immediately frozen in liquid nitrogen and kept at -80°C. The frontal cortex (anterior pieces) and cerebellum were powdered in liquid nitrogen and lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors. Brain extracts were vigorously vortexed, ground with a plastic pestle, and rocked for 30 min at 4°C. Tissue brain lysates were cleared by centrifugation at 10,000 × g. For immunofluorescence studies of colocalization of Aβ42 and phospho-Akt, perfused brain tissue from 9-month-old mice was cut (40 μm thick) on a vibrating microtome. The free-floating sections were labeled with Chemicon Aβ42 and phospho-Akt Ser473 587F11 antibodies and then incubated with secondary antibodies in 0.1 m Tris-0.1 m NaCl and 0.1% BSA. Images were collected using a Zeiss LSM510 laser scanning confocal microscope (Zeiss Microimaging, Thornwood, NY).
Synthetic Aβ peptide preparation. Human Aβ42, Aβ25-35, and reverse Aβ35-25 peptides were obtained from the Biopolymer Laboratory at Harvard Medical School (Boston, MA). They were solubilized in 5% DMSO and 25 mm Tris-HCl, pH 7.4, incubated for 3 d at 37°C, and then applied to the cultures at a final concentration of 10 μm for 24 h.
Measurement of luciferase activity. Cells were infected with Ad-HSP-Luc (10 m.o.i.) for 24 h and then infected with either Ad-LacZ control or Ad-myrAkt (10 m.o.i.) and harvested 24 h later. Cell extracts were prepared and luciferase activity was measured with the luciferase assay system (Promega) according to the instructions of the manufacturer.
Small-interfering RNA design and RNA interference. For RNA interference, 6 d in vitro cortical neurons and SH-SY5Y cells (10,000 cells per well in an eight-well chamber) were transfected with duplex RNA by using TransMessenger transfection reagent (Qiagen, Valencia, CA) according to the instructions of the manufacturer. Briefly, small-interfering RNA (siRNA) (2-3 μg per well in six-well plates) was condensed with Enhancer R and mixed with TransMessenger reagent (1:5 siRNA to TransMessenger ratio). The transfection complex was diluted in 900 μl of medium without serum and added to the cells for 3 h. Cells were analyzed 72 h later. All siRNA duplexes were chemically synthesized by Xeragon-Qiagen (Germantown, MD). Only sense sequences are provided. To inhibit Akt1 synthesis, we used 5′-UGCCCUUCUACAACCAGGA-3′ siRNA (Katome et al., 2003). siRNA against rat and human Hsp70 mRNA were designed as described previously (Elbashir et al., 2001). Hsp70 was targeted with each of four different sequences: Hsp70.1 5′-GCCCAAGGUGCAGGUGAAC-3′, Hsp70.2 5′-GGGCGAGAACCGGUCGUUC-3′, Hsp70.3 5′-CGUGCUGCGGAUCAUCAAC-3′, and Hsp70.4 5′-GGUGGAGAUCAUCGCCAAC-3′. A rhodamine-labeled (nonsilencing) siRNA was used for transfection condition optimization and also as a negative control (Xeragon-Qiagen). Silencing of targeted genes was confirmed in three independent experiments. When indicated, cultures were infected with AdTet-On and AdTRE viral vectors 24 h after siRNA transfection, and transgene expression was induced 18 h later with Dox for 24 h. When described, cells were also infected at the time of induction with 10 m.o.i. of Ad-APP751, Ad-myrAkt, or Ad-Hsp70 and analyzed 24 h later.
Western blot analysis. Cells were washed once in PBS and harvested in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.25% Na-deoxycholate, 1% NP-40) containing protease inhibitor mixture (Roche Biochemicals, Indianapolis, IN), 1 mm sodium vanadate, and 1 mm sodium fluoride. Lysis occurred on ice for 30 min, and the total lysates were then cleared by centrifugation at 10,000 × g. Protein concentration was measured with the bicinchoninic acid protein assay reagent (Pierce, Rockford, IL) protocol and 40 μg of samples were electrophoresed in 7.5% SDS-PAGE gels under reducing conditions. Proteins were transferred onto polyvinylidine difluoride membranes (Millipore, Billerica, MA) and immunoblotted for the indicated proteins as described previously (Magrane et al., 2004). The immunoreaction was visualized by a chemiluminescence system (Amersham Biosciences, Piscataway, NJ). Band intensities were quantified by using the Fluor-S MultiImager (Bio-Rad, Richmond, CA).
For immunoprecipitation studies, cells were harvested in STEN buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 0.2% BSA) containing protease and phosphatase inhibitors, lysed, and cleared by centrifugation. Soluble proteins (50 μg of protein) were incubated with anti-Akt or anti-Hsp70 antibody overnight at 4°C followed by 2-3 h of incubation with 40 μl of protein-G-Sepharose or protein-A-Sepharose (Amersham Biosciences), respectively. The beads were washed three times in STEN buffer, once in PBS, and then heated at 70°C for 10 min in loading buffer.
Immunofluorescence. Cells were grown on coated coverslips, washed once in PBS, and fixed in 4% paraformaldehyde in phosphate buffer as described previously (Magrane et al., 2004). The percentage of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells (in situ cell death detection kit; Roche Biochemicals) and pyknotic nuclei (Hoechst 33342 staining) were obtained as described previously (Magrane et al., 2004). To measure immunofluorescence intensity, cells were imaged using an Olympus IX70 inverted microscope. Images were taken using a 40× lens, with equal exposure times for specific antibodies. MetaMorph software (Universal Imaging, West Chester, PA) was used for quantitative analysis. To quantify phospho-Akt levels (phospho-Akt Ser473 IHC Specific antibody), 5-10 neurons were randomly picked from each of three independent experiments, and integrated intensity values were measured from cell bodies and their processes (40 μm of neurite). Phospho-Akt intensity was plotted against Aβ42 or β-galactosidase control levels. To quantify Aβ immunofluorescence (Aβ 6E10 antibody), average intensities were measured in selected areas. To assess the relative distribution of Aβ, the intensities of processes were divided by the intensities of cell bodies. The ratio was compared between Aβ-positive neurons and Aβ-positive neurons with green fluorescent protein (GFP) or myrAkt coexpression. To measure Akt and Hsp70 immunofluorescence, at least 20 random fields each containing 8-10 cells were analyzed for every experiment performed, and integrated intensity values were similarly quantified. The mean fluorescence value for Ad-myrAkt or Ad-Hsp70-infected cultures was calculated and compared with the mean fluorescence value of noninfected cells.
Statistical analysis. All quantified results are expressed as the mean ± 1 SE. Statistical significance was determined by the Student's two-tailed, unpaired t test, and a p value <0.05 was considered indicative of a significant difference.
Results
Intracellular, but not extracellular, accumulation of Aβ42 decreases Akt activation
We developed a doxycycline-inducible adenoviral system and have shown that intraneuronal Aβ42 was cytotoxic in neuronal cultures (Magrane et al., 2004). Here, we investigate the effects of intracellular Aβ accumulation on neuronal signaling pathways. The serine/threonine protein kinase B, also known as Akt, is an essential mediator of neuronal survival (Brunet et al., 2001). Because phosphorylation of serine 473 is critical for full function, Akt activation was assessed by immunoblot analysis using an antibody specific for Ser(P)473-Akt. Total Akt levels were measured using a nonphosphorylation dependent anti-Akt antibody. We infected rat primary cortical neurons with 100 m.o.i. of the adenoviral constructs AdTet-On and AdTRE-transgene (1:5 ratio) and induced transgene expression with 1 μg/ml Dox for 24 h. Phospho-Akt levels significantly decreased following induction of Aβ42 expression (∼30% decline). In contrast, expression of control β-galactosidase in parallel cultures had no effect on phospho-Akt levels. Total Akt levels were similar for all culture conditions (Fig. 1A).
Figure 1.
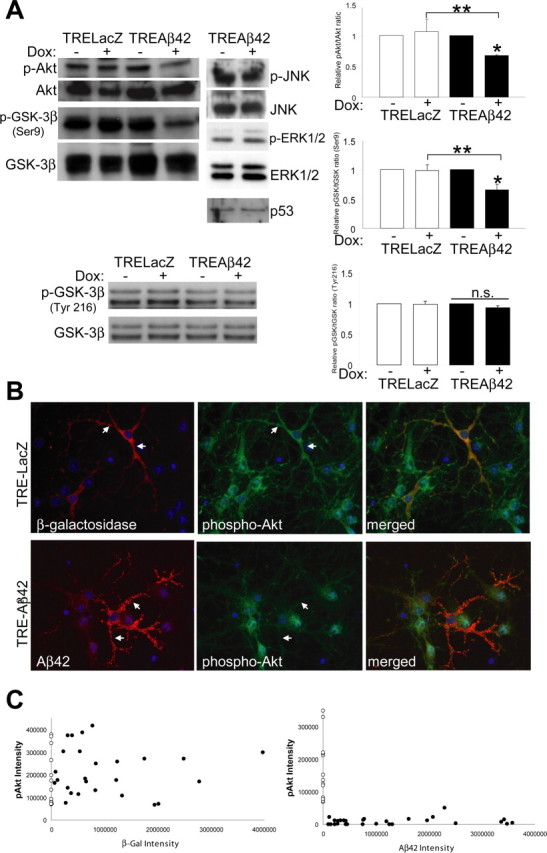
Intracellularly expressed Aβ42 downregulates the Akt survival pathway. Primary rat cortical neurons were infected with AdTet-On and either AdTRE-LacZ or AdTRE-Aβ42 as indicated, and transgene expression was induced by 1 μg/ml Dox for 24 h (+, top). A, Phospho-protein levels of Akt, GSK-3 β (Ser9 and Tyr216), members of the MAPK pathway, and levels of p53 were analyzed by Western blot. Corresponding total protein levels showed equivalent protein loading. Densitometric quantitation expressed as the relative phospho-protein to total protein ratio (n = 3; *p < 0.005; **p < 0.05; n.s., nonsignificant). Error bars represent 1 SE. B, Immunofluorescent images from primary neurons infected with AdTet-On and AdTRE-LacZ (top) or AdTRE-Aβ42 (bottom). Cells were immunostained for transgene expression (red) and phospho-Akt levels (green); merged images are on the right. C, Quantification of phospho-Akt fluorescence relative to transgene expression. Closed circles, Phospho-Akt intensity in either Aβ42 or β-galactosidase (β-gal)-expressing neurons; open circles, phospho-Akt intensity in noninfected cells.
We next tested whether the decrease in Akt activation was associated with altered signaling downstream of Akt in neurons. GSK-3β is an in vivo substrate of Akt (Cross et al., 1995) and has been shown to promote apoptosis in a number of neurodegenerative conditions including AD, in which it is important for the hyperphosphorylation of tau and loss of β-catenin stability (Lucas et al., 2001). Because Akt negatively regulates GSK-3β activity by phosphorylating serine 9, cell extracts were probed with antibodies for Ser(P)9-GSK3β and total GSK-3β. Consistent with a loss of Akt activity, phospho-GSK-3β levels were also significantly decreased by Aβ42 (∼35% decline) but not in controls (Fig. 1A). To further explore the specificity of GSK-3β activation under Aβ conditions, we analyzed the phosphorylation of tyrosine residue 216, which proceeds from autophosphorylation or other kinases distinct from Akt (Hartigan et al., 2001; Cole et al., 2004). Phosphorylation at this site has been shown to activate GSK-3β and to be increased during apoptosis (Bhat et al., 2000). However, in our studies, levels of Tyr(P)216-GSK3β remained unchanged by the expression of Aβ42 (Fig. 1A), suggesting that activation of GSK-3β is directly related to downregulation of the Akt pathway. We also explored the effects of Aβ42 expression on p53 levels and on mitogen-activated protein kinase (MAPK) family members, including ERK1/2 and JNK, which play important roles in signaling responses to stress stimuli. Unlike Akt-GSK-3β, phosphorylation of MAPK pathway components was not altered by intraneuronal Aβ42 expression (Fig. 1A). The ratio of phosphorylated to total protein for JNK and ERK1/2 was not significantly different between Aβ42-expressing cells and controls, suggesting that the Akt survival pathway was specifically affected by intraneuronal Aβ42 expression. p53 levels also remained unchanged (Fig. 1A). To further confirm the decrease in Akt activation, cortical cultures were infected, transgene expression was induced, and Ser(P)473-Akt levels were analyzed by immunofluorescence (Fig. 1B). In accordance with the results obtained by Western blot analysis, quantification of fluorescence intensities by image morphometric analysis showed a robust reduction in Ser(P)473-Akt levels in Aβ42-expressing neurons, but not in β-galactosidase-expressing controls, compared with noninfected neurons (Fig. 1C).
To compare the effects of extracellular Aβ peptide with intracellular Aβ expression, we applied synthetic Aβ peptides (10 μm) to neuronal cultures for 24 h. As expected, Aβ42 and Aβ25-35 peptides increased TUNEL-positive neurons, whereas the reverse-sequence Aβ35-25 control peptide had no effect (Fig. 2A). However, Aβ synthetic peptides did not appear to contribute to cell death through downregulation of Akt. Immunoblot of neuronal extracts for levels of activated Akt showed no significant changes at 6 or 24 h time points using either Aβ42 or Aβ25-35. Only a transient increase in Akt phosphorylation was observed after 30 min of treatment. The above data indicate different mechanisms of toxicity for intracellular and extracellular Aβ peptides and a specific effect of intraneuronal Aβ accumulation on the Akt signaling pathway.
Figure 2.
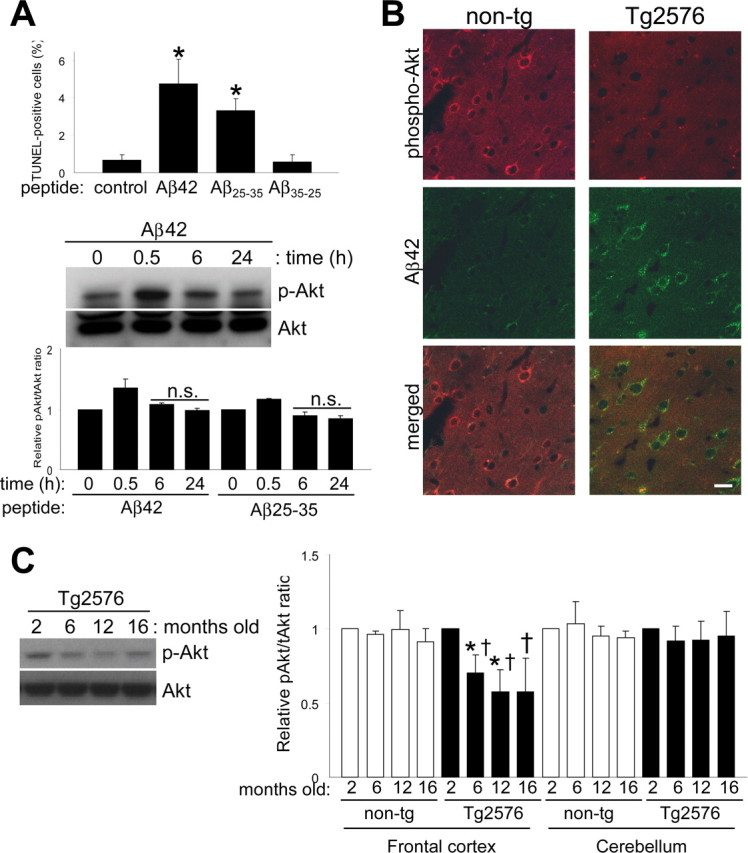
Effects of extracellularly applied Aβ peptide in vitro and intraneuronal accumulation of Aβ42 in vivo. A, Extracellularly applied Aβ peptides result in apoptosis through a mechanism independent of the Akt pathway. Different Aβ peptides were added to the culture medium at a final concentration of 10 μm for 24 h. Quantification of TUNEL staining data from four independent experiments is shown (comparison of treated cultures vs control; *p < 0.0005). Levels of phospho-Akt after Aβ42 peptide treatment over time. Densitometric quantitation expressed as the relative phospho-Akt to total Akt ratio for Aβ42 or Aβ25-35 peptide-treated cultures. n.s., Nonsignificant compared with untreated controls. B, Cortical sections of nontransgenic (non-tg) and Tg2576 mice were analyzed for intraneuronally accumulated Aβ42 and phospho-Akt by confocal microscopy. Scale bar, 20 μm. C, Phospho-Akt levels decreased with age in frontal cortex extracts from Tg2576 mice. Equal amounts of proteins were loaded. Quantitative data on relative changes of phospho-Akt levels in the frontal cortex and cerebellum of nontransgenic (non-tg) and Tg2576 mice (n = 3; *p < 0.05, relative to 2 months; †p < 0.05, relative to its companion age of nontransgenic). Error bars represent 1 SE.
To confirm these effects in vivo and to further explore the difference between intracellular or extracellular Aβ, we analyzed phospho-Akt levels in the brains of the Tg2576 mouse model for AD (Hsiao et al., 1996), which expresses an amyloid precursor protein (APP) harboring the Swedish familial AD mutation that leads to high Aβ levels in the brain. In this model, intraneuronal Aβ accumulation is detectable at 6 months of age, well before any extracellular plaque deposition is apparent (Takahashi et al., 2002). We first compared levels of phospho-Akt in the cortex of nontransgenic control mice and Tg2576, in which intraneuronally accumulated Aβ can be observed by immunofluorescence. A reduction in phospho-Akt immunoreactivity was observed in Tg2576 brains compared with controls (Fig. 2B). We then analyzed the frontal cortex and cerebellum from Tg2576 and age-matched controls at ages corresponding to no Aβ deposition (2 months), intracellular Aβ accumulation (pre-plaque stage; 6 months), and extracellular Aβ deposition (postplaque stage, 12 and 16 months) (Fig. 2C). Ser(P)473-Akt levels from the frontal cortex of Tg2576 mice were already significantly reduced at 6 months. The reduction in phospho-Akt levels coincided with the appearance of intracellular Aβ accumulation (Takahashi et al., 2002). No changes were observed in age-matched controls or in the cerebellum (Fig. 2C). These results confirm the reductions we observed in vivo by immunofluorescence.
Constitutively active Akt protects against intracellular Aβ-induced toxicity
To address whether the impaired Akt phosphorylation in response to Aβ42 accumulation could contribute to cell death, we cotransduced neuronal cultures with adenoviral vectors expressing a constitutively active form of Akt (myrAkt) or a dominant-negative form (dnAkt) (Fig. 3A). Primary cultures were infected with 100 m.o.i. of AdTet-On and AdTRE-Aβ42 (1:5 ratio) and induced with Dox and infected with 10 m.o.i. of Ad-myrAkt, Ad-dnAkt, or Ad-LacZ for 24 h. Strikingly, overexpression of myrAkt protected against the toxic effects of Aβ42 accumulation, as shown by significant reductions in the number of TUNEL-positive cells in Aβ42-expressing cultures. Overexpression of β-galactosidase had no such beneficial effect on Aβ42 toxicity. As expected, overexpression of dnAkt did not reverse Aβ42-induced neurotoxicity, and cell death occurred at levels similar to those produced by Aβ42 alone (Fig. 3A). To ascertain whether myrAkt neuroprotection resulted in changes in the levels or distribution of intraneuronal Aβ42, we examined Aβ levels in those neurons coexpressing myrAkt or GFP control (Fig. 3B). We used immunofluorescence and image morphometric analysis to quantify fluorescence intensities in both cell bodies and processes. As expected, overexpression of myrAkt resulted in no changes in the level or distribution of intraneuronal Aβ, compared with controls (Fig. 3C). We also analyzed the effects of GFP and myrAkt overexpression on β-galactosidase-expressing cells and observed no differences (data not shown). These data implicate the Akt pathway as a target in Aβ-induced toxicity in neuronal cultures.
Figure 3.
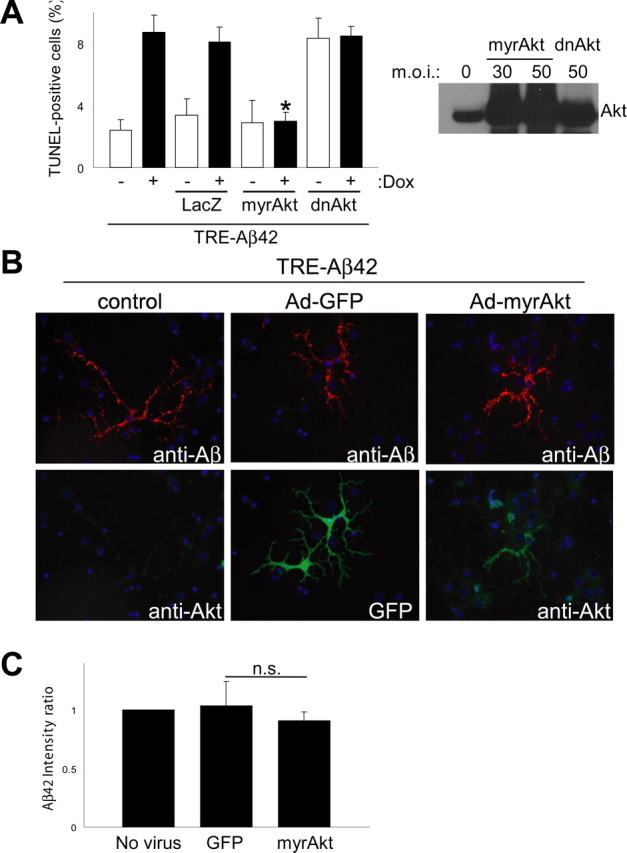
Akt overexpression rescues Aβ42-induced toxicity without affecting levels of intraneuronal Aβ. A, Cortical neurons were infected with AdTet-On and AdTRE-Aβ42. Transgene expression was induced by 1 μg/ml Dox for 24 h (+, bottom), and cells were coinfected with Ad-LacZ, Ad-myrAkt (constitutively active Akt), or Ad-dnAkt (inactive Akt) as indicated. Ad-myrAkt fully protected against Aβ42-induced neuronal death, as measured by TUNEL staining (*p < 0.005). Coinfection with Ad-dnAkt or Ad-LacZ control did not affect Aβ42 toxicity. Values from three independent experiments are shown. Immunoblot analysis of Akt overexpression in primary neurons (right). B, Immunofluorescent images from primary neurons infected with AdTet-On and AdTRE-Aβ42. Cells were coinfected with Ad-GFP or Ad-myrAkt, as indicated. No apparent changes in levels or localization of Aβ were observed. C, Quantification of Aβ immunofluorescence from neurons with or without coexpression of GFP control or myrAkt. Ratio of Aβ42 intensity in processes divided by that in cell bodies was calculated, with the ratio standardized to 1.0 for cells without GFP or myrAkt coexpression. Error bars represent 1 SE. Results from three independent experiments are shown. n.s., Nonsignificant.
Akt overexpression induces Hsp70
Akt is known to act on several anti- and pro-apoptotic molecules to achieve its survival function (Brunet et al., 2001). We previously described the cellular stress response as an important protective modulator against intracellular Aβ42 toxicity in neurons (Magrane et al., 2004). To explore whether the protective function of Akt is mediated through the neuronal stress response, we analyzed the effect of Akt expression on Hsp promoter activity (Smith et al., 2002). We infected neuroblastoma cells with 10 m.o.i. of Ad-Hsp-Luc, followed by superinfection with 10 m.o.i. of Ad-myrAkt or Ad-LacZ as a control, and performed a luciferase activity assay on cellular lysates (Fig. 4A). We demonstrated that Akt overexpression, but not β-galactosidase, induced a robust stress response (a fourfold increase in luciferase activity).
Figure 4.
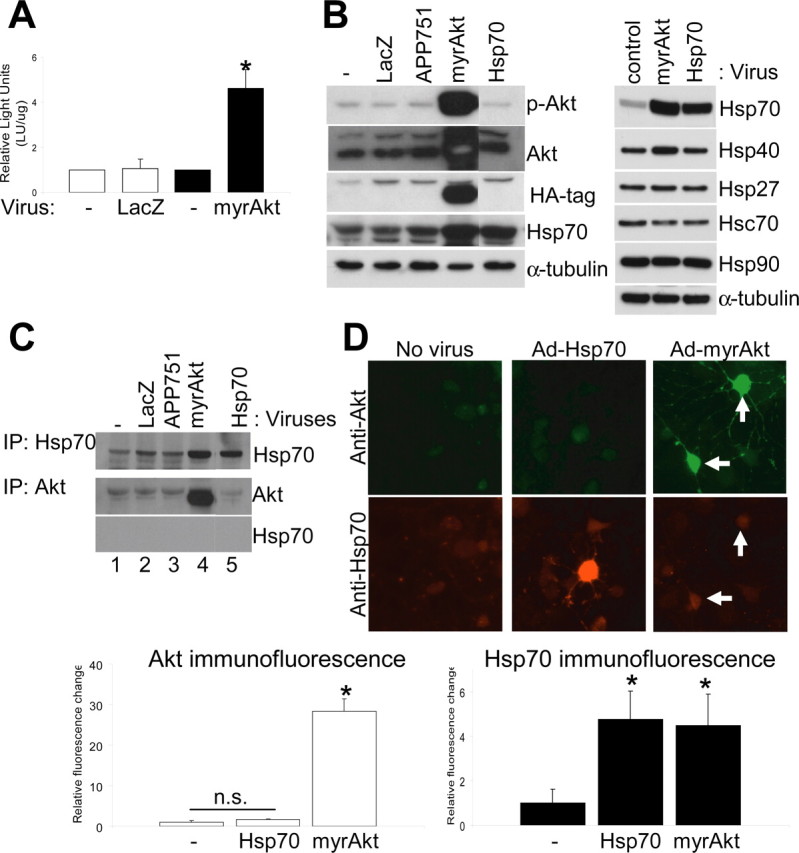
Akt overexpression induces the neuronal stress response. A, Luciferase assay performed on neuroblastoma cells first infected with Ad-HSP-Luc reporter and then with the indicated viruses (see Materials and Methods). MyrAkt overexpression, but not controls, activated the Hsp70B promoter and induced luciferase activity. Values are presented as x-fold increase compared with control (n = 5; p < 0.0005). B, Immunoblot of proteins extracted from primary neuronal cultures that were uninfected or infected with different viral vectors, as indicated. Equal amounts of protein were loaded. Overexpression of Akt (HA-tagged), but not of other transgenes, induced Hsp70 expression (left). Analysis of the induction of the stress response in neuroblastoma cells (right). C, Neuroblastoma cells were infected with viruses (top), and cell extracts were immunoprecipitated with anti-Hsp70 or anti-Akt antibodies and probed for the indicated proteins. Akt overexpression increased the amount of Hsp70 available for immunoprecipitation from extracts (top, lane 4), but Hsp70 overexpression had no effect on Akt levels (middle, lane 5). Increased recovery of Hsp70 did not occur in Akt-Hsp70 coimmunoprecipitations (bottom, lane 4). D, Immunofluorescent images from primary neurons infected with no virus, Ad-Hsp70, or Ad-myrAkt and immunostained for total Akt (green) and Hsp70 (red). Akt overexpression in neurons correlated with increased Hsp70 immunostaining, whereas Hsp70 had no effects on Akt levels. Quantification of fluorescence for Akt and Hsp70 expression from several independent experiments is shown. Error bars represent 1 SE. n.s., Nonsignificant. *p < 0.01.
We next analyzed the stress response by infecting primary neuronal cultures and neuroblastoma cells with vectors expressing putative regulatory proteins to study induction of specific heat shock proteins. Control adenoviral infections with Ad-LacZ and Ad-APP751 did not change activated Akt or steady-state levels of any of the Hsps examined. However, Akt overexpression (Ad-myrAkt) increased Hsp levels, including mainly Hsp70 but also Hsp40 (Fig. 4B). In contrast, Hsp27, Hsc70, and Hsp90 levels did not increase. Furthermore, in using a second approach, we observed that more Hsp70 was immunoprecipitated when Akt was overexpressed, whereas levels of immunoprecipitated Akt only increased in lysates when Akt was overexpressed (Fig. 4C). Conversely, Hsp70 overexpression (Ad-Hsp70) had no effect on Akt levels, suggesting that its neuroprotective role is independent of Akt activation (Fig. 4B,C).
To confirm the upregulation of Hsp70, we infected rat cortical cultures and used immunofluorescence and image morphometric analysis to quantify fluorescence intensities. As described above, Hsp70-expressing cells did not show significant increases in Akt staining; however, Akt-positive cells exhibited a fourfold increase in Hsp70 expression compared with controls (Fig. 4D). These findings suggest that Akt may serve to activate the stress response and, by this means, protect neurons against Aβ42-induced toxicity.
Hsp70 protective effect acts downstream of Akt activation
To further explore the link between Akt activation and Hsp70 induction, we used siRNA to deplete neurons of the targeted proteins and examined the effects. siRNA has been used previously to decrease the expression of both endogenous and transfected genes in cultured neurons (Krichevsky and Kosik, 2002). First, we transfected primary neurons with rhodamine-labeled control siRNA and determined the transfection efficiency by fluorescence, obtaining between 35 and 50% rhodamine-positive cells (data not shown). We then used siRNA to reduce Akt1 protein levels in primary neurons and observed that Akt1 levels decreased ∼20% at 72 h after transfection (Fig. 5A). Neither rhodamine-labeled control nor Hsp70 siRNA had any effect on Akt protein levels. α-Tubulin levels were similar under all conditions, and this degree of depletion of total Akt1 did not alter the steady-state levels of JNK, a proapoptotic MAPK family member (Fig. 5A). Remarkably, Akt siRNA transfected cultures showed a significant increase in cell death compared with non-transfected or rhodamine-labeled control siRNA-treated cultures (Fig. 5B). Similar results using TUNEL staining were obtained with neuroblastoma cells treated with Akt siRNA (data not shown).
Figure 5.
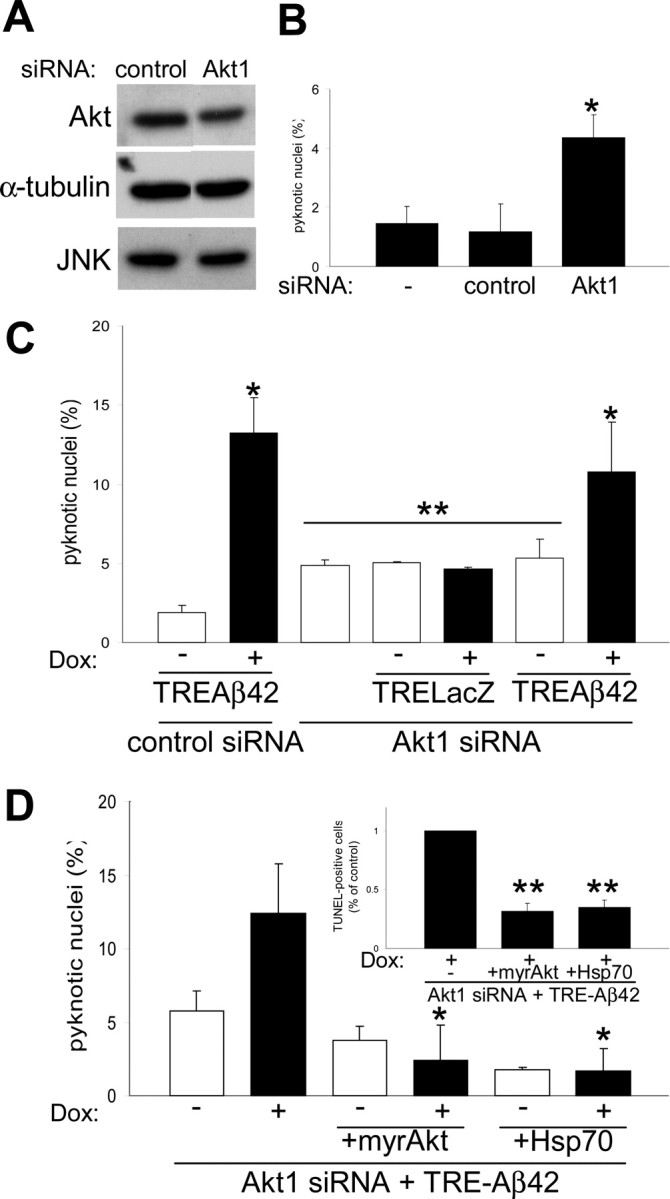
Akt and Hsp70 overexpression can reverse Aβ42-induced cell death in Akt1-depleted neurons. A, Primary neurons were treated for 72 h with rhodamine-control or Akt1 siRNA, and cell lysates were analyzed using the indicated antibodies. Total Akt1 levels were reduced ∼20%, whereas JNK and α-tubulin levels were unaffected (representative blots are shown). B, Suppression of endogenous Akt1 protein in primary neurons produced a threefold increase in neuronal death under basal conditions, as measured by nuclear pyknosis (n = 3; *p < 0.005). C, Primary neurons were treated with rhodamine-control or Akt1 siRNA and infected with either AdTRE-LacZ or AdTRE-Aβ42 viruses. Induction of transgene expression with Dox (+) increased cell death in Aβ42-expressing cells (n = 3; *p < 0.05, compared with noninduced cultures) but not in β-galactosidase-expressing controls. Suppression of Akt1 caused an overall increase in neuronal death (**p < 0.05, compared with noninduced control siRNA). D, Primary neurons and neuroblastoma cells (inset) were treated with Akt1 siRNA and infected with AdTRE-Aβ42 viruses. Transgene expression was then induced with Dox (+), and cells were infected with either Ad-myrAkt or Ad-Hsp70, as indicated (see Materials and Methods for details). Apoptotic death increased in Akt1-depleted Aβ42-expressing cultures. Coinfection with either Ad-myrAkt or Ad-Hsp70 reversed the observed toxic effects of Aβ42 expression in both cell types (n = 3; *p < 0.05 and **p < 0.005, compared with Akt1-depleted Aβ42-expressing cells). Error bars represent 1 SE.
We then analyzed the combined effects of Akt1 depletion and Aβ42 expression on survival in both primary neurons and neuroblastoma cells. Primary neurons were treated with rhodamine-control or Akt1 siRNA, infected with AdTRE-LacZ or AdTRE-Aβ42 viruses, and transgene expression induced with Dox. Akt1-depleted cultures showed an overall increase in cell death (compared with rhodamine-control siRNA) under all conditions, whereas induction of Aβ42 expression, but not β-galactosidase, increased toxicity in Akt1 siRNA-treated cultures (Fig. 5C). Coinfection of Akt1-depleted cultures with either Ad-myrAkt or Ad-Hsp70 fully relieved the toxic effects in both primary neurons and neuroblastoma cells (Fig. 5D, bottom). These and the above findings suggest that Hsp70 exerts its neuroprotective effects by acting downstream of Akt signaling.
Because Hsp70 was neuroprotective against Aβ42-induced toxicity even when Akt1 levels were reduced, we decided to explore the effects of depleting Hsp70 from cells. Cortical neurons were transfected with each of four different Hsp70 siRNA, and changes in Hsp70 protein levels were determined. Immunoblot analysis showed 30-70% reduction depending on the siRNA used. Levels of the constitutive form of Hsp70, Hsc70, and Akt did not change (Fig. 6A). Suppression of Hsp70 protein alone in primary neurons resulted in increased cell death to levels comparable with that produced by Akt siRNA (Figs. 6B, 5B). Next, we infected Hsp70-depleted, TREAβ42-infected cultures with either Ad-myrAkt or Ad-Hsp70. Hsp70 overexpression reversed the toxicity caused by Aβ42 expression; however, Akt overexpression did not. These results suggest that Hsp70 acts downstream of the Akt pathway and that the neuroprotective role of Akt in preventing Aβ42-induced cell death occurs through a mechanism involving Hsp70 induction.
Figure 6.

Hsp70 acts downstream of Akt. A, Immunoblot analysis of primary neuronal cultures treated with no siRNA (-), rhodamine-control siRNA, or different Hsp70 siRNA (1-4). Hsp70 protein levels were reduced 30-70%; however, levels of noninducible Hsc70, Akt, and α-tubulin were not affected. B, Depletion of endogenous Hsp70 (siRNA Hsp70.1 and Hsp70.2) from cortical neurons induced cell death, as measured by nuclear pyknosis (n = 3; *p < 0.05). C, Neuroblastoma cells were treated with Hsp70.2 siRNA, infected with AdTet-On and AdTRE-Aβ42 viruses, and transgene expression induced with Dox (+). Overexpression of Hsp70 was shown to reverse TUNEL labeling caused by Hsp70 depletion alone and in combination with Aβ42 accumulation (*p < 0.0005, compared with Aβ42-expressing cultures; **p < 0.05, compared with noninduced cultures). Coinfection with Ad-myrAkt had a nonstatistically significant effect. Error bars represent 1 SE. Values from four independent experiments are shown.
Discussion
A pathogenic role for intracellular Aβ has been suggested for AD, Down's syndrome, and the human muscle disorder inclusion body myopathy (Tseng et al., 2004). Intraneuronal accumulation of Aβ42 has been detected in AD brains and in transgenic mice overexpressing familial AD mutations even before plaque deposition occurs (Gouras et al., 2000; D'Andrea et al., 2001; Wirths et al., 2001; Takahashi et al., 2002; Oddo et al., 2003; Casas et al., 2004). Aβ42 accumulates in endosomes and multivesicular bodies (Takahashi et al., 2002; Cataldo et al., 2004). This intracellular pool of Aβ may contribute to neuronal dysfunction and degeneration.
Observations made when using synthetic Aβ peptides applied to cell cultures do not necessarily reflect what happens intracellularly when Aβ accumulates. Our results and those obtained by others suggest that intracellular and extracellular Aβ may have independent effects (Zhang et al., 2003). Interestingly, we only observed a reduction of phospho-Akt levels when Aβ42 was intracellularly expressed but not when it was applied extracellularly (Figs. 1, 2). These results are consistent with those of other studies in which synthetic Aβ peptides were used to identify various roles for MAPK family members (most notably JNK activation), but not Akt, in transducing cell death after peptide application (Dineley et al., 2001; Morishima et al., 2001; Troy et al., 2001; Wei et al., 2002).
Several reports using transgenic animals (LaFerla et al., 1995; Link, 1995; Iijima et al., 2004) or cultured cells (Johnstone et al., 1996; Querfurth et al., 2001; Zhang et al., 2002; Suhara et al., 2003) have provided direct observations on the effects of intracellular Aβ42 expression or injection. However, our inducible adenoviral vector system (Magrane et al., 2004) has allowed us to test molecular mechanisms of intracellular Aβ42 toxicity following controlled expression within cultured neurons and other primary cell types. Accumulation of intracellular Aβ42 may affect a variety of signal transduction pathways, including Akt and MAPK family members that have important roles in neuronal function (Yuan and Yankner, 2000). Here, we report that induced Aβ42 expression disrupts signaling through the Akt pathway. Our in vivo observations using the Tg2576 mouse model for AD further supports the correlation between intracellular Aβ deposition and downregulation of Akt (Figs. 1, 2). The mechanisms linking the effects of Aβ42 to disruption of Akt signaling are not completely clear; however, we have shown previously that Aβ42 may disrupt the interaction between Akt and its activator, PDK-1 (phosphoinositide-dependent kinase-1) (Suhara et al., 2003). Although we observe that Aβ42 interferes with the Akt survival pathway, there is evidence that the MAPK pathway involving ERK2 may also be affected in an in vivo model for intracellular Aβ accumulation (Echeverria et al., 2004). However, in our cell culture-based work, we did not find evidence for an effect on ERK2 or JNK. It has also been reported that injected or transfected Aβ42 can promote p53-dependent cell death possibly through p53 promoter activation (Zhang et al., 2002; Ohyagi et al., 2005). However, we did not observe an increase in p53 levels. Importantly, our Aβ42 construct was engineered with a signal peptide sequence to express the transgene product in the secretory pathway and, thus, mimic the in vivo sites of Aβ production (Takahashi et al., 2002).
Akt is essential to neuronal cell survival (Dudek et al., 1997), and we have shown that Aβ42 expression impacts the Akt-GSK-3β axis in neurons (Fig. 1) and endothelial cells (Suhara et al., 2003). Akt downregulation increases GSK-3β activity, which in turn may favor conditions that promote neuronal death, tau aggregation, and β-catenin destabilization (Hanger et al., 1992; Aberle et al., 1997; Crowder and Freeman, 2000; Hetman et al., 2000; Alvarez et al., 2004), all of them reported to be important features in AD pathogenesis. GSK-3β also phosphorylates and inactivates heat shock factor-1 (HSF-1), which regulates the cellular stress response (Chu et al., 1996). In addition, FOXO3 (Forkhead box O transcription factor; FKHRL1), an Akt-mediated transcription factor, has been shown to regulate Hsp70 in endothelial cells (Kim et al., 2005). Furthermore, deficient insulin receptor signaling through deactivated PI3K-Akt increases Aβ generation and retards its degradation in the brain (Ho et al., 2004; Zhao et al., 2004). Inhibition of the PI3K-Akt pathway may also affect synaptic plasticity and long-term potentiation expression (Daw et al., 2002; Sanna et al., 2002; Wang et al., 2003). Thus, alterations in signal transduction pathways resulting from Aβ effects on Akt plausibly result in both neuronal dysfunction and cell loss observed in AD.
Several studies have shown that Akt activation can be neuroprotective in several neurodegenerative disease models. For instance, activation of Akt kinase activity by insulin growth factor 1 (IGF-1) protected against mutant huntingtin-induced toxicity, and Akt was altered in Huntington's disease patients (Humbert et al., 2002). Viral administration of IGF-1 or BDNF delayed disease progression in an amyotrophic lateral sclerosis mouse model, presumably by activating the Akt signaling pathway (Kaspar et al., 2003). Our results demonstrating that constitutively active Akt expression conferred protection against Aβ42-induced cell death (Fig. 3) suggest that the activation of the Akt pathway could have therapeutic value in AD and supports a model in which Akt activation contributes to neuronal survival.
Several neurodegenerative diseases appear to have an impaired stress response, further compromising both neuronal function and survival (Bruening et al., 1999; Kobayashi et al., 2000; Auluck et al., 2002; Batulan et al., 2003; Shimura et al., 2004). It is therefore important to understand the intracellular signaling mechanisms affected by the accumulation of toxic proteins in the context of the cellular stress response. The heat shock response is initiated by the activation of HSF-1, a transcription factor that induces the expression of Hsp. GSK-3β is an important component in the repression of HSF-1 activity. GSK-3β phosphorylates HSF-1 within an essential region of its transcriptional regulatory domain, thereby preventing induction of the heat shock response (Chu et al., 1996). PI3K activation inhibits its downstream target GSK-3β and prevents the phosphorylation of HSF-1 (Bijur and Jope, 2000). We previously reported that intracellular Aβ42 accumulation in neurons also produced a modest induction of the stress response but only to a degree still insufficient to prevent cell death (Magrane et al., 2004). Overexpression of Hsp70 was used to boost the endogenous stress response and reversed Aβ toxicity, as presented again in more detail in Figure 5D. We now show that this deficiency of Hsp 70 induction is exacerbated or possibly caused by the Aβ-dependent downregulation of the Akt pathway. The evidence in Figures 4, 5, 6 suggests that Akt plays a significant role in positively controlling the gain of the induction in heat shock proteins to misfolded proteins in neurons. Based on the combined data, we believe that Akt downregulation compromises the corrective but ultimately feeble, neuronal stress response against intracellular Aβ toxicity. We believe this represents the first report to show functional significance of the Akt pathway in the regulation of the stress response in neurons.
Our results suggest that downregulation of the Akt pathway may be an early event in the disease process, leading to both suppression of the stress response and decreased neuronal survival. While acknowledging that intraneuronal Aβ may also activate proapoptotic pathways, our results demonstrate that inhibition of survival signaling is at least an equally critical event in the progression of the disease. Modulation of the Akt pathway or the neuronal stress response may provide a potential treatment for AD and other neurodegenerative diseases characterized by misfolded amyloidogenic proteins.
Footnotes
This work was supported by National Institutes of Health Grants NS41371 (H.W.Q), AG17241 (K.W.), and AG009464 (G.K.G.). We thank Anna Krichevsky for helpful discussions on siRNA design.
Correspondence should be addressed to Dr. Henry W. Querfurth, Caritas Saint Elizabeth's Medical Center, Department of Neurology, 736 Cambridge Street, Boston, MA 02135. E-mail: henry.querfurth@tufts.edu.
J. Magrané's present address: Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York, NY 10021.
R. C. Smith's present address: New England OncoTherapeutics, Cambridge, MA 02142.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510960-10$15.00/0
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16: 3797-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC (2004) Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res 297: 186-196. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295: 865-868. [DOI] [PubMed] [Google Scholar]
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK (2004) PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J 23: 2586-2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z, Shinder GA, Minotti S, He BP, Doroudchi MM, Nalbantoglu J, Strong MJ, Durham HD (2003) High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 23: 5789-5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA 97: 11074-11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS (2000) Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3beta in the regulation of HSF-1 activity. J Neurochem 75: 2401-2408. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM (2005) Intraneuronal abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45: 675-688. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Park J, Hemmings BA (2002) PKB binding proteins. Getting in on the Akt. Cell 111: 293-303. [DOI] [PubMed] [Google Scholar]
- Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD (1999) Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem 72: 693-699. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11: 297-305. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165: 1289-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 25: 1263-1272. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY (2003) Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113: 457-468. [DOI] [PubMed] [Google Scholar]
- Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK (1996) Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J Biol Chem 271: 30847-30857. [DOI] [PubMed] [Google Scholar]
- Chui DH, Dobo E, Makifuchi T, Akiyama H, Kawakatsu S, Petit A, Checler F, Araki W, Takahashi K, Tabira T (2001) Apoptotic neurons in Alzheimer's disease frequently show intracellular Abeta42 labeling. J Alzheimers Dis 3: 231-239. [DOI] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P (2004) Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J 377: 249-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS (2000) Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem 275: 34266-34271. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38: 120-134. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bortolotto ZA, Saulle E, Zaman S, Collingridge GL, Isaac JT (2002) Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nat Neurosci 5: 835-836. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 21: 4125-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Dowd E, Janne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC (2004) Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 129: 583-592. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494-498. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375-387. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA (2004) Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet 36: 131-137. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Walsh K (1999) Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem 274: 16349-16354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME (2003) Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-d-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem 278: 50514-50522. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: 15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Prevec L (1995) Methods for construction of adenovirus vectors. Mol Biotechnol 3: 207-220. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH (1992) Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 147: 58-62. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Xiong WC, Johnson GV (2001) Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun 284: 485-489. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM (2004) Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J 18: 902-904. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2: 831-837. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y (2004) Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc Natl Acad Sci USA 101: 6623-6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone EM, Babbey LE, Stephenson D, Paul DC, Santerre RF, Clemens JA, Williams DC, Little SP (1996) Nuclear and cytoplasmic localization of the beta-amyloid peptide (1-43) in transfected 293 cells. Biochem Biophys Res Commun 220: 710-718. [DOI] [PubMed] [Google Scholar]
- Kashour T, Burton T, Dibrov A, Amara FM (2003) Late Simian virus 40 transcription factor is a target of the phosphoinositide 3-kinase/Akt pathway in anti-apoptotic Alzheimer's amyloid precursor protein signalling. Biochem J 370: 1063-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH (2003) Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301: 839-842. [DOI] [PubMed] [Google Scholar]
- Katome T, Obata T, Matsushima R, Masuyama N, Cantley LC, Gotoh Y, Kishi K, Shiota H, Ebina Y (2003) Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. J Biol Chem 278: 28312-28323. [DOI] [PubMed] [Google Scholar]
- Kim HS, Skurk C, Maatz H, Shiojima I, Ivashchenko Y, Yoon SW, Park YB, Walsh K (2005) Akt/FOXO3a signaling modulates the endothelial stress response through regulation of heat shock protein 70 expression. FASEB J 19: 1042-1044. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G (2000) Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275: 8772-8778. [DOI] [PubMed] [Google Scholar]
- Kowall NW, McKee AC, Yankner BA, Beal MF (1992) In vivo neurotoxicity of beta-amyloid [beta(1-40)] and the beta(25-35) fragment. Neurobiol Aging 13: 537-542. [DOI] [PubMed] [Google Scholar]
- Krichevsky AM, Kosik KS (2002) RNAi functions in cultured mammalian neurons. Proc Natl Acad Sci USA 99: 11926-11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G (1995) The Alzheimer's A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet 9: 21-30. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Hall CK, Ngo L, Jay G (1996) Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Invest 98: 1626-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD (1995) Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans Proc Natl Acad Sci USA 92: 9368-9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD, Cypser JR, Johnson CJ, Johnson TE (1999) Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones 4: 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD, Taft A, Kapulkin V, Duke K, Kim S, Fei Q, Wood DE, Sahagan BG (2003) Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer's disease model. Neurobiol Aging 24: 397-413. [DOI] [PubMed] [Google Scholar]
- Liolitsa D, Powell J, Lovestone S (2002) Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry 73: 261-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed β-amyloid in neurons. J Neurosci 24: 1700-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME (2001) β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci 21: 7551-7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409-421. [DOI] [PubMed] [Google Scholar]
- Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T (2005) Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer's disease. FASEB J 19: 255-257. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW (1993) Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 13: 1676-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, Suhara T, Rosen KM, McPhie DL, Fujio Y, Tejada G, Neve RL, Adelman LS, Walsh K (2001) Beta-amyloid peptide expression is sufficient for myotube death: implications for human inclusion body myopathy. Mol Cell Neurosci 17: 793-810. [DOI] [PubMed] [Google Scholar]
- Ryder J, Su Y, Ni B (2004) Akt/GSK3beta serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer's disease mutations. Cell Signal 16: 187-200. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W (2002) Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci 22: 3359-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Miura-Shimura Y, Kosik KS (2004) Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 279: 17957-17962. [DOI] [PubMed] [Google Scholar]
- Smith RC, Machluf M, Bromley P, Atala A, Walsh K (2002) Spatial and temporal control of transgene expression through ultrasound-mediated induction of the heat shock protein 70B promoter in vivo. Hum Gene Ther 13: 697-706. [DOI] [PubMed] [Google Scholar]
- Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H (2003) Abeta42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3beta signaling-dependent mechanism. Neurobiol Aging 24: 437-451. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161: 1869-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK (2004) Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24: 3592-3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA (2001) Beta-amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem 77: 157-164. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Kitazawa M, LaFerla FM (2004) Amyloid β-peptide: the inside story. Curr Alz Res 1: 231-239. [DOI] [PubMed] [Google Scholar]
- Wagey R, Pelech SL, Duronio V, Krieger C (1998) Phosphatidylinositol 3-kinase: increased activity and protein level in amyotrophic lateral sclerosis. J Neurochem 71: 716-722. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT (2003) Control of synaptic strength, a novel function of Akt. Neuron 38: 915-928. [DOI] [PubMed] [Google Scholar]
- Wei W, Wang X, Kusiak JW (2002) Signaling events in amyloid beta-peptide-induced neuronal death and insulin-like growth factor I protection. J Biol Chem 277: 17649-17656. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP (1999) Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci 19: 5360-5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306: 116-120. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Bayer TA (2004) A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide-the first step of a fatal cascade. J Neurochem 91: 513-520. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res 52: 691-698. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250: 279-282. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802-809. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A (2002) Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156: 519-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hong Y, Bounhar Y, Blacker M, Roucou X, Tounekti O, Vereker E, Bowers WJ, Federoff HJ, Goodyer CG, LeBlanc A (2003) p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid beta peptide cytotoxicity. J Neurosci 23: 7385-7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM (2004) Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci 24: 11120-11126. [DOI] [PMC free article] [PubMed] [Google Scholar]