

Abstract
The presynaptic protein α-synuclein has a central role in Parkinson's disease (PD). However, the mechanism by which the protein contributes to neurodegeneration and its normal function remain unknown. α-Synuclein localizes to the nerve terminal and interacts with artificial membranes in vitro but binds weakly to native brain membranes. To characterize the membrane association of α-synuclein in living neurons, we used fluorescence recovery after photobleaching. Despite its enrichment at the synapse, α-synuclein is highly mobile, with rapid exchange between adjacent synapses. In addition, we find that α-synuclein disperses from the nerve terminal in response to neural activity. Dispersion depends on exocytosis, but unlike other synaptic vesicle proteins, α-synuclein dissociates from the synaptic vesicle membrane after fusion. Furthermore, the dispersion of α-synuclein is graded with respect to stimulus intensity. Neural activity thus controls the normal function of α-synuclein at the nerve terminal and may influence its role in PD.
Keywords: α-synuclein, membrane association, synaptic vesicle, neural activity, Parkinson's disease, synapsin
Introduction
Genetic studies have implicated the protein α-synuclein in the pathogenesis of Parkinson's disease (PD). Mutations in α-synuclein cause a rare autosomal-dominant form of PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004). However, the protein also accumulates in Lewy bodies and dystrophic neurites of idiopathic PD, suggesting an important pathogenic role for α-synuclein even in the absence of inherited mutations (Spillantini et al., 1998a,b; Galvin et al., 1999). The recent demonstration that a simple increase in dosage of the α-synuclein gene can cause PD further supports a role for the wild-type protein in the idiopathic disorder (Singleton et al., 2003). However, the circumstances that trigger a pathogenic role for wild-type α-synuclein and indeed its normal function remain unclear.
Considerable work has suggested a role for α-synuclein in neurotransmitter release. Neurons deficient in α-synuclein show a reduction in the reserve pool of synaptic vesicles required for the response to repeated stimulation (Murphy et al., 2000; Cabin et al., 2002). Other studies using knock-out mice have not detected changes in hippocampal synaptic physiology (Abeliovich et al., 2000; Chandra et al., 2004) but rather, increases in dopamine release (Abeliovich et al., 2000; Yavich et al., 2004).
Consistent with a role in neurotransmitter release, α-synuclein localizes to the nerve terminal (Jakes et al., 1994; Iwai et al., 1995; Withers et al., 1997). Originally identified as a result of its association with synaptic vesicles (Maroteaux et al., 1988), α-synuclein lacks a transmembrane domain or lipid anchor and has been considered a peripheral membrane protein. Indeed, α-synuclein binds to artificial membranes in vitro by adopting an α-helical conformation (Davidson et al., 1998; Jo et al., 2000; Eliezer et al., 2001; Chandra et al., 2003). α-Synuclein also associates with axonal transport vesicles, lipid droplets, and yeast membranes (Jensen et al., 1998; Cole et al., 2002; Outeiro and Lindquist, 2003). However, α-synuclein behaves almost entirely as a soluble protein in brain extracts (Maroteaux and Scheller, 1991; Jakes et al., 1994; Iwai et al., 1995), raising questions about the mechanism of synaptic localization in vivo. We found recently that an interaction with membrane microdomains known as lipid rafts is required for the localization of α-synuclein to the nerve terminal (Fortin et al., 2004). The PD-associated A30P mutation specifically disrupts both raft association and the synaptic localization of α-synuclein (Fortin et al., 2004). All of these studies have, however, relied on biochemical approaches or steady-state localization in fixed cells, limiting the scope of analysis and conclusions.
To characterize the kinetic behavior of α-synuclein in a physiological context, we used live cell imaging of green fluorescent protein (GFP)-tagged proteins expressed in dissociated hippocampal neurons. Fluorescence recovery after photobleaching (FRAP) experiments show that the localization of α-synuclein at the nerve terminal is highly dynamic, with rapid exchange of the protein between neighboring synapses. However, the mobility of wild-type α-synuclein is distinctly slower than that of GFP alone and the PD-associated A30P mutant, which is not enriched at the nerve terminal (Fortin et al., 2004). The slowed recovery of wild-type α-synuclein indicates that transient, rapidly reversible interactions account for the steady-state localization of α-synuclein to the synapse. We also find that neural activity results in the graded dispersion of α-synuclein from synapses, and dispersion requires synaptic vesicle exocytosis, not simply calcium entry. Unlike other synaptic vesicle proteins (Sankaranarayanan and Ryan, 2000; Li and Murthy, 2001), however, α-synuclein does not accumulate in the perisynaptic region after membrane fusion, indicating that it dissociates from synaptic membranes after exocytosis. Thus, neural activity controls both the synaptic localization and membrane association of α-synuclein.
Materials and Methods
Plasmids. The plasmid encoding GFP-synaptic vesicle protein 2 (GFP-SV2) was the generous gift from C. Waites (Stanford University, Palo Alto, CA). Subcloning of wild-type and mutant α-synuclein as well as synapsin I into enhanced GFP vectors (Clontech, Palo Alto, CA) has been described previously (Chi et al., 2001; Fortin et al., 2004).
Hippocampal cultures and transfections. Dissociated hippocampal cultures containing glia were prepared from embryonic day 18.5 rats, plated on glass coated with 50 μg/ml poly-d-lysine, and maintained in Neuro-basal medium supplemented with B27, Glutamax I (Invitrogen, San Diego, CA), penicillin, and streptomycin (Higgins and Banker, 1998). Hippocampal neurons were transfected at 7 d in vitro using Effectene (Qiagen, Valencia, CA). One-half of the culture medium was replaced at the time of transfection and then 1 week later. Cells were imaged at 15-21 d in vitro. Where indicated, neurons were treated with 10 nm tetanus toxin (List Biologicals, Campbell, CA) for 16 h before imaging and with 5 μm epoxomicin for 15 h before imaging (Ehlers, 2003).
Immunofluorescence. At 15-21 d in vitro, neuronal cultures were fixed in PBS containing 4% paraformaldehyde. Cells were permeabilized, blocked, and stained in PBS containing 0.05% saponin, 5% cosmic calf serum (Hyclone, Logan, UT) with antibodies to α-synuclein (BD Biosciences, San Diego, CA), synapsin I (Synaptic Systems, Goettingen, Germany), and vesicular glutamate transporter 1 (VGLUT1) (Chemicon, Temecula, CA). Secondary antibodies conjugated to FITC, cyanine 3 (Cy3), or Cy5 were obtained from Jackson ImmunoResearch (West Grove, PA). Fluorescence was visualized and images acquired on a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope using a 63× oil objective [numerical aperture (NA), 1.4].
Fluorescence recovery after photobleaching. Neurons were imaged at 18-21 d in vitro with a Zeiss LSM510 confocal microscope and a 63× oil immersion objective (NA, 1.4) in Tyrode's solution (in mm: 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES-NaOH, pH 7.4, 30 glucose) containing 10 μm CNQX and 50 μm AP-5. Physiological temperature was maintained at 37°C on a heated stage, and individual cultures were imaged for ≤2 consecutive hours. Transfected neurons were identified under epifluorescence and imaged using the 488 nm line of the argon laser. Emitted light was collected using a 505 long-pass filter. To increase the depth of field and collect light from the entire synapse, the pinhole was set to 2.5 μm. During image collection, laser power was maintained at 0.3-2% to reduce photobleaching. After collecting the first image, laser power was increased to 100% and a single synapse bleached by five consecutive scans. Laser power was then restored to 0.3-2%, and 40 images were collected every 400 ms. Fluorescence in the bleached area was quantified using the Zeiss LSM510 software and converted to relative fluorescence, with the initial fluorescence defined as 100% and that after bleaching as 0%. Data are shown are mean ± SEM.
Electrical stimulation and epifluorescence microscopy. Transfected neurons were imaged at 15-21 d in vitro with an inverted epifluorescence Nikon (Tokyo, Japan) microscope and 60× oil objective (NA, 1.4) at room temperature in a closed, laminar flow chamber containing platinum electrodes (Warner Instruments, Hamden, CT). Action potentials were elicited using an A310 Accupulser (World Precision Instruments, Sarasota, FL) at 10 Hz with 1 ms bipolar current pulses yielding fields of 5-10 V/cm across the chamber after acquisition of the fifth image. Samples were illuminated using a Xenon lamp (Sutter Instruments, Novato, CA) with a 492/18 nm excitation filter and 513-547 nm bandpass emission filter (Chroma, Rockingham, VT), and images were acquired every 6 s on a CCD camera (Hamamatsu, Bridgewater, NJ) using on-chip 2 × 2 pixel binning. Stimulation, filter wheels, shutter, and camera were controlled by MetaMorph software (Universal Imaging, Downington, PA). Fluorescence in 4 × 4 pixels regions, manually placed on the center of individual synapses, was quantified, baseline values averaged from the first five frames (before stimulation), and the dynamics of GFP-tagged proteins expressed as fractional fluorescence change. Data shown are mean ± SEM.
Results
Transient, rapidly reversible interactions localize α-synuclein to the synapse
At steady-state, α-synuclein exhibits synaptic enrichment similar in extent to that observed for polytopic membrane proteins of the synaptic vesicle such as VGLUT1 (Fig. 1A) (Withers et al., 1997; Fortin et al., 2004; Specht et al., 2005). To understand how this protein, which does not contain a transmembrane domain or known lipid anchor, localizes to the nerve terminal, we studied the dynamics of α-synuclein in live hippocampal neurons. Transfection of hippocampal neurons with human α-synuclein fused at its N terminus to enhanced GFP (GFP-α-synuclein) leads to a modest ∼50% overexpression compared with untransfected cells as determined by quantitative immunofluorescence using an antibody that recognizes both rat and human proteins: 1545 ± 39 arbitrary fluorescence units (AFU) in transfected cells (n = 320 boutons from eight different neurons) versus 1050 ± 21 AFU in untransfected cells (n = 409 boutons from the same eight fields). This level of overexpression does not perturb the enrichment of either human or endogenous rat α-synuclein at the nerve terminal of transfected neurons (Fig. 1B). In addition, the overexpression does not appear to affect the development of transfected neurons or the localization of other presynaptic proteins including synaptophysin, SV2, synapsin, and VGLUT1 (data not shown). The synaptic localization of GFP-α-synuclein also strongly suggests that the GFP fusion does not influence the behavior of α-synuclein or its interactions at the nerve terminal. Other N-terminal tags such as glutathione S-transferase or the calmodulin-binding protein CBP do not interfere with the membrane binding of α-synuclein in vitro and do not change the requirement for acidic phospholipid headgroups or the α-helical transition of synuclein during membrane binding (Perrin et al., 2000; Kubo et al., 2005). Although we cannot completely exclude some difference from the untagged protein that has eluded detection in vitro and in vivo, GFP-α-synuclein thus appears to serve as an accurate, nondisruptive reporter for the endogenous protein.
Figure 1.

Steady-state enrichment of α-synuclein at the synapse. A, Endogenous α-synuclein colocalizes with VGLUT1 at the synapses of cultured hippocampal neurons. In the overlay images, α-synuclein and VGLUT1 are pseudocolored green and red, respectively. Colocalized α-synuclein and VGLUT1 appear yellow in the overlay. Scale bars: top, 20 μm; bottom, 4 μm. B, GFP-α-synuclein (GFP-αsyn) localizes to the synapses of transfected rat hippocampal neurons. Transfection leads to a modest ∼50% increase in the expression of total α-synuclein (synuclein) detected using an antibody that recognizes both rat and human proteins. GFP-α-synuclein is pseudocolored green in the overlay, whereas total α-synuclein (transfected and endogenous) is red. Scale bar, 6 μm.
To assess the mobility of α-synuclein in live cells, we used FRAP (Lippincott-Schwartz et al., 2001). After photobleaching GFP-α-synuclein at a single synapse, we observed rapid fluorescence recovery in the bleached region (Fig. 2A), suggesting that wild-type human α-synuclein is highly mobile in hippocampal neurons, an unexpected finding given its strong synaptic enrichment. No fluorescence recovery was observed after photobleaching fixed neurons, confirming that the conditions used here irreversibly bleach GFP-α-synuclein and that recovery results from the movement of unbleached molecules into the bleached area (data not shown). In addition, fluorescence recovery at the bleached synapse was typically accompanied by a decrease in fluorescence at neighboring boutons (24.3 ± 0.8%; n = 57 boutons), consistent with adjacent synapses acting as the source of mobilized GFP-α-synuclein (Fig. 2A). Recovery after photobleaching was best fit with a single exponential, supporting the behavior of GFP-α-synuclein as a homogeneous population. GFP-α-synuclein thus exchanges rapidly between neighboring synapses.
Figure 2.

α-Synuclein interacts transiently with synaptic components. A, A single, isolated hippocampal bouton expressing GFP-α-synuclein was photobleached at high laser power, and recovery was monitored every 400 ms thereafter. GFP-α-synuclein recovers rapidly after photobleaching. The two synapses outside the bleached box show a reduction in fluorescence during recovery of the bleached region. The color scale is shown to the right. Scale bar, 2 μm. B, The intensity of fluorescence at the bleached synapse was quantified and expressed as a percentage of initial fluorescence, with fluorescence after the bleach defined as zero. GFP (green) and GFP-A30P (blue) recover with similar, rapid kinetics after photobleaching, consistent with their behavior as soluble proteins. In contrast, GFP-α-synuclein (red) recovers more slowly, indicating a distinct but rapidly reversible interaction with synaptic components. GFP-synapsin (GFPsyp; purple) recovers more slowly than α-synuclein, consistent with a higher affinity for synaptic vesicles. GFP-SV2 (black) does not recover after photobleaching, indicating that synaptic vesicles do not exchange between synapses, at least during this time scale. Data shown are averages ± SEM of 30-40 boutons per construct.
Rapid recovery after photobleaching makes it unlikely that α-synuclein remains tightly associated with synaptic vesicles. Synaptic vesicles can exhibit mobility within a single terminal under certain conditions (Henkel et al., 1996), but there is no precedent for their movement between terminals at rest (Henkel et al., 1996; Kraszewski et al., 1996). However, spontaneous, sustained network activity can lead to the mixing of synaptic vesicles between terminals (Li and Murthy, 2001). To exclude activity-dependent vesicle mobility as a cause for the rapid recovery of α-synuclein, we used the glutamate receptor antagonists CNQX and AP-V in the FRAP experiments described above. In addition, we measured the recovery after photobleaching of a GFP fusion to SV2, an integral membrane protein of synaptic vesicles (Lowe et al., 1988). The fluorescence of GFP-SV2 does not recover from photobleaching in the time course of these experiments (Fig. 2B), consistent with the relative immobility of synaptic vesicles. The rapid recovery of α-synuclein after FRAP thus excludes the tight association of α-synuclein with synaptic vesicles. A fusion of GFP to synapsin I, another peripheral membrane protein associated with synaptic vesicles, also recovers more slowly from photobleaching than α-synuclein. The FRAP recovery kinetics thus indicates that α-synuclein binds more weakly to synaptic vesicles than synapsin (Fig. 2B) (Phair and Misteli, 2001).
Despite its rapid recovery after photobleaching, wild-type α-synuclein recovers more slowly than GFP alone. The slower recovery of GFP-α-synuclein does not reflect its larger mass, because a fusion of GFP to the PD-associated A30P α-synuclein (GFP-A30P), which has essentially the same molecular weight as GFP-α-synuclein, recovers as rapidly as GFP alone (Fig. 2B). The recovery kinetics of GFP-A30P is consistent with its lack of synaptic enrichment and its behavior as a soluble protein in neurons (Fortin et al., 2004). The synaptic localization of wild-type α-synuclein thus reflects rapidly reversible interactions that enable exchange of the protein between adjacent synapses. Nonetheless, these transient interactions are sufficient for the steady-state enrichment of wild-type α-synuclein at the nerve terminal, and their disruption by the A30P mutation eliminates synaptic localization.
To determine whether a small fraction of α-synuclein might be immobile, we have assessed the extent as well as the rate of recovery after photobleaching. GFP and GFP-A30P do not recover fully after photobleaching, presumably reflecting the limited local pool of protein from which recovery can occur (Elson and Qian, 1989). The extent of their recovery (∼80%) was therefore considered the maximal recovery for a soluble protein. The reduced recovery of GFP-α-synuclein (∼70%) relative to that of GFP and GFP-A30P suggests that a small proportion (∼10%) of the protein may be immobile (Fig. 2B). To assess more accurately the existence of an immobile fraction, we repeatedly photobleached the same synapse. In the case of a true immobile fraction, a second photobleach will be followed by more complete recovery than the first photobleach, because the fluorescence is normalized to that observed at the start of each bleach, thereby eliminating the contribution of the immobile fraction to recovery from the second bleach (Lippincott-Schwartz et al., 2003). In the case of GFP-synapsin, recovery is substantially greater after the second photobleach (Fig. 3), suggesting the presence of a significant immobile fraction. In contrast, the fluorescence of GFP-α-synuclein recovers to the same extent after the first and second photobleach (Fig. 3), excluding a significant immobile fraction and indicating that the entire pool of protein is mobile and can exchange between adjacent synapses.
Figure 3.

GFP-α-synuclein has no immobile fraction. A, To assess a possible immobile fraction of α-synuclein, individual boutons were bleached consecutively, and the extent of recovery normalized to the fluorescence observed at the start of each bleach. The extent of fluorescence recovery for GFP-α-synuclein (αsyn) remains the same after the second bleach, excluding an immobile fraction. In contrast, the recovery of synapsin (syp) increases after the second bleach, consistent with an immobile fraction. B, The extent of recovery for GFP-α-synuclein after bleach 1 and 2 (filled) falls on a line with slope ∼1, confirming a similar extent of recovery after both bleaches and no immobile fraction. In the case of synapsin (open), however, the ratio of recovery exceeds 1, indicating a larger recovery after the second bleach and hence an immobile fraction.
Neural activity disperses α-synuclein from synaptic boutons
Neural activity modulates the localization of multiple synaptic proteins. For example, synapsin disperses from the synapse early in the course of stimulation, suggesting dissociation from synaptic vesicles before their fusion with the plasma membrane (Chi et al., 2001, 2003). Similarly, depolarization of hippocampal neurons with high K+ influences the distribution of α-synuclein observed in fixed cells. Before stimulation, endogenous α-synuclein concentrates at synaptic boutons where it colocalizes with synapsin and VGLUT1 (Fig. 4A, rest). Depolarization with high K+ results in the loss of α-synuclein from synaptic boutons (Fig. 4A, stimulated, arrowheads). Unlike synapsin, however, α-synuclein does not increase substantially in the axon after stimulation (Fig. 4A, stimulated, arrows), and synaptic α-synuclein does not recover within 10 min after cessation of the stimulus (Fig. 4A, recovery, arrowheads).
Figure 4.
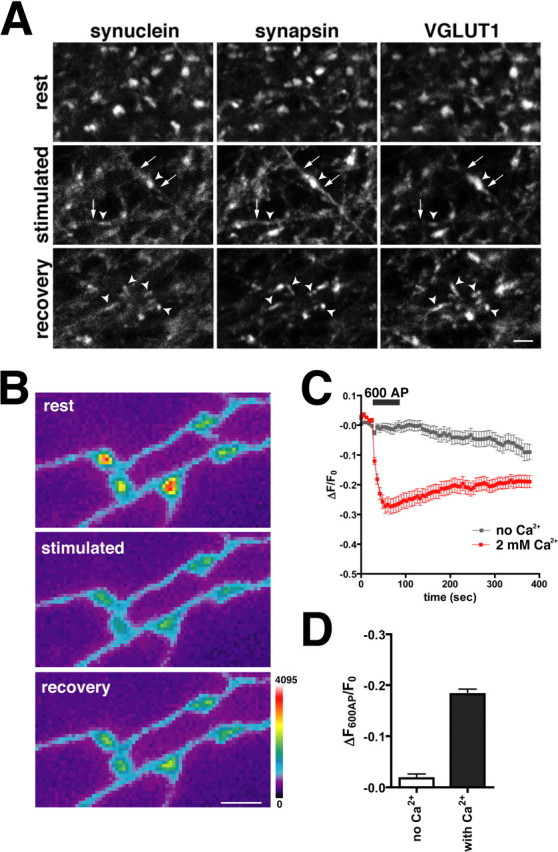
α-Synuclein disperses in response to depolarization. A, Neurons were fixed (rest), depolarized with 45 mm KCl and immediately fixed (stimulated), or depolarized followed by a 10 min recovery (recovery) before fixation. Synaptic boutons were identified by VGLUT1 staining and are indicated with arrowheads. Similar to synapsin, endogenous α-synuclein disperses from boutons after depolarization. Unlike synapsin, however,α-synuclein does not accumulate in the axon (arrows) after stimulation. Ten minutes after recovery, synapsin has reclustered in the synaptic terminal and colocalizes with VGLUT1. α-Synuclein does not reaccumulate at the synapse in this time frame. Scalebar, 2 μm. B, At rest, GFP-α-synuclein is enriched at synaptic boutons and disperses during stimulation with 600 action potentials (AP) delivered at 10Hz. Minimal reclustering of the protein has occurred by 5 min (recovery). The color scale is shown to the right. Scalebar, 2 μm. C, No dispersion of α-synuclein occurs during stimulation with 600 AP in calcium-free medium (gray). After stimulation in the absence of calcium, the cells were washed in calcium-containing medium for 10 min and were then restimulated (red). The dispersion of α-synuclein thus depends on calcium entry. The traces are the average ± SEM dispersion at 30 synapses from one representative cell. D, Average dispersion of GFP-α-synuclein during sequential stimulation in calcium-free and calcium-containing medium. Medium was exchanged during a 10 min rest separating the two stimulation rounds. Error bars indicate SEM. p<0.0001, Student's ttest; n = 138 synapses from three cells.
To characterize the activity-dependent dispersion of α-synuclein, we imaged the GFP fusion in living hippocampal neurons. Similar to the endogenous protein, the amount of GFP-α-synuclein at the synapse decreases by ∼20% after a 600 action potentials stimulus (Fig. 4B-D). GFP-α-synuclein does not appear to accumulate in the axon, and very little returns to the boutons after the stimulus has stopped (Fig. 4B,C). The loss of synaptic GFP-α-synuclein produced by activity depends on external calcium (Fig. 4C,D), indicating a requirement for calcium entry. GFP and GFP-A30P show no change in fluorescence after stimulation, consistent with their behavior as soluble proteins and supporting the significance of the dispersion observed for GFP-α-synuclein (Fig. 5A). The PD-associated A53T mutant, which localizes normally to the nerve terminal (Fortin et al., 2004), exhibits the same activity-dependent dispersion as wild-type α-synuclein (Fig. 5B).
Figure 5.

Synaptic vesicle proteins disperse with different kinetics after stimulation. A, GFP-α-synuclein (GFP αsyn) and GFP-A53T-α-synuclein (data not shown) disperse more slowly than GFP-synapsin (GFP-syp) after 600 action potentials (AP) delivered at 10 Hz. In contrast, GFP and GFP-A30P-α-synuclein (GFPA30P) do not disperse after 600 AP, consistent with their behavior as soluble proteins. The kinetics of α-synuclein dispersion most closely resembles that of GFP-SV2. The traces shown are averages ± SEM of 22-54 boutons from one representative cell for each construct. B, Average maximal dispersion of GFP-tagged proteins immediately after 600 AP. Error bars indicate SEM (n > 300 boutons from 6-9 cells for each construct).
The kinetics of dispersion differs for the two peripheral membrane proteins synapsin and α-synuclein (Fig. 5A). The activity-dependent dispersion of α-synuclein appears somewhat slower than that of synapsin (Fig. 5A) (Chi et al., 2001). α-Synuclein may thus disperse at a later step during the evoked release of neurotransmitter. In addition, the synaptic fluorescence of GFP-synapsin rapidly recovers to prestimulation levels, whereas GFP-α-synuclein recovers only in part, resulting in the net loss of GFP-α-synuclein from the nerve terminal (Fig. 5A).
Exocytosis triggers the dispersion of α-synuclein
What triggers the dispersion of α-synuclein from the nerve terminal? The stringent requirement for calcium indicates that dispersion of α-synuclein occurs after action potential invasion in the terminal (Fig. 4C,D). The dispersion of synapsin also requires calcium entry and results from phosphorylation by calcium/calmodulin-dependent protein kinase II before exocytosis (Chi et al., 2001). However, the delay in movement of α-synuclein relative to synapsin suggests that α-synuclein may disperse at a later step in the exocytic process. We therefore assessed dispersion of α-synuclein in neurons pretreated with tetanus toxin, which blocks the regulated exocytosis of synaptic vesicles without affecting calcium influx. Because the dispersion of synaptic vesicle proteins requires fusion of synaptic vesicles with the plasma membrane and subsequent diffusion along the surface of the axon (Sankaranarayanan and Ryan, 2000; Li and Murthy, 2001), tetanus toxin substantially reduces the dispersal of GFP-SV2 (Fig. 6A). Tetanus toxin treatment also impairs the dispersion of α-synuclein (Fig. 6A), indicating that the exocytic event itself triggers the movement of α-synuclein. The similar dependence on exocytosis presumably accounts for the similar kinetics of SV2 and α-synuclein dispersion from the synapse (Fig. 5A). In contrast, the dispersion of synapsin is unaffected by tetanus toxin (Fig. 6A), consistent with its release from synaptic vesicles before exocytosis (Chi et al., 2001).
Figure 6.

The dispersion of α-synuclein depends on exocytosis and shows a graded response to increasing numbers of action potentials (AP). A, The average maximal dispersion after 600 AP in cells expressing GFP-α-synuclein (GFP αsyn), GFPSV2, or GFP-synapsin (GFPsyp) either with or without tetanus toxin pretreatment. Tetanus toxin greatly reduces the activity-dependent dispersion of α-synuclein and GFP-SV2. In contrast, tetanus toxin does not affect the dispersion of synapsin, consistent with its dissociation before synaptic vesicle exocytosis. Error bars indicate SEM. *p < 0.0001, Student's t test; n > 140 boutons from four to nine cells for each construct and treatment. B, Average maximal dispersion of α-synuclein increases with the number of action potentials, all delivered at 10 Hz. Error bars indicate SEM (n > 80 boutons from 3-5 cells for each of the different stimulus durations). C, Frequency histograms of GFP-α-synuclein dispersion. The number of boutons exhibiting different levels of dispersion at the end of the stimulus is plotted as a function of initial fluorescence binned in 5% increments. Increasing AP number produces greater dispersion, and the distributions show a graded rather than all-or-none, bimodal response.
The extent of synaptic vesicles exocytosis, the time course of endocytosis, and the reclustering of synaptic vesicles are regulated by the duration of a stimulus (Sankaranarayanan and Ryan, 2000; Li and Murthy, 2001). To determine whether the extent of α-synuclein dispersion from the terminal or the time course of recovery also depends on stimulus duration, we varied the length of the 10 Hz pulse, in effect delivering different numbers of action potentials. Stimulation with a larger number of action potentials increases the average extent of α-synuclein dispersion from synaptic boutons (Fig. 6B). This may reflect the increasing probability of all-or-none dispersion from individual boutons. Alternatively, the extent of α-synuclein dispersion at individual boutons may be graded, reflecting a fixed amount of dispersion per action potential. Figure 6C indeed shows that the dispersion of α-synuclein within individual terminals is graded rather than bimodal. The amount of α-synuclein at the nerve terminal thus reflects the level of previous synaptic activity.
α-Synuclein dissociates from the synaptic vesicle membrane after fusion
Where does α-synuclein go after exocytosis? The failure to observe a significant increase in axonal α-synuclein after stimulation raised the possibility that the protein is degraded in an activity-dependent manner. However, Western analysis showed that the total amount of α-synuclein does not change after depolarization of untransfected neurons (data not shown). Arguing further against a role for protein degradation, pretreatment of neurons with the proteasome inhibitor epoxomicin does not affect the activity-dependent loss of GFP-α-synuclein from the nerve terminal. Treated boutons show a decrease in fluorescence of 24.8 ± 1.1% (ΔF/F0) after 600 action potentials (n = 107 boutons from three cells), in the same range as the decrease in fluorescence observed in untreated cells (Figs. 4, 5).
Over the entire field, quantification of GFP-α-synuclein fluorescence indicates a modest 1.9 ± 0.4% reduction in fluorescence (Fig. 7B), similar in extent to that observed for stimulation of neurons expressing GFP and GFP-A30P (6.4 ± 0.4 and 6.6 ± 0.5% decrease, respectively) but significantly smaller than the stimulation-induced dispersion of GFP-α-synuclein observed at boutons (18.3 ± 0.5% decrease) (Figs. 4, 7). Thus, the total amount of GFP-α-synuclein does not change with stimulation, supporting rapid diffusion away from the synapse rather than degradation or secretion.
Figure 7.

α-Synuclein dissociates from synaptic vesicles during exocytosis. A, The activity-dependent dispersion of GFP-SV2 (SV2; filled squares) from synaptic boutons is accompanied by an increase in fluorescence in the perisynaptic area (filled circles). After endocytosis and the reclustering of synaptic vesicles, the fluorescence of GFP-SV2 returns to the center of the bouton. In contrast, GFP-α-synuclein (αsyn) does not accumulate perisynaptically (open circles) during dispersion from the bouton (open squares), consistent with its dissociation from the membrane after synaptic vesicle exocytosis. A small, brief increase in perisynaptic fluorescence follows the onset of stimulation and may reflect the rapid diffusion of GFP-α-synuclein away from the bouton. The inset indicates the regions used for quantification of fluorescence at the center of the bouton and in the perisynaptic area. The traces are averages ± SEM from one representative cell for each construct (n = 30 boutons for SV2 and 75 boutons for α-synuclein). B, Average integrated fluorescence intensity of GFP-α-synuclein over the entire field declines by 1.8 ± 0.4% after 600 action potentials (AP). Please note the difference in scale from A. The trace shows average ± SEM fluorescence (n = 5 fields).
Because the dispersion of α-synuclein requires synaptic vesicle exocytosis and follows kinetics similar to that of SV2, α-synuclein may behave like a typical membrane protein of the synaptic vesicle, inserting into the plasma membrane after exocytosis and spreading laterally into the perisynaptic area before recycling into newly formed synaptic vesicles that eventually recluster in the bouton (Sankaranarayanan and Ryan, 2000; Li and Murthy, 2001). During a train of 600 action potentials, the fluorescence of both SV2 and α-synuclein in the center of the bouton declines to a similar extent (21.6 ± 0.9% decrease for SV2, n = 105 boutons from three cells; 24.3 ± 0.8% decrease for α-synuclein, n = 163 boutons from four cells) and with similar kinetics (Fig. 7A). In the case of SV2, this decrease in fluorescence over the bouton is accompanied by an equivalent increase in fluorescence over the perisynaptic region (13.5 ± 0.9% increase for each of the two perisynaptic areas surrounding a bouton; n = 184 perisynaptic areas from three cells) (Fig. 7A). Synaptic vesicle recycling then redistributes the accumulated perisynaptic fluorescence back to the center of the bouton (Fig. 7A). In contrast, the activity-dependent dispersion of α-synuclein is not accompanied by an equivalent accumulation in the perisynaptic area (0.38 ± 0.52% increase; n = 308 perisynaptic areas from four cells), suggesting that the protein does not remain associated with the plasma membrane after synaptic vesicle exocytosis (Fig. 7A). Rather, α-synuclein appears to dissociate from its binding site after exocytosis, behaving like a soluble protein that rapidly diffuses away from the bouton. We regularly observed a transient, small, stimulation-induced increase in the perisynaptic fluorescence of α-synuclein (Fig. 7A), presumably reflecting the rapid movement of soluble α-synuclein into the axonal cytoplasm.
Discussion
α-Synuclein exhibits enrichment at the nerve terminal equivalent to that of an integral membrane protein of synaptic vesicles, but the kinetic analysis in live neurons using FRAP indicates that it is extremely mobile, exchanging rapidly between adjacent synapses. We cannot detect even a small fraction of immobile α-synuclein by sequential photobleaching, whereas the peripheral membrane protein synapsin exhibits a significant immobile fraction. Nonetheless, the recovery of wild-type α-synuclein from photobleaching is slower than that of the A30P mutant. Because A30P-α-synuclein behaves as a soluble protein not enriched at the nerve terminal (Fortin et al., 2004), we conclude that the slowed recovery of wild-type α-synuclein reflects the interactions that underlie its synaptic localization. Thus, transient interactions are sufficient for the precise steady-state localization of α-synuclein to the nerve terminal.
Live cell imaging enables us to reconcile previous conflicting reports about the membrane interactions of α-synuclein. α-Synuclein binds to a number of artificial membranes in vitro, in particular those containing phospholipids with an acidic head group (Davidson et al., 1998; Jo et al., 2000; Eliezer et al., 2001). However, α-synuclein shows very little if any membrane association in native brain extracts (Maroteaux and Scheller, 1991; Jakes et al., 1994; Iwai et al., 1995). Although we cannot exclude some difference between the behavior of GFP-α-synuclein and the untagged protein, our demonstration that GFP-α-synuclein exhibits only transient interactions in live neurons is consistent with these observations. Presumably, the conditions used in vitro, for example the absence of other protein, drive the binding of α-synuclein to artificial membranes, whereas the high concentration of other cytoplasmic proteins present in cells make these interactions transient and difficult to preserve during biochemical fractionation of native extracts. In contrast, FRAP experiments can detect low-affinity interactions that mediate the synaptic localization of α-synuclein in vivo.
Can transient interactions account for the observed enrichment of α-synuclein at the synapse? Rapid association and dissociation has been demonstrated previously to underlie the steady-state localization of histones to stable compartments within the nucleus (Phair and Misteli, 2000). Low-affinity interactions between cytoplasmic domains of proteins are also sufficient to form and maintain stable compartments of the endoplasmic reticulum (Snapp et al., 2003). Thus, readily reversible, low-affinity interactions can create apparently stable cellular structures. However, they also confer potential for rapid regulation. Indeed, we find that neural activity regulates the synaptic localization of α-synuclein.
Although several synaptic proteins have been reported to redistribute into the axon in response to stimulation, our results show that the behavior of α-synuclein is unique. The activity-dependent dispersion of α-synuclein is slower than that of another peripheral membrane protein, synapsin. Although calcium entry is required for the dispersion of both α-synuclein and synapsin, tetanus toxin only blocks the dispersion of α-synuclein. Thus, in contrast to synapsin, which disperses after calcium entry but before synaptic vesicle fusion (Chi et al., 2001), α-synuclein disperses during or after fusion. Integral membrane proteins of the synaptic vesicle such as synaptobrevin (Sankaranarayanan and Ryan, 2000; Li and Murthy, 2001) and SV2 (this study) also disperse after exocytosis, diffusing along the axonal membrane, but unlike these proteins, α-synuclein does not accumulate in the perisynaptic region or the axon. Rather, the disappearance of α-synuclein from the bouton and its absence from the perisynaptic region indicate that it dissociates from the membrane after fusion and rapidly diffuses away. Indeed, stimulation does not alter the localization of GFP or A30P-α-synuclein, confirming that dispersion of α-synuclein involves a disruption of membrane binding.
Unlike synapsin and integral synaptic vesicle membrane proteins, α-synuclein does not reaccumulate at the nerve terminal within 10 min after stimulation. Synaptic vesicles reform within at least 20-60 s after exocytosis (Gandhi and Stevens, 2003; Fernandez-Alfonso and Ryan, 2004), indicating that their disappearance cannot account for the failure to recover synaptic α-synuclein. Interestingly, α-synuclein is considered to behave as a natively unfolded protein in solution (Weinreb et al., 1996) and may not require ubiquitination for degradation by the proteasome (Tofaris et al., 2001; Snyder et al., 2003), raising the possibility that rapid proteolysis accounts for the disappearance of synaptic α-synuclein after stimulation. However, we cannot detect a decrease in the level of the endogenous protein by Western analysis after neuronal stimulation. In addition, inhibition of the proteasome does not block the stimulation-induced dispersion of GFP-α-synuclein or promote its reclustering after stimulation. Furthermore, the analysis of total GFP-α-synuclein fluorescence (over axons as well as boutons) shows no significant decline with stimulation, indicating that the loss of α-synuclein from synapses is balanced by its appearance elsewhere. Thus, the lack of recovery after stimulation presumably reflects rapid diffusion to distant sites. The low affinity of α-synuclein for synaptic components may contribute to this relatively unimpeded diffusion and also to the lack of recovery. In addition, we do not know whether α-synuclein remains α-helical or unfolds after dissociation from the membrane, and this may influence its ability to rebind synaptic vesicles and hence to recluster.
We have observed the dispersion of α-synuclein from the bouton after strong stimulation (600 action potentials). However, smaller numbers of action potentials also produce easily detectable dispersion. The loss of α-synuclein from the terminal thus correlates with the extent of stimulation, suggesting a physiological role for the activity-dependent dispersion. Consistent with this, α-synuclein inhibits refilling of the readily releasable synaptic vesicle pool, and this effect diminishes with repeated stimulation (Abeliovich et al., 2000; Yavich et al., 2004). In particular, α-synuclein knock-out mice exhibit immediate, strong facilitation in response to high-frequency stimulation that does not appear in wild-type animals until after one or two previous trains of 600 action potentials each (Yavich et al., 2004). Because we have demonstrated that this amount of stimulation is sufficient to disperse α-synuclein, the observation that wild-type animals begin to behave like α-synuclein knock-outs after stimulation may simply reflect the loss of α-synuclein from wild-type terminals. The effects of α-synuclein on transmitter release presumably reflect the amount of protein remaining at the nerve terminal.
Activity may influence the role of α-synuclein in PD. Because α-synuclein has a central role in PD but inherited mutations are rare, activity-dependent changes in α-synuclein have the potential to influence triggering of the sporadic disorder. For example, membrane association has been reported to modulate the tendency of α-synuclein to aggregate (Narayanan and Scarlata, 2001; Perrin et al., 2001; Cole et al., 2002; Lee et al., 2002). The dissociation of α-synuclein from the membrane after exocytosis may thus regulate its aggregation and toxicity. Interestingly, mice lacking the DJ-1 gene implicated in familial PD exhibit a defect in dopamine release (Goldberg et al., 2005), which may influence the predisposition to degeneration by affecting the dispersion of α-synuclein. In addition, smoking protects against PD, presumably through the action of nicotine (Gorell et al., 1999; Tanner et al., 2002; Quik, 2004). Nicotine activates ionotropic receptors at the nerve terminal to cause the exocytic release of dopamine (McGehee et al., 1995; Zhou et al., 2001). Thus, nicotine may protect against PD by causing the dispersion of α-synuclein.
Footnotes
This work was supported by the Hillblom Foundation (D.L.F), the University of California, San Francisco Medical Scientist Training Program grant from the National Institutes of Health (V.M.N.), the Howard Hughes Medical Institute (S.M.V.), a Society for Neuroscience Minority Fellowship (M.D.A.), the Sandler Neurogenetics Program, and the Valley Foundation (R.H.E.). We thank S. Finkbeiner and K. Svoboda for helpful discussion and W. Yan, C. Tran, J. Park, and Y. Dobryy for technical assistance.
Correspondence should be addressed to R. H. Edwards, Departments of Neurology and Physiology, University of California, San Francisco School of Medicine, 600 16th Street, San Francisco, CA 94143-2140. E-mail: edwards@itsa.ucsf.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510913-09$15.00/0
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239-252. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci 22: 8797-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC (2003) A broken alpha-helix in folded alpha-synuclein. J Biol Chem 278: 15313-15318. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC (2004) Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA 101: 14966-14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2001) Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci 4: 1187-1193. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2003) Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38: 69-78. [DOI] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL (2002) Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem 277: 6344-6352. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273: 9443-9449. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231-242. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell Jr R, Browne G (2001) Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 307: 1061-1073. [DOI] [PubMed] [Google Scholar]
- Elson EL, Qian H (1989) Interpretation of fluorescence correlation spectroscopy and photobleaching recovery in terms of molecular interactions. Methods Cell Biol 30: 307-332. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA (2004) The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron 41: 943-953. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH (2004) Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci 24: 6715-6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ (1999) Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci USA 96: 13450-13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF (2003) Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423: 607-613. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45: 489-496. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Rybicki BA, Johnson CC, Peterson EL (1999) Smoking and Parkinson's disease: a dose-response relationship. Neurology 52: 115-119. [DOI] [PubMed] [Google Scholar]
- Henkel AW, Simpson LL, Ridge RM, Betz WJ (1996) Synaptic vesicle movements monitored by fluorescence recovery after photobleaching in nerve terminals stained with FM1-43. J Neurosci 16: 3960-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D, Banker GA (1998) Primary dissociated cell cultures. In: Culturing nerve cells, Ed 2 (Banker GA, Goslin K, eds) pp 37-78. Cambridge, MA: MIT.
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T (1995) The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14: 467-475. [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345: 27-32. [DOI] [PubMed] [Google Scholar]
- Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M (1998) Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J Biol Chem 273: 26292-26294. [DOI] [PubMed] [Google Scholar]
- Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE (2000) Alpha-synuclein membrane interactions and lipid specificity. J Biol Chem 275: 34328-34334. [DOI] [PubMed] [Google Scholar]
- Kraszewski K, Daniell L, Mundigl O, De Camilli P (1996) Mobility of synaptic vesicles in nerve endings monitored by recovery from photobleaching of synaptic vesicle-associated fluorescence. J Neurosci 16: 5905-5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18: 106-108. [DOI] [PubMed] [Google Scholar]
- Kubo SI, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, Fortin DL (2005) A combinatorial code for the interaction of alpha-synuclein with membranes. J Biol Chem 280: 31664-31672. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Choi C, Lee SJ (2002) Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem 277: 671-678. [DOI] [PubMed] [Google Scholar]
- Li Z, Murthy VN (2001) Visualizing postendocytic traffic of synaptic vesicles at hippocampal synapses. Neuron 31: 593-605. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A (2001) Studying protein dynamics in living cells. Nat Rev Mol Cell Biol 2: 444-456. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Altan-Bonnet N, Patterson GH (2003) Photobleaching and photoactivation: following protein dynamics in living cells. Nat Cell Biol [Suppl]: S7-S14. [PubMed]
- Lowe AW, Madeddu L, Kelly RB (1988) Endocrine secretory granules and neuronal synaptic vesicles have three integral membrane proteins in common. J Cell Biol 106: 51-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH (1991) The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 11: 335-343. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8: 2804-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269: 1692-1696. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20: 3214-3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan V, Scarlata S (2001) Membrane binding and self-association of alpha-synucleins. Biochemistry 40: 9927-9934. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302: 1772-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM (2000) Interaction of human alpha-synuclein and Parkinson's disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem 275: 34393-34398. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM (2001) Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem 276: 41958-41962. [DOI] [PubMed] [Google Scholar]
- Phair RD, Misteli T (2000) High mobility of proteins in the mammalian cell nucleus. Nature 404: 604-609. [DOI] [PubMed] [Google Scholar]
- Phair RD, Misteli T (2001) Kinetic modelling approaches to in vivo imaging. Nat Rev Mol Cell Biol 2: 898-907. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Quik M (2004) Smoking, nicotine and Parkinson's disease. Trends Neurosci 27: 561-568. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA (2000) Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol 2: 197-204. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) Alpha-synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Snapp EL, Hegde RS, Francolini M, Lombardo F, Colombo S, Pedrazzini E, Borgese N, Lippincott-Schwartz J (2003) Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol 163: 257-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B (2003) Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem 278: 11753-11759. [DOI] [PubMed] [Google Scholar]
- Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R (2005) Subcellular localisation of recombinant alpha- and gamma-synuclein. Mol Cell Neurosci 28: 326-334. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998a) Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95: 6469-6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998b) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 251: 205-208. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Goldman SM, Aston DA, Ottman R, Ellenberg J, Mayeux R, Langston JW (2002) Smoking and Parkinson's disease in twins. Neurology 58: 581-588. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG (2001) Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509: 22-26. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury Jr PT (1996) NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 35: 13709-13715. [DOI] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF (1997) Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res 99: 87-94. [DOI] [PubMed] [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P (2004) Role of α-synuclein in presynaptic dopamine recruitment. J Neurosci 24: 11165-11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: 164-173. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4: 1224-1229. [DOI] [PubMed] [Google Scholar]