

Abstract
Neurofibrillary tangles (NFTs), neuropil threads, and neuritic elements of senile plaques predominantly comprise hyperphosphorylated tau protein and represent pathological characteristics of Alzheimer's disease (AD). These lesions occur before the presentation of clinical symptoms and correlate with the severity of dementia. In vivo detection of these lesions would thus prove useful for preclinical diagnosis of AD and for tracking disease progression. The present study introduces three novel compounds, 4-[2-(2-benzoimidazolyl)ethenyl]-N,N-diethylbenzenamine (BF-126), 2-[(4-methylamino)phenyl]quinoline (BF-158), and 2-(4-aminophenyl)quinoline (BF-170), as candidate probes for in vivo imaging of tau pathology in the AD brain. When solutions of these compounds are injected intravenously into normal mice, these agents exhibit excellent brain uptake and rapid clearance from normal brain tissue. These compounds display relatively lower binding affinity to β-amyloid fibrils and higher binding affinity to tau fibrils, compared with previously reported probe BF-168. In neuropathological examination using AD brain sections, BF-126, BF-158, and BF-170 clearly visualize NFTs, neuropil threads, and paired helical filament-type neuritis. Autoradiography using 11C-labeled BF-158 further demonstrated labeling of NFTs in AD brain sections. These findings suggest the potential usefulness of quinoline and benzimidazole derivatives for in vivo imaging of tau pathology in AD.
Keywords: imaging, neuropathology, Aβ peptide, tau, neurofibrillary tangles, paired helical filaments
Introduction
The deposition of senile plaques (SPs) and neurofibrillary tangles (NFTs) represents a pathological hallmark of Alzheimer's disease (AD). Definitive diagnosis of AD relies on the postmortem assessment of these pathological changes. SPs are extracellular deposits containing β-amyloid (Aβ) peptide cleaved from a longer amyloid precursor protein to produce a 40-43 aa peptide. NFTs comprise bundles of paired helical filaments (PHFs) that result from the abnormal aggregation of tau protein (Lee et al., 1991). PHFs accumulate in the neuronal cytoplasm and form NFT with age. Initial lesions of NFTs occur in the transentorhinal cortex, followed by involvement of the entorhinal cortex and hippocampus, progressing to the neocortex (Braak and Braak, 1991). Severity of neurofibrillary pathology correlates with severity of cognitive impairment (Dickson, 1997). However, these pathological changes can be observed in elderly individuals with normal cognition. The pathological process of AD must therefore begin before the presentation of the clinical symptoms of dementia (Price and Morris, 1999). Recent drug development has been aimed at preventing the accumulation of SPs and NFTs in presymptomatic AD patients. The ability to measure levels of these lesions in the living human brain is thus desirable for presymptomatic diagnosis of AD.
Many attempts have been made to visualize AD-specific pathological changes in the living brain (Nordberg, 2004). Currently, the most practical methods for this purpose are to measure the distribution of intravenously administered radiotracers that selectively bind to SPs or NFTs using positron emission tomography (PET) or single photon emission computed tomography. Such radiotracers require sufficient permeability to the blood-brain barrier (BBB). Several researchers have focused on developing lipophilic radiotracers for imaging AD-specific pathology (Shoghi-Jadid et al., 2002; Klunk et al., 2004; Kung et al., 2004; Mathis et al., 2004; Okamura et al., 2004a). To obtain a better understanding of the pathophysiology of AD, individual evaluation of the distributions of Aβ pathology and tau pathology is desirable. However, no surrogate markers exist that allow evaluation of the severity of neurofibrillary pathology in AD brains, because of the difficulty in developing an NFT-specific imaging probe (Small et al., 2002).
We screened >2000 small molecules to develop novel agents for use in PET with a high affinity for SPs and NFTs. This process has identified novel quinoline and benzimidazole derivatives with a preference to bind NFTs rather than SPs and readily cross the BBB. The present study evaluated whether these compounds may offer a good indicator of tau pathology in AD patients.
Materials and Methods
Preparation of BF compounds. Three novel compounds, 4-[2-(2-benzoimidazolyl)ethenyl]-N,N-diethylbenzenamine p-toluenesulfonate (BF-126), 2-[(4-methylamino)phenyl]quinoline (BF-158), and 2-(4-aminophenyl)quinoline (BF-170), were originally designed by us and custom-synthesized by Tanabe R&D Service (Osaka, Japan) (see Fig. 1A). [11C]BF-158 was synthesized from N-Boc-protected BF-170 and [11C]methyl triflate (see Fig. 1B). Briefly, to a solution of N-Boc-protected BF-170 and NaH in dry acetone was bubbled thorough [11C]methyl triflate at room temperature, followed by heating at 80°C for 1 min. The reaction was then acidified with a dioxane solution of hydrochloric acids and heated for an additional 5 min. After neutralizing the reaction with sodium phosphate buffer, the crude mixture was purified with semipreparative reverse-phase HPLC to give [11C]BF-158 with a radiochemical purity of >95% and a specific activity of 11-15 GBq/μmol at the end of synthesis. Isolated [11C]BF-158 was solubilized into 40% ethanol solution for in vitro autoradiography of AD brain sections and saline with polysorbate 80 for in vivo brain-uptake study using normal mice.
Figure 1.
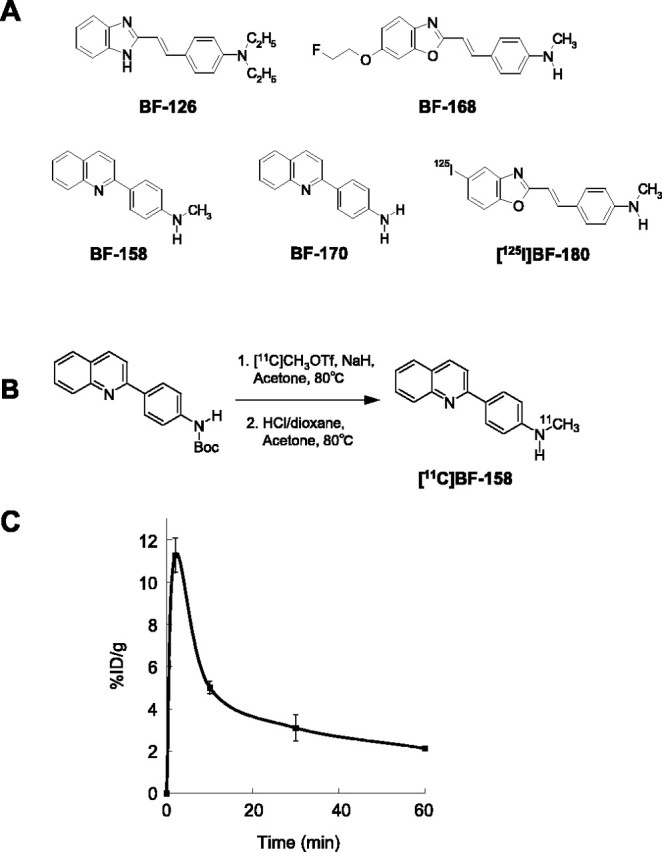
A, Chemical structures of BF-126, BF-158, BF-168, BF-170, and [125I]BF-180. B, Radiolabeling of BF-158. C, The %ID/g obtained in the brains of mice after intravenous administration of [11C]BF-158.
Biodistribution of BF compounds in normal mice. Brain uptake after intravenous injection of BF-126 and BF-170 in mice was analyzed using HPLC with a fluorescence detector, as described previously (Okamura et al., 2004b). Briefly, each compound (1 mg/kg), dissolved in diluted HCl, was administered into the tail vein of male Institute of Cancer Research (ICR) mice (7 weeks of age; body weight, 30-40 g; n = 3). At 2 and 30 min after the injection of compounds, the brain was removed. Brain homogenates were centrifuged at 14,000 rpm for 10 min, and the supernatant was used for extraction. The mobile phase was 20 mm phosphate buffer, pH 6.5, and acetonitrile at a ratio of 2:3 for BF-126 and 1:1 for BF-170, at a flow rate of 1 ml/min. An FS-8020 fluorescence detector (Tosoh, Tokyo, Japan) was operated at excitation/emission wavelengths of 420/525 nm for BF-126 and 280/500 nm for BF-170. Brain uptake of BF-158 was measured using 11C-labeled compound. [11C]BF-158 (2.6-3.3 MBq) was administered into the tail vein of ICR mice (n = 12; male; average weight, 24 g). The mice were then killed by decapitation at 2, 10, 30, and 60 min after injection. The brains were removed and weighted, and the radioactivity was counted with an automatic γ-counter (Wizard 1480; PerkinElmer Wallac, Turku, Finland). The percentage injected dose per gram (%ID/g) was calculated by comparison of tissue count to tissue weight. Each %ID/g value is an average ± SEM of three separate experiments.
In vitro binding assays. The dissociation constant (Kd) and maximum specific binding of BF-180 were determined as described previously (Okamura et al., 2004a). For inhibition studies, binding studies were performed using aggregates of synthetic Aβ1-42 (Peptide Institute, Osaka, Japan). A mixture containing 50 μl of BF-126, BF-158, and BF-170, 50 μl of 0.05 nm [125I]BF-180, 100 μl of 100 nm Aβ1-42, and 800 μl of 8% ethanol was incubated at room temperature for 4 h. The mixture was then filtered, and filters containing bound 125I ligand were counted using the γ-counter. Values for half-maximal inhibitory concentration (IC50) were determined from displacement curves of three independent experiments using GraphPad Prism software (GraphPad Software, San Diego, CA), and those for the inhibition constant (Ki) were determined using the Cheng-Prusoff equation.
In addition, fluorescence assay of the compounds with Aβ1-42 and tau fibrils was performed. The 412 aa isoform of human tau was expressed from cDNA clone htau46 in Escherichia coli and purified as described previously (Hasegawa et al., 1998). Aβ1-42 (0.2 mg/ml; Peptide Institute) in 50 mm potassium phosphate buffer, pH 7.4, was incubated at 37°C for 72 h with gentle shaking. Tau protein (1 mg/ml) was incubated in the presence of 0.1 mg/ml heparin for 72 h with continuous shaking. Fluorescence spectra of a mixture of the same amount of 0.2 mg/ml Aβ1-42 or 0.1 mg/ml tau and 10, 3, 1, 0.3, 0.1, 0.03, and 0.01 μm concentrations of each compound were measured using a microplate spectrofluorometer (Gemini XS; Molecular Devices, Sunnyvale, CA). Optimal excitation wavelength for the mixture of four BF compounds and Aβ1-42 or tau fibrils were determined. Then, EC50 values (effective concentrations to achieve 50% maximal fluorescence intensity at optimal excitation wavelength) were calculated using GraphPad Prism. All measurements were performed in duplicate.
Measurement of octanol/water partition coefficients. 1-Octanol (Wako Pure Chemicals, Osaka, Japan) and PBS was saturated with PBS and 1-octanol before use, respectively. Test compounds were dissolved in 1-octanol and shaken with equal amounts of PBS for 30 min at room temperature. After centrifugation at 2000 per min for 15 min, the absorbency of 1-octanol layer was measured at the peak wavelength of the absorbance spectrum of each compound using a microplate reader (Molecular Devices Spectra Max 190). The octanol/water partition coefficients were determined by comparing the absorbency with that before shaking with PBS. Each data point was performed in duplicate.
Neuropathological staining. Stainability of tested compounds were examined using postmortem brain tissues from cases of autopsy-confirmed AD (78-year-old woman), Pick's disease (75-year-old woman), and progressive supranuclear palsy (PSP) (78-year-old man). Brain sections were obtained from Fukushimura Hospital. Experiments were performed in accordance with the regulations of the ethics committee of the BF Research Institute. Serial sections (6 μm thick) from paraffin-embedded blocks of hippocampus were used for staining. After deparaffinization, quenching of autofluorescence was performed as described previously (Okamura et al., 2004a). Quenched tissue sections were immersed in 100 μm compound or 0.125% thioflavin-S solution containing 50% ethanol for 10 min. Finally, sections were dipped briefly into water, rinsed in PBS for 60 min, coverslipped with FluorSave Reagent (Calbiochem, Darmstadt, Germany), and examined using a Nikon (Tokyo, Japan) Eclipse microscope equipped with a violet filter (excitation, 380-420 nm; dichroic mirror, 430 nm; barrier filter, 450 nm) and a blue filter (excitation, 450-490 nm; dichroic mirror, 505 nm; barrier filter, 520 nm). For the comparison of stainability between BF compounds and thioflavin-S, a blue-violet filter (excitation, 400-440 nm; dichroic mirror, 455 nm; long-pass filter, 470 nm) was used. Staining was also performed in AD brain sections pretreated with 90% formic acid for 5 min. Sections stained with BF compounds were subsequently immunostained with AT8 anti-tau antibody (diluted 1:20; Innogenetics, Ghent, Belgium) to detect tau pathology. An adjacent section was also immunostained with 6F/3D anti-Aβ antibody (diluted 1:50; Dako, High Wycombe, UK). Sections were placed in blocking buffer for 30 min and then incubated at 4°C with primary antibodies for 18 h (AT8) or 1 h (6F/3D). After washing, sections were incubated with biotinylated anti-mouse IgG (Wako Pure Chemicals, Tokyo, Japan) for 60 min, followed by Texas Red-conjugated avidin (Vector Laboratories, Burlingame, CA) for 60 min. After cover-slipping, sections were examined using a Nikon Eclipse microscope with a green filter set (excitation, 510-560 nm; dichroic mirror, 575 nm; barrier filter, 590 nm).
In vitro autoradiography. A 6-μm-thick section from paraffin-embedded blocks of hippocampus was used for autoradiographic study. After deparaffinization, the section was labeled using 4.7 MBq/ml of [11C]BF-158 at room temperature for 10 min, and then washed briefly with H2O and 50% ethanol. After drying, the labeled section was exposed to a BAS-III imaging plate (Fujifilm, Tokyo, Japan) for 1 h. Autoradiographic images were obtained using a BAS-5000 phosphorimaging instrument (Fujifilm). Adjacent brain sections were immunostained with anti-tau antibody (AT8) and anti-Aβ antibody (6F/3D), as described previously (Okamura et al., 2004a).
Results
We investigated whether tested compounds would enter the brain in amounts sufficient for in vivo brain imaging using PET (Table 1, Fig. 1C). The octanol/water partition coefficients of BF-126, BF-158, BF-168, and BF-170 were 12.5, 47.4, 62.6, and 70.3, respectively. These values indicate that these compounds were highly lipophilic. High uptake of these compounds was apparent in the brain after intravenous administration of each compound, with 7.2%ID/g for BF-126 and 9.1%ID/g for BF-170 at 2 min. These values were higher than those reported previously for compound BF-168 (Okamura et al., 2004a). In addition, brain uptake at 30 min after injection was 0.16%ID/g for BF-126 and 0.25%ID/g for BF-170. Brain uptake of BF-158 was measured using 11C-labeled compound. Brain uptake at 2, 10, 30, and 60 min after intravenous injection of [11C]BF-158 was 11.3 ± 0.81, 5.0 ± 0.31, 3.1 ± 0.61, and 2.1 ± 0.097%ID/g, respectively (Fig. 1C). In vitro binding of tested compounds to Aβ was examined using synthetic Aβ fibrils. In competitive binding assays with 125I-labeled styrylbenzoxazole compound BF-180, BF-126 displayed high binding affinity to Aβ1-42 fibrils, comparable with BF-168. In contrast, Ki for BF-158 and BF-170 was >5000 nm, suggesting a difference in binding sites between quinoline derivatives and benzoxazole derivatives (Table 1). In the fluorescence binding assay of the mixture of tested compounds and Aβ1-42 fibrils, EC50 value was lowest in BF-168, suggesting higher affinity to Aβ compared with the other agents. In contrast, in the assay of the mixture of compounds with tau fibrils, EC50 values of BF-170, BF-158, and BF-126 was lower than that of BF-168, suggesting relatively higher binding affinity to tau fibrils.
Table 1.
Octanol/water partition coefficients, brain uptakes at 2 and 30 min after injection in normal mice, Ki values for Aβ 1-42 fibrils, EC50 values for Aβ 1-42, and tau fibrils from fluorescence assay of the compounds
|
|
Octanol/water partition coefficient |
Brain uptakes (%ID/g) |
Ki values for Aβ fibrils (nm) |
Fluorescence assay |
|
||
|---|---|---|---|---|---|---|---|
|
|
2 min |
30 min |
Ec50 for Aβ fibrils (nm) |
EC50 for τ fibrils (nm) |
|||
| BF-126 | 12.5 | 7.2 ± 0.49 | 0.16 ± 0.030 | 1.2 ± 0.68 | 1280 ± 1.64 | 583 ± 2.07 | |
| BF-158 | 47.4 | 11.3 ± 0.81 | 3.1 ± 0.61 | >5000 | 659 ± 2.04 | 399 ± 2.41 | |
| BF-170 | 70.3 | 9.1 ± 1.2 | 0.25 ± 0.027 | >5000 | 786 ± 1.79 | 221 ± 2.31 | |
| BF-168 |
62.6 |
3.9 ± 0.22a
|
1.6 ± 0.0071a
|
6.4 ± 1.0a
|
346 ± 2.21 |
1010 ± 1.72 |
|
Brain uptake and EC50 values are shown as the mean ± SEM. Ki values for Aβ fibrils are shown as the mean ± SD.
From Okamura et al., 2004a.
Subsequently, neuropathological staining was performed using AD brain samples to examine the in vitro binding characteristics of tested compounds for AD pathology. BF-126, BF-158, and BF-170 clearly stained NFTs and neuropil threads in the hippocampal section of AD brain (Fig. 2A-C). In the comparison with tau immunostaining in the same sections, binding of these compounds with tau pathology was confirmed. In contrast, SPs were faintly stained with BF-126, BF-158, and BF-170, as opposed to clear staining of SPs with thioflavin-S (Fig. 2A-C). We further investigated the stainability of neuritic changes using these compounds and BF-168. BF-168 clearly visualized both the central core and peripheral zone of core plaques under violet and blue filters (Fig. 2D). The staining pattern for BF-168 did not coincide with that for tau immunostaining in the same section but closely resembled Aβ immunostaining in the adjacent section (Fig. 2D). In contrast, BF-126, BF-158, and BF-170 predominantly stained neuritic elements under a blue filter and coincided well with tau immunostaining (Fig. 2E-G). Under a violet filter, the central core and peripheral halo were barely visible. These findings suggest that BF-126, BF-158, and BF-170 prefer to bind to PHF-type neurites rather than Aβ fibrils. Pretreatment with formic acid eliminated the staining of NFTs with BF-170 (Fig. 2H, I), BF-158, and BF-126 (data not shown), suggesting that both compounds recognize the β-sheet structure of NFTs rather than monomeric tau protein. To investigate whether these compounds detect neuropathological lesions in non-AD tauopathy, staining was performed in brain sections from Pick's disease and PSP patients. As a result, BF-126, BF-170, and BF-158 (data not shown) were unable to identify Pick bodies (Fig. 2J), globose tangles (Fig. 2K), and glial pathology in PSP brain (data not shown).
Figure 2.
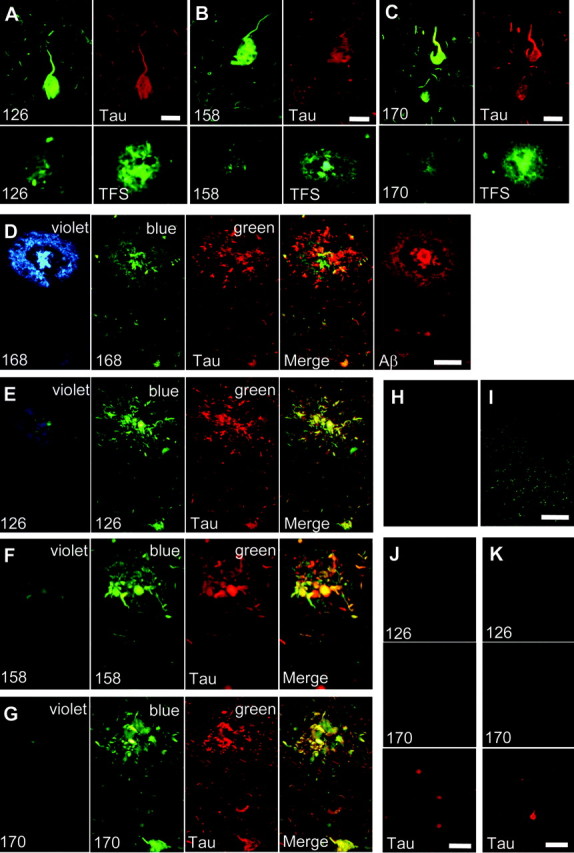
A-J, Neuropathological staining of brain sections from cases of AD (A-I), Pick's disease (J), and progressive supranuclear palsy (K). BF-126 (A), BF-158 (B), and BF-170 (C) clearly stained neurofibrillary tangles and neuropil threads. These stainings were consistent with tau immunostaining. In contrast, these compounds faintly stained amyloid plaques, which were clearly stained with thioflavin-S (TFS) in adjacent sections. D-G, Double staining with BF compounds and anti-tau antibody was performed in hippocampal brain sections. D, Cored plaque is clearly visualized by BF-168 using violet and blue filters. A merged image of BF-168 fluorescence using a blue filter and tau immunostaining (green) indicates that BF-168 does not recognize PHF-type neurites. The image of Aβ immunostaining in an adjacent section closely resembles BF-168 staining. BF-126 (E), BF-158 (F), and BF-170 (G) faintly stain the core and halo of amyloid plaque using a violet filter. However, images using a blue filter closely resemble tau immunostaining. E-I, A merged image indicates that these three compounds preferentially bind to PHF-type neurites (E-G). BF-170 (H), BF-158, and BF-126 (data not shown) did not stain any pathological structures in the section pretreated with formic acid, in contrast to the clear visualization of NFTs in non-pretreated brain sections (I). J, K, Staining of Pick bodies and globose tangles were compared with tau immunostaining in brain sections from cases of Pick's disease (J) and progressive supranuclear palsy (K). Neither Pick bodies (G; green) nor globose tangles (H; green) are detected in brain sections treated with BF-126 and BF-170. Scale bars: A-C, 25 μm; D-G, 50 μm; H-I, 400 μm; J-K, 100 μm
Finally, in vitro autoradiography using [11C]BF-158 was performed in AD brain section. Accumulation of tracer was observed primarily in the gray matter region of the brain. The distribution of labeling with [11C]BF-158 correlated well with tau immunostaining in the adjacent section (Fig. 3A,B). In particular, high levels of tracer accumulation were observed in NFT-rich brain regions (Fig. 3D,E, arrows). This finding indicates the binding of [11C]BF-158 for NFTs in the AD brain.
Figure 3.

A-C, Autoradiography of AD hippocampal brain section using [11C]BF-158 (B) and immunostaining by anti-tau (A) and anti-Aβ (C) antibodies in adjacent sections. D-F, Close-up images of A, B, and C, respectively. D, E, Accumulation of [11C]BF-158 (E, arrows) correlates well with tau immunostaining (D).
Discussion
Although the present research did not confirm whether the studied compounds can visualize tau pathology in the living brain, the results strongly suggest that quinoline and benzimidazole derivatives represent potential candidates for an in vivo tau-imaging agent in AD patients. Requirements for the ideal tau-imaging probe include the following: (1) high BBB and cell membrane permeability; (2) rapid clearance from normal brain tissue; (3) high affinity for NFTs, neuropil threads, and PHF-type neurites; and (4) low nonspecific binding. Our results indicate that BF-126, BF-158, and BF-170 display sufficient BBB permeability for use as PET imaging tracers. Permeability of the neuronal membrane represents another important factor for in vivo imaging of intracellular tau aggregates (Small et al., 2002). High lipophilicity of these compounds has the advantage in this factor. In vivo imaging using a tangle-laden mouse model that accurately reflects AD pathology will elucidate this issue. Furthermore, three tested compounds demonstrated fast clearance from normal brain tissue. This property will contribute to improving the sensitivity of detecting NFT accumulation by minimizing residual background levels of tracer.
The most intriguing point of this study was that BF-126, BF-158, and BF-170 have a preference for tau pathology in AD brain. Results from fluorescence binding assay suggest relatively higher binding affinity to tau fibrils of these three compounds. These compounds also show excellent stainability for tau pathology and little background fluorescence in AD brain. An autoradiographic study using [11C]BF-158 confirmed the accumulation of this tracer in tangle-rich brain regions, emphasizing the binding preference of these compounds to tau pathology in AD brains. However, fluorescence assay data show the binding ability of these agents to Aβ fibrils. In neuropathological staining, these agents faintly stained amyloid plaques. Thus, these compounds are not completely specific to tau pathology. Nevertheless, our data suggest that these three agents have relatively higher binding affinity to tau pathology rather than to Aβ pathology in AD brain.
Previous neuropathological research suggests that the deposition of NFTs occurs before the presentation of clinical symptom in AD. Even in the very early stages of AD, patients display considerable numbers of NFTs in the entorhinal cortex and hippocampus, sufficient for the neuropathological diagnosis of AD. Thus, in vivo imaging of NFTs in conjunction with imaging SPs could prove useful for the early and accurate diagnosis of AD. Quantitative evaluation of tau pathology could also be helpful for tracking severity of dementia, because the formation of neuritic pathology correlates well with clinical severity of dementia (Dickson, 1997). NFT deposition in the entorhinal cortex is closely associated with neuronal loss in very early AD patients (Gomez-Isla et al., 1996). Prevention of NFT formation is thus an important target of anti-dementia drugs. If novel treatments that prevent the pathological formation of neurofibrillary pathology could be turned into clinical applications, this imaging technique would be applicable for the evaluation of treatment efficacy.
The general mechanism of abnormal protein aggregation leading to disease is β-sheet formation. AD is accompanied by the formation of two distinct β-sheet aggregates, Aβ fibrils and PHFs. As is the case with previously reported imaging agents, BF-126, BF-158, and BF-170 recognize β-pleated sheet structure, because staining in AD brain sections was disturbed by pretreatment with formic acid (Kitamoto et al., 1987). Our results suggest that tested compounds display relatively higher binding affinities to PHFs than to Aβ fibrils. Previous reports describe 2-(1-{6-[(2-fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile (FDDNP) as a candidate of tau imaging agent (Agdeppa et al., 2001). This compound is a highly lipophilic agent and specifically labels NFTs as well as SPs in AD brain sections. [18F]FDDNP, the first PET probe to see clinical use for AD patients, reportedly detects the combined signals of SPs and NFTs (Shoghi-Jadid et al., 2002). FDDNP shows high binding affinity to Aβ fibrils; however, binding affinity to tau fibrils is unknown. Pittsburgh compound B, another promising PET probe, preferentially detects the accumulation of SPs rather than NFTs (Klunk et al., 2003). BF-145 and TZDM (2-[4′-(dimethylamino)phenyl]-6-iodobenzothiazole) also reportedly preferentially recognize SPs (Kung et al., 2003; Okamura et al., 2004b). Why these β-sheet binding agents show such a variety of binding characteristics with SPs and NFTs is not well understood. The diameter of a PHF is 18-20 nm at the widest portion and 8-10 nm at the narrowest portion (Wisniewski et al., 1984), whereas Aβ fibrils are 7-12 nm in diameter (Sunde et al., 1997). Although the nature of PHF structure remains contentious, both structures share a common pattern, with 4.76 Å meridional spacing and 10.6 Å equatorial spacing, characteristic of a cross-β structure (Berriman et al., 2003; Barghorn et al., 2004). A recent study indicated the presence of multiple ligand-binding sites on Aβ fibrils (Lockhart et al., 2005). If PHF also includes multiple binding sites for β-sheet binding agents, this factor will contribute to the difference in ligand-binding affinity to SPs and NFTs. The manner of binding between β-sheet binding agents and both NFTs and SPs is not well clarified. However, differences in conformation between SPs and NFTs and differences in ligand-binding sites may result in the diversity of binding properties in these compounds to SPs and NFTs.
Tau lesions are not only the hallmark of AD but are also characteristic of non-AD tauopathy such as PiD and PSP (Goedert, 2004). However, tested compounds did not show any significant binding to pathological lesions in these diseases, implying that both compounds are highly specific to PHFs in AD. Accordingly, these probes are potentially useful for differentiating between AD and other neurodegenerative disorders. For future clinical applications, optimization of the compounds is necessary to assure compound safety, facilitate radiolabeling, and reduce nonspecific binding. We are currently investigating the in vivo binding characteristics of these optimized probes.
Footnotes
This study was supported by the Organization for Pharmaceutical Safety and Research of Japan, the New Energy and Industrial Technology Development Organization, and the Novartis Foundation for Gerontological Research.
Correspondence should be addressed to Yukitsuka Kudo, Tohoku University Biomedical Engineering Research Organization, 2-1, Seiryo-machi, Aoba-ku, Sendai 980-8575, Japan. E-mail: kudoyk@tubero.tohoku.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510857-06$15.00/0
References
- Agdeppa ED, Kepe V, Liu J, Flores-Torres S, Satyamurthy N, Petric A, Cole GM, Small GW, Huang SC, Barrio JR (2001) Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer's disease. J Neurosci 21: RC189(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Davies P, Mandelkow E (2004) Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 43: 1694-1703. [DOI] [PubMed] [Google Scholar]
- Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA (2003) Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc Natl Acad Sci USA 100: 9034-9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82: 239-259. [DOI] [PubMed] [Google Scholar]
- Dickson DW (1997) Neuropathological diagnosis of Alzheimer's disease: a perspective from longitudinal clinicopathological studies. Neurobiol Aging 18 [4 Suppl]: S21-S26. [DOI] [PubMed] [Google Scholar]
- Goedert M (2004) Tau protein and neurodegeneration. Semin Cell Dev Biol 15: 45-49. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437: 207-210. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57: 230-236. [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA (2003) The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23: 2086-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, et al. (2004) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 55: 306-319. [DOI] [PubMed] [Google Scholar]
- Kung MP, Skovronsky DM, Hou C, Zhuang ZP, Gur TL, Zhang B, Trojanowski JQ, Lee VM, Kung HF (2003) Detection of amyloid plaques by radioligands for Aβ40 and Aβ42: potential imaging agents in Alzheimer's patients. J Mol Neurosci 20: 15-24. [DOI] [PubMed] [Google Scholar]
- Kung MP, Hou C, Zhuang ZP, Skovronsky D, Kung HF (2004) Binding of two potential imaging agents targeting amyloid plaques in postmortem brain tissues of patients with Alzheimer's disease. Brain Res 1025: 98-105. [DOI] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos Jr L, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251: 675-678. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong G, Gee AD, Brown J (2005) Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer's disease PET imaging agents on beta-amyloid peptide fibrils. J Biol Chem 280: 7677-7684. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Klunk WE (2004) Imaging beta-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr Pharm Des 10: 1469-1492. [DOI] [PubMed] [Google Scholar]
- Nordberg A (2004) PET imaging of amyloid in Alzheimer's disease. Lancet Neurol 3: 519-527. [DOI] [PubMed] [Google Scholar]
- Okamura N, Suemoto T, Shimadzu H, Suzuki M, Shiomitsu T, Akatsu H, Yamamoto T, Staufenbiel M, Yanai K, Arai H, Sasaki H, Kudo Y, Sawada T (2004a) Styrylbenzoxazole derivatives for in vivo imaging of amyloid plaques in the brain. J Neurosci 24: 2535-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Suemoto T, Shiomitsu T, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Arai H, Sasaki H, Yanai K, Staufenbiel M, Kudo Y, Sawada T (2004b) A novel imaging probe for in vivo detection of neuritic and diffuse amyloid plaques in the brain. J Mol Neurosci 24: 247-255. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 45: 358-368. [DOI] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR (2002) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10: 24-35. [PubMed] [Google Scholar]
- Small GW, Agdeppa ED, Kepe V, Satyamurthy N, Huang SC, Barrio JR (2002) In vivo brain imaging of tangle burden in humans. J Mol Neurosci 19: 323-327. [DOI] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC (1997) Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol 273: 729-739. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Merz PA, Iqbal K (1984) Ultrastructure of paired helical filaments of Alzheimer's neurofibrillary tangle. J Neuropathol Exp Neurol 43: 643-656. [DOI] [PubMed] [Google Scholar]