

Abstract
Long-term depression (LTD) in sensory cortices depends on the activation of NMDA receptors. Here, we report that in visual cortical slices, the induction of LTD (but not long-term potentiation) also requires the activation of receptors coupled to the phospholipase C (PLC) pathway. Using immunolesions in combination with agonists and antagonists, we selectively manipulated the activation of α1 adrenergic, M1 muscarinic, and mGluR5 glutamatergic receptors. Inactivation of these PLC-coupled receptors prevents the induction of LTD, but only when the three receptors were inactivated together. LTD is fully restored by activating any one of them or by supplying intracellular d-myo-inositol-1,4,5-triphosphate (IP3). LTD was also impaired by intracellular application of PLC or IP3 receptor blockers, and it was absent in mice lacking PLCβ1, the predominant PLC isoform in the forebrain. We propose that visual cortical LTD requires a minimum of PLC activity that can be supplied independently by at least three neurotransmitter systems. This essential requirement places PLC-linked receptors in a unique position to control the induction of LTD and provides a mechanism for gating visual cortical plasticity via extra-retinal inputs in the intact organism.
Keywords: adrenergic, cholinergic, glutamatergic, LTD, plasticity, neuromodulation
Introduction
Selective synaptic weakening driven by sensory experience is essential for the postnatal maturation of cortical circuits and also for learning and memory in the adult brain (Kandel and O'Dell, 1992; Singer, 1995; Katz and Shatz, 1996; Bear and Rittenhouse, 1999). Long-term depression (LTD) of synaptic strength can be elicited virtually throughout all cortical regions, although the induction mechanisms are diverse and may vary in different systems (Malenka and Bear, 2004). Two distinct forms of LTD coexist in the hippocampus: one depends on the activation of NMDA receptors (NMDARs) (Dudek and Bear, 1992; Mulkey and Malenka, 1992), whereas another type is linked to the activation of type I metabotropic glutamate receptors (mGluRs) (Oliet et al., 1997; Palmer et al., 1997; Huber et al., 2000). Other types of LTD that require coactivation of mGluRs and NMDARs have been described in the perirhinal cortex (Cho et al., 2000) and in frontal cortex LTD (Otani et al., 1999).
The role of NMDARs in LTD in the supragranular layers of sensory cortices is well established (Kirkwood and Bear, 1994; Castro-Alamancos et al., 1995; Allen et al., 2003; Rao and Daw, 2004), whereas the possible involvement of mGluRs is still debated (Haruta et al., 1994; Huber et al., 1998; Sawtell et al., 1999). Indeed, the observation that application of NMDAR agonists can induce LTD in the hippocampus and visual cortex in vitro support the supposition that activation of NMDARs is not only necessary but also sufficient for the induction of NMDAR-dependent LTD in these cortical regions (Kandler et al., 1998; Lee et al., 1998; Dodt et al., 1999). In contrast, in vivo studies indicate that developmental plasticity in the sensory cortex not only requires NMDARs (Kleinschmidt et al., 1987; Fox et al., 1996; Roberts et al., 1998; Daw et al., 1999; Iwasato et al., 2000) but also depends on the integrity of cholinergic, noradrenergic, and serotonergic innervation (Kasamatsu and Pettigrew, 1976; Bear and Singer, 1986; Gu and Singer, 1995; Siciliano et al., 1997; Gu, 2002). However, in vitro studies have led to the suggestion that these extra-retinal inputs act as neuromodulators that, like changes in the inhibitory tone (Steele and Mauk, 1999), facilitate or promote NMDAR-dependent plasticity, but without being essential for the induction mechanism (Brocher et al., 1992; Kirkwood et al., 1999; Kirkwood, 2000; Kojic et al., 2000).
Mechanistically, the distinction between an essential component and a regulatory factor of plasticity is not trivial (Sanes and Lichtman, 1999). The importance of neuromodulators for plasticity in vivo prompted us to re-examine their role in the induction of LTD in cortical slices. Our results indicate that activation of receptors coupled to the phospholipase C (PLC) pathway, which include subtypes of cholinergic, noradrenergic, and metabotropic glutamatergic receptors, is essential for the induction of NMDAR-dependent LTD in the visual cortex. Importantly, each of the receptors examined, the metabotropic glutamate receptor mGluR5, the α1 adrenoreceptor, and the M1 muscarinic receptor, could independently enable the induction of LTD, indicating that each pathway is sufficient to support this function and that the critical requirement is a minimal level of PLC activity.
Materials and Methods
Subjects. Three- to 5-week-old male Long-Evans rats and PLCβ1 knockout mice (Kim et al., 1997) served as subjects. Three- to 4-week-old animals were used for intracellular recordings, and 4- to 5-week-old animals were used for extracellular recordings. Wild-type and homozygous mice were obtained as the F1 of heterozygous mating pairs (F1 hybrid crosses from 129 PLCβ1+/- and C57BL/6J PLCβ1+/-).
Coronal slices (300-400 μm) from the visual cortex and hippocampus were prepared as described previously (Kirkwood and Bear, 1994) in ice-cold dissection buffer (in mm: 212.7 sucrose, 5 KCl, 1.25 NaH2PO4, 3 MgCl2, 1 CaCl2, 26 NaHCO3, and 10 dextrose, 95% O2/5% CO2) and transferred to normal artificial CSF (ACSF) for at least 1 hour before recording. Normal ACSF is similar to the dissection ACSF, except that sucrose is replaced by 124 mm NaCl, MgCl2 is lowered to 1 mm, and CaCl2 is raised to 2 mm. In hippocampal experiments, MgCl2 was 2 mm, and CaCl2 was 2.5 mm.
Electrophysiology. Synaptic responses were recorded at 30°C in layer II/III and evoked with 0.2 ms current pulses delivered with a bipolar stimulating electrode (200 μm diameter; FHC, Bowdoinham, ME) placed approximately in the border between layers IV and V. Microelectrodes filled with ACSF (1-2 MΩ) were used for extracellular recordings. Visualized whole-cell recordings were done with an infrared-differential interference contrast microscope (Zeiss, Thornwood, NY). Patch pipettes (2-4 MΩ) were filled with an internal solution consisting of the following (in mm): 130 K-gluconate, 8 KCl, 10 EGTA, 10 HEPES, 3 ATP, and 0.3 GTP, pH 7.4, 275-285 mOsm. In voltage-clamp experiments, KCl and K-gluconate were replaced by the Cs salts, and 5 mm QX314 (lidocaine-N-ethyl-bromide) was also included. The junction potential (typically <5 mV) was not compensated. Only cells with membrane potentials more negative than -65 mV and an input resistance >70 MΩ (103.17 ± 4.48 MΩ) were studied. NMDAR-dependent and AMPA receptor (AMPAR)-dependent responses were discriminated based on their distinct kinetics and voltage dependence as described previously (Hayashi et al., 2004): the NMDAR-mediated currents were measured at +40 mV, 100 ms after the response onset, whereas the AMPAR-mediated currents were taken as the peak amplitude response recorded at -80 mV. Isolated NMDAR-mediated response currents were recorded at -40 mV in the presence of 10 μm CNQX with an internal solution containing 19.5 μm CsCl.
Synaptic responses were quantified as the initial slope of the intracellularly recorded EPSP and the extracellularly recorded EPSP in CA1, or as the amplitude of the maximum negative field potential (FP) in layer III. Changes in the amplitude of the FP correlate with changes in the initial slope of EPSPs recorded in layer III neurons (Kirkwood and Bear, 1994). LTD was induced by delivering 900 pulses at 1 Hz, or by pairing 200 pulses at 1 Hz with postsynaptic depolarization to -40 mV in rat slices. In mice, we used 900 paired-pulses (40 ms apart) at 1 Hz, which is much more effective in inducing LTD in animals >3 weeks of age. Only data from slices with stable recordings (<5% change over the baseline period) were included in the analysis. All data are presented as average ± SEM normalized to the preconditioning baseline. For statistical comparisons, the LTD magnitude was taken as the average of the last 5 min recorded. Statistical significance was assessed using t test or ANOVA, followed by the Newman-Keuls post hoc tests. Most drugs, including 6-methyl-2-(phenylethynyl)pyridine (MPEP), d,l-2-amino-5-phosphonovaleric acid (APV), 4-[N-(3-chlorophenyl) carbamoyloxy]-2-butynyltrimethyl-ammonium chloride (McN), 2-aminoethyl diphenyl borate (2-APB), heparin, staurosporine, d-myo-inositol-1,4,5-triphosphate (IP3), cyclopiazonic acid (CPA), GF109203X (GFX), prazosin, sulpiride, ketanserine, U73122, and U73343, were purchased from Sigma (St. Louis, MO). 2-Methyl-2-(carboxycyclopropyl)glycine (MCCG), CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), and LY341495 were purchased from Tocris (Ellisville, MO). U73122, U73343, and prazosin were dissolved in 0.01% dimethylsulfoxide (DMSO); corresponding controls were done in 0.01% DMSO. Stock drug solutions were made at a 1000× final concentration, aliquoted, and stored at -20°C.
Surgical lesions. Selective cholinergic lesions of the nucleus basalis magnocellularis (nBM) and substantia innominata (SI) were made with injections of 192 IgG-saporin, as described previously (Baxter et al., 1995). Selective lesions of noradrenergic neurons in the locus ceruleus (LC) were made with anti-dopamine β hydroxylase (DBH)-saporin. The 192 IgG-saporin and DBH-saporin (Chemicon, Temecula, CA) were dissolved in sterile 0.1 m PBS at a concentration of 0.25 μg/μl and 1 μg/μl, respectively. Anesthesia was induced and maintained by isoflurane (Isovet; Mallinckrodt, Mundelein, IL) inhalation. Injections of 192 IgG-saporin (0.4 μl; n = 9 rats) were made through a 30 gauge Hamilton syringe at eight sites [stereotaxic coordinates (in mm): anteroposterior (AP), -0.40; mediolateral (ML), ±1.5 and 2.5; dorsoventral (DV), -8.0; and AP, -1.20; ML, ±1.6 and ±2.6; DV, -8.0], according to the atlas of Paxinos and Watson (1986). Injections of sterile PBS (n = 12) served as control in sham-operated animals. DBH-saporin (0.2 μl) was injected at two sites (AP, -10 mm; ML, ±1.2 mm; DV, -7.4 mm). Injections were made at the rate of 0.05 μl/min, and the needle was left in place 3-4 min after each injection. Two weeks of recovery were allowed before the experiments. In some cases, reserpine (5 mg/kg, i.p., in 10% propylene glycol plus 100 μl of glacial acetic acid) was given the day before the experiment to deplete cathecolamine pools.
Histological confirmation of immunolesions. After the rats were killed, the brains were removed rapidly and cut coronally into two blocks slightly posterior to the optic chiasm. The posterior block was used to obtain visual cortical slices. The anterior block and brainstem were stored in 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, for 3 d, cryoprotected in PB containing 20% sucrose for 24 h, frozen on powdered dry ice, and stored at -80°C until further processing. Coronal sections through the basal forebrain (40 μm) and brainstem were processed for the immunohistochemical localization of either choline acetyltransferase (ChAT) or DBH. Additional adjacent coronal sections were processed for acetycholinesterase (AChE) histochemistry to confirm cholinergic depletion in the cortex (Karnowsky and Roots, 1964). The extent of the noradrenergic and cholinergic depletions in the visual cortex was directly assessed in four rats.
Endogenous peroxidase was blocked by a 30 min wash with 3% H2O2/10% MeOH in PBS, followed by two 5 min washes in PBS with 0.3% Triton-X (PBST). To block background staining, the sections were incubated for 1 h in PBST with 10% the appropriate serum. The sections were then incubated with the appropriate antibody (ChAT, 1:200 dilution; DBH, 1:5000 dilution; Chemicon) overnight at 4°C, rinsed twice in PBST, incubated for 1 h at room temperature in the appropriate biotinylated secondary (1:200 dilution), rinsed twice again in PBST, and incubated for 1 h at room temperature in ExtrAvidin peroxidase conjugate (1:1000 dilution; Sigma). After two 5 min rinses in PBS, tissue was reacted using a Vector SG substrate kit for peroxidase (Vector Laboratories, Burlingame, CA).
Results
Previous studies using bath application of agonists have established that neuromodulators can act as regulators that promote or facilitate an NMDAR-dependent form of LTD (Kirkwood et al., 1999; Kojic et al., 2000; Blond et al., 2002). However, a more essential role might have been overlooked for at least two reasons. First, by having a common target, multiple receptors coupled to PLC might substitute each other in supporting LTD. Second, background levels of endogenous agonists can be very high in slices (Kirkwood et al., 1999), and electrical stimulation might recruit cholinergic and noradrenergic axons. Thus, under standard recording conditions, blocking the activation of a single receptor might be insufficient to prevent the induction of LTD. For that reason, we studied the induction of LTD under conditions aimed to reduce or prevent the activation of three receptors coupled to PLC: α1 adrenergic receptors, M1 muscarinic receptors, and the metabotropic glutamate receptor mGluR5.
Antagonists against neurotransmitter receptors coupled to PLC block the induction of LTD. We first tested whether antagonists against PLC-coupled receptors could block the induction of LTD. The antagonists, atropine (5 μm), urapidil (10 μm), and MPEP (10 μm), which, respectively, target the M1 muscarinic, the α1 adrenergic, and the mGluR5 receptors, were bath applied throughout the experiments. At these concentrations, atropine and urapidil block the facilitation of LTD (Kirkwood et al., 1999), and MPEP blocks the effects of mGluR5 activation on cell excitability and plasticity in brain slices (Mannaioni et al., 2001; Ireland and Abraham, 2002; Lee et al., 2002). LTD was attempted with a low-frequency stimulation (LFS) protocol (see Materials and Methods) that reliably induces LTD in the visual cortex (Kirkwood and Bear, 1994). As shown in Figure 1A, when the three antagonists were applied together, LFS induced very little LTD (94.6 ± 3.5% of baseline response at 1 h after LFS; n = 8 rats; 14 slices) compared with the interleaved controls without the drugs (76.2 ± 3.3%; n = 8 and 16). LTD was also blocked when atropine was replaced by the M1-specific antagonist pirenzepine (10 μm; 95.5 ± 5.6%; n = 3 and 7) in the antagonist mixture (Fig. 1B), when urapidil was replaced by the α1 antagonist prazosin (10 μm; 97.4 ± 2.2; n = 6 and 13), and when MPEP was replaced by LY341495 [100 μm, which blocks type I/III and type II mGluRs at this concentration (Fitzjohn et al., 1998; Kingston et al., 1998), also blocked LTD (92.4 ± 2.8%; n = 3 and 7)] (Fig. 1B). In contrast, robust LTD was observed when only two antagonists were applied in any combination (urapidil plus atropine: 79.5 ± 2.7%, n = 4 and 8; urapidil plus MPEP: 76 ± 4.3%, n = 5 and 9; atropine plus MPEP: 77.3 ± 3.7%, n = 5 and 11) (Fig. 1B). An ANOVA test indicated that the differences in LTD among these conditions were significant (F(6,63) = 5.796; p < 0.0001), and Newman-Keuls post hoc analysis confirmed that LTD was blocked only when three antagonists were combined. Together, the results indicate that simultaneous blockade of several PLC-linked neurotransmitters is necessary to block LTD. This is consistent with the concept that a minimum of PLC-linked receptor activation is required to support LTD.
Figure 1.

Antagonists of PLC-coupled receptors block the induction of LTD. A, Bath application of 10 μm MPEP, 5 μm atropine, and 10 μm urapidil blocks the induction of LTD. The graph shows the effects of LFS (1 Hz, 15 min) on the FP amplitude in normal ACSF (open circles) or in the presence of the drugs (filled circles). The magnitude of LTD at the end of each individual experiment is indicated on the right of the graph. Examples of responses recorded 1 min before (thin traces) or 60 min after (thick traces) LFS are shown above. B, LTD (expressed as percentage of decrease from baseline) is not blocked when only two antagonists are present in the bath. Asterisks denote statistical significance from control. C, Effects of bath-applied antagonists (MUA) on baseline synaptic responses (top, open symbols) and after the induction of LTD (bottom, filled symbols). D, Application of antagonists (MUA) does not affect the FP amplitude during LFS (1 Hz, 900 pulses). Each data point (open circles, control; filled circles, MUA) is an average of four consecutive responses. E, Application of antagonists does not affect NMDAR responses. Left, Representative synaptic currents recorded at -80 and +40 mV with and without the antagonist mixture (MUA). Right, NMDAR/AMPAR ratios and AMPAR amplitude in the presence and absence of MUA. Calibration: 100 ms, 0.2 nA. F, Application of antagonists (MUA) do not affect the isolated NMDA responses (recorded at -40 mV; see Materials and Methods) during conditioning stimulation (200 pulses, 1 Hz). A, Atropine (5 μm); M, MPEP (10 μm); U, urapidil (10 μm); L, LY341495 (100 μm); Pr, pirenzepine (10 μm); Pz, prazosin (10 μm); Cont, control. Error bars represent SEM.
Subsequently, we tested the possibility that the absence of LTD could be attributed to changes in synaptic transmission and/or NMDAR-mediated responses. As shown in Figure 1C, the antagonist mixture (in μm: 5 atropine, 10 urapidil, and 10 MPEP) produced only a minor and reversible increase in the naive responses (107.0 ± 1.5% at 20 min; n = 2 and 8) and barely affected synaptic responses if applied after LTD was established (from 73.0 ± 4.9% at 40 min to 80 ± 5.3% at 60 min; n = 3 and 6). We also tested the possibility that the mixture affected the responses to conditioning stimulation (Fig. 1D). The decay of the FP amplitude during LFS was similar (repeated-measures ANOVA; F(1,5250) = 0.05; p = 0.945) in the absence (n = 4 and 15) and in the presence (n = 4 and 17) of the mixture. The NMDAR-mediated responses, in contrast, were also unaffected by the mixture. The ratio of NMDAR/AMPAR responses, measured as described (see Materials and Methods) and shown in Figure 1E, were similar (p = 0.65) in the presence or absence of the mixture [MPEP-urapidil-atropine (MUA), 0.31 ± 0.04, n = 6 and 19; control, 0.33 ± 0.04, n = 9 and 23]. Similarly, the mixture did not affect the rate of decay of the NMDA responses (repeated-measures ANOVA; F(1,3980) = 0.16; p = 0.9011; control, n = 4 and 12; MUA, n = 4 and 10) during 1 Hz conditioning stimulation (Fig. 1F). These results suggest that the antagonist mixture blocks the induction, but not the expression, of LTD, probably by acting at a step subsequent to the activation of NMDARs.
PLC-coupled receptors are required specifically for the induction of LTD. It was important to determine whether blockade of PLC-coupled receptors also affects long-term potentiation (LTP). In these experiments, we attempted LTD and LTP by pairing a brief epoch of LFS (1 Hz, 200 pulses) with postsynaptic depolarization to -40 mV to induce LTD, to 0 mV to induce LTP (see Materials and Methods), to an intermediate potential of -20 mV, and to -60 mV. As shown in Figure 2, in the presence of the antagonists, the induction of LTD with pairing at -40 mV was virtually eliminated (94.9 ± 8.4%; n = 6 and 8) compared with controls (73.3 ± 6.0%; n = 6 and 8). In contrast, pairing at -20 mV, which normally evokes a modest potentiation (106.9 ± 4.6%; n = 10 and 22), induced robust LTP in the presence of the antagonists (144.8 ± 1.2%; n = 7 and 14). In these last experiments (pairing at -20 mV), the experimenter was blind to the ACSF composition. Finally, pairing at 0 mV induced LTP of comparable magnitude (p = 0.83) in the presence (148.5 ± 8.7%; n = 7 and 10) or absence (146.2 ± 6.5%; n = 5 and 12) of the mixture, whereas pairing at -60 mV induced no change in both cases (control: 101.6 ± 2.2, n = 3 and 6; MUA, 101.5 ± 2.4, n = 3 and 8). An ANOVA test confirmed the significance of these differences. Figure 2D depicts how blocking PLC-coupled receptors affects the dependence of synaptic plasticity on the membrane potential (Vm) during pairing. The antagonist mixture caused an upward displacement on the voltage dependence of plasticity. Vertical shifts, as opposed to lateral shifts, of this type of plasticity dependence curve are interpreted as arising from specific changes in LTD (Ikegaya et al., 2002; Philpot et al., 2003). Thus, the results are consistent with the idea that PLC-coupled receptors are selectively required for the induction of LTD only.
Figure 2.
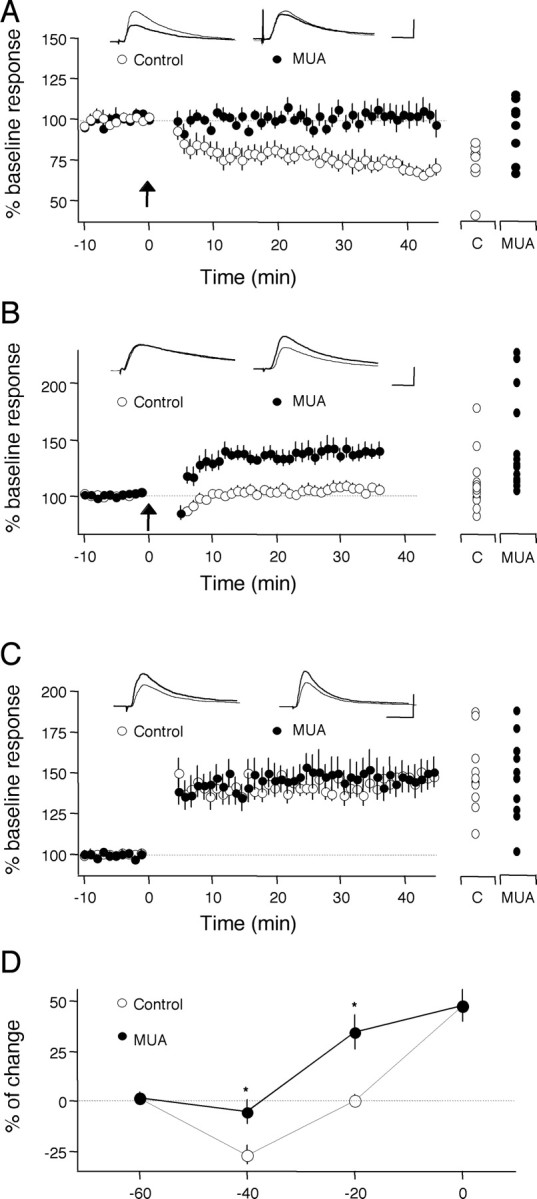
Antagonists against PLC-coupled receptors block pairing-induced LTD and promote pairing-induced LTP. A-C, Changes in the EPSP slope induced by pairing in normal ACSF (open circles) in the presence of the drugs (MUA; filled circles) are shown. The pairings were 1 Hz, 200 pulses plus depolarization to -40 mV (A), to -20 mV (B), and to 0 mV (C). C, Control. D, Blockade of PLC-coupled receptors causes an upward shift in the voltage dependence of pairing-induced LTD. Asterisks denote statistical significance from control. Individual experiments are shown on the right of each graph. Superimposed traces are average of four consecutive responses recorded 1 min before (thin traces) or 30 min after (thick traces) pairing. Calibration: 10 ms, 5 mV.
Elimination of cholinergic and noradrenergic inputs to the cortex alters the induction of LTD. The observation that LTD is not prevented by blocking two of the three receptors examined suggests that background levels of endogenous agonists are sufficient to support LTD in slices. To test this idea, we studied LTD in slices deprived of extra-cortical cholinergic and noradrenergic inputs. Cholinergic neurons in the nBM and SI were selectively lesioned with injections of 192 IgG-saporin, and selective lesions of noradrenergic neurons in the LC were made with anti-DBH-saporin (see Materials and Methods). These combined lesions were done at 3 weeks of age, and the experiments were conducted 2 weeks later. Immunohistological analysis of the visual cortex revealed a pronounced absence of ChAT-positive staining in 192 IgG-saporin-lesioned rats and also a loss of DBH-positive (noradrenergic) fibers after DBH-saporin lesions (Fig. 3). Synaptic responses (quantified as the half-maximal FP) were comparable in slices from lesioned (1.19 ± 0.13 mV) and sham-operated animals (1.23 ± 0.13 mV). Also comparable was the magnitude of LTD induced with LFS in slices prepared from sham-operated animals (81.1 ± 1.4%; n = 5 and 12) and lesioned animals (75.4 ± 2.4%; n = 5 and 7) (Fig. 4A). In contrast, adding the mGluR5 antagonist MPEP (10 μm) to the bath blocked the induction of LTD in the slices prepared from these lesioned animals (94.9 ± 2.54%; n = 5 and 7), but it did not affect LTD in sham-operated animals (MPEP: 80.5 ± 5.0%, n = 3 and 8) (Fig 4A). These differences were robust and statistically significant (ANOVA; F(3,29) = 7.8; p = 0.0006). Similar results were obtained with animals in which the double neurochemical depletion was attempted in a different way, by combining the cholinergic lesion with injection of reserpine 1 d before the experiment (Fig. 4B) (see Materials and Methods). Again, LTD was robust in slices from treated animals (treated: 77.3 ± 3.2%, n = 4 and 8; sham: 77.0 ± 2.5%, n = 4 and 10) and blocked by the addition of MPEP (94.8 ± 3.1%; n = 4 and 11) when the experimenter was blind to the presence of MPEP in the bath (Fig. 4B). A two-way ANOVA confirmed the similarities between the two depletion approaches (F(1,46) = 0.139; p = 0.711). Together, these results indicate that, in the absence of endogenous acetylcholine and norepinephrine, the induction of LTD becomes critically dependent on the activation of mGluR5 receptors.
Figure 3.
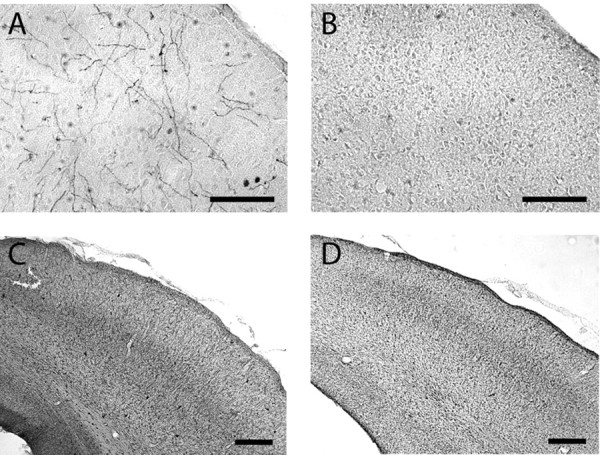
Immunohistochemical confirmation of cholinergic and noradrengic lesions. A, B, Photomicrographs of the visual cortex show abundant DBH-immunopositive noradrenergic fibers in a control brain (A) and sparse density in a lesioned brain (B). C, D, Photomicrographs of the visual cortex show AChE staining in a control brain (C), which is substantially depleted in an immunolesioned brain (D). Scale bars: A, B, 100 μm; C, D, 200 μm. Immunostaining for the subcortical cell bodies of these projection systems also showed marked removal in lesioned brains (data not shown).
Figure 4.
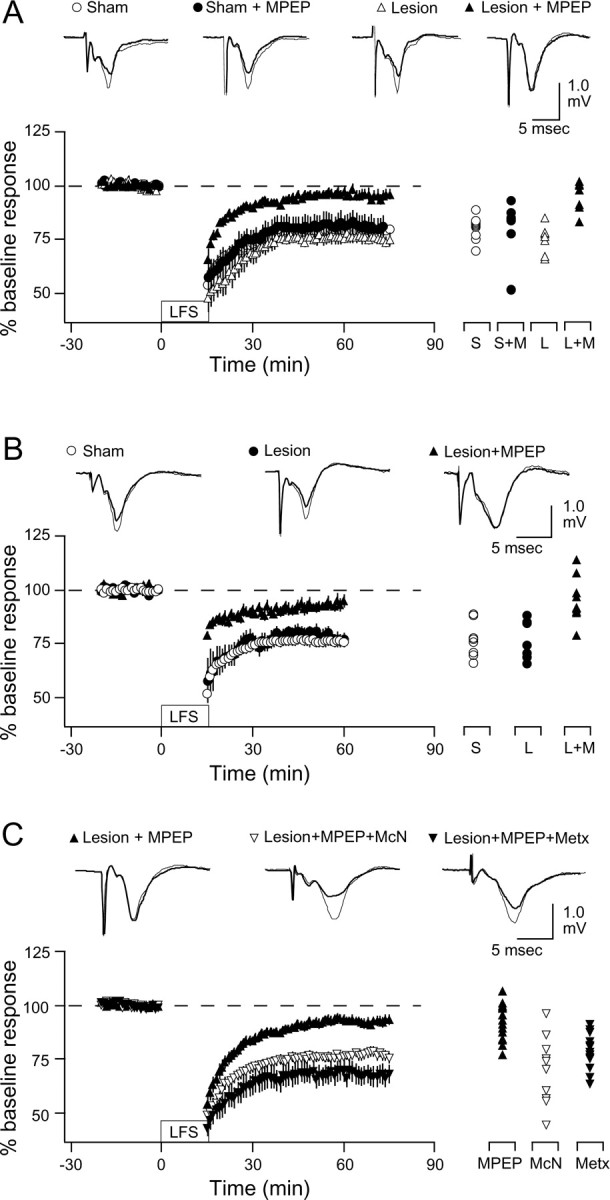
LTD in immunolesioned slices. A, Bath application of the mGluR5 antagonist MPEP blocks LTD in slices from lesioned animals (192 IgG-saporin and DBH-saporin). The graph shows LTD induced in slices prepared from sham-operated animals (circles) and lesioned animals (triangles) superfused with normal ACSF (open symbols) or with 10 μm MPEP (filled symbols). B, Bath application of the mGluR5 antagonist MPEP also blocks LTD in slices from animals lesioned with 192 IgG-saporin and reserpine. C, Bath application of the M1 agonist McN or the α1 agonist methoxamine rescues LTD in slices from lesioned animals treated with MPEP. The graph shows LTD induced in slices from lesioned animals in the presence of 10 μm MPEP (filled triangles) and in the presence of 10 μm MPEP plus 5 mm McN (open triangles) or 40 μm methoxamine (filled triangles). Results from individual experiments (as percentage of LTD at 60 min after LFS) are shown on the right; representative recordings (before and after LFS) are shown at the top of each graph. S, Sham; M, MPEP; L, LY341495; Metx, methoxamine.
Cholinergic or adrenergic activation restores LTD in “depleted” slices. We wanted to test whether the block of LTD by the mGluR5 antagonist MPEP in the slices from lesioned animals resulted from insufficient PLC activation. Previous studies reported substantial M1 receptor functionality in the visual cortex after 192 IgG-saporin lesions (Rossner et al., 1995; Schliebs et al., 1996; Kuczewski et al., 2005). Therefore, we attempted to rescue LTD by adding the M1 muscarinic agonist McN A-343 (McN, 3 μm) to the bath. These experiments, shown in Figure 4C, were done in slices from six double-lesioned animals in which MPEP blocked LTD (93.3 ± 2.3%; n = 6 and 14). In slices from five of these lesioned animals, inclusion of McN allowed the induction LTP in the presence of MPEP (76.3 ± 2.7%; n = 5 and 12). In slices from two of these lesioned animals, we also rescued the MPEP block of LTD (68.0 ± 4.5%; n = 2 and 13) by adding the α1 adrenergic agonist methoxamine (10 μm) in the bath. These differences were significant (F(2,36) = 14.7; p < 0.001). Application of either McN or methoxamine alone without conditioning stimulation, in contrast, had little effect on the response amplitude (McN: 93.51 ± 5.1%, n = 2 and 5; methoxamine: 93.9 ± 2.8, n = 3 and 6). Together, these results indicate that activation of either cholinergic, noradrenergic, or glutamatergic metabotropic receptors is sufficient to permit the induction of LTD.
Type II mGluRs are not essential for NMDAR-dependent LTD in the visual cortex. Our results indicate that PLC-coupled receptors are essential for the induction of LTD in the visual cortex. Type II mGluRs, which are not primarily coupled to the PLC pathway, have also been implicated in the induction of LTD in other neocortical regions (Otani et al., 1999, 2002; Cho et al., 2000; Cho and Bashir, 2002). However, the role of type II mGluRs in visual cortical LTD remains unclear. In one study, the potent antagonist LY341495 was found ineffective in preventing LTD (Sawtell et al., 1999), whereas a complete block of LTD with the antagonist MCCG was reported recently (Renger et al., 2002). We examined the effects of 10 μm MCCG (to selectively block type II mGluRs) and 100 μm LY341495 (to block both, type I and type II mGluRs) on LTD under our experimental conditions. As shown in Figure 5, neither 10 μm MCCG nor 100 μm LY341495 affected the magnitude of LTD (control: 80.5 ± 6.5%, n = 3 and 7; MCCG: 78.4 ± 3.9%, n = 3 and 8; LY341495: 77.5 ± 4.3%, n = 3 and 7; F(2,19) = 0.093; p = 0.92). Moreover, robust LTD was also observed when MCCG was coapplied with muscarinic (5 μm atropine) and α1 adrenergic (10 μm urapidil; LTD: 78.6 ± 3.6%, n = 4 and 10) antagonists. Thus, under our experimental conditions, blockade of type II mGluRs, alone or in conjunction with other receptor blockade, does not prevent LTD. Although we cannot rule out that activation of type II mGluRs might facilitate LTD under certain conditions, our results clearly indicate that type II mGluRs are not essential for NMDAR-dependent LTD in the visual cortex.
Figure 5.

Type II mGluRs are not essential for NMDAR-dependent LTD in the visual cortex. The average effects of LFS on the FP amplitude in slices superfused with normal ACSF (C; open circles), in the presence of 100 μm MCCG (MC; filled circles), 100 μm LY341495 (LY; filled triangles), and 100 μm MCCG plus 10 μm atropine and 10 μm urapidil (MAU; open triangles) are shown. The results (as percentage of LTD 60 min after LFS) of all individual experiments are shown to the right of the graph. Representative experiments are shown at the top of the graphs.
Postsynaptic PLC is required for LTD induction. The results described above indicate that activation of receptors linked to the PLC second-messenger pathway is essential for the induction of LTD. To directly test the involvement of PLC in LTD, we used slices prepared from mice lacking the β1 isoform of PLC (PLCβ1). PLCβ1 is the major PLC isoform in the forebrain, and PLCβ1 knock-out mice exhibit little increment of IP3 turnover in the cortex in response to cholinergic (Kim et al., 1997) and glutamatergic (Hannan et al., 2001) activation. We compared the magnitude of pairing induced LTD in age-matched PLCβ1 knock-out, wild-type, and heterozygote 4-week-old littermates (Fig. 6A). LTD was substantially and significantly (F(2,28) = 8.4; p = 0.0014) reduced in the PLCβ1 knock-out (99.5 ± 5.9%; n = 4 and 9) and in the heterozygotes (102.4 ± 3.6% in knock-out; n = 4 and 13) compared with wild types (76.5 ± 3.4%; n = 4 and 10). The lack of LTD in the PLCβ1 knock-out mice was confirmed in “blind” experiments in which LTD was induced with LFS (knock-out: 95.5 ± 2.0%, n = 6 and 17; wild type: 82.4 ± 2.3%, n = 6 and 19; p = 0.00017; data not shown.). These results strongly suggest that a minimum level of PLCβ1 is required for the induction of LTD.
Figure 6.

Pairing-induced LTD requires postsynaptic PLC. A, Impaired LTD in PLCβ1 knock-out mice. The averaged effects of pairing in cells from homozygotes (hom and Homo; filled circles), heterozygotes (het and Hetero; filled triangles), and their age-matched wild type (wt and Wt; open circles) and littermates. B, Postsynaptic application of the PLC blocker U73122 impairs LTD. The averaged effects of pairing in cells recorded with electrodes filled with control 0.001% DMSO solution (C; open circles), 10 μm U73122 (U.. .2; filled circles), or the inactive isomer 10 μm U73343 (U.. .3; filled triangles) are shown. Results from individual experiments are shown on the right. Traces are averages of four consecutive responses recorded 1 min before (thin traces) and 40 min after (thick traces) pairing.
Additional studies using slices from rat brain were done to examine the PLC dependency of LTD. A common limitation of knock-out studies is the possibility that the observed changes result from adaptations to compensate for the deleted genes. To further confirm the involvement of PLC in LTD induction, we studied the effects of the specific PLC blocker U73122, which has been reported to prevent LTD in other brain areas (Reyes-Harde and Stanton, 1998; Otani et al., 2002). In addition, because LTD induction is initiated by postsynaptic events (Kirkwood and Bear, 1994; Torii et al., 1995; Yasuda and Tsumoto, 1996), it was important to determine whether PLC is also required postsynaptically. LTD was attempted with the pairing paradigm (see Materials and Methods) under whole-cell current-clamp conditions, and 10 μm U73122 dissolved in 0.01% DMSO was included in the recording pipette to block PLC postsynaptically. In control experiments, pipettes contained 0.01% DMSO or the inactive isomer U73343 (10 μm). The results, shown in Figure 6B, indicate that the pairing paradigm induced robust LTD when the pipette contained control solutions (DMSO only: 74.1 ± 5.3%, n = 9; U73343: 71.7 ± 5.5%, n = 12) but no depression when it contained U73122 (101.1 ± 4.7%; n = 14; F(2,23) = 10.917; p = 0.0002). These results are consistent with the idea that postsynaptic PLC activity is essential for the induction of LTD.
The effects of U73122 on synaptic responses were assessed with extracellular recordings. Bath application of 10 μm U73122 severely reduced the magnitude of LTD induced with LFS (91.5 ± 2.7%, n = 10% compared with 75.8 ± 2.5%, n = 18 in control slices; data not shown). The block of LTD was reversible: in those cases in which LTD was nearly absent (95.0 ± 0.9%; n = 4), we were able to induce substantial (p = 0.045) LTD 20-30 min after washing out the drug (78.6 ± 4.7%) (Fig. 7A). In contrast, a 20 min application of U73122 did not affect baseline responses, nor did it affect previously induced LTD, suggesting that the drug does not compromise the expression of LTD (Fig. 7B).
Figure 7.

U73122 blocks the induction, not the expression, of LTD. A, Reversibility of LTD block. The effects of LFS applied in the presence of 10 μm U73122 (shaded area; left) and 30-40 min after washing out the drug (right) are shown. B, Average effects of the FP amplitude of U73122 (shaded area) applied in naive slices (top, open circles) and 20 min after the induction of LTD (bottom, filled circles). Representative traces in A and B are averages of four consecutive responses recorded before (thin traces) and 30-60 min after (thick traces) LFS.
LTD induction requires activation of IP3 receptors. A simple interpretation of the data presented above is that M1, α1, and mGluR5 receptors can enable LTD induction by activating PLC. To further test this idea, we sought to restore LTD in slices treated with the antagonist mixture (Fig. 1) by activating steps down-stream from PLC. PLC activity generates two second messengers: DAG, which in turn activates protein kinase C (PKC), and IP3, which promotes Ca2+ release from intracellular stores. We first used a variety of specific blockers to assess the contribution of each pathway to the induction of LTD. As shown in Figure 8, A and C, bath application of neither of the two PKC blockers tested, GFX (0.5 μm) or staurosporine (1 μm), affected the induction of LFS LTD, suggesting that this pathway is not necessary for LTD (GFX: 80.0 ± 6.1%, n = 6 and 8; control: 81.2 ± 3.6%; p = 0.67; staurosporine: 83.8 ± 2.5%, n = 10 and 11; control: 83.8 ± 5.3%, n = 8 and 9; p = 0.86). In contrast, as shown in Figure 8, B and C, intracellular application of IP3 receptor blockers, 1 μm 2-APB or 5 ml of heparin, significantly reduced pairing-induced LTD (2-APB: 96.9 ± 8.6%, n = 5 and 10; control 0.001% DMSO: 64.5 ± 3.7%, n = 7 and 9; p = 0.002; heparin: 89.6 ± 3.6%, n = 8 and 14; control: 70.3 ± 4.4, n = 12 and 18; p = 0.001). Similarly, bath application of 5 μm CPA, a manipulation aimed to deplete the intracellular Ca2+ stores by blocking the Ca2+ pump also reduced LTD (data not shown; CPA: 90.6 ± 1.1%, n = 7 and 9; control: 78.8 ± 3.5%, n = 9 and 12; p = 0.018). Thus, only manipulations targeting the IP3 pathway affected the induction of LTD. Therefore, we tested whether IP3 can restore pairing-induced LTD when the antagonist mixture (MUA) was present in the bath. In these experiments, 200 μm IP3 was included in the recording pipette, and pairing was applied at 20 min after breaking the seal to facilitate the diffusion of IP3. As shown in Figure 8D, the blockage of LTD by the antagonist mixture (MUA: 102.3 ± 4.9%, n = 6 and 13; control: 68.8 ± 2.5%, n = 10 and 17) was virtually eliminated by inclusion of IP3 (73 ± 5.5%, n = 4 and 13) in the pipette. These differences were significant (F(2,40) = 17.184; p < 0.0001), supporting the idea that PLC-coupled receptors permit LTD by providing a minimum level of postsynaptic IP3 receptor activation.
Figure 8.

LTD induction requires activation of IP3 receptors. A, Bath application of the PKC blocker GF109203X (GFX; 0.5 μm) does not affect LFS-induced LTD. Open circles, Control; filled circles, GFX. B, Intracellular application of the IP3 receptor blocker 2-APB (0.5 μm) blocks pairing-induced LTD of the EPSCs (recorded under voltage clamp at -80 mV). Open circles, Control DMSO; filled circles, 2-APB, DMSO control. C, Summary of the effects on LTD of PKC blockers [GFX and staurosporine (Stau.)] and IP3 receptor blockers [2-APB and heparin (Hep.)]. LTD is expressed as a decrease from baseline. Asterisks indicate significant difference (p < 0.05) from control. D, Postsynaptic application of IP3 relieves the block of pairing-induced LTD by antagonists against PLC-coupled receptors (MUA: 10 μm MPEP, 5 μm atropine, and 10 μm urapidil). The averaged effects of pairing in cells recorded in control ACSF (Con; open circles), in MUA (filled circles), or with MUA with IP3 (200 μm) in the pipette (filled triangles) are shown. Results from individual experiments are shown on the right of each panel. Traces are averages of four consecutive responses recorded 1 min before (thin traces) and 40 min after (thick traces) attempting LTD. Con and Cont., Control.
LTD induction in the temporal cortex and CA1 requires PLC-coupled receptors. LTD mechanisms have been described throughout the brain. We examined whether the activation of PLC-coupled receptors is required for LFS-induced LTD in two other brain regions: the temporal cortex, as a model of the associative cortex (Hayashi et al., 2004), and the CA1 region, where NMDAR-dependent plasticity is best understood. As shown in Figure 9A, an antagonist mixture (MAP: 10 μm MPEP, 5 μm atropine, and 10 μm prazozin) significantly reduced the magnitude of LTD induced by LFS in the layer IV→II/III pathway of the temporal cortex (control: 73 ± 2.6%, n = 7 and 14; MAP: 91.1 ± 1.6%, n = 6 and 17; p < 0.001). Similarly, LTD in the Schaffer collateral→CA1 pathway was also reduced by the mixture, but not when any one of the antagonists was omitted from the mixture (Fig. 9B,C) (control: 75.0 ± 1.6, n = 9 and 21; MAP: 89.7 ± 1.9, n = 5 and 13; MA: 77.6 ± 2.6, n = 4 and 10; MP: 74.8 ± 3.8, n = 4 and 8; AP: 74.5 ± 2.0%, n = 4 and 7). The suppression of LTD by MAP (89.7 ± 1.9%, n = 5 and 13) was not as complete as compared with that obtained with the NMDAR antagonist APV (97.8 ± 2.2%; n = 4 and 18). Therefore, we tested antagonists against 5-HT2 serotonergic receptors (10 μm ketanserine) and against D2 dopaminergic receptors (10 μm sulpiride), which are also coupled to PLC. Adding sulpiride or sulpiride plus ketanserine to the mixture did not cause a further block of LTD (MAPS: 88.4 ± 3%, n = 5 and 10; MAPSK: 88.05 ± 2.5%, n = 5 and 11). In contrast, both sulpiride and ketanserin were capable of reducing LTD when administrated together with MPEP and atropin (MAS: 89.8 ± 2.6%, n = 6 and 12; MAK: 91.2 ± 2.9%, n = 6 and 14). An ANOVA test confirmed the significance of these findings (F(9,118) = 14.133; p < 0.0001). Although we did not exhaust all antagonist combinations, the results are consistent with the notion that a portion of LTD in CA1 requires the activation of at least two of the five PLC-coupled receptors examined. Whether the PLC-dependent and -independent fractions of CA1 LTD represent independent mechanisms remains to be investigated.
Figure 9.
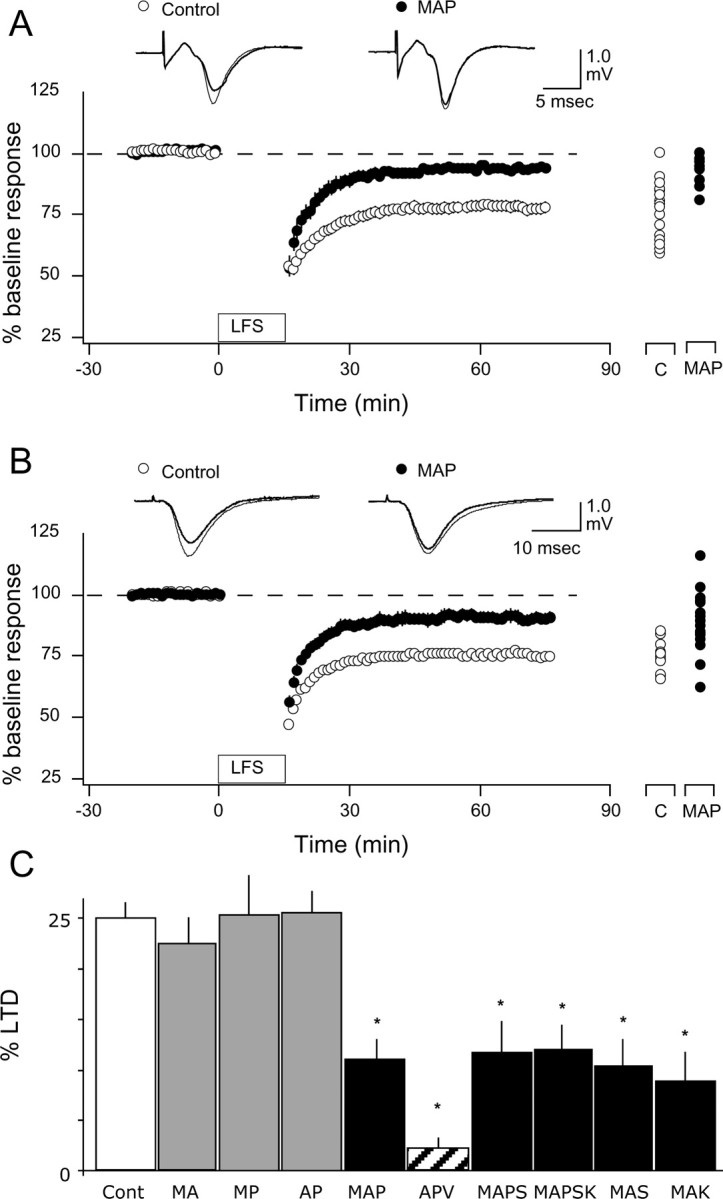
Antagonists of PLC-coupled receptors reduce the induction of LTD in the temporal cortex and CA1. A, B, Bath application of 10 μm MPEP, 5 μm atropine, and 10 μm prazosin reduces the induction of LTD of the layer IV-III pathway in the temporal cortex (A) and the Schaffer collateral→CA1 pathway in the hippocampus (B). Individual experiments are shown on the right of each graph. Traces are examples of responses recorded 1 min before (thin traces) or 60 min after (thick traces) LFS. C, LTD in CA1 is not affected when only two antagonists are present in the bath. C, Control; A, atropine (5 μm); M, MPEP (10 μm); P, prazosin (10 μm); S, sulpiride (10 μm); K, ketanserin (10 μm). Asterisks denote statistical significance from control.
Discussion
In most settings, the induction of LTD with patterned synaptic stimulation requires the activation of NMDARs, mGluRs, or both (Bear and Abraham, 1996; Huber et al., 2000; Kemp and Bashir, 2001). In the visual cortex, nonglutamatergic receptors have been primarily regarded as modulators that promote or facilitate the induction of LTP and LTD with suboptimal stimulation (Brocher et al., 1992; Kirkwood et al., 1999; Kirkwood, 2000; Kojic et al., 2000). Contrary to this notion, we found that the activation of receptors coupled to PLC is essential for the induction of LTD in the layer IV→III pathway of the visual cortex. Activating any one of the receptors studied (α1 adrenoreceptor, M1 muscarinic receptor, and mGluR5) was sufficient to permit the induction of LTD, indicating that PLC-coupled receptors can substitute for each other in supporting LTD. Based on these results, we propose that the activation of postsynaptic PLC is an obligatory step in the induction of NMDR-dependent LTD in the visual cortex. Such an essential requirement of PLC activation could provide a mechanism to regulate cortical plasticity by extra-retinal inputs.
The involvement of PLC in the induction of LTD has been explored previously in the hippocampus (Reyes-Harde and Stanton, 1998) and prefrontal cortex (Otani et al., 2002). However, these studies relayed on the use of the PLC blocker U73122, a drug of limited specificity (Taylor and Broad, 1998). Moreover, U73122 can promote LTD in slices treated with BDNF in the hippocampus (Ikegaya et al., 2002). Several lines of evidence in the current studies underscore the critical importance of PLC-coupled receptors in the induction of LTD in the layer IV→III pathway of the visual cortex. The induction of LTD was not only prevented by the blocker U73122, but it was virtually absent in mice lacking the PLCβ1 isoform. In addition, LTD was not induced when the activation of three PLC-coupled receptors (α1, M1, and mGluR5) was prevented either with antagonists or by a combination of antagonists and immunolesions of cholinergic and noradrenergic nuclei. It seems unlikely that the blockade of LTD could be attributed to nonspecific effects, because it was specific to LTD rather than LTP, and converging results were obtained both with drugs and immunolesions: LTD was blocked only when the three systems/receptors were targeted together. When any one of these was spared, the induction of LTD was also spared. Moreover, when the three antagonists were present, LTD was restored by including IP3 in the recording pipette. Similarly, after eliminating all three neuromodulatory systems, adding one agonist was sufficient to restore LTD (Fig. 4), suggesting that the combined antagonists/immunolesions did not irreversibly disable the LTD machinery. Thus, with all of these lines of evidence, it seems reasonable to conclude that activation of PLC is a required step for the induction of LTD in the layer IV→III pathway of the visual cortex.
The induction mechanisms of LTD vary considerably from synapse to synapse (Bear and Abraham, 1996; Kemp and Bashir, 2001), and different forms of LTD (mGluR and NMDAR dependent) can be induced in different cortical layers (Rao and Daw, 2004) and even induced at the same synapses (Oliet et al., 1997; Fitzjohn et al., 1999; Huber et al., 2000; Montgomery and Madison, 2002). An essential role for PLC has also been proposed for LFS-induced LTD in CA1 (Reyes-Harde and Stanton, 1998) and for mGluR agonist-induced LTD in the prefrontal cortex (Otani et al., 2002). In addition, activation of M1 muscarinic receptors (coupled to PLC) alone is sufficient to induce an NMDAR-independent form of LTD in the perirhinal cortex (Massey et al., 2001). The requirement for PLC activity may not be universal, however, because LTD induced with type I mGluR agonists in the CA1 region may occur independent of the PLC pathway (Schnabel et al., 1999; Cho et al., 2002).
LTD in the layer IV→III pathway of the visual cortex requires the activation of NMDARs, followed by an increase in postsynaptic Ca2+ (Kirkwood and Bear, 1994; Yasuda and Tsumoto, 1996). It remains to be determined how the signals (IP3 and PKC) generated by the activation of PLC interact with the NMDAR-dependent pathway. PKC inhibitors did not block LTD, arguing against the involvement of this pathway in the visual cortex. Our results seem more consistent with the idea that Ca2+ release from internal stores induced by IP3 boosts the Ca2+ signal initiated by the activation of NMDARs, as proposed for the CA1 region of the hippocampus (Reyes-Harde and Stanton, 1998), and perirhinal (Cho and Bashir, 2002) and frontal (Otani et al., 2002) cortices. However, such a scenario would not account for the finding that the antagonist mixture does not affect pairing-induced LTP and even promote LTP at -20 mV (Fig. 2). Alternatively, postsynaptic PLC activity might be required for the release of endocannabinoids (Chevaleyre and Castillo, 2003; Hashimotodani et al., 2005), recently implicated in the induction of presynaptic forms of LTD (Sjostrom et al., 2003). This scenario would account better for the specificity of the effects of the antagonists on LTD.
An essential role of PLC in LTD is seemingly at odds with the observation that application of exogenous glutamatergic agonists is sufficient to induce LTD in both hippocampal and visual cortical slices (Kandler et al., 1998; Lee et al., 1998; Dodt et al., 1999). One must bear in mind, however, that the background levels of endogenous agonists in slices, which can be high (Kirkwood et al., 1999), could be sufficient to support LTD (Fig. 1B). Thus, if under standard slice recording conditions, sufficient cholinergic and/or noradrenergic activity exists, then the induction of LTD might seem to require only the activation of ionotropic glutamate receptors. In that case, antagonism of mGluRs might not block the induction of LTD. Indeed, some of the discrepancies concerning effects of mGluR antagonists on LTD (Haruta et al., 1994; Selig et al., 1995; Huber et al., 1998; Sawtell et al., 1999) may relate to differing levels of endogenous acetylcholine and norepinephrine.
The notion of different neurotransmitter systems acting together to induce LTD is not restricted to the visual cortex. For example, in perirhinal cortex LTD is triggered by the coactivation of NMDAR and mGluR type I and II (Cho et al., 2000), and in the prefrontal cortex, the induction of LTD requires the simultaneous activation of dopamine receptors and mGluRs (Otani et al., 1999). Although the corequirement of neuromodulators may emerge as a common feature across cortical regions, the underlying mechanisms seem to differ. In the prefrontal and perirhinal cortex, the neuromodulatory systems interact in a cooperative manner such that antagonists against any receptor disrupt the induction of LTD. In contrast, in the visual cortex, at least three different PLC-linked receptors, M1, α1, and mGluR5, can independently enable the induction of LTD. Interestingly, the magnitude of LTD is independent of the number of receptor systems activated. This suggests that LTD requires a threshold of PLC activity and that the three PLC-coupled receptors examined can substitute for each other in this function. Whether other routes for PLC activation, like TrkB (Mizoguchi and Nabekura, 2003) and serotonergic receptors (Kojic et al., 2000), or increases in intracellular Ca2+ (Thore et al., 2004), can also enable LTD remains an open question.
From a functional perspective, the essential requirement for neurotransmitters linked to PLC in LTD may serve to make the induction of NMDAR-dependent plasticity conditional on the behavioral state of the animal. NMDARs are thought to act as coincident detectors that can translate correlation of presynaptic and postsynaptic activity into Ca2+ signals. NMDAR-dependent LTD is a particularly attractive model for developmental plasticity in the sensory cortex because it is the best known mechanism to selectively weaken synapses when the presynaptic and postsynaptic activity is uncorrelated (Song and Abbott, 2001; Shouval et al., 2002). We propose an additional level of coincidence detection that is required for that NMDAR-dependent plasticity. Synaptic modifications (LTD) will occur only when NMDARs are activated in conjunction with activation of PLC-linked neuromodulators. In this context, the activation of NMDARs will convey information about specific external inputs, whereas neuromodulators could provide information about the behavioral state of the organism, reflecting such variables as attention and arousal.
There is ample evidence in the intact organism that neuromodulatory systems are required for normal development (Hohmann et al., 1991; Siciliano et al., 1999; Hannan et al., 2001) and experience-dependent plasticity in the sensory cortices of juvenile animals (Kasamatsu and Pettigrew, 1976; Bear and Singer, 1986; Sachdev et al., 1998; Zhu and Waite, 1998). Moreover, stimulation of the basal forebrain (cholinergic) or infusion of norepinephrine allows the experience-dependent cortical rearrangement in adult sensory cortices (Kasamatsu and Pettigrew, 1979; Kilgard and Merzenich, 1998; Shulz et al., 2000; Ego-Stengel et al., 2001; Manunta and Edeline, 2004). In most cases, however, the mechanisms and receptors involved remain to be identified. We previously interpreted the role of neuromodulators in terms of facilitating plasticity, such that otherwise subthreshold input activity becomes adequate to enable cortical reorganization. Although the in vitro data in the current report suggest that activators of PLC, including glutamate, can substitute for one another, it is conceivable that the levels of PLC activity in vivo are insufficient to support LTD in the absence of cholinergic and noradrenergic inputs. In either case, behavioral states that promote activation of extra-retinal input to the cortex play a very significant role in cortical plasticity by direct gating or enhancement of NMDAR-dependent plasticity. These actions of neuromodulators provide a basis for understanding more integrative functions in cortical plasticity at a cellular level.
Footnotes
This work was supported by National Institutes of Health Grants R01-EY12124 and PO1-AG09973. G.-H.S. was partially supported by the Korea Science and Engineering Foundation. We thank Dr. C. Bonansco, L. Karelias, and M. McTague for conducting pilot studies and Dr. H.-K. Lee for help with genotyping and valuable discussions and comments.
Correspondence should be addressed to Dr. Alfredo Kirkwood, Mind/Brain Institute and Department of Neurosciences, Johns Hopkins University, 3400 North Charles Street, Baltimore, MD 21218. E-mail: kirkwood@jhu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511433-11$15.00/0
References
- Allen CB, Celikel T, Feldman DE (2003) Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci 6: 291-299. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Bucci DJ, Gorman LK, Wiley RG, Gallagher M (1995) Selective immunotoxic lesions of basal forebrain cholinergic cells: effects on learning and memory in rats. Behav Neurosci 109: 714-722. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC (1996) Long-term depression in hippocampus. Annu Rev Neurosci 19: 437-462. [DOI] [PubMed] [Google Scholar]
- Bear MF, Rittenhouse CD (1999) Molecular basis for induction of ocular dominance plasticity. J Neurobiol 41: 83-91. [DOI] [PubMed] [Google Scholar]
- Bear MF, Singer W (1986) Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature 320: 172-176. [DOI] [PubMed] [Google Scholar]
- Blond O, Crepel F, Otani S (2002) Long-term potentiation in rat prefrontal slices facilitated by phased application of dopamine. Eur J Pharmacol 438: 115-116. [DOI] [PubMed] [Google Scholar]
- Brocher S, Artola A, Singer W (1992) Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res 573: 27-36. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW (1995) Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci 15: 5324-5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38: 461-472. [DOI] [PubMed] [Google Scholar]
- Cho K, Bashir Z (2002) Cooperation between mglu receptors: a depressing mechanism. Trends Neurosci 25: 405-411. [DOI] [PubMed] [Google Scholar]
- Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI (2000) A new form of long-term depression in the perirhinal cortex. Nat Neurosci 3: 150-156. [DOI] [PubMed] [Google Scholar]
- Cho K, Brown MW, Bashir ZI (2002) Mechanisms and physiological role of enhancement of mGlu5 receptor function by group II mGlu receptor activation in rat perirhinal cortex. J Physiol (Lond) 540: 895-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw N, Gordon B, Fox K, Flavin H, Kirsch J, Beaver C, Ji Q, Reid S, Czepita D (1999) Injection of MK-801 affects ocular dominance shifts more than visual activity. J Neurophysiol 81: 204-215. [DOI] [PubMed] [Google Scholar]
- Dodt H, Eder M, Frick A, Zieglgansberger W (1999) Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science 286: 110-113. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF (1992) Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA 89: 4363-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ego-Stengel V, Shulz DE, Haidarliu S, Sosnik R, Ahissar E (2001) Acetylcholine-dependent induction and ixpression of functional plasticity in the barrel cortex of the adult rat. J Neurophysiol 86: 422-437. [DOI] [PubMed] [Google Scholar]
- Fitzjohn S, Bortolotto ZA, Palmer M, Doherty A, Ornstein P, Schoepp D, Kingston A, Lodge D, Collingridge G (1998) The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology 37: 1445-1458. [DOI] [PubMed] [Google Scholar]
- Fitzjohn S, Kingston A, Lodge D, Collingridge G (1999) DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterization and sensitivity to novel mGlu receptor antagonists. Neuropharmacology 38: 1577-1583. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O'Leary DDM (1996) Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci USA 93: 5584-5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q (2002) Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 111: 815-835. [DOI] [PubMed] [Google Scholar]
- Gu Q, Singer W (1995) Involvement of serotonin in developmental plasticity in kitten visual cortex. Eur J Neurosci 7: 1146-1153. [DOI] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC (2001) PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci 4: 282-288. [DOI] [PubMed] [Google Scholar]
- Haruta H, Kamishita T, Hicks TP, Takahashi MP, Tsumoto T (1994) Induction of LTD but not LTP through metabotropic glutamate receptors in visual cortex. NeuroReport 5: 1829-1832. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M (2005) Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45: 257-268. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S (2004) Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron 42: 773-787. [DOI] [PubMed] [Google Scholar]
- Hohmann CF, Wilson L, Coyle JT (1991) Efferent and afferent connections of mouse sensory-motor cortex following cholinergic deafferentation at birth. Cereb Cortex 1: 158-172. [DOI] [PubMed] [Google Scholar]
- Huber K, Kayser M, Bear M (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254-1257. [DOI] [PubMed] [Google Scholar]
- Huber KM, Sawtell NB, Bear MF (1998) Effects of the metabotropic glutamate receptor antagonist MCPG on phosphoinositide turnover and synaptic plasticity in visual cortex. J Neurosci 18: 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Ishizaka Y, Matsuki N (2002) BDNF attenuates hippocampal LTD via activation of phospholipase C: implications for a vertical shift in the frequency-response curve of synaptic plasticity. Eur J Neurosci 16: 145-148. [DOI] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC (2002) Group I mGluRs increase excitability of hippocampal CA1 pyramidal neurons by a PLC-independent mechanism. J Neurophysiol 88: 107-116. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S (2000) Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406: 726-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel E, O'Dell T (1992) Are adult learning mechanisms also used for development? Science 258: 243-245. [DOI] [PubMed] [Google Scholar]
- Kandler K, Katz LC, Kauer JA (1998) Focal photolysis of caged glutamate produces long-term depression of hippocampal glutamate receptors. Nat Neurosci 1: 119-123. [DOI] [PubMed] [Google Scholar]
- Karnowsky M, Roots L (1964) A “direct-coloring” thiocholine method for cholinesterase. J Histochem Cytochem 12: 219-221. [DOI] [PubMed] [Google Scholar]
- Kasamatsu T, Pettigrew JD (1976) Depletion of brain catacholamines: failure of ocular dominance shift after monocular occlution in kittens. Science 194: 206-209. [DOI] [PubMed] [Google Scholar]
- Kasamatsu T, Pettigrew JD (1979) Restoration of visual cortical plasticity by local microperfusion of norepinephrine. J Comp Neurol 185: 163-182. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133-1138. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI (2001) Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol 65: 339-365. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM (1998) Cortical map reorganization enabled by nucleus basalis activity. Science 279: 1714-1718. [DOI] [PubMed] [Google Scholar]
- Kim D, Jun K, Lee S, Kang N, Min D, Kim Y, Ryu S, Suh P, Shin H (1997) Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature 389: 290-293. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD (1998) LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37: 1-12. [DOI] [PubMed] [Google Scholar]
- Kirkwood A (2000) Serotonergic control of developmental plasticity. Proc Natl Acad Sci USA 97: 1951-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF (1994) Homosynaptic long-term depression in the visual cortex. J Neurosci 14: 3404-3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF (1999) Modulation of long-term depression in visual cortex by acetylcholine and norepinephrine. J Neurosci 19: 1599-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt A, Bear MF, Singer W (1987) Blockade of “NMDA” receptors disrupts experience-dependent modifications of kitten striate cortex. Science 238: 355-358. [DOI] [PubMed] [Google Scholar]
- Kojic L, Dyck RH, Gu Q, Douglas RM, Matsuraba J, Cynader MS (2000) Columnar distribution of serotonin-dependent plasticity within kitten striate cortex. Proc Natl Acad Sci USA 97: 1841-1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Aztiria E, Leanza G, Domenici L (2005) Selective cholinergic immunolesioning affects synaptic plasticity in developing visual cortex. Eur J Neurosci 21: 1807-1814. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151-1162. [DOI] [PubMed] [Google Scholar]
- Lee O, Lee CJ, Choi S (2002) Induction mechanisms for L-LTP at thalamic input synapses to the lateral amygdala: requirement of mGluR5 activation. NeuroReport 13: 685-691. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5-21. [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001) Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21: 5925-5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manunta Y, Edeline JM (2004) Noradrenergic induction of selective plasticity in the frequency tuning of auditory cortex neurons. J Neurophysiol 92: 1445-1463. [DOI] [PubMed] [Google Scholar]
- Massey PV, Bhabra G, Cho K, Brown MW, Bashir ZI (2001) Activation of muscarinic receptors induces protein synthesis-dependent long-lasting depression in the perirhinal cortex. Eur J Neurosci 14: 145-152. [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Nabekura J (2003) Sustained intracellular Ca2+ elevation induced by a brief BDNF application in rat visual cortex neurons. NeuroReport 14: 1481-1483. [DOI] [PubMed] [Google Scholar]
- Montgomery J, Madison D (2002) State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron 33: 765-777. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9: 967-975. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969-982. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin MP, Crepel F (1999) Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci 19: 9788-9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F (2002) Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci 22: 3434-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer M, Irving A, Seabrook G, Jane D, Collingridge G (1997) The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 36: 1517-1532. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Perth, Australia: Academic Press Australia.
- Philpot BD, Espinosa JS, Bear MF (2003) Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci 23: 5583-5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Daw NW (2004) Layer variations of long-term depression in rat visual cortex. J Neurophysiol 92: 2652-2658. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Hartman KN, Tsuchimoto Y, Yokoi M, Nakanishi S, Hensch TK (2002) Experience-dependent plasticity without long-term depression by type 2 metabotropic glutamate receptors in developing visual cortex. Proc Natl Acad Sci USA 99: 1041-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Stanton PK (1998) Postsynaptic phospholipase C activity is required for the induction of homosynaptic long-term depression in rat hippocampus. Neurosci Lett 252: 155-158. [DOI] [PubMed] [Google Scholar]
- Roberts EB, Meredith MA, Ramoa AS (1998) Suppression of NMDA receptor function using antisense DNA blocks ocular dominance plasticity while preserving visual responses. J Neurophysiol 80: 1021-1032. [DOI] [PubMed] [Google Scholar]
- Rossner S, Schliebs R, Hartig W, Bigl V (1995) 192IGG-saporin-induced selective lesion of cholinergic basal forebrain system: neurochemical effects on cholinergic neurotransmission in rat cerebral cortex and hippocampus. Brain Res Bull 38: 371-381. [DOI] [PubMed] [Google Scholar]
- Sachdev RN, Lu SM, Wiley RG, Ebner FF (1998) Role of the basal forebrain cholinergic projection in somatosensory cortical plasticity. J Neurophysiol 79: 3216-3228. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Can molecules explain long-term potentiation? Nat Neurosci 2: 597-604. [DOI] [PubMed] [Google Scholar]
- Sawtell NB, Huber KM, Roder JC, Bear MF (1999) Induction of NMDA receptor-dependent long-term depression in visual cortex does not require metabotropic glutamate receptors. J Neurophysiol 82: 3594-3597. [DOI] [PubMed] [Google Scholar]
- Schliebs R, Rossner S, Bigl V (1996) Immunolesion by 192IgG-saporin of rat basal forebrain cholinergic system: a useful tool to produce cortical cholinergic dysfunction. Prog Brain Res 109: 253-264. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick I, Collingridge G (1999) An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology 38: 1585-1596. [DOI] [PubMed] [Google Scholar]
- Selig DK, Lee H-K, Bear MF, Malenka RC (1995) Reexamination of the effects of MCPG on hippocampal LTP, LTD, and depotentiation. J Neurophysiol 74: 1075-1082. [DOI] [PubMed] [Google Scholar]
- Shouval H, Bear M, Cooper L (2002) A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci USA 99: 10831-10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulz DE, Sosnik R, Ego V, Haidarliu S, Ahissar E (2000) A neuronal analogue of state-dependent learning. Nature 403: 549-553. [DOI] [PubMed] [Google Scholar]
- Siciliano R, Fontanesi G, Casamenti F, Berardi N, Bagnoli P, Domenici L (1997) Postnatal development of functional properties of visual cortical cells in rats with excitotoxic lesions of basal forebrain cholinergic neurons. Vis Neurosci 14: 11-123. [DOI] [PubMed] [Google Scholar]
- Siciliano R, Fornai F, Bonaccorsi I, Domenici L, Bagnoli P (1999) Cholinergic and noradrenergic afferents influence the functional properties of the postnatal visual cortex in rats. Vis Neurosci 16: 1015-1028. [DOI] [PubMed] [Google Scholar]
- Singer W (1995) Development and plasticity of cortical processing architectures. Science 270: 758-759. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641-654. [DOI] [PubMed] [Google Scholar]
- Song S, Abbott L (2001) Cortical development and remapping through spike timing-dependent plasticity. Neuron 32: 339-350. [DOI] [PubMed] [Google Scholar]
- Steele PM, Mauk MD (1999) Inhibitory control of LTP and LTD: stability of synapse strength. J Neurophysiol 81: 1559-1566. [DOI] [PubMed] [Google Scholar]
- Taylor CW, Broad LM (1998) Pharmacological analysis of intracellular Ca2+ signalling: problems and pitfalls. Trends Pharmacol Sci 19: 370-375. [DOI] [PubMed] [Google Scholar]
- Thore S, Dyachok O, Tengholm A (2004) Oscillations of phospholipase C activity triggered by depolarization and Ca2+ influx in insulin-secreting cells. J Biol Chem 279: 19396-19400. [DOI] [PubMed] [Google Scholar]
- Torii N, Kamishita T, Otsu Y, Tsumoto T (1995) An inhibitor for calcineurin, FK506, blocks induction of long-term depression in rat visual cortex. Neurosci Lett 185: 1-4. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Tsumoto T (1996) Long-term depression in rat visual cortex is associated with a lower rise of postsynaptic calcium than long-term potentiation. Neurosci Res 24: 265-274. [DOI] [PubMed] [Google Scholar]
- Zhu XO, Waite PM (1998) Cholinergic depletion reduces plasticity of barrel field cortex. Cereb Cortex 8: 63-72. [DOI] [PubMed] [Google Scholar]