

Abstract
AMPA and GABAA receptors mediate most of the fast signaling in the CNS. However, the retina must, in addition, also convey slow and sustained signals. Given that AMPA and GABAA receptors desensitize quickly in the continuous presence of agonist, how are sustained excitatory and inhibitory signals transmitted reliably across retinal synapses? Reciprocal synapses between bipolar and amacrine cells in the retina are thought to play a fundamental role in tuning the bipolar cell output to the dynamic range of ganglion cells. Here, we report that glutamate release from goldfish bipolar cell terminals activates first AMPA receptors, followed by fast and transient GABAA-mediated feedback. Subsequently, prolonged NMDA receptor activation triggers GABAA and a slow, sustained GABAC-mediated reciprocal inhibition. The synaptic delay of the NMDA/GABAC-mediated feedback showed stronger dependence on the depolarization of the bipolar cell terminal than the fast AMPA/GABAA-mediated response. Although the initial depolarization mediated by AMPA receptors was important to prime the NMDA action, NMDA receptors could trigger feedback by themselves in most of the bipolar terminals tested. This AMPA-independent feedback (delay ≈ 10 ms) was eliminated in 2 mm external Mg2+ and reduced in some terminals, but not eliminated, by TTX. NMDA receptors on amacrine cells with depolarized resting membrane potentials therefore can mediate the late reciprocal feedback triggered by continuous glutamate release. Our findings suggest that the characteristics of NMDA receptors (high agonist affinity, slow desensitization, and activation/deactivation kinetics) are well suited to match the properties of GABAC receptors, which thus provide part of the prolonged inhibition to bipolar cell terminals.
Keywords: neurotransmission, amacrine cells, GABAC receptor, retinal bipolar cell, NMDA receptor, GABAA receptor, AMPA receptor
Introduction
Glutamatergic synapses mediate the transfer of visual information from photoreceptors to bipolar cells (BCs) (Copenhagen and Jahr, 1989) and from BCs to ganglion and amacrine cells (ACs) (Slaughter and Miller, 1983; Wässle, 2005). Most of the ACs are GABAergic and provide inhibition to other ACs and ganglion cells and inhibitory feedback to BC terminals. A portion of this feedback is reciprocal, in which a BC terminal excites an AC bouton that provides direct inhibitory feedback to the same BC terminal (Dowling and Boycott, 1966; Witkovsky and Dowling, 1969; Marc and Liu, 2000). Here, we examined the temporal characteristics of this reciprocal synapse in the inner retina and evaluated the role of AMPA receptors (AMPARs) and NMDA receptors (NMDARs). In the CNS, the majority of fast excitatory transmission is mediated by non-NMDA glutamate receptors, whereas the NMDARs play a critical role in long-term synaptic plasticity (Collingridge and Lester, 1989). In the retina, AMPA and NMDARs are involved in mediating light responses in ACs (Dixon and Copenhagen, 1992). However, although AMPARs seem to be essential, NMDARs may play a lesser role, at least in transient ACs (Matsui et al., 2001; Vigh and Witkovsky, 2004). The role of NMDARs in mediating reciprocal feedback has also been questioned, because CNQX or 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonoamide (NBQX) completely blocked the reciprocal inhibition recorded in BCs (Dong and Werblin, 1998; Hartveit, 1999; Singer and Diamond, 2003). In contrast, NMDARs play a major role in the olfactory bulb at the mitral cell ↔ granule cell reciprocal synapse (Isaacson and Strowbridge, 1998; Schoppa et al., 1998; Chen et al., 2000).
In slices of goldfish retina, depolarization of a single mixed-input (Mb) BC terminal triggers exocytosis and prolonged glutamate release that evokes GABAergic feedback (Hull and von Gersdorff, 2004; Vigh et al., 2005). Here, we studied how AMPA and NMDARs control GABA release from ACs. We found that block of the AMPARs eliminates an initial fast GABA-mediated feedback peak, whereas blocking NMDARs reduces exclusively the later GABAergic events. AMPA and NMDARs thus contribute to reciprocal communication with distinct temporal properties. The initial GABAergic feedback transient triggered by AMPAR activation was mostly GABAA receptor mediated. After reducing desensitization of AMPARs with cyclothiazide, the boosted AMPAR action resulted in larger activation of both GABAA and GABAC receptors on the BC terminals. AMPAR block with NBQX or GYKI 53655 did not eliminate the reciprocal inhibition in most of the terminals tested; however, the remaining inhibition was blocked by d-AP-5, an NMDAR antagonist. This remaining NMDAR-mediated feedback (1) had longer delay compared with the onset of AMPAR-mediated feedback, (2) was sometimes reduced but not blocked by TTX, and (3) consisted of both GABAA and GABAC receptor-mediated inhibition. Late and prolonged excitatory signals from BCs are thus conveyed to ACs via NMDARs, and they can be matched by continuous inhibitory reciprocal feedback that is signaled to BCs via both GABAA and GABAC receptors.
Materials and Methods
Retinal slice preparation. Retinal slices (200-250 μm) were prepared from goldfish (Carassius auratus; 8-14 cm) as described previously (Palmer et al., 2003a). Slices were transferred to the recording chamber and perfused continuously (2-3 ml/min) with Ringer's solution comprising the following (in mm): 100 NaCl, 2.5 KCl, 1.0 MgCl2, 2.5 CaCl2, 25 NaHCO3, and 12 glucose, pH 7.45 (set with NaOH). The Ringer's solution was gassed continuously with 95% O2 and 5% CO2. Osmolarity was kept constant at 260 mOsm by changing the NaCl concentration in the experiments using 0 or 2 mm MgCl2. Drugs were bath applied in the perfusing medium. NBQX, CNQX, (S)-3,5-dihydroxyphenylglycine (DHPG), 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid (SR95531), and AP-5 were obtained from Tocris (Bristol, UK). All other chemicals and salts were obtained from Sigma (St. Louis, MO).
Slice preparation and recordings were performed at room temperature (21-23°C) in daylight conditions. Slices were viewed with infrared differential interference contrast optics through a 40× water-immersion objective coupled with a 2× premagnification (Optovart; Zeiss, Oberkochen, Germany) and a CCD camera (C79; Hamamatsu, Tokyo, Japan). BC terminals were identified by their size (6-10 μm), shape, and position in the slice, as well as depolarization-evoked Ca2+ currents and capacitance responses. A subset of isolated terminals was obtained by severing the BC axon during the slicing procedure, and the terminals were identified via their single-exponential capacitative current response to a short, 10 mV hyperpolarizing voltage step from -60 mV (Palmer et al., 2003a). The Mb terminal baseline membrane capacitance was 3-7 pF. Only isolated terminals (axon severed) were used for this study.
Electrophysiology. Whole-cell voltage-clamp recordings were obtained using 7-11 MΩ patch pipettes pulled from thick-walled borosilicate glass (World Precision Instruments, Sarasota, FL) using either a Narishige (Tokyo, Japan; model PP-830) or a Sutter (Novato, CA; model P-97) puller. Pipettes were coated with regular dental wax (Cavex, West Chester, PA) to reduce pipette capacitance and electrical noise and filled with solution comprising the following (in mm): 95 Cs-gluconate, 25 HEPES, 10 TEA-Cl, 3 Mg-ATP, 0.5 Na-GTP, and 0.5 EGTA, adjusted to pH 7.2 with CsOH. Methylamine HCl (10 mm) was also routinely included to buffer vesicular pH (Cousin and Nicholls, 1997; Vigh et al., 2005). The osmolarity was adjusted to 257 ± 2 mOsm. Cells with Rs > 30 MΩ (or leak current >50 pA at a holding potential of -60 mV) were excluded from any further evaluation. Data acquisition was controlled by Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany), and signals were recorded via a double EPC-9 (HEKA Elektronik) patch-clamp amplifier. Sampling rates and low-pass filter settings were 10 and 3 kHz, respectively. Capacitance measurements were performed by the “sine + DC” method, in which a 1 kHz sinusoidal voltage command (30 mV peak to peak) was added to the holding potential of -60 mV, and the resulting current was analyzed at two orthogonal phase angles by the EPC-9 lock-in amplifier (Gillis, 2000). When indicated, P/4 leak subtraction was performed by the Pulse software, by applying four leak pulses starting with a 10 ms delay after the termination of the test depolarization. Current responses for the leak pulses were then averaged, and the calculated leak current was subtracted from the total current trace.
Analysis. Off-line analysis of the data was performed with IgorPro software (Wavemetrics, Lake Oswego, OR) and SigmaPlot (SPSS, Chicago, IL). The increase in membrane capacitance, ΔCm, evoked by membrane depolarization, was measured as ΔCm = Cm(response) - Cm (baseline), where Cm(baseline) was the average Cm value during the 100 ms before the depolarizing step, and Cm(response) was the average Cm value measured during >100 ms after the step, starting 350-400 ms after repolarization to allow time for all evoked conductances to have decayed.
Results
Reciprocal feedback is evoked by glutamate release
We applied whole-cell voltage clamp on large, ON-type Mb BC terminals in goldfish retinal slice preparation. Choosing BC terminals with their axons cut (Palmer et al., 2003a) allowed us to precisely control the membrane potential of the BC terminal and also to accurately record membrane capacitance increases (ΔCm jumps) associated with exocytosis (von Gersdorff and Matthews, 1999). When we depolarized the BC terminals from the holding potential of -60 to 0 mV in the presence of a Cs+-based internal solution (see Materials and Methods), inward current associated with Ca2+ influx (ICa) and Cm jumps were observed (Fig. 1A, black trace). We also observed an outward current during the depolarizing pulse that overlapped with the Ca2+ current (Fig. 1A, black arrow) and an inward current (Fig. 1A, double black arrow) after the pulse. Most of the former current is likely to be GABAergic feedback current (ECl = -41 mV), because we blocked a proton-mediated inhibition of the Ca2+ current with 10 mm methylamine in the patch pipette (Vigh et al., 2005). The inward tail current (Fig. 1A, double black arrow) after the termination of the depolarizing pulse was not reduced by the excitatory amino acid transporter blocker d,l-threo-β-benzyloxyaspartate (TBOA; 50-100 mm) (Palmer et al., 2003a) (control, 37.9 ± 10.4 pA; TBOA, 38.3 ± 8.3 pA; p < 0.7; paired Student's t test; n = 10) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Therefore, this tail current appears to be mostly a Ca2+-dependent Cl- [ICl(Ca)] current (Okada et al., 1995; Hull and von Gersdorff, 2004).
Figure 1.

The reciprocal feedback at the BC terminal strongly depends on the voltage-gated Ca influx into the BC terminal and on the consequent exocytosis. A, Depolarization of the BC terminal from the holding potential of -60 to 0 mV for 200 ms activated calcium inflow through voltage-gated calcium channels (ICa), which triggers glutamate release, as evidenced by the jump in the Cm. The protocol used is demonstrated at the bottom trace. The fast voltage sinewave used to measure Cm was not delivered during the depolarization. This also applies to subsequent figures showing Cm traces. The inhibitory feedback to the presynaptic terminal is expressed as a flurry of outward IPSCs superimposed on ICa (black trace, arrow). The times indicated were measured from break-in throughout this report. Note the ICl(Ca) tail current (double arrow). Including 10 mm BAPTA (red trace) in the pipette completely blocked the exocytosis as evidenced by the lack of Cm jump in response to the depolarization, the inhibitory feedback, and the ICl(Ca) tail current without affecting the ICa. Note the unaffected spontaneous events in the presence of BAPTA (red arrow). The resting Cm of this terminal was 5.4 pF. B, Dissection of evoked versus spontaneous feedback. Strong reciprocal feedback occurs during the depolarization of the BC. The hypothesis is that reciprocal feedback is preceded by presynaptic Ca2+ inflow and glutamate release. To increase the IPSC size, the membrane was then hyperpolarized to -120 mV to generate greater electrochemical driving force for Cl- (ECl =-41 mV). Most terminals showed a low rate of sIPSCs (arrowheads), and those terminals showing spontaneous activity during the hyperpolarized period comparable to that during depolarization were excluded from our analysis. Note the small endocytosis (i.e., drop in the Cm; red arrow) occurring 1 s after the termination of the depolarizing step. C, Reversal of the DHPG-enhanced sIPSCs in the presence of CNQX, AP-5, and TTX. The sIPSCs reversed at -30 mV instead of the Nernst reversal potential for chloride (ECl =-41 mV for this particular cell), and the offset matched exactly the calculated liquid junction potential (+9.8 mV). The series resistance-induced voltage error was < 1 mV at -30 mV. HP, Holding potential.
We first wanted to determine whether this GABAergic feedback current results from the spontaneous activity from ACs during the depolarization of the BC terminal or whether it is evoked by glutamate release via exocytosis. Including 10 mm BAPTA in the recording pipette did not change the amplitude of the ICa, but it completely blocked exocytosis from the BC terminal (Singer and Diamond, 2003) as reflected by the elimination of the ΔCm jump 5 min after break (when the terminals are fully loaded with BAPTA; n = 7) (Fig. 1A, red trace). In concert with this, the reciprocal feedback and the ICl(Ca)-mediated tail current were eliminated. Note that spontaneous events remained after the BAPTA exerted its effect on the reciprocal feedback (Fig. 1A, red arrow). However, when the same cell was depolarized for at least 800 ms, the incoming Ca2+ saturated the BAPTA and resulted in a Cm jump (i.e., glutamate release), as well as a reappearance of the GABAergic feedback and the ICl(Ca)-mediated tail current (data not shown). Therefore, the fact that 10 mm BAPTA eliminated the ΔCm jump evoked by a 200 ms step was not attributable to rundown of exocytosis from BCs. This result suggests that glutamate release from BC terminals is necessary to evoke significant GABA feedback.
To determine the frequency of IPSCs and the degree to which they may contaminate the evoked IPSC recordings during a depolarizing pulse, we used the following protocol. After termination of the depolarizing step from -60 to 0 mV for 200 ms, we delivered a 2-s-long hyperpolarization to -120 mV. IPSCs recorded at -120 mV reflect the degree of spontaneous activity and will have larger amplitudes (because of the larger driving force), making them more easily detectable. Consistent with our previous study of spontaneous IPSCs (sIPSC) (Vigh et al., 2005), we found that the degree of spontaneous activity (Fig. 1B, inset) is very low compared with that occurring during the depolarizing pulse. This suggests that the sIPSC activity detected in Mb terminals does not contaminate significantly the GABAergic feedback evoked during the depolarizing pulse.
Note also that IPSCs change polarity when recorded at 0 or -120 mV (Fig. 1B), suggesting that IPSCs were Cl- mediated (ECl =-41 mV). To test this further, we recorded sIPSCs in the presence of CNQX (25 μm), AP-5 (50 μm), and TTX (1 μm) to block ionotropic glutamate receptors and action potentials of ACs. We then added the group I metabotropic glutamate receptor tor agonist DHPG (100 μm) to enhance the frequency of IPSCs (Vigh et al., 2005), so that we could measure their reversal potential. Under these conditions, the liquid junction potential (+9.8 mV) corrected reversal potential of the sIPSCs was -40 ± 2mV (n = 5), matching the calculated ECl (Fig. 1C). Based on the above findings, we conclude that most of the outward current overlapping the ICa during the depolarizing pulse was attributable to evoked, reciprocal IPSCs rather than sIPSCs. Nevertheless, in some rare cases, we observed terminals that had an unusually large spontaneous activity, and we excluded them from additional analysis.
Temporal characteristics of the reciprocal feedback
Increasing the amplitude of BC depolarizing steps (from -30 to 0 mV) increased the magnitude of the reciprocal feedback and of the Ca2+-dependent Cl--mediated tail current (n = 10) (Fig. 2Ai). This enhancement of the feedback by depolarization can be explained partly by the scaling effect generated by the simultaneous increase in the electrochemical driving force for Cl- as one steps away from its reversal potential (ECl =-41 mV). However, more depolarized membrane potentials also increased the amplitude of the ICa and the corresponding exocytosis (i.e., ΔCm is smaller for -30 mV than for 0 mV pulses) (Fig. 2Ai). The degree of GABAergic feedback and its kinetics varied greatly from terminal to terminal. However, close inspection of the feedback often revealed two components: (1) a transient, fast component (Fig. 2Aii) and (2) a slower, more sustained component (Fig. 2Aiii). These two components had variable amplitudes among different terminals, but the first transient component peaked with a short delay of 3.4 ± 1.2 ms (n = 10) at 0 mV. This delay was very consistent at all membrane potentials tested (3.75 ± 1.8 ms at -10 mV, 3.08 ± 0.88 ms at -20 mV, and 2.86 ± 0.85 ms at -30 mV) (Fig. 2Aii,Aiv). In contrast, the peak delay of the slower, more sustained portion of the feedback showed a stronger voltage dependence, peaking earlier with more presynaptic depolarization. This peak also showed a greater variability in amplitude and delay, so to determine its delay, we filtered the current traces with a 10 Hz cutoff (Fig. 2Aiii). Nevertheless, in some cells it was impossible to determine the peak delay even after filtering. Those terminals were excluded from the analysis shown in Figure 2Aiv.
Figure 2.
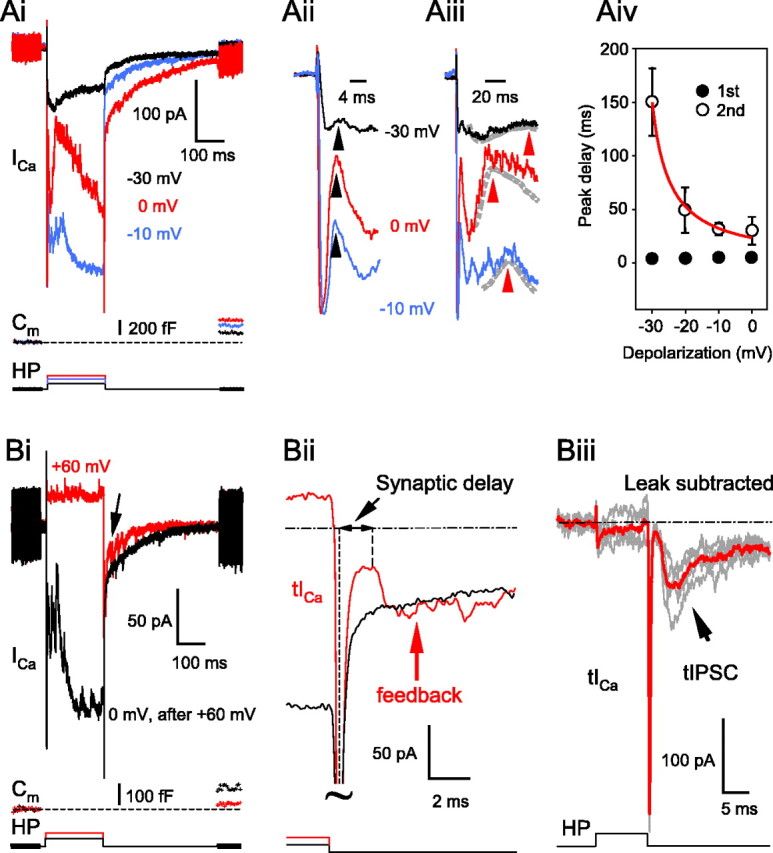
Temporal characteristics of the reciprocal feedback depend on the depolarization of the BC terminal. Ai, Changing the level of depolarization of the BC terminal membrane changed the magnitude of the ICa, with corresponding changes in the Ca2+-dependent Cl- tail current as well as in the Cm jumps in a graded manner. Note the two components of the inhibitory transmitter-mediated feedback (i.e., a fast, short portion and a later, more sustained part). The resting Cm of this terminal was 6.9 pF. Aii, An extended time scale of Ai focusing on the fast, transient part of the reciprocal feedback. The arrowheads point to where the peak delay measurements were made (see Aiv). Aiii, An extended time scale of Ai focusing on the peaks of the second, more sustained portion of the feedback. After filtering the traces at 10 Hz (dashed lines), local maximums were determined (red arrowheads). Aiv, Summary diagram of the delay of both components of the reciprocal feedback. The delay was measured from the peak of the ICa to the peak of the first or the second portion of GABAergic feedback at different depolarization levels. Data were fit with an exponential to guide the eye. Bi, Depolarization of the terminals to +60 mV resulted in very small reciprocal activity during the pulse, but apparently more feedback triggered by the tail current (arrow), compared with the depolarization to 0 mV. The resting capacitance of this terminal was 5.2 pF. Note the smaller Cm jump (exocytosis) in response to the faster, but smaller tail Ca2+ influx (red Cm trace). Bii, An extended time scale of Bi focusing on the tail Ca2+ current (tICa; red trace). The delay of the feedback triggered by the tail current was measured from the peak of the ICa tail to the onset of the IPSC (first local maximum after the tICa peak). Tilde, Truncated traces. Biii, Leak-subtracted reciprocal tIPSCs evoked by tICa after a 10 ms depolarization from -60 to +60 mV. A P/4 leak-subtraction protocol was used to reveal the presynaptic tICa. In this particular cell, five individual traces (gray) were averaged (red trace) before the onset delay of the IPSC was measured. HP, Holding potential.
When we depolarized the BC terminals to +60 mV (n = 6), which is close to the reversal potential for Ca2+, there was very little activity during the depolarizing step compared with what could be seen in the same cell in response to a depolarization to 0 mV (Fig. 2Bi), although sIPSC events are more detectable at +60 mV (ECl = -41 mV). However, the ICa tail current (tICa) after strong depolarizations in BCs should trigger some glutamate exocytosis (von Gersdorff et al., 1998; Singer and Diamond, 2003) and thereby reciprocal activity during the tail current (Hartveit, 1999). Indeed, the early portion of the tail current after the +60 mV step carried several GABAergic IPSCs that may have been evoked by the rapid glutamate release triggered by the tail current (Fig. 2Bi, arrow). Driving release with the brief “impulse” stimulus provided by tICa is a particularly good way to estimate the maximal speed of the glutamate release (von Gersdorff et al., 1998) and, consequently, the maximal speed of the reciprocal synapse. The tICa evoked by a 200 ms depolarization to +60 mV triggered only 31.2 ± 8.9% (n = 6) of the exocytosis (note the small Cm jump in Fig. 2Bi, red trace) compared with that when the same bipolar terminals were depolarized to 0 mV (black Cm trace). Nevertheless, the glutamate released by the tICa was sufficient to evoke significant feedback in four of the six terminals (all six had sizeable reciprocal feedback during the depolarizing step to 0 mV). The delay for the tICa-evoked feedback was 1.3 ± 0.2 ms (n = 4), measured between the tICa peak and the first local maximum (Fig. 2Bii). Further addressing this question, in a separate set of experiments, we subtracted the leak to get a more precise timing of the tICa peak after a 10 ms depolarization to +60 mV. Under these conditions, we evoked reciprocal feedback in 6 of 11 bipolar terminals. In those cells, up to seven traces were averaged to measure the onset of the small feedback responses (Fig. 2Biii), which was 1.4 ± 0.2 ms (n = 6), similar to what was found for the traces without P/4 leak subtraction. Our data are in good agreement with measurements showing that ∼0.7 ms is needed for releasing glutamate from voltage-clamped BC terminals (von Gersdorff et al., 1998). GABA release from a depolarized AC bouton may thus occur with a synaptic delay of ∼0.7 ms. Although this tICa-driven reciprocal feedback has little physiological relevance, its short delay indicates that the di-synaptic event we are studying probably occurs via local microcircuit interactions (i.e., it does not involve events occurring in the distant AC cell somas). Considering the narrow synaptic cleft between the BC and AC terminals (≤20 nm) [Marc and Liu (2000), their Fig. 3C,D], the speed of tICa-evoked reciprocal communication suggests that transmitter action can be very fast and probably involves receptors directly facing the release sites.
Figure 3.

Pharmacology of the reciprocal feedback: GABA receptors on the BC terminal. Ai, Application of the specific GABAA antagonist SR95531 (SR; 25 μm) completely eliminated the fast, transient part of the feedback and revealed the slowly rising portion (red trace) without affecting the presynaptic glutamate release, as evidenced by the overlapping Cm traces. The slow feedback was eliminated after the addition of PTX (100 μm; blue trace), suggesting it was mediated by GABAC receptors. Traces were recorded at the following times after break-in: control: 2 min, 50 s; SR: 5 min, 20 s; SR+PTX: 8 min, 20 s,. The resting Cm of this cell was 4.5 pF. Aii, An extended time scale of Ai. Note the small, residual proton-mediated inhibition of the ICa in the presence of SR95531 and PTX (pH; blue trace) unblocked by intracellular methylamine (10 mm). Aiii, The pure GABAA and GABAC components of the reciprocal feedback, obtained by trace subtraction. GABAC = SR trace - (SR+PTX) trace; GABAA= (control - SR+PTX trace) - GABAC. ICa peaks were scaled to overlap perfectly before subtraction. Note that fast (GABAA) IPSC components were present during the entire length of depolarization (arrow, black trace). More importantly, the subtraction method revealed that the onset of the GABAC component (red arrow) had much longer synaptic delay than that of GABAA (∼12 vs ∼2 ms, respectively).
GABAA and GABAC receptors contribute to reciprocal feedback with different time courses
We reported recently that both GABAA and GABAC receptors are involved in the reciprocal communication at the Mb goldfish BC terminal (Hull and von Gersdorff, 2004). The GABAA feedback consisted of fast, transient peaks, the earliest peak occurring with a 2-3 ms delay, whereas the GABAC feedback was slower, more sustained, and took ∼75 ms to peak, although the amplitude of these different components showed great variability from terminal to terminal, as well as the ratio of their relative contribution to the reciprocal feedback (Vigh et al., 2005). In the present set of experiments, we focused on the onsets of these feedback components. Application of the GABAA receptor blocker SR95531 (n = 13) markedly reduced the reciprocal feedback (Fig. 3Ai, red trace) evoked by depolarization of the Mb terminals from -60 to 0 mV. In particular, it blocked the first “3 ms peak-delay” part and uncovered a slowly activating outward current component, which was blocked by (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid (TPMPA) (100-200 μm; n = 9 of 9) or picrotoxin (PTX) (100 μm; n = 4 of 4) (Fig. 3Ai, blue trace), indicating that it was mediated by GABAC receptors. Depolarization-evoked glutamate release at retinal ribbon synapses is followed by acidification of the synaptic cleft, because of the coreleased protons (DeVries, 2001; Palmer et al., 2003b). This proton-mediated inhibition of the ICa overlaps with the transmitter-mediated reciprocal feedback and makes the measurements of feedback onset very difficult. However, the vesicular pH can be buffered reasonably well with intracellular methylamine without affecting either the glutamate release from BC terminals or the reciprocal feedback (Vigh et al., 2005). Although we included methylamine (10 mm; see Materials and Methods) in all of our experiments, detailed examination of the feedback onset still revealed a small, residual proton-mediated inhibition of the ICa in the presence of PTX (pH) (Fig. 3Aii, blue trace), which could not be eliminated even when we doubled the methylamine concentration in the pipette. It suggests that methylamine could not buffer intravesicular pH completely, most likely because it is too weak of a base (pKa = 10.6) (Johnson, 1987) or its diffusion into the vesicles is slower than the accumulation of protons. Therefore, to more precisely determine the onsets, the pure GABAC current trace was calculated by subtracting the trace recorded in the presence of GABAA and GABAC blockers (PTX) (Fig. 3Aii, blue trace) from the trace recorded in the presence of a GABAA blocker (SR95531) (Fig. 3Aii, red trace). Likewise, the pure GABAA current trace could be calculated by subtracting the calculated GABAC trace from the control (Fig. 3Aiii). The inhibitory feedback onset was then measured from the peak of the ICa. We found that the GABAA-mediated inhibition had a much faster onset compared with that of GABAC (1.6 ± 0.4 vs 10 ± 4.5 ms, respectively; n = 6). This finding was somewhat puzzling, considering that both GABAA and GABAC receptors are thought to be synaptic (Koulen et al., 1998) and GABAC receptors have ∼40 times higher affinity for GABA than GABAA receptors (Amin and Weiss, 1994). However, it is important to point out that the GABAC single-channel conductance is much smaller than that for GABAA channels and GABAC currents are slow to activate (Amin and Weiss, 1994), making an accurate determination of the GABAC-mediated synaptic delay difficult. Furthermore, GABAA and GABAC mediated feedback are not completely independent. Blocking GABAA receptors in a slice preparation likely disinhibited the ACs, resulting in an increased GABA release from ACs (Zhang et al., 1997; Watanabe et al., 2000; Vigh et al., 2005). Therefore, the GABAC component recorded in SR95531 is probably an overestimate of the true amount of GABAC fraction of the control. Consequently, the GABAC onset delay in SR95531 might actually be an underestimate. Nevertheless, these findings suggest that GABAA and GABAC receptors form synapses with very different kinetics and synaptic strengths.
Desensitizing AMPARs trigger a fast GABAA feedback
Amacrine cells possess AMPARs and NMDARs, and both contribute to their light response (Dixon and Copenhagen, 1992). A mixture of NBQX and AP-5 (25 and 50 μm, respectively) completely eliminates the reciprocal feedback at the goldfish Mb terminal (Hull and von Gersdorff, 2004; Vigh et al., 2005). How much do these receptors contribute separately to the GABAergic feedback? When the NMDAR-specific antagonist d-AP-5 (50 μm) was applied alone, the late phase of the feedback was markedly reduced without any reduction in the glutamate release (ΔCm jump) (Fig. 4Ai, red trace), but the initial fast portion remained intact (Fig. 4Aii, inset). Similar results were obtained in 31 cells tested. The initial fast feedback peak was, however, reversibly eliminated after the addition of 1 μm NBQX (n = 5 of 5 terminals) (Fig. 4Bi,Bii). Both AMPA and kainate receptors are present on ACs postsynaptic to bipolar terminals (Grunert et al., 2002; Brandstatter et al., 1997). Nevertheless, because the NBQX is approximately threefold more potent against AMPA-preferring glutamate receptors than against kainate-preferring ones (Wilding and Huettner, 1996) and the IC50 value of NBQX at kainate-type glutamate receptors is ∼5.75 μm (Pook et al., 1993), this finding strongly indicates that the AP-5-independent feedback at the Mb terminal was mediated by AMPA and not by kainate receptors. Accordingly, GYKI 53655 (100 μm), a specific noncompetitive AMPAR blocker (Donevan et al., 1994; Wilding and Huttener, 1995; Balannik et al., 2005) reversibly eliminated the AP-5-independent feedback in every cell tested (n = 5) (Fig. 4Ci,Cii).
Figure 4.
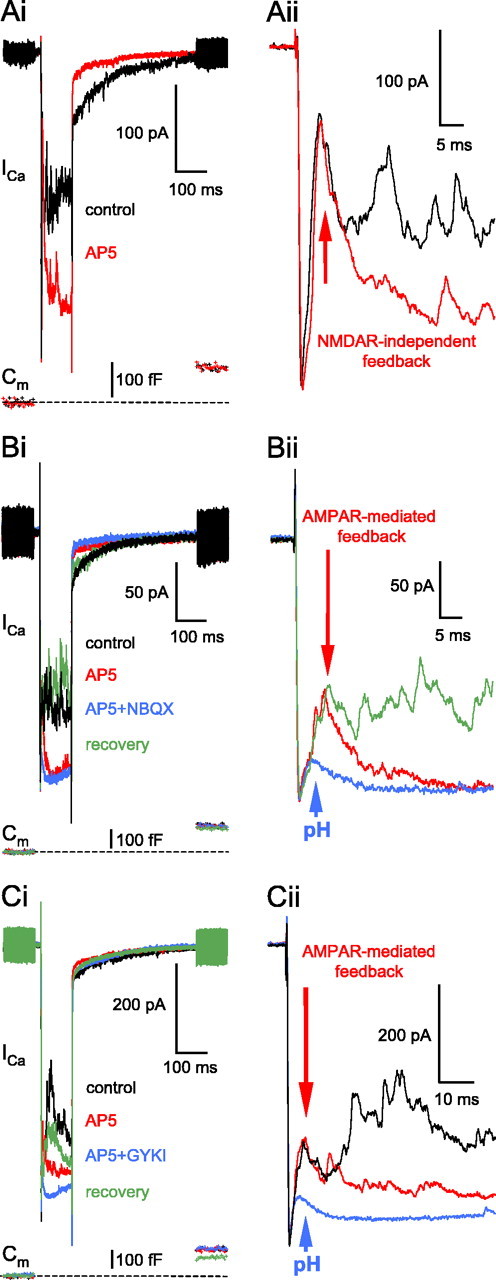
Pharmacology of the reciprocal feedback: the role of AMPARs on ACs. Ai, Application of the NMDAR blocker AP-5 (50μm) markedly inhibited the reciprocal feedback (red trace; 6 min, 41 s) without changing the presynaptic glutamate release or affecting the fast 3 ms peak (Aii, inset; red arrow). The resting Cm of this terminal was 4.3 pF. Bi, The NMDAR-independent reciprocal feedback in the presence of AP-5 (50 μm; red trace) was reversibly eliminated after the addition of NBQX (1μm; blue trace). Note the very small rundown in the glutamate release (i.e., Cm jump) over the course of the experiment and the small, residual proton-mediated inhibition of the ICa in the presence AP-5 and NBQX (pH; blue trace). Traces were recorded at the following times: control: 2 min, 11 s; AP-5: 5 min, 11 s; AP-5+NBQX: 8 min, 12 s; recovery: 30 min, 11 s. The resting Cm of this terminal was 4.2 pF. Bii, An extended time scale of Bi. The control trace is not shown for clarity. Ci, The NMDAR-independent reciprocal feedback in the presence of AP-5 (50 μm; 3 min, 11 s; red trace) was reversibly eliminated after the addition of the specific AMPAR blocker GYKI53655 (GYKI; 100 μm; 5 min, 41 s; blue trace). Traces were recorded at the following times: control: 1 min, 1 s; recovery: 20 min 11 s. The resting Cm of this terminal was 6.3 pF. Cii, An extended time scale of Ci. The recovery trace is not shown for clarity.
In the presence of AP-5, the specific GABAA blocker SR95531 (25 μm) eliminated the AMPAR-evoked feedback almost completely (n = 7 of 7) (Fig. 5Ai) apart from a small initial inflection in the ICa plateau (Fig. 5Aii, blue trace). Because neither the addition of the specific GABAC receptor antagonist TPMPA (200 μm) or PTX (100 μm) affected this small inflection, we considered it residual proton-mediated inhibition of ICa unbuffered by methylamine (pH) (Fig. 5Aii, green trace). In a separate set of experiments, when SR95531 (25 μm) was present first, the remaining late GABAC portion of the reciprocal feedback was mostly blocked by the NMDAR blocker AP-5 (50 μm; n = 4) (Fig. 5Bi,Bii).
Figure 5.
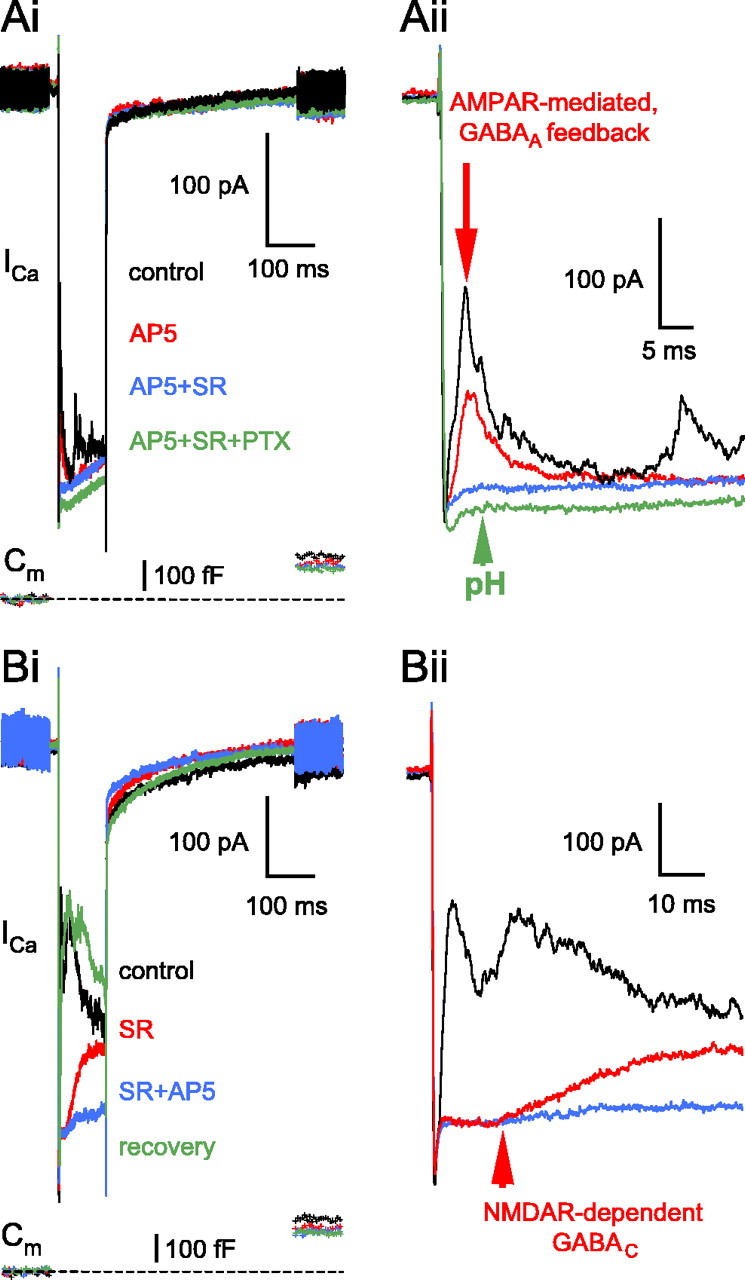
Interaction between glutamatergic and GABAergic receptors at the reciprocal feedback synapse. Ai, After dissecting the AMPAR activation-evoked feedback (AP-5; 50 μm; red trace), the specific GABAA receptor blocker SR95531 (25 μm; 9 min, 41 s; blue trace) eliminated the reciprocal IPSCs peaking at ≈3 ms (Aii on an extended time scale; red arrow). The addition of PTX (100 μm; 12 min, 50 s; green trace) slightly reduced the leak current, thereby increasing the ICa because of the elevated input resistance, but did not change otherwise the ICa, indicating no additional reciprocal feedback components in the presence of AP-5 and SR95531. Note that a slight reduction in the peak amplitude of the AMPAR-mediated feedback (Cii, red vs black current trace) coincides with the small rundown in the glutamate release (Ci, red vs black Cm trace). The resting Cm of this terminal was 6.0 pF. Bi, The GABAC portion of the reciprocal feedback isolated by application of the GABAA blocker SR95531 (25 μm; 3 min, 23 s; red trace) was blocked after the addition of AP-5 (50 μm; 5 min, 34 s; blue trace), indicating that it was triggered by NMDAR activation. Note the long onset delay of the GABAC component (Bii, red arrow). The reciprocal feedback was recovered after 19 min of wash with normal external solution (green trace). The resting Cm of this terminal was 6.6 pF. SR, SR95531.
Thus, the initial peak of the reciprocal feedback (Fig. 2Aii) seemed to be produced by AMPA and primarily GABAA receptor activation (Fig. 3Aiii). To test this, we compared the inhibitory charge transfer mediated by AMPA and GABAA receptors during the first 10 ms, starting from the peak of the ICa. To do that, we subtracted the control current traces from the corresponding ones recorded in the presence of AMPA or GABAA receptor antagonists (25 μm NBQX/CNQX or SR95531, respectively; data not shown). Although the amplitude of the initial peak appeared the least variable component of the reciprocal feedback, the inhibitory charge transfer still showed substantial variation across the terminals tested: it ranged between 0.49 and 2.24 pC (0.99 ± 0.34 pC, on average; n = 10) for the AMPAR-triggered feedback transient and between 0.19 and 1.21 pC (0.87 ± 0.32 pC, on average; n = 10) for the GABAAR-mediated feedback transient. A small contribution from GABAC receptor activation could account for the slightly larger AMPAR-triggered feedback; however, when compared, the difference was not statistically significant (Student's t test; p < 0.2).
The previous results suggest that AMPAR-mediated depolarization of the AC boutons cannot trigger a large and prolonged GABAC-mediated reciprocal feedback. This may be caused by the transient action of a rapidly desensitizing AMPAR and the slow kinetics of GABAC receptors. To test this hypothesis, we used cyclothiazide to reduce the desensitization of the AMPARs on ACs (Matsui et al., 2001; Singer and Diamond, 2003), although it can also increase release probability at some synapses (Diamond and Jahr, 1995), and block GABAA receptors (Deng and Chen, 2003). In our experiments, cyclothiazide (50 μm) caused a robust enhancement in the reciprocal feedback at the Mb terminals (Fig. 6Ai, blue trace) even if the NMDARs were blocked (n = 14 of 14). Note that cyclothiazide did not enhance the glutamate release from the BC terminal (Fig. 6Ai, compare blue and red Cm traces) (von Gersdorff et al., 1998). Under these conditions, the addition of SR95531 (25 μm) markedly reduced the feedback (n = 6 of 9) but, apart from one case, could not block it completely (Fig. 6Aii, green trace). The remaining large, slowly rising, and decaying current was blocked after the addition of PTX (50 μm; n = 3 of 3) (Fig. 6Aii,Aiii, purple trace) or TPMPA (200 μm; n = 3 of 3; data not shown), indicating that it was mediated by GABAC receptors. Note also the substantial GABAA receptor-mediated feedback in the presence of cyclothiazide, which indicates that in our preparation, the potentiating effect of cyclothiazide on the AMPAR function was stronger than any possible block of GABAA receptors by cyclothiazide (Deng and Chen, 2003).
Figure 6.

AMPAR desensitization limits the AMPAR-mediated reciprocal feedback to mostly GABAA receptors on the BC terminal. Ai, After dissecting the AMPAR activation-evoked feedback (AP-5; 50 μm; red trace), the addition of the AMPAR desensitization blocker cyclothyazide (50 μm; blue trace) markedly increased both the amplitude and the duration of the AMPAR-mediated feedback. Traces were recorded at the following times after break-in: control, 10 min; AP-5, 13 min;AP-5+CTZ, 16 min. Aii, Same cell as in Ai; consecutive traces divided to two panels for better visibility. In the presence of AP-5 and cyclothyazide (CTZ+AP-5; 50μm each; blue trace), the specific GABAA receptor blocker SR95531 (SR; 25μm; 24 min, 48 s; green trace) revealed the slowly activating GABAC receptor-mediated reciprocal feedback (Aiii, arrow), which was eliminated after the addition of PTX (100μm; 28 min, 17 s; purple trace). Note that there was very little rundown either in the ICa or in the Cm jump amplitude over the course of the experiment. The resting capacitance of this cell was 5.3 pF. CTZ, Cyclothyazide.
These results suggest that when AMPAR desensitization is reduced, AMPAR activation can trigger substantial GABAC receptor-mediated feedback. It thus appears that under physiological conditions, in which retinal AMPARs do rapidly desensitize (Lukasiewicz et al., 1995), NMDAR activation might be the sole coupling between prolonged AC excitation and GABAC receptor-mediated feedback inhibition.
NMDAR activation alone can evoke both GABAA and GABAC feedback
AMPAR block either with the CNQX or NBQX (25 μm each) markedly reduced the feedback inhibition (n = 27 and n = 7, respectively) (Fig. 7Ai) and even completely eliminated it in some instances (n = 8 of 27 terminals with CNQX and n = 3 of 7 with NBQX; data not shown). Nonetheless, in most of the terminals, there was a significant reciprocal feedback with AMPARs blocked, even in the presence of our standard external Mg2+ (1 mm). Close inspection of the data revealed that NBQX/CNQX affected in particular the first fast feedback component (Fig. 7Aii). The onset of AMPAR-independent reciprocal feedback did not differ in response to a 100 or 200 ms depolarization from -60 to 0 mV [i.e., it was 10 ± 1.5 ms, n = 4 with NBQX (Fig. 7Aii, red arrow) and 12.1 ± 3.8 ms, n = 8 with CNQX for 100 ms depolarizations; and 8.3 ± 2.6 ms, n = 19 with CNQX for a 200 ms depolarization], but it was much longer than that of AMPAR-evoked feedback (Fig. 4Aii). In the presence of NBQX (25 μm), GABA released from ACs activated both GABAA and GABAC receptors on the BC terminals (n = 4) (Fig. 7Bi). Note that application of SR95531 (25 μm) not only reduced the magnitude of the NBQX-insensitive feedback (Fig. 7Bi, compare red and blue traces) but also further increased the delay (Fig. 7Bii, compare red and blue arrow positions) by eliminating the faster GABAA portion of the inhibition.
Figure 7.
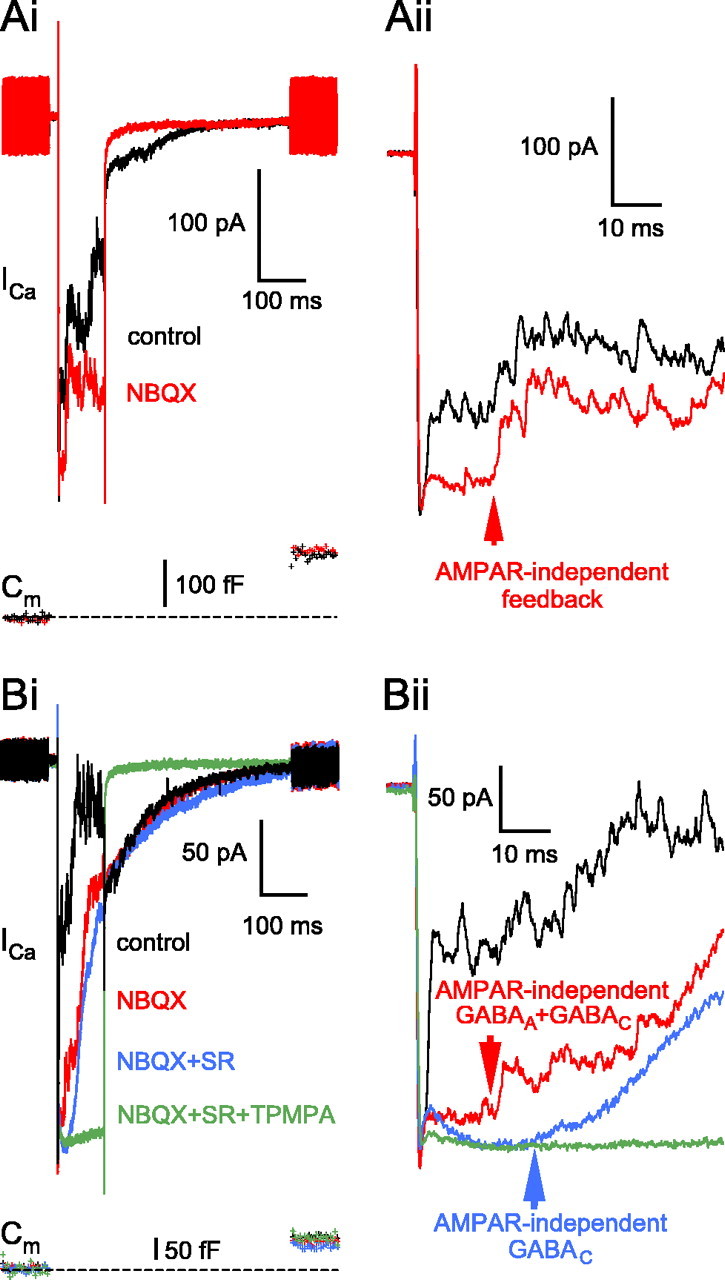
Pharmacology of the reciprocal feedback: the role of NMDARs on the ACs. A, In this example, the specific AMPAR antagonist NBQX (25 μm), in the presence of 1 mm Mg2+, drastically reduced the reciprocal feedback (red trace; 8 min, 37 s) compared with control (black trace; 1 min, 31 s) but did not block it. In particular, NBQX eliminated the initial fast 3 ms portion (Aii, inset). The AMPAR-independent synaptic delay, measured from the peak of the Ca2+ current, was 14.1 ms (Aii, inset, red arrow). The resting Cm of this terminal was 4.4 pF. Bi, In the presence of NBQX (25 μm; 4 min, 39 s; red trace), GABAA block by SR95531 (SR; 25 μm; 9 min, 38 s; blue trace) revealed the slowly activating GABAC feedback, which was sensitive to TPMPA (200 μm; 14 min, 19 s; green trace). The resting Cm of this terminal was 4.2 pF. Bii, Inset of Bi focusing on the delay of the feedback components. The delay of AMPAR-independent feedback, consisting of both GABAA and GABAC, was 11.9 ms (red arrow). In the presence of NBQX and SR95531, the GABAC onset was ∼21 ms (blue arrow).
Our standard extracellular solution contained 1 mm Mg2+. In the presence of NBQX (25 μm), the AMPAR-independent reciprocal feedback was enhanced after removal of the external Mg2+ (Fig. 8Ai, blue trace), whereas increasing the Mg2+ to 2 mm reversibly eliminated the feedback (Fig. 8Aii, green trace). Note that this concentration of Mg2+ did not reduce the ICa amplitude or the glutamate release (ΔCm jump). This block of feedback with 2 mm Mg2+ was observed in 17 terminals (n = 5 of 5 with NBQX; n = 12 of 13 with CNQX), suggesting that the NMDARs participating in the reciprocal feedback are very sensitive to Mg2+ block.
Figure 8.
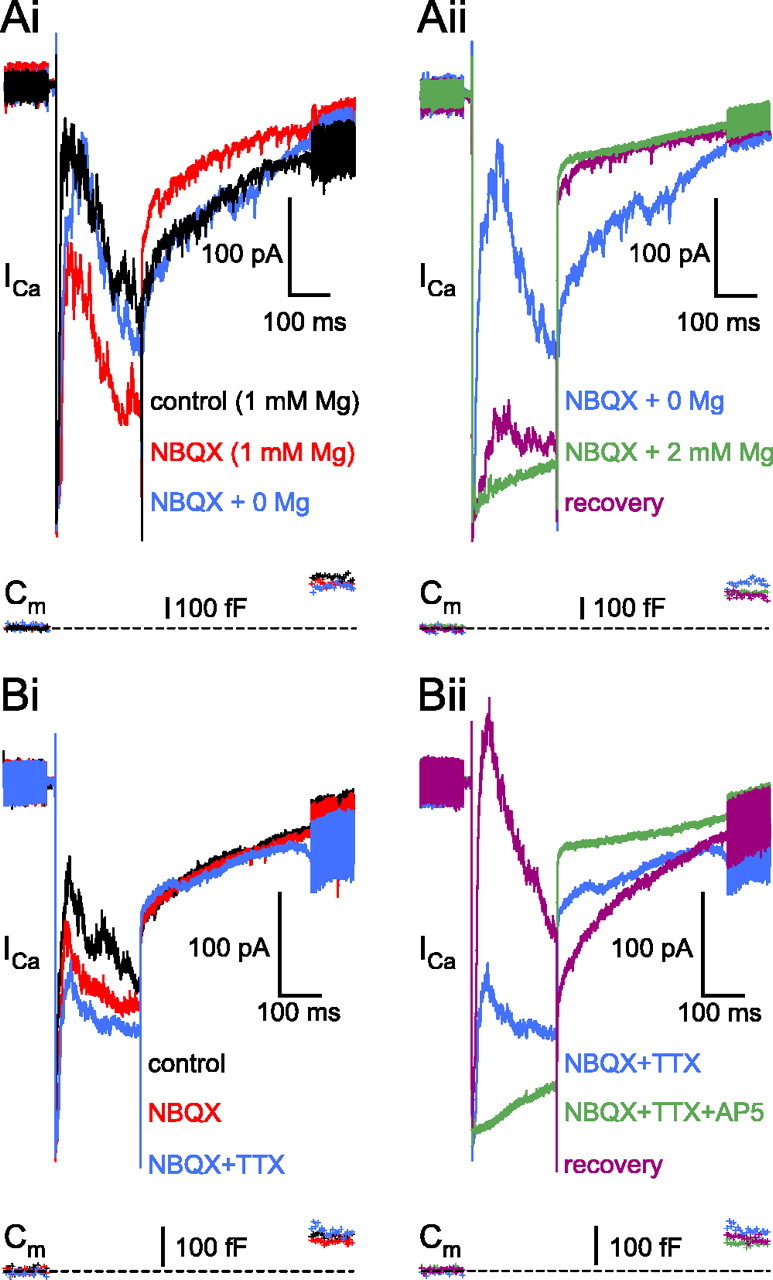
NMDARs involved in reciprocal feedback were sensitive to external Mg2+ and could detect the action potential. Ai, The AMPAR-independent feedback in the presence of NBQX (25 μm) and 1 mm Mg2+ (red trace; 1 min, 18 s) was enhanced after removal of Mg2+ (blue trace; 4 min, 17 s) without increasing either the ICa amplitude or the glutamate release (compare red and blue Cm traces). Aii, Same cell as in Ai; consecutive traces are divided into two panels for better visibility. The small elevation of Mg2+ in the external solution (to 2 mm) in the presence of NBQX reversibly blocked the feedback (green trace; 7 min, 18 s), without markedly reducing the glutamate release. Note that partial recovery of the feedback occurs after wash despite the additional reduction in the exocytosis (purple trace; 14 min, 18 s). The resting Cm of this terminal was 5.2 pF. Bi, In the presence of NBQX (25μm; 4 min; red trace) and 1 mm Mg2+, the AMPAR-independent reciprocal feedback is reduced but not blocked by TTX (1 μm; 7 min; red trace). The resting Cm of this terminal was 4.5 pF. Bii, Same cell as in Bi; consecutive traces are divided into two panels for better visibility. The addition of AP-5 (50 μm) completely blocked the NBQX- and TTX-independent feedback (green trace; 12 min). Recovery of the reciprocal feedback was achieved in the control external solution (purple trace; 33 min). Note that there was very little change in either the ICa or Cm jump amplitude over the course of the experiment.
Spontaneous spiking in goldfish ACs (Watanabe et al., 2000; Yamada et al., 2002) may remove the Mg2+ block of the NMDARs, making them capable of participating independently of AMPARs in the reciprocal communication, as was shown at the mitral cell ↔ granule cell reciprocal synapse in the olfactory bulb (Chen et al., 2000). Therefore, we investigated whether putative action potentials fired by the ACs providing reciprocal feedback to the recorded Mb terminal could influence the NMDA-mediated feedback in the presence of NBQX (25 μm) and 1 mm Mg2+. The NBQX-resistant feedback (Fig. 8Bi, red trace) was reduced by the addition of TTX (1 μm) (Fig. 8Bi, blue trace) but was never blocked completely (n = 5). Complete, reversible block of the feedback was achieved after the addition of AP-5 (50 μm) (Fig. 8Bii, green trace). The above results suggest that NMDARs on ACs can be activated in the presence of 1 mm external Mg2+ without the help of AMPAR activation or Na+-dependent action potentials to relieve the Mg2+ block. Nonetheless, action potentials can further increase the NMDAR contribution to the GABA release from the ACs during reciprocal communication.
Onset delay of the reciprocal feedback at physiological membrane potentials
Under physiological conditions in response to light, the membrane potential of the ON-type Mb BC can reach about -20 mV (Saito and Kujiraoka, 1982; Protti et al., 2000) from the dark resting level of approximately -53 mV (Wong et al., 2005). Therefore, we compared the onset of the AMPAR- and NMDAR-mediated feedback at different command potentials between -30 and 0 mV, in the presence of AP-5 (50 μm; n = 5) or CNQX (25 μm; n = 8), respectively (Fig. 9Ai,Bi). NMDA-receptor mediated feedback responses were studied in the absence of external Mg2+ to increase their amplitude. Cells were excluded from evaluation if testing at three or more command potentials was not possible. Increasing depolarization elicited larger ICa amplitudes and evoked greater glutamate release, resulting in stronger reciprocal feedback. Note that the amplitudes of the ICl(Ca) generally correlated with an increased ICa and feedback. Feedback onset delay was measured from the ICa peak to the inflection point caused by the outward Cl- conductance. It is important to point out that the small residual proton inhibition of the ICa overlapped with the onset of the fast AMPA-mediated component and made the measurements particularly difficult at weak depolarizations (-30 and -25 mV) (Fig. 9Aii). Therefore, we measured by eye the onset delay under our standard extracellular buffering conditions (25 mm bicarbonate bubbled continuously with 95% O2 and 5% CO2) only at three holding potentials (0, -10, and -20 mV), at which it was particularly clear (Fig. 9Aii, arrows). Note also that the initial slope of the small residual ICa protonmediated inhibition becomes less steep at less depolarized potentials (Fig. 9Aii), in accordance with less evoked exocytosis at less depolarized potentials (Palmer et al., 2003b).
Figure 9.
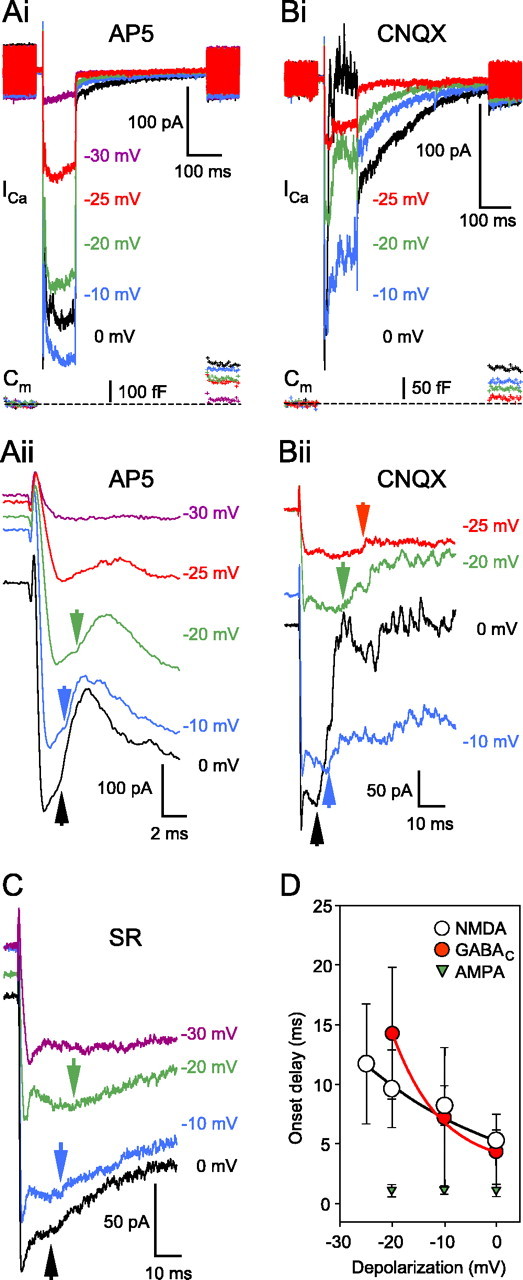
Measuring the delay of the reciprocal feedback at the BC terminal. Ai, Increasing the depolarization of the presynaptic terminal elevated the glutamate release and the magnitude of feedback in the presence of AP-5 (50μm). The resting Cm of this terminal was 5.4 pF. Aii, An extended time scale of Ai inspecting the delay of the fast portion in the presence of AP-5 (50 μm). In this case, we measured the delay between the ICa peak and the inflection point indicating the beginning of the feedback response (Aii, inset, arrows). Note that the onset delay changed little with the depolarizing voltage, measured ∼3 ms at every depolarization level. At -25 mV, it was impossible to discriminate between the transmitter- and leftover proton-mediated inhibition of the ICa. Traces were offset for better visibility. Bi, Detailed examination of the delay of the NMDAR-mediated portion of the reciprocal feedback at various depolarization levels, in the presence of CNQX (25μm) but in the absence of external Mg2+. The resting Cm of this terminal was 3.9 pF. Bii, An extended time scale of Bi. The delay was measured with the time between the peak ICa and the beginning of the response (arrowheads). Note the inversely proportional correlation between the depolarization and the delay. Traces were shifted for better visibility. C, Measuring the delay of the GABAC receptor-mediated feedback at various levels of presynaptic depolarization in the presence of SR95531 (SR; 25 μm). The delay was measured as the time between the ICa peak and the inflection point, indicating the beginning of the feedback response. Similar to the NMDA component, the depolarization and the delay were inversely proportional. D, Summary diagram of the onset delay for the AMPA, NMDA, and GABAC receptor-mediated feedback. The plotted points are averages of n ≥ 5 and for the NMDA and GABAC receptors were fitted with an exponential to guide the eye. Every cell included was tested at three or more depolarization levels.
Detailed examination on a slower time scale revealed that the NMDAR fraction of the feedback had a long onset delay (or slowly rising current), particularly at lower membrane potential levels (Fig. 9Bii). The delay was inversely proportional to the depolarization of the BC terminal. However, the faster AMPAR onset did not show a very strong dependence on the BC membrane potential (Fig. 9D). We also tested the delay of the GABAC feedback onset after blocking the GABAA receptors with SR95531 (25 μm). A representative cell is shown in Figure 9C, and the summarized data are plotted in Figure 9D. As for the NMDA portion, the GABAC delay was inversely proportional to the depolarization of the BC terminal, in concert with the notion that GABAC receptor-mediated feedback was triggered predominantly by NMDAR activation as demonstrated above. This might be an indication of an underlying slow transmitter diffusion preceding NMDA and, in turn, GABAC activation. In other words, NMDA and/or GABAC receptors may be located farther away from the glutamate release, at extrasynaptic sites. However, the timing of transmitter release from the Mb terminal strongly depends on the amplitude and activation kinetics of the ICa (i.e., smaller and slower ICa rises produce slower evoked glutamate release with longer delays) (von Gersdorff et al., 1998). This, in addition to the slow NMDAR kinetics (Lester et al., 1990), may account in part for the unusually long depolarization-dependent delay we found for the NMDA and GABAC receptors.
Discussion
The ionotropic glutamate receptors of reciprocal synapses
The relative contribution of AMPARs and NMDARs to glutamatergic synaptic transmission varies among different synapses, but this ratio is thought to be important for determining the temporal properties of the excitatory synaptic transmission (Dingledine et al., 1999). This may be especially true for different subtypes of retinal ACs (Dixon and Copenhagen, 1992). Here, we report the interplay between AMPARs and NMDARs located on ACs that feed back inhibitory signals directly to the BC terminal. First, AMPAR activation resulted in an initial bout of GABA release that elicited a transient burst of GABAA-mediated IPSCs. This was followed by significant NMDAR activation, which resulted in a continuous flurry of GABAA-mediated IPSCs superimposed on a more sustained and slower GABAC-mediated IPSC. Depolarization-evoked glutamate release from BC terminals thus activated both AMPARs and NMDARs on the postsynaptic ACs, conferring different temporal characteristics to the reciprocal feedback.
Sustained excitatory signals: AMPARs are insufficient
Block of the NMDARs on ACs resulted in a fast and transient GABAergic feedback peak. Therefore, AMPARs by themselves cannot mediate the signaling of continuous glutamate release from BCs. The quick desensitization of the AMPARs on the ACs in the continuous presence of glutamate (Tran et al., 1999; Matsui et al., 2001) thus shapes AC output. Cyclothiazide, which reduces the desensitization and increases the glutamate affinity of the AMPARs (Yamada et al., 2002), resulted in a marked increase in the glycinergic feed-forward inhibition of ganglion cells in tiger salamander retina (Tran et al., 1999). In our experiments in the presence of the NMDAR blocker AP-5, cyclothiazide not only enhanced the GABAA feedback to BC terminals but also increased GABAC receptor-mediated currents, without increasing glutamate release (or ΔCm jumps) (von Gersdorff et al., 1998). In contrast, no GABAC receptor activation was observed at the rod BC ↔ A17 reciprocal synapse in rat retina without cyclothiazide (Singer and Diamond, 2003). We found that selective AMPAR block did not eliminate the reciprocal feedback in every cell, although it did block the very fast GABAA portion completely in every cell tested. This suggests that NMDAR activation without AMPAR priming is sufficient to trigger significant GABA release from ACs. However, although the NMDAR activation evoked both GABAA and GABAC currents on the BC terminal, their amplitudes under these conditions was markedly reduced, indicating the importance of AMPAR priming for NMDAR activation at reciprocal synapses (Isaacson, 2001).
NMDAR activation evokes prolonged feedback
Interestingly, NMDAR activation alone can evoke GABA release from ACs. The depolarization provided by NMDAR activation thus probably opens voltage-gated N- and/or L-type Ca2+ channels that trigger the release of GABA from ACs (Gleason et al., 1994; Vigh and Lasater, 2004). In addition, the high Ca2+ permeability of NMDARs raises the possibility that their Ca2+ influx contributes to transmitter release or may provide enough Ca2+ to mediate release by itself, bypassing the voltage-gated Ca2+ channels (Isaacson and Strowbridge, 1998; Chen et al., 2000). This intriguing possibility at the BC ↔ AC synapse has yet to be addressed. Here, we show that NMDARs on ACs can be activated in an external solution that contains 1 mm Mg2+. NMDARs thus mediate the reciprocal feedback without the requirement for previous AMPAR priming via a relief of Mg2+ block. NMDARs are composed of a common NR1 subunit (Moriyoshi et al., 1991) and one or more of four different NR2(A-D) subunits (Ishii et al., 1993), the primary determinants of the Mg2+ sensitivity of the NMDARs (Monyer et al., 1994; Qian et al., 2005). The fact that 2 mm external Mg2+ can almost eliminate the feedback mediated by NMDARs suggests that the participating receptors were sensitive to Mg2+. Nonetheless, the dark resting level of ACs in the fish retina can be depolarized enough (approximately -36 mV) to remove the Mg2+ block without the help of AMPAR-mediated depolarizations (Djamgoz et al., 1996). Alternatively, glutamate may evoke a large enough inward current in ACs through NMDARs even at -70 mV to cause depolarization as in ganglion cells (Gottesman and Miller, 1992). TTX did not eliminate the solely NMDAR-evoked feedback, suggesting NMDAR opening without the need for voltage-gated sodium currents (action potentials). However, TTX sometimes reduced the NMDAR-triggered feedback, indicating that certain NMDARs on ACs may act as coincidence detectors sensing both lateral inhibition carried by action potentials (Cook and Werblin, 1994; Shields and Lukasiewicz, 2003) and local glutamate release. Therefore, lateral inhibition might boost the reciprocal feedback via NMDAR action. An alternative explanation for the TTX effect is that TTX-sensitive persistent sodium currents in AC boutons contribute to the reciprocal feedback (Koizumi et al., 2001).
Complementary properties of glutamate and GABA receptors
We showed that synaptically released glutamate can activate both AMPARs and NMDARs on ACs. This is similar to what was reported in retinal ganglion cells (Mittman et al., 1990) and in granule cells in the olfactory bulb (Isaacson and Strowbridge, 1998). NMDARs do not contribute to the spontaneous EPSCs in ganglion cells, although they are activated during light stimuli (Taylor et al., 1995; Chen and Diamond, 2002). Furthermore, in the mouse retina, NMDARs were activated during the light response of transient ON-type ACs only when glutamate release was increased by blocking GABAC receptors on BCs (Matsui et al., 2001). Based on this pattern, a spatial segregation of the AMPARs and NMDARs was suggested on both ganglion cells (Chen and Diamond, 2002) and ACs (Matsui et al., 2001), so that AMPARs are close to the glutamate release sites and NMDARs are somewhat farther away (extrasynaptic).
The temporal pattern of the reciprocal feedback suggests a complementary “match-up” between functionally coupled excitatory and inhibitory receptors with similar characteristics: lowagonist-affinity, fast, and transient receptors that desensitize quickly (AMPA and GABAA) and high-affinity, slowly activating and deactivating, sustained receptors that desensitize slowly (NMDA and GABAC). The terminal of an Mb BC receives ≈350 distinct AC synapses: 98% are GABAergic, and 59% of these GABAergic synapses are reciprocal (Marc and Liu, 2000). Different AC boutons may face either GABAA or GABAC receptors, because these are suggested not to be colocalized [rat retina (Koulen et al., 1998)]. AMPARs and NMDARs are thought to be colocalized on the same AC bouton, although the relevant double-labeling immunohistochemical data published so far is not very extensive (Fletcher et al., 2000; Grunert et al., 2002). Moreover, electrophysiological studies have recorded NMDAR-mediated responses only in ACs that possessed AMPARs as well (Dixon and Copenhagen, 1992; Hartveit and Veruki, 1997; Matsui et al., 2001).
In our working model (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), some AC boutons have AMPARs that must be located in the immediate vicinity of the glutamate release sites and GABAA receptors on the opposing surface of the bipolar terminal, so that the di-synaptic delay is very short. Because of their intrinsic slow response to glutamate (Lester et al., 1990), NMDARs may be in the synaptic cleft, or they may be further away from glutamate release sites, being thus activated by glutamate spillover (Matsui et al., 2001). This may increase the slowness of the GABA feedback mediated by NMDARs. In addition, the late GABAA/C IPSCs may be generated by “trans-synaptic” GABA spillover during and/or after NMDAR activation. This model contrasts with the notion that GABAC receptors are not involved in reciprocal feedback (Singer and Diamond, 2003) but does not rule out an additional spillover possibility in GABAC activation. Moreover, the reciprocal feedback between A17 amacrines and rod bipolars seems different in that NMDARs are not involved (Hartveit, 1999; Singer and Diamond, 2003), which may reflect species differences between mammalian and non-mammalian rod circuitry.
Our results support the notion that inhibitory synaptic interactions in the inner retina are highly specialized for different visual processing tasks (Zhang and Slaughter, 1995; Roska and Werblin, 2001; Ichinose and Lukasiewicz, 2005). The membrane potential of BCs varies in a graded manner with light intensity, and we showed that the amount and delay of the inhibitory feedback is dependent on the membrane potential of the cell. As a consequence, glutamate release from a BC terminal may have a wider dynamic range of signaling that enables it to fully encode the degree of excitation represented by graded potentials. We propose that both the fast but transient AMPARs and the slow but more prolonged NMDARs are necessary to faithfully translate the temporal pattern of glutamate release from ON-type BCs to ACs. GABAA and GABAC receptors similarly equip BC terminals for the reception of fast and slow components of feedback inhibition. Controlling the BC output by a diverse set of reciprocal inhibitory synapses may increase their capacity for differential processing of visual information along distinct temporal and spatial channels in the retina.
Footnotes
This work was supported by a National Institutes of Health-National Eye Institute RO1 grant and a Human Frontier Science Program grant. We thank Ko Matsui (The Vollum Institute) for a critical reading of this manuscript. The GYKI 53655 was a kind gift from Craig Jahr (The Vollum Institute), who obtained it from the IVAX Drug Research Institute (Budapest, Hungary).
Correspondence should be addressed to Dr. Henrique von Gersdorff, The Vollum Institute, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: vongersd@ohsu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511412-12$15.00/0
References
- Amin J, Weiss DS (1994) Homomeric rho 1 GABA channels: activation properties and domains. Receptors Channels 2: 227-236. [PubMed] [Google Scholar]
- Balannik V, Menniti SF, Paternain VA, Lerma J, Stern-Bach Y (2005) Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron 48: 279-288. [DOI] [PubMed] [Google Scholar]
- Brandstatter JH, Koulen P, Wässle H (1997) Selective synaptic distribution of kainate receptor subunits in the two plexiform layers of the rat retina. J Neurosci 17: 9298-9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Diamond JS (2002) Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci 22: 2165-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WR, Xiong W, Shepherd GM (2000) Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron 25: 625-633. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Lester RA (1989) Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev 41: 143-210. [PubMed] [Google Scholar]
- Cook PB, Werblin FS (1994) Spike initiation and propagation in wide field transient amacrine cells of the salamander retina. J Neurosci 14: 3852-3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhagen DR, Jahr CE (1989) Release of endogenous excitatory amino acids from turtle photoreceptors. Nature 341: 536-539. [DOI] [PubMed] [Google Scholar]
- Cousin MA, Nicholls DG (1997) Synaptic vesicle recycling in cultured cerebellar granule cells: role of vesicular acidification and refilling. J Neurochem 69: 1927-1935. [DOI] [PubMed] [Google Scholar]
- Deng L, Chen G (2003) Cyclothiazide potently inhibits gammaaminobutyric acid type A receptors in addition to enhancing glutamate responses. Proc Natl Acad Sci USA 100: 13025-13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH (2001) Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron 32: 1107-1117. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1995) Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15: 1097-1107. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Dixon DB, Copenhagen DR (1992) Two types of glutamate receptors differentially excite amacrine cells in the tiger salamander retina. J Physiol (Lond) 449: 589-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamgoz MB, Low JC, Saito T, Toyoda JI, Yamada M (1996) Voltage dependency of light-evoked on-off transient amacrine cell responses in carp retina. Neurosci Lett 214: 127-130. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Yamaguchi S, Rogawski MA (1994) Non-N-methyl-d-aspartate receptor antagonism by 3-N-substituted 2,3-benzodiazepines: relationship to anticonvulsant activity. J Pharmacol Exp Ther 271: 25-29. [PubMed] [Google Scholar]
- Dong CJ, Werblin FS (1998) Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. J Neurophysiol 79: 2171-2180. [DOI] [PubMed] [Google Scholar]
- Dowling JE, Boycott BB (1966) Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci 166: 80-111. [DOI] [PubMed] [Google Scholar]
- Fletcher EL, Hack I, Brandstatter JH, Wässle H (2000) Synaptic localization of NMDA receptor subunits in the rat retina. J Comp Neurol 420: 98-112. [PubMed] [Google Scholar]
- Gillis KD (2000) Admittance-based measurement of membrane capacitance using the EPC-9 patch-clamp amplifier. Pflügers Arch 439: 655-664. [DOI] [PubMed] [Google Scholar]
- Gleason E, Borges S, Wilson M (1994) Control of transmitter release from retinal amacrine cells by Ca2+ influx and efflux. Neuron 13: 1109-1117. [DOI] [PubMed] [Google Scholar]
- Gottesman J, Miller RF (1992) Pharmacological properties of N-methyl-d-aspartate receptors on ganglion cells of an amphibian retina. J Neurophysiol 68: 596-604. [DOI] [PubMed] [Google Scholar]
- Grunert U, Haverkamp S, Fletcher EL, Wässle H (2002) Synaptic distribution of ionotropic glutamate receptors in the inner plexiform layer of the primate retina. J Comp Neurol 447: 138-151. [DOI] [PubMed] [Google Scholar]
- Hartveit E (1999) Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. J Neurophysiol 81: 2923-2936. [DOI] [PubMed] [Google Scholar]
- Hartveit E, Veruki ML (1997) AII amacrine cells express functional NMDA receptors. NeuroReport 8: 1219-1223. [DOI] [PubMed] [Google Scholar]
- Hull C, von Gersdorff H (2004) Fast endocytosis is inhibited by GABA-mediated chloride influx at a presynaptic terminal. Neuron 44: 469-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Lukasiewicz P (2005) Inner and outer retinal pathways both contribute to surround inhibition of salamander ganglion cells. J Physiol (Lond) 565: 517-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS (2001) Mechanisms governing dendritic gamma-aminobutyric acid (GABA) release in the rat olfactory bulb. Proc Natl Acad Sci USA 98: 337-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW (1998) Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron 20: 749-761. [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M (1993) Molecular characterization of the family of the N-methyl-d-aspartate receptor subunits. J Biol Chem 268: 2836-2843. [PubMed] [Google Scholar]
- Johnson Jr RG (1987) Proton pumps and chemiosmotic coupling as a generalized mechanism for neurotransmitter and hormone transport. Ann NY Acad Sci 493: 162-177. [DOI] [PubMed] [Google Scholar]
- Koizumi A, Watanabe SI, Kaneko A (2001) Persistent Na+ current and Ca2+ current boost graded depolarization of rat retinal amacrine cells in culture. J Neurophysiol 86: 1006-1016. [DOI] [PubMed] [Google Scholar]
- Koulen P, Brandstatter JH, Enz R, Bormann J, Wässle H (1998) Synaptic clustering of GABAC receptor rho-subunits in the rat retina. Eur J Neurosci 10: 115-127. [DOI] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346: 565-567. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz PD, Lawrence JE, Valentino TL (1995) Desensitizing glutamate receptors shape excitatory synaptic inputs to tiger salamander retinal ganglion cells. J Neurosci 15: 6189-6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Liu W (2000) Fundamental GABAergic amacrine cell circuitries in the retina: nested feedback, concatenated inhibition, and axosomatic synapses. J Comp Neurol 425: 560-582. [DOI] [PubMed] [Google Scholar]
- Matsui K, Hasegawa J, Tachibana M (2001) Modulation of excitatory synaptic transmission by GABAC receptor-mediated feedback in the mouse inner retina. J Neurophysiol 86: 2285-2298. [DOI] [PubMed] [Google Scholar]
- Mittman S, Taylor WR, Copenhagen DR (1990) Concomitant activation of two types of glutamate receptors mediates excitation of salamander retinal ganglion cells. J Physiol (Lond) 428: 175-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529-540. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S (1991) Molecular cloning and characterization of the rat NMDA receptor. Nature 354: 31-37. [DOI] [PubMed] [Google Scholar]
- Okada T, Horiguchi H, Tachibana M (1995) Ca2+-dependent Cl- current at the presynaptic terminals of goldfish retinal bipolar cells. Neurosci Res 23: 297-303. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H (2003a) Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci 23: 4831-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J, von Gersdorff H (2003b) Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci 23: 11332-11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pook P, Brugger F, Hawkins NS, Clark KC, Watkins JC, Evans RH (1993) A comparison of the actions of agonists and antagonists at non-NMDA receptors of C fibres and motoneurones of the immature rat spinal cord in vitro. Br J Pharmacol 108: 179-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H (2000) Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron 25: 215-227. [DOI] [PubMed] [Google Scholar]
- Qian A, Buller AL, Johnson JW (2005) NR2 subunit-dependence of NMDA receptor channel block by external Mg2+ J Physiol (Lond) 562: 319-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roska B, Werblin F (2001) Vertical interactions across ten parallel, stacked representations in the mammalian retina. Nature 410: 583-587. [DOI] [PubMed] [Google Scholar]
- Saito T, Kujiraoka T (1982) Physiological and morphological identification of two types of on-center bipolar cells in the carp retina. J Comp Neurol 205: 161-170. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL (1998) Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci 18: 6790-6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields CR, Lukasiewicz PD (2003) Spike-dependent GABA inputs to bipolar cell axon terminals contribute to lateral inhibition of retinal ganglion cells. J Neurophysiol 89: 2449-2458. [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS (2003) Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci 23: 10923-10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter MM, Miller RF (1983) Bipolar cells in the mudpuppy retina use an excitatory amino acid neurotransmitter. Nature 303: 537-538. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Chen E, Copenhagen DR (1995) Characterization of spontaneous excitatory synaptic currents in salamander retinal ganglion cells. J Physiol (Lond) 486: 207-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Higgs MH, Lukasiewicz PD (1999) AMPA receptor kinetics limit retinal amacrine cell excitatory synaptic responses. Vis Neurosci 16: 835-842. [DOI] [PubMed] [Google Scholar]
- Vigh J, Lasater EM (2004) L-type calcium channels mediate transmitter release in isolated, wide-field retinal amacrine cells. Vis Neurosci 21: 129-134. [DOI] [PubMed] [Google Scholar]
- Vigh J, Witkovsky P (2004) Neurotransmitter actions on transient amacrine and ganglion cells of the turtle retina. Vis Neurosci 21: 1-11. [DOI] [PubMed] [Google Scholar]
- Vigh J, Li GL, Hull C, von Gersdorff H (2005) Long-term plasticity mediated by mGluR1 at a retinal reciprocal synapse. Neuron 46: 469-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G (1999) Electrophysiology of synaptic vesicle cycling. Annu Rev Physiol 61: 725-752. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Sakaba T, Berglund K, Tachibana M (1998) Submillisecond kinetics of glutamate release from a sensory synapse. Neuron 21: 1177-1188. [DOI] [PubMed] [Google Scholar]
- Wässle H (2005) Parallel processing in the mammalian retina. Nat Rev Neurosci 5: 747-757. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Koizumi A, Matsunaga S, Stocker JW, Kaneko A (2000) GABA-mediated inhibition between amacrine cells in the goldfish retina. J Neurophysiol 84: 1826-1834. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE (1995) Differential antagonism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol Pharmacol 47: 582-587. [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE (1996) Antagonist pharmacology of kainate- and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring receptors. Mol Pharmacol 49: 540-546. [PubMed] [Google Scholar]
- Witkovsky P, Dowling JE (1969) Synaptic relationships in the plexiform layers of carp retina. Z Zellforsch Mikrosk Anat 100: 60-82. [DOI] [PubMed] [Google Scholar]
- Wong KY, Cohen ED, Dowling JE (2005) Retinal bipolar cell input mechanisms in giant danio. II. Patch-clamp analysis of ON bipolar cells. J Neurophysiol 93: 94-107. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Koizumi A, Iwasaki E, Watanabe S, Kaneko A (2002) Propagation of action potentials from the soma to individual dendrites of cultured rat amacrine cells is regulated by local GABA input. J Neurophysiol 87: 2858-2866. [DOI] [PubMed] [Google Scholar]
- Zhang J, Slaughter MM (1995) Preferential suppression of the ON pathway by GABAC receptors in the amphibian retina. J Neurophysiol 74: 1583-1592. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jung CS, Slaughter MM (1997) Serial inhibitory synapses in retina. Vis Neurosci 14: 553-563. [DOI] [PubMed] [Google Scholar]