

Abstract
Although the induction of persistent behavioral alterations by drugs of abuse requires the regulation of gene transcription, the precise intracellular signaling pathways that are involved remain mainly unknown. Extracellular signal-regulated kinase (ERK) is critical for the expression of immediate-early genes in the striatum in response to cocaine and Δ9-tetrahydrocannabinol and for the rewarding properties of these drugs. Here we show that in mice a single injection of cocaine (10 mg/kg) activates mitogen- and stress-activated protein kinase 1 (MSK1) in dorsal striatum and nucleus accumbens. Cocaine-induced phosphorylation of MSK1 threonine 581 and cAMP response element-binding protein (CREB) serine 133 (Ser133) were blocked by SL327, a drug that prevents ERK activation. Cocaine increased the acetylation of histone H4 lysine 5 and phosphorylation of histone H3 Ser10, demonstrating the existence of drug-induced chromatin remodeling in vivo. In MSK1 knock-out (KO) mice CREB and H3 phosphorylation in response to cocaine (10 mg/kg) were blocked, and induction of c-Fos and dynorphin was prevented, whereas the induction of Egr-1 (early growth response-1)/zif268/Krox24 was unaltered. MSK1-KO mice had no obvious neurological defect but displayed a contrasted behavioral phenotype in response to cocaine. Acute effects of cocaine and dopamine D1 or D2 agonists were unaltered. Sensitivity to low doses, but not high doses, of cocaine was increased in the conditioned place preference paradigm, whereas locomotor sensitization to repeated injections of cocaine was decreased markedly. Our results show that MSK1 is a major striatal kinase, downstream from ERK, responsible for the phosphorylation of CREB and H3 and is required specifically for the induction of c-Fos and dynorphin as well as for locomotor sensitization.
Keywords: drug addiction, extracellular signal-regulated kinase, CREB phosphorylation, histone modification, immediate-early genes, long-term behavioral responses
Introduction
The complex behavioral alterations leading to drug addiction are thought to result from molecular and cellular adaptations in specific brain neurons and, consequently, in neuronal circuits in which these neurons operate. A central feature of most drugs of abuse is to cause the induction of immediate-early genes (IEGs) in the striatum, a major reward center in the brain (Berke and Hyman, 2000; Nestler, 2001). However, the signaling pathways responsible for this induction remain poorly characterized.
Extracellular signal-regulated kinase (ERK) plays multiple roles in the activity-dependent regulation of neuronal function, including long-term synaptic plasticity (Thomas and Huganir, 2004), and ERK activation is crucial in various models of learning and memory (Atkins et al., 1998; Sweatt, 2004). The two isoforms of ERK (1 and 2) may have distinct properties in this context (Mazzucchelli et al., 2002), although these differences are yet to be characterized fully. We showed previously that the pharmacological blockade of the ERK pathway prevents some of the transcriptional and rewarding effects of cocaine and Δ9-tetrahydrocannabinol administration (Valjent et al., 2000, 2001b). Thus, the ERK signaling pathway seems to play a central role in the neuronal adaptations that underlie behavioral alterations leading to drug addiction, as it does in learning and memory formation in other brain structures.
Little is known about the substrates of ERK that mediate its function in neuronal plasticity and drug addiction. In most eukaryotic cells, ERK controls gene expression by phosphorylating transcription factors and components of chromatin, both directly and via the action of downstream kinases (Yang et al., 2003). The transcription factor cAMP response element-binding protein (CREB), which plays an important role in the expression of genes involved in synaptic plasticity and drug addiction (Carlezon et al., 1998) (for review, see Nestler, 2004), is one of the nuclear targets of ERK in the striatum in response to the stimulation of glutamate and dopamine D1 and D2 receptors (Vanhoutte et al., 1999; Yan et al., 1999; Zanassi et al., 2001). The phosphorylation of CREB serine 133 (Ser133) is not catalyzed directly by ERK but by protein kinases activated by ERK, including two isoforms of mitogen-activated protein kinase-activated protein kinase 1 (MAPK-APK1; also called ribosome S6 kinases, RSKs) (Xing et al., 1996; De Cesare et al., 1998) and mitogen- and stress-activated protein kinases (MSKs) (Deak et al., 1998; Arthur and Cohen, 2000). Activation of MSKs is also implicated in histone H3 phosphorylation (Soloaga et al., 2003) and thus in chromatin reorganization (Nowak and Corces, 2004).
Because both chromatin remodeling and gene regulation may be critical for long-term neuronal alterations leading to drug addiction, we analyzed the regulation of MSKs and RSKs in response to cocaine and found a strong activation of MSK1. The use of MSK1 knock-out (MSK1-KO) mice demonstrated the critical role of this kinase for CREB and histone H3 phosphorylation and the induction of c-Fos and dynorphin, but not early growth response-1 (Egr-1), in response to cocaine. The study of acute and long-term behavioral responses to cocaine in these mice revealed a selective impairment of locomotor sensitization, showing that MSK1 governs specific cocaine-induced transcriptional events and behavioral alterations.
Materials and Methods
Animals and drugs. Mice C57BL/6 were purchased from Janvier (Le Genest-St.-Isle, France). MSK1-KO mice (Wiggin et al., 2002) were back-crossed with C57BL/6 for nine generations. MSK1-KO mice have normal size and weight, with no apparent health problems, and show normal fasting glucose levels and T-cell development (J. Darragh and A. Simon, unpublished data). Cocaine hydrochloride (Sigma-Aldrich, St. Quentin Fallavier, France) dissolved in a 0.9% NaCl (w/v) aqueous solution (saline) was administrated by intraperitoneal injection. The mitogen-activated and extracellular-regulated protein kinase (MEK) inhibitor α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)-benzene acetonitrile (SL327) was dissolved in DMSO, diluted twice in sterile water, and injected (50 mg/kg) 1 h before cocaine administration.
Tissue preparation and immunohistochemistry. At the indicated times after drug treatments, the mice were anesthetized rapidly with pentobarbital (30 mg/kg, i.p.; Sanofi-Aventis, Paris, France) and perfused transcardially with a fixative solution containing 4% paraformaldehyde (w/v) in 0.1 m Na2HPO4/NaH2PO4 buffer, pH 7.5. Brains were postfixed overnight in the same solution and stored at 4°C. Sections (30 μm thick) were cut with a vibratome (Leica, Nussloch, Germany) and kept at -20°C in a solution containing 30% ethylene glycol (v/v), 30% glycerol (v/v), and 0.1 m phosphate buffer. For the detection of phosphorylated proteins, 50 mm NaF was included in all buffers and incubation solutions, as described previously (Sgambato et al., 1998). Floating sections were saturated for 1 h with 3% bovine serum albumin (BSA; w/v) in Tris-buffered saline (25 mm Tris-Cl, 150 mm NaCl, pH 7.4) for phospho-MSK1 (P-MSK1) and dynorphin; 3% BSA, 0.2% Triton X-100 (v/v) in PBS [containing (in mm) 4 Na2HPO4, 1.7 KH2PO4, 150 NaCl, pH 7.4] for P-CREB; 10% normal goat serum (NGS; v/v) in TBS for acetylated lysine 14 H3 (AcLys14 H3) antibody; and without saturation for phosphorylated histone H3 (P-H3), phosphorylated and acetylated histone H3 (P-AcH3), and Egr-1. Then, the sections were rinsed three times in TBS and incubated with one of the following polyclonal antibodies: phosphothreonine 581-MSK1 (P-Thr581-MSK1; 1:750; Cell Signaling Technology, Beverly, MA), dynorphin-1-13 (1:200; Bachem, Essex, UK), Egr-1 (1:500; Santa Cruz Biotechnology, Tebu, France), P-Ser133-CREB (1: 750; Upstate Biotechnology, Lake Placid, NY), P-Ser10 H3 (1:1000; Upstate Biotechnology), or P-Ser10 or AcLys14 H3 (1:500; Upstate Biotechnology). Incubations were performed overnight at 4°C in TBS with 3% BSA for P-MSK1 and dynorphin, in PBS with 1% BSA and 0.1% Triton X-100 for P-CREB, or in TBS with 3% NGS for Egr-1. As for secondary antibodies, we used anti-rabbit Cy3-conjugated antibody (Amersham Biosciences, Les Ulis, France) (1:3000 for AcH3; 1:1000 for P-MSK1, dynorphin, and P-CREB; 1:500 for P-H3, P-AcH3, and AcH4). For double immunofluorescence, a polyclonal antibody anti-P-MSK1 was incubated simultaneously with a monoclonal antibody anti-P-ERK (P-Thr202/P-Tyr204 p44/42 MAPK) (1:200; Sigma, Lyon, France). Sections were then incubated sequentially with a Cy3-conjugated anti-rabbit antibody (1:750; Amersham Biosciences) and a FITC-conjugated antimouse antibody (1:500; Sigma) for 2 h each. For Egr-1 immunostaining, a biotinylated goat anti-rabbit antibody was used (1:250; Vector Laboratories, Paris, France), and floating sections were incubated with avidin-biotin-peroxidase complex, revealed with 3,3′-diaminobenzidine/H2O2. For immunofluorescence, the tissue sections were mounted in Vectashield (Vector Laboratories). Quantification of immunoreactive cells was performed with image analyzer software (Image-Pro Plus; Media Cybernetics, Silver Spring, MD), taking into account the cells with nuclear immunofluorescence above basal background (see Fig. 1a). In each case two brain sections per animal were counted bilaterally, with at least three animals per group.
Figure 1.
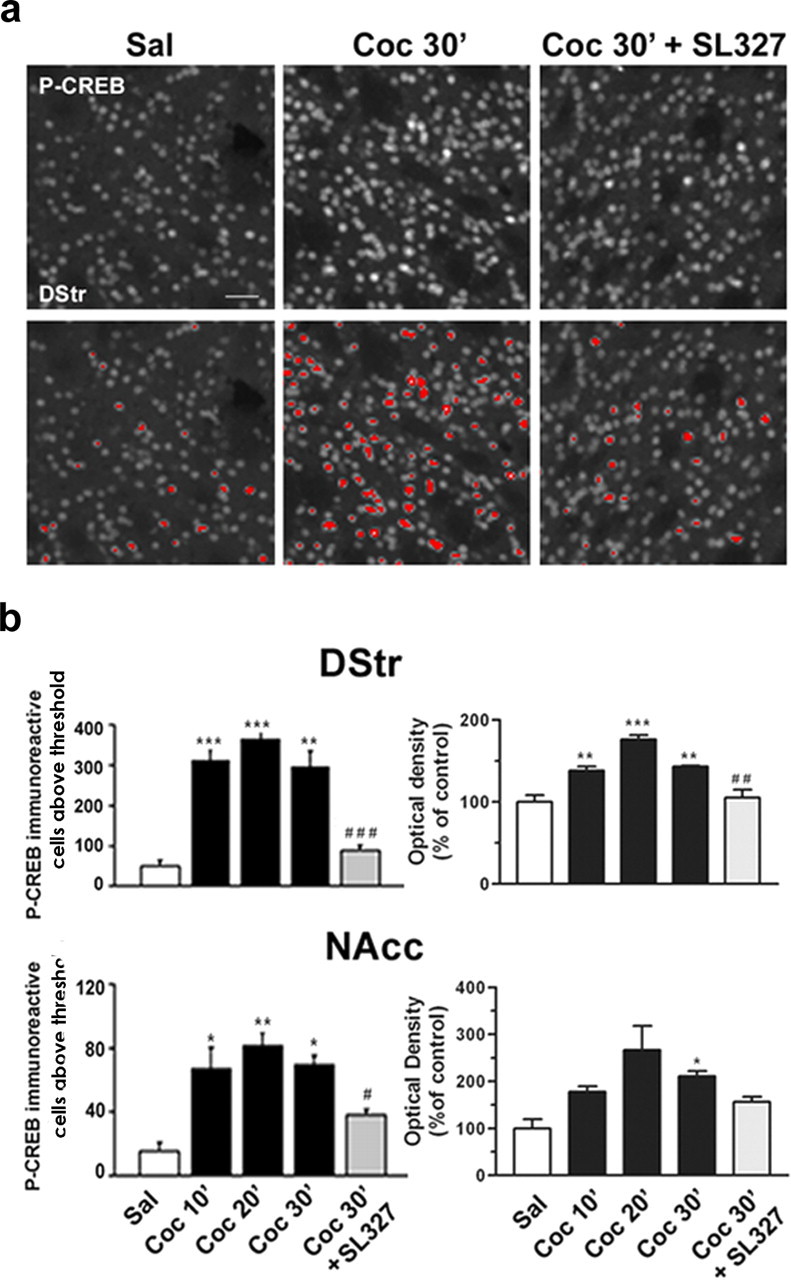
Cocaine-induced CREB phosphorylation is ERK dependent in the striatum. Mice were injected intraperitoneally with saline or cocaine (10 mg/kg) in the absence or presence of SL327 (50 mg/kg) before the cocaine. a, Top panels present immunocytochemical detection of phospho-Ser133-CREB in striatal neurons from mice injected with saline, cocaine, and SL327 before the cocaine. The bottom panels are a computerized representation of P-CREB-positive cells after the application of a threshold determined from saline-treated striatal sections. Cells with fluorescence above this threshold were counted. Scale bar, 40 μm. b, P-CREB immunoreactivity above a threshold (left panels) or global P-CREB immunofluorescence from the striatal sections (right panels) were analyzed in the dorsal striatum (DStr) and in the nucleus accumbens (NAcc) at 10, 20, or 30 min after cocaine injection (Coc 10′, Coc 20′, Coc 30′) in the absence or in the presence of SL327 (50 mg/kg; Coc 30′ plus SL327). Statistical comparisons in the left panels (means ± SEM; 4 mice per group) include one-way ANOVA (in DStr, F(4, 14) = 30.04 and p < 0.001; in NAcc, F(4, 14) = 11.90 and p < 0.001), followed by post hoc comparison (Scheffé's test; *p < 0.05, **p < 0.01, and ***p < 0.001 Sal vs Coc; #p < 0.05 and ###p < 0.001 Coc 30′ vs Coc 30′ plus SL327). Statistical comparisons in the right panels (means ± SEM; 3-4 mice per group) include one-way ANOVA (in DStr, F(4, 14) = 22.36 and p < 0.001; in NAcc, F(4, 14) = 5.82 and p < 0.05), followed by post hoc comparison (Scheffé's test; *p < 0.05, **p < 0.01, and ***p < 0.001 Sal vs Coc; and ##p < 0.001 Coc 30′ vs Coc 30′ plus SL327).
Kinase assays. Frozen brain samples were lysed in the following (in mm): 50 Tris-HCl, pH 7.5, 1 EGTA, 1 EDTA, 1 sodium orthovanadate, 50 sodium fluoride, 1 sodium pyrophosphate, plus 0.27 m sucrose, 1% Triton X-100 (v/v), 0.1% 2-mercaptoethanol (v/v), and complete protease inhibitor mixture (Roche, Mannheim, Germany). The lysates were centrifuged at 13,000 rpm for 5 min at 4°C, and the pellet was discarded. Lysates were precleared with 10 μl of protein G-Sepharose for 60 min, and then MSK1 was immunoprecipitated from 0.5 mg of soluble protein with the use of an anti-peptide MSK1 antibody coupled to protein G-Sepharose. After the immunoprecipitation of MSK1, MSK2 and then RSK2 were immunoprecipitated with anti-peptide antibodies against these kinases. Immunoprecipitation and kinase assays were performed as described previously (Williams et al., 2000). The peptide GRPRTSS-FAEG was used as a substrate for all three kinases. One unit of activity was that amount of enzyme that incorporates 1 nmol of phosphate into the peptide substrate in 1 min. Antipeptide antibodies that recognize MSK1 (residues 384-402) and MSK2 (residues 753-772) have been described previously (Williams et al., 2000).
Immunoblot analysis. Mice were injected with saline or cocaine and decapitated; immediately, their brains were dissected out from the skull and frozen on dry ice. Microdisks of tissue were punched out from frozen slices (500 μm thick) within the striatum with a stainless steel cylinder (1.4 mm in diameter). Samples were homogenized in 1% SDS, equalized for their content in protein, and analyzed by Western blot. The antibody dilutions were 1:1000, 1:2000 for antibodies against P-Thr581-MSK1 and MSK1, respectively. Antibodies were revealed by the peroxidase/chemiluminescence method (ECL; Amersham Biosciences) and autoradiography. Antibodies for measuring protein levels in the striatum of wild-type and MSK1-KO and procedures for 7-chloro-8-hydroxy-3-methyl-5-phenyl-2,3,4,5,-tetrahydro-1H-3-benzazepine ([3H]-SCH23390) binding were as described previously (Corvol et al., 2001; Valjent et al., 2005).
Locomotor activity and sensitization. Locomotor activity was measured by using a circular corridor with four infrared beams placed at every 90° (Imetronic, Pessac, France) in a low-luminosity environment. Locomotor activity was counted as travels through one-fourth of the circular corridor as detected by consecutive interruption of two adjacent beams. Mice were placed in the activity box for 30 min during 3 d consecutively (days 1-3) for habituation before the actual experiments were performed. The acute locomotor effects of cocaine (0, 10, 20, and 30 mg/kg, i.p.) were evaluated as a recording of locomotor activity during 15 min before and 30 min after the injection of saline or cocaine. Locomotor activity was measured in 5 min intervals. In cocaine sensitization experiments, the mice were treated repeatedly with saline or cocaine (10 or 20 mg/kg, i.p., every day) for 5 d consecutively. This repeated exposure was followed by 9 d of withdrawal and, on day 14, by a challenge injection of cocaine (10 or 20 mg/kg). Locomotion was measured as described above on days 1-5 and 14.
Conditioned place preference. The conditioned place preference (CPP) was evaluated in a Plexiglas Y-shaped apparatus (Imetronic) located in a soundproof testing room with low luminosity. Two of the three compartments were used for the conditioning and distinguished by different patterns on floors and walls, separated by a central neutral area. The apparatus was connected to an electronic interface for data collection. Entries and time spent in each compartment were measured, as well as the locomotor activity of the mice during the experiments. The CPP protocol included three different phases. (1) In preconditioning (habituation), the mice were placed in the middle of the neutral central area and allowed to explore both compartments. The time spent in each compartment was measured during 18 min. (2) In conditioning, the treatments were counter-balanced between compartments to use an unbiased procedure. Mice were treated for 6 d consecutively with alternate injections of cocaine or saline. After injection, the mice were confined to a given chamber for a period of 25 min. Cocaine was administrated on days 1, 3, and 5 and saline on days 2, 4, and 6. Control mice received saline every day. (3) The postconditioning (test) phase was conducted exactly as the preconditioning phase with free access to both compartments. The time spent in each compartment was measured during 18 min. Data were expressed as the time spent in the drug-paired compartment minus time spent in the saline-paired compartment.
Statistical analysis. Statistical analysis for immunohistochemistry was performed with one-way ANOVA, followed by Scheffé's test. Locomotor sensitization and CPP were analyzed with repeated measures ANOVA, using Bonferroni post hoc test.
Results
Cocaine-induced CREB phosphorylation in the striatum is ERK-dependent
Acute cocaine administration induces ERK activation in the striatum of mice (Valjent et al., 2000). This activation is transient and culminates within the nucleus, where ERK is thought to control the phosphorylation of transcription factors, including CREB. The precise contribution of the ERK pathway to the activation of CREB in response to cocaine has not yet been determined. We analyzed cocaine-induced CREB phosphorylation in striatal sections by immunofluorescence with the use of a phosphorylation state-specific antibody (Fig. 1a) previously demonstrated to recognize phospho-Ser133-CREB specifically by Western blot analysis of striatal tissues (Vanhoutte et al., 1999). Although a basal level of P-CREB immunoreactivity was observed in striatal neurons from saline-treated mice, this immunoreactivity increased after cocaine treatment (Fig. 1a, top panel). The increase in P-CREB immunoreactivity was clearly apparent after application of a threshold determined from saline-treated striatal sections (Fig. 1b, bottom panel). Thus, this threshold was applied for quantifying P-CREB immunoreactivity after cocaine administration (Fig. 1b, left panels). This showed a rapid and sustained increase in CREB phosphorylation by cocaine, which was reduced dramatically when SL327 (50 mg/kg, i.p.), a selective inhibitor of MEK1/2 (Atkins et al., 1998), the kinase upstream from ERKs, was injected before cocaine. Similar results were found when global immunofluorescence from striatal sections was measured (Fig. 1b, right panels). This indicates that the ERK pathway plays a major role in the control of CREB phosphorylation in response to cocaine administration.
Cocaine induces ERK-dependent activation of MSK1 in the striatum
ERK cannot phosphorylate CREB directly at Ser133 because it is not a proline-directed phosphorylation site. Both RSK2 (Xing et al., 1996; De Cesare et al., 1998) and MSKs (Simon et al., 2004) have been proposed as possible intermediates between ERK and CREB. We therefore investigated the activation of MSK1, MSK2, and RSK2 in striatal extracts from saline- and cocaine-treated mice, using an in vitro kinase assay. The cocaine-induced activation was more pronounced for MSK1 (almost threefold) than for MSK2 or RSK (less than twofold) (Fig. 2a). Thr581 is a residue located in the C-terminal kinase domain of MSK1 and critical for its activation (McCoy et al., 2005). Phosphorylation of this residue was increased 20 min after the injection of cocaine, as shown by immunoblotting with an affinity-purified antiserum that specifically recognizes the phosphorylated form of Thr581-MSK1 (Fig. 2b). Phosphorylation of Thr581 in response to cocaine also was demonstrated by immunofluorescence with this antibody (Fig. 2c). Although Thr581 can be phosphorylated by both ERKs and p38-MAP kinases (Deak et al., 1998), SL327 treatment totally abolished P-MSK1 immunoreactivity (Fig. 2c), indicating that MSK1 is activated downstream from ERKs, but not p38-MAP kinases, in response to cocaine. These data are in agreement with previous work showing that p38-MAP kinases are not activated by cocaine in vivo (Zhang et al., 2004). The time course of cocaine-induced MSK1 activation was monitored by immunofluorescence and quantified with an image analyzer. A rapid but transient increase in P-MSK1-immunoreactive cells was observed in the dorsal striatum and in the nucleus accumbens, which peaked 20 min after cocaine injection, and returned close to basal levels at 30 min (Fig. 2d). Quantification also confirmed the total inhibition of P-MSK1 immunoreactivity by SL327 pretreatment. Thus MSK1 is a kinase activated by ERK in response to cocaine.
Figure 2.
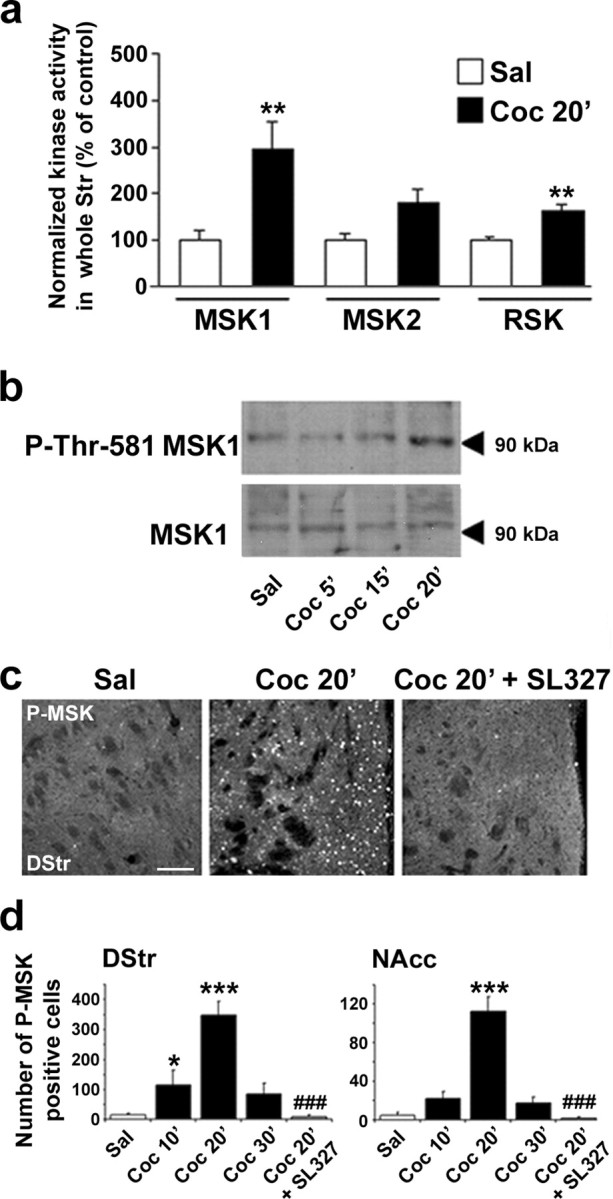
Cocaine-induced MSK1 activation is ERK-dependent in the striatum. a, Kinase activity of MSK1, MSK2, and RSK was measured in striatal extracts from mice treated with saline (Sal) or 10 mg/kg cocaine (Coc 20′) 20 min before death. Data (means ± SEM; 6 mice per group) were analyzed by Student's t test (for MSK1, t10 = 3.33 and **p < 0.01; for MSK2, t10 = 2.145, not significant; for RSK, t10 = 4.619 and **p < 0.01). b, Immunoblot analysis of striatal extracts obtained 5, 15, or 20 min after cocaine injection (Coc 5′, Coc 15′, Coc 20′), using antibodies reacting with phospho-MSK1 (P-Thr581-MSK1) or MSK1 independently of its phosphorylation. A 90 kDa immunoreactive band with the expected molecular mass of MSK1 was observed with both antibodies. Similar results were obtained in three independent experiments. c, Phospho-MSK1 (P-MSK) immunohistochemical detection from dorsal striatum (DStr) of saline-treated (Sal) or cocaine-treated mice in the absence (Coc 20′) or presence of SL327 (50 mg/kg; Coc 20′ plus SL327). Scale bar, 80 μm. d, P-MSK1-immunoreactive cells were counted as described for P-CREB in the legend for Figure 1. Data (means ± SEM; 3-4 mice per group) were analyzed with one-way ANOVA (in DStr, F(4,14) = 18.48 and p < 0.001; in NAcc, F(4,14) = 36.65 and p < 0.001), followed by post hoc comparison (Scheffé's test; *p < 0.05 and ***p < 0.001 Sal vs Coc; ###p < 0.001 Coc 30′ vs Coc 30′ plus SL327).
MSK1 is a major kinase of CREB in response to cocaine
To address specifically the role of MSK1 in cocaine-induced CREB phosphorylation, we used homozygous knock-out mice for the MSK1 gene (MSK1-KO) (Arthur and Cohen, 2000; Wiggin et al., 2002). Given the important role played by CREB in neuronal differentiation and survival (Lonze and Ginty, 2002), it was important to analyze whether the brain was developed normally in adult MSK1-KO mice. When we compared cresyl violet-stained sections of wild-type and MSK1-KO mice, the overall anatomy of brain appeared normal (Fig. 3a), and no evident alteration of specific brain regions was found at higher magnification, including in the accumbens nucleus, hippocampus, and cerebellum (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We also analyzed by Western blotting the expression levels of striatal proteins important for cell signaling after cocaine administration, including CREB, RSK1, Ets-like kinase-1 (Elk-1), ERK1 and 2, the glutamate receptor Glu-R1 subunit, dopamine- and cAMP-regulated phosphoprotein, Mr = 32,000 (DARPP-32), Gαolf, and tyrosine hydroxylase (TH), and we found no alteration in MSK1-KO mice (Fig. 3b,c). Similarly, D1 receptor-binding activity measured by autoradiography was normal in the dorsal striatum and nucleus accumbens of MSK1-KO mice (Fig. 3d).
Figure 3.
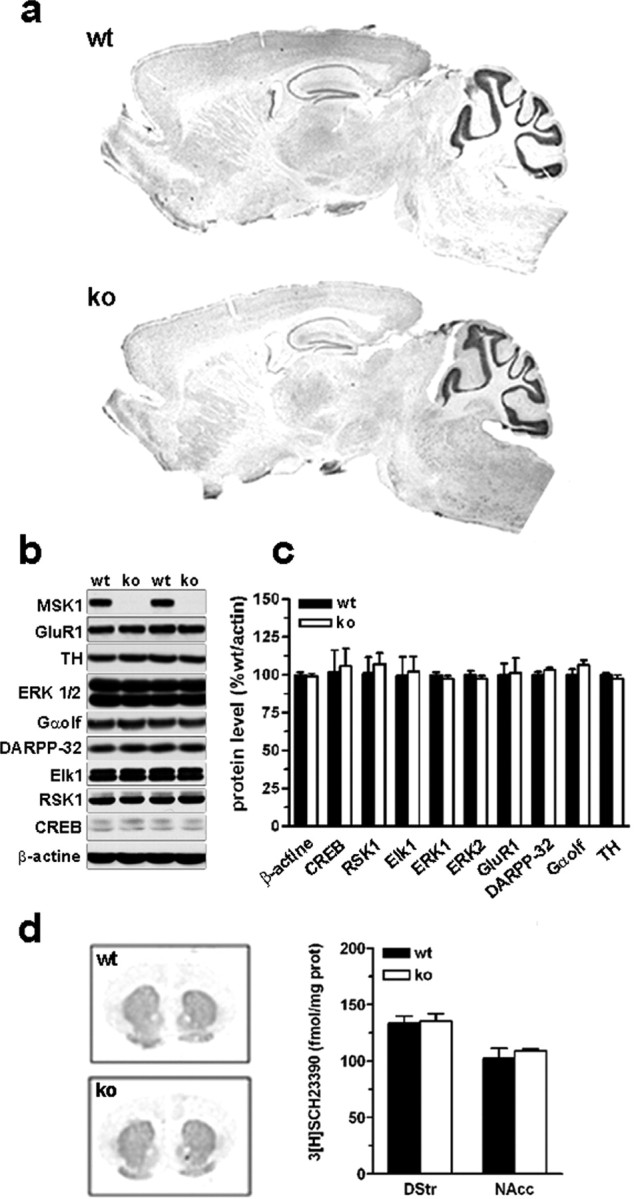
Brain morphology and protein expression in MSK1 knock-out mice. a, Brain morphology was analyzed by using cresyl violet staining in wild-type (wt) and MSK1-KO (ko) mice. b, Expression levels of some important proteins for cell signaling studied by immunoblotting in striatal extracts. MSK1 is expressed strongly in the striatum of wild-type mice. None of the analyzed proteins was modified in MSK1-KO mice. c, Quantification of protein expression levels in wt and ko mice (means ± SEM; n = 7 mice per group). d, D1 receptor binding was studied and quantified by using [3H]SCH23390 as a radioligand (means ± SEM; n = 3 mice per group).
In the absence of any obvious brain alteration in mutant mice, we analyzed cocaine-induced CREB phosphorylation in MSK1-KO mice compared with their wild-type littermates. At 30 min after cocaine administration CREB phosphorylation was decreased dramatically in MSK1-KO mice as compared with wild-type mice (Fig. 4a). Virtually no cocaine-induced P-CREB immunoreactivity was observed in the dorsal striatum (Fig. 4a,b) of mutant mice. Because the nucleus accumbens core and shell have been reported to control cocaine-induced behavioral alterations differentially, these two areas were distinguished, and in both areas the number of P-CREB-immunoreactive neurons remained at basal levels in MSK1-KO mice (Fig. 4a,b). Thus our data strongly indicate that MSK1 is a major CREB kinase in striatal neurons in response to cocaine.
Figure 4.
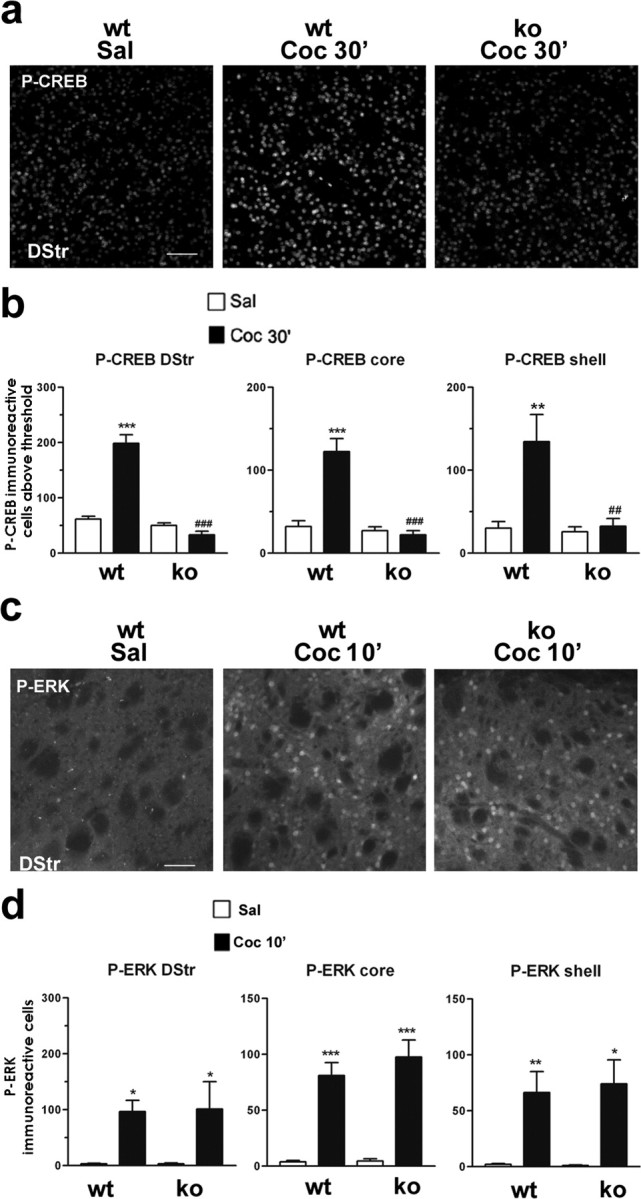
Cocaine-induced CREB phosphorylation is impaired in MSK1 knock-out mice. a, Illustration of P-CREB immunoreactivity 30 min after saline (Sal) or cocaine (10 mg/kg; Coc 30′) injection in the dorsal striatum of wild-type (wt) or MSK1-KO (ko) mice. Scale bar, 80 μm. b, P-CREB-immunoreactive cells below the threshold were counted in the dorsal striatum (DStr) and in the nucleus accumbens core and shell 30 min after saline or cocaine injection in wt and ko mice. Data (means ± SEM; 5 mice per group) were analyzed with one-way ANOVA (in DStr, F(5, 21) = 63.82 and p < 0.00 1; in core, F(5, 21) = 24.54 and p < 0.0001; in shell, F(5, 21) = 7.743 and p < 0.005), followed by post hoc comparison (Scheffé's test; **p < 0.01 and ***p < 0.001 Sal vs Coc in wt mice; ##p < 0.01 and ###p < 0.001 wt vs ko mice). c, Cocaine-induced phospho-ERK (P-ERK) is unaltered in ko mice. P-ERK immunoreactivity was detected 10 min after saline (Sal) or cocaine (10 mg/kg; Coc 10′) injection in wt or ko mice. Scale bar, 80 μm. d, P-ERK-immunoreactive cells were counted from wt and ko mice 10 min after cocaine administration in the dorsal (DStr) and ventral parts (core and shell) of the striatum. Data (means ± SEM; 3-4 mice per group) were analyzed with one-way ANOVA (in DStr, F(3,12) = 6.21 and p < 0.05; in core, F(3,12) = 37.38 and p < 0.0001; in shell, F(3,12) = 11.02 and p < 0.005), followed by post hoc comparison (Scheffé's test; *p < 0.05, **p < 0.01, and ***p < 0.001 Sal vs Coc in wt and ko mice).
To test whether the signaling pathway upstream from MSK1 was functional in mutant mice, we analyzed ERK activation in response to cocaine by using immunocytochemical detection of its active diphosphorylated form (P-ERK) (Vanhoutte et al., 1999). P-ERK was induced by cocaine to similar levels in the dorsal and ventral parts (core and shell) of the striatum in MSK1 wild-type (MSK1-WT) and MSK1-KO mice (Fig. 4c,d). Moreover, the nuclear translocation of activated ERK was similar in MSK1-WT and MSK1-KO mice (data not shown). Altogether, these results in MSK1-KO mice reveal the essential role of MSK1 downstream from ERK in the activation of CREB by cocaine.
Cocaine-induced histone modifications in wild-type and MSK1 knock-out mice
The remodeling of chromatin structure is a crucial step in controlling the transcription of eukaryotic genes (Nowak and Corces, 2004). Nucleosomal alterations result from a combination of modifications of histones at their N-terminal tails, modifications which are required to induce changes in the chromatin environment. Both acetylation of Lys14 and phosphorylation of Ser10 in histone H3 have been associated with activation of transcription in yeast, mammals, and Drosophila. More recently, these observations were extended to nonproliferating hippocampal neurons in which phosphorylation of histone H3 Ser10, but not Lys14 acetylation, was induced by stimulation of various neurotransmitter receptors in correlation with c-fos induction (Crosio et al., 2003).
Using an antibody that specifically recognizes the N-terminal tail of H3 phosphorylated on Ser10, we found a clear induction of H3 phosphorylation in the striatum 20 min after the injection of cocaine. Detailed investigation of the time course of H3 phosphorylation showed a persistent increase 30 min (Fig. 5) after cocaine injection and a return to basal levels at 60 min (data not shown). The cocaine-induced phosphorylation of H3 Ser10 was blocked by pretreatment of the mice with a selective D1 antagonist (SCH23390), an NMDA antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d]cyclohepten-5,10-imine maleate (MK801), or a MEK inhibitor (SL327) (Fig. 5). Interestingly, P-H3 immunoreactivity was restricted to the dorsal striatum and nucleus accumbens (Fig. 6a) (data not shown). The cocaine-induced phosphorylation of H3 was abolished totally in MSK1-KO mice (Fig. 6a). These results show that the control of H3 phosphorylation by cocaine is mediated by the ERK/MSK pathway and that it requires the coincident activation of dopamine D1 and glutamate NMDA receptors, as we have demonstrated previously for the activation of ERK itself (Valjent et al., 2000, 2005).
Figure 5.

Cocaine-induced histone H3 phosphorylation depends on D1 and NMDA receptor stimulation and ERK activation. Histone modification was analyzed 30 min after an injection of saline (Sal) or cocaine (10 mg/kg; Coc 30′) by immunofluorescence, using a phospho-Ser10 H3 (P-H3) antibody. These experiments were performed in the absence of (-) or after pretreatment with a selective D1 receptor antagonist (SCH23390; 0.1 mg/kg 15 min before cocaine), NMDA receptor antagonist (MK801; 0.15 mg/kg 30 min before cocaine), or an MEK inhibitor (SL327; 50 mg/kg 60 min before cocaine), as indicated. Scale bar, 20 μm.
Figure 6.

Cocaine-induced histone modifications in MSK1 knock-out mice. Histone modifications were analyzed 30 min after cocaine injection (10 mg/kg; Coc 30′) in wild-type (wt) and MSK1-KO (ko) mice. a, Immunofluorescence labeling of phospho-Ser10 H3 (P-H3), acetylated Lys14 H3 (AcH3), and phospho-Ser10/acetylated Lys14 H3 (P-AcH3). Note that histone H3 phosphorylation and phosphoacetylation were restricted to the striatum (Str; right panels). b, Data (means ± SEM; 3 mice per group) were analyzed in the dorsal striatum (DStr) with one-way ANOVA (for P-H3, F(2,6) = 571.84 and p < 0.001; for AcH3, F(2,6) = 1.79, not significant; for P-AcH3, F(2, 6) = 11194.95 and p < 0.001), followed by post hoc comparison (Scheffé's test; ***p < 0.001 Sal vs Coc; ###p < 0.001 wt vs ko). c, Immunohistochemical detection of acetylated Lys5 H4 (AcH4) in the DStr. d, Data (means ± SEM; 3 mice per group) were analyzed in the DStr with one-way ANOVA (for AcH4, F(2, 6) = 8.91 and p < 0.05), followed by post hoc comparison (Scheffé's test; *p < 0.05 Sal vs Coc).
Although H3 phosphorylation can occur independently of acetylation in neurons (Crosio et al., 2003), it is admitted generally that acetylation is a crucial event for gene transcription, and it has been proposed that the two modifications have a synergistic role (Nowak and Corces, 2004). It was thus important to analyze the levels of H3 acetylation in saline- and cocaine-treated mice. We examined AcH3 on Lys14 in the striatum and found high levels in saline-treated animals with no modification in response to cocaine (Fig. 6a,b). AcH3 immunoreactivity was similar in cocaine-treated MSK1-KO mice and in their wild-type littermates (Fig. 6a,b). In contrast, immunoreactivity for phosphoacetylated H3 antibodies, which label H3 modified on both Ser10 and Lys14, was induced strongly by cocaine, and this induction was abolished in MSK1-KO mice (Fig. 6a,b). Thus in striatal neurons H3 phosphorylation appears to be stimulated in response to cocaine independently of Lys14 acetylation.
To assess whether the lack of effect of cocaine on H3 acetylation in wild-type mice resulted from a general failure of this drug to alter histone acetylation, we also examined histone H4 acetylation. Acetylation of Lys5 is one of the major post-translational events that govern the role of H4 in chromatin remodeling during transcription in hippocampal neurons (Tsankova et al., 2004). We analyzed H4 Lys5 acetylation in response to cocaine and found it was increased significantly in the striatum (Fig. 6c,d). Acetylation of H4 was not altered in MSK1-KO mice. Altogether, our data strongly suggest that cocaine-induced activation of MSK1 is involved specifically in one of the events needed for the nucleosomal response, the phosphorylation of histone H3.
Cocaine-induced expression of c-Fos and dynorphin, but not Egr-1, is abolished in MSK1 knock-out mice
Transient induction of IEGs, including c-fos and Egr-1 (also termed zif268, Krox24, or NGFI-A), in the striatum after acute administration of cocaine is thought to play a key role in the neuronal alterations leading to addiction (Berke and Hyman, 2000). Because most of these IEGs encode transcription factors, their induction governs a vast program of gene expression (Berke et al., 1998). Furthermore, drug-induced c-Fos and Egr-1 expression is blocked by MEK inhibitors, demonstrating the requirement of the ERK pathway for their expression (Valjent et al., 2000; Derkinderen et al., 2003). c-Fos and Egr-1 expression was analyzed by immunocytochemistry 1 h after administration of cocaine (10 mg/kg) in MSK1-KO mice and wild-type littermates. Although a strong induction of c-Fos was found in wild-type mice, no c-Fos immunoreactivity was detected in the dorsal striatum and nucleus accumbens of MSK1-KO mice (Fig. 7a,b), including both the core and shell regions (data not shown). At a higher dose of cocaine (20 mg/kg) c-Fos induction was observed in MSK1-KO mice (data not shown), suggesting that other kinases downstream from ERK (for example MSK2 or RSK2) or other signaling pathways can bypass MSK1 to control gene transcription. In contrast, cocaine-induced c-Fos expression was not altered in MSK2-KO mice (data not shown), a result consistent with the lack of significant activation of MSK2 by cocaine (Fig. 2a). In contrast to the results for c-Fos, cocaine-induced Egr-1 expression was unchanged in the dorsal and ventral striatum of MSK1-KO mice (Fig. 7a,b), including core and shell (data not shown), when compared with their wild-type littermates. These results reveal a specific requirement of MSK1 for the transcription of some IEGs, but not others.
Figure 7.

Cocaine-induced expression of c-Fos and dynorphin, but not Egr-1, is impaired in MSK1 knock-out mice. a, Immunocytochemical detection of c-Fos and Egr-1 expression in the striatum of wild-type (wt) and MSK1-KO (ko) mice 60 min after the administration of saline (Sal) or cocaine (10 mg/kg; Coc 60′). Scale bars, 40 μm. b, Quantification of c-Fos- and Egr-1-immunoreactive cells in the dorsal striatum (DStr) and in the nucleus accumbens (NAcc). Data (means ± SEM; 3-6 mice per group) were analyzed with one-way ANOVA: for c-Fos (in DStr, F(3, 12) = 8.16 and p < 0.001; in NAcc, F(3, 12) = 31.20 and p < 0.001); for Egr-1 (in DStr, F(3, 12) = 40.53 and p < 0.001; in NAcc, F(3, 12) = 16.87 and p < 0.01). c, Immunocytochemical detection of dynorphin expression was performed as in a. Scale bar, 40 μm. d, Quantification of dynorphin-immunoreactive cells. Data (means ± SEM; 3-6 mice per group) were analyzed with one-way ANOVA (in DStr, F(2, 12) = 8.62 and p < 0.001; in NAcc, F(2, 12) = 9.87 and p < 0.001). Post hoc comparisons were made with Scheffé's test: *p < 0.05, **p < 0.01, and ***p < 0.001 Sal versus Coc; ##p < 0.01 and ###p < 0.001 wt versus ko.
Cocaine administration increases the striatal mRNA levels of preprodynorphin, which encodes dynorphin, a neuropeptide thought to play an important role in cocaine-induced behavioral alterations (Berke and Hyman, 2000). Importantly, the preprodynorphin gene has been identified as a target gene for CREB (Carlezon et al., 1998). We investigated the expression levels of dynorphin in the striatum of mice receiving cocaine or not. At 60 min after cocaine administration, the levels of dynorphin immunoreactivity increased in a significant proportion of striatal neurons, and this induction was blocked totally in MSK1-KO mice (Fig. 7c,d). Thus, MSK1 controls molecular events that are necessary for the transcription and translation of both c-Fos and dynorphin, but not Egr-1.
Selective alteration of delayed cocaine-induced behaviors in MSK1-deficient mice
Given that some molecular responses to cocaine, but not others, were altered in MSK1-KO mice, it was particularly interesting to examine which behavioral responses to cocaine were modified in these mice so that we might gain some insights on their mechanisms. MSK1-KO mice displayed a normal horizontal and vertical activity throughout the light/dark cycle (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and showed spontaneous locomotor activity and habituation to a new environment similar to those of wild-type littermates, as illustrated by the decreased locomotion on days 2 and 3 in both groups of animals (Fig. 8a). A single injection of psychostimulant drugs, such as cocaine, induces locomotor hyperactivity in rodents. As illustrated in Figure 8b, cocaine administration (10, 20, and 30 mg/kg, i.p.) induced an identical dose-dependent hyperlocomotion in wild-type and MSK1-KO mice. Thus acute behavioral responses to cocaine were not altered in the absence of MSK1. The functional activity of dopamine receptors in mutant mice was tested by examining their acute locomotor responses to D1 and D2 agonists. These responses were virtually identical to those observed in wild-type littermates (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), providing strong evidence, in addition to the biochemical results described above, that the acute responsiveness of the dopamine system is normal in MSK1 mutant mice.
Figure 8.
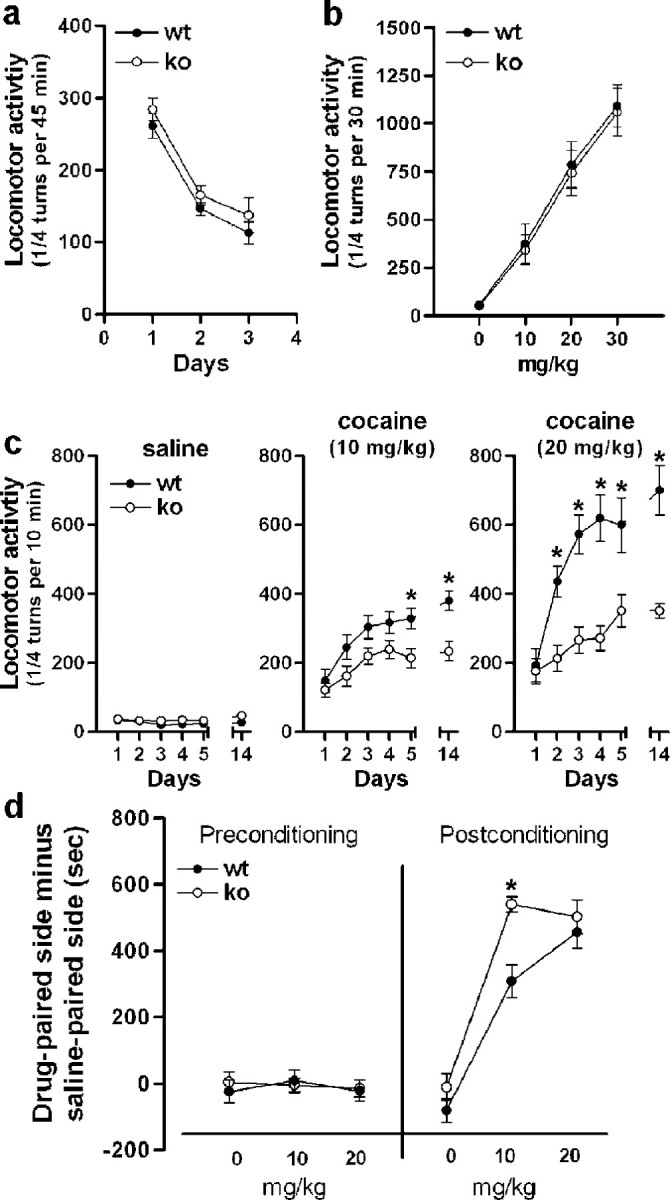
Locomotor and rewarding effects of cocaine in MSK1 knock-out mice. a, Spontaneous locomotor activity of wild-type (wt) and MSK1-KO (ko) mice for 3 d consecutively. b, Locomotor activity induced by a first intraperitoneal injection of cocaine at the indicated doses. c, Locomotor sensitization to repeated cocaine injections (10 and 20 mg/kg; n = 8-10 per group). After habituation, the wild-type and MSK1-KO mice were injected daily for 5 d consecutively with cocaine or saline, as indicated. They also were injected with saline or cocaine on day 14 after a 10 d withdrawal. Data were analyzed with a mixed factor ANOVA (repeated measure over time): for saline (effect of time, F(5, 54) = 2.561, not significant; effect of genotype, F(1, 54) = 1.012, not significant); for 10 mg/kg cocaine (effect of time, F(5, 54) = 12.87 and p < 0.01; effect of genotype, F(1, 54) = 9.748 and p < 0.01); for 20 mg/kg cocaine (effect of time, F(5,36) = 39.68 and p < 0.01; effect of genotype, F(1,36) = 27.31 and p < 0.01). In the posthoc comparison (Bonferroni test), *p < 0.01 wt versus ko. d, Conditioned place preference induced by cocaine (n = 7-8 mice per group). The left side of the graph (preconditioning) shows the lack of initial preference for either side by any experimental group (effect of treatment, F(3, 53) = 0.184, not significant; effect of genotype, F(1, 53) = 0.065, not significant) (note that the abscissa labels indicate the doses of cocaine used for each group in the later phase of the test). The right side of the graph shows that MSK1-KO mice displayed an enhanced preference for the side paired with 10 mg/kg cocaine as compared with wild-type mice. At 20 mg/kg of cocaine both wt and ko displayed an identical preference for the cocaine-paired side (effect of treatment, F(3, 53) = 83.75 and p < 0.01; effect of genotype, F(1, 53) = 10.05 and p < 0.01). In the post hoc comparison (Bonferroni test) *p < 0.01 wt versus ko.
Because the known functions of MSK1 are related to the control of gene transcription, we examined in MSK1-KO mice those long-term behavioral changes induced by repeated administration of cocaine that are likely to involve transcriptional regulation. Repeated exposure to cocaine triggers a progressive and persistent enhancement of locomotor responses, termed behavioral sensitization. As expected, after repeated cocaine administration (10 or 20 mg/kg) a robust locomotor sensitization was observed in wild-type mice (day 14). In contrast, although the acute hyperlocomotion induced by cocaine was unchanged in MSK1-KO mice, the locomotor sensitization was impaired strongly in these mice at doses of cocaine of either 10 or 20 mg/kg (Fig. 8c).
In a different set of animals, we analyzed the CPP to cocaine, which reflects a preference for a context repeatedly associated with drug injection and commonly is used to evaluate the rewarding effects of drugs. Preconditioning day data revealed no initial preference for either side (Fig. 8d). After being conditioned with doses of 10 or 20 mg/kg of cocaine, wild-type mice showed a robust CPP, as illustrated by the increased time spent in the drug-associated compartment (Fig. 8d). MSK1-KO mice did not show any decrease in CPP (Fig. 8d). In contrast, mutant mice conditioned at the lowest dose that was tested (10 mg/kg) spent significantly more time in the cocaine-associated side, as compared with wild-type littermates (Fig. 8d). This apparent increase in CPP was not observed at 20 mg/kg, suggesting that the behavioral response may have reached a maximum level in this experimental paradigm. The measurement of locomotor activity during the conditioning phases revealed no difference in the number of ambulations between wild-type and MSK1-KO mice (data not shown), indicating that the greater sensitivity of the mutant mice to the rewarding effects of cocaine is not attributable to an increased locomotor activity in the CPP paradigm. Altogether, our results show that in the absence of MSK1 the spontaneous behavior and acute responses to cocaine were unaltered, whereas the long-lasting behavioral modifications induced by repeated injections of cocaine were altered differentially. The markedly decreased locomotor sensitization of mutant mice in response to repeated injections of cocaine contrasted with the apparent enhanced sensitivity to the rewarding effects of this drug.
Discussion
Our results reveal that MSK1 is an important component of an intracellular signaling cascade that drives the regulation of specific genes in response to cocaine in striatal neurons (Fig. 9). MSK1 is activated downstream from ERK and is essential for phosphorylation of both CREB Ser133 and histone H3 Ser10. Importantly, in the absence of MSK1 the behavioral responses to repeated, but not acute, cocaine exposure were impaired. Moreover, this impairment was differential, with a decreased locomotor sensitization and increased sensitivity to cocaine CPP. These data provide new insights into the molecular mechanisms that specifically underlie the various cocaine-induced long-term behavioral alterations.
Figure 9.

Schematic representation of the role of MSK1 in neuronal adaptation in response to cocaine. Both dopamine D1 and glutamate NMDA receptors are involved in cocaine-induced ERK activation and immediate-early gene regulation (Valjent et al., 2000, 2005). D1 receptor stimulation increases intracellular levels of cAMP, which activates PKA. NMDA receptor stimulation causes increases in intracellular calcium (Ca2+) levels. Both events converge on the MEK/ERK signaling cascade (P-MEK and P-ERK). Phosphorylated ERKs (P-ERKs) translocate to the nucleus, where they phosphorylate MSK1 (P-MSK1), which controls gene expression via its dual (P-MEK and P-ERK) role on histone H3 and CREB phosphorylation. (1) Histone H3 phosphorylation (P-H3) by MSK1 leads to chromatin remodeling, facilitating DNA accessibility. (2) Phosphorylation of CREB (P-CREB) by MSK1 induces transcription of the immediate-early genes c-fos or preprodynorphin (Dyn) by RNA polymerase II (Pol II).
A number of results support a role of ERK in learning and memory as well as in synaptic plasticity (Thomas and Huganir, 2004). We and others showed a rapid and transient activation of ERK in the striatum in response to cocaine and other drugs of abuse (Valjent et al., 2000, 2001b, 2004; Zhang et al., 2004). With activation by cocaine, phosphorylated ERK is observed first within the cytoplasm, where it possibly controls a number of substrates, including ion channels, cytoskeletal proteins, and proteins involved in neurotransmitter release such as synapsins (for review, see Valjent et al., 2001a). The phosphorylation of these substrates could contribute to the neuronal responses underlying hyperlocomotion induced by acute cocaine administration and, thus, be involved in “immediate” functions of ERK (Valjent et al., 2000, 2003). Activation of ERK in response to drugs of abuse occurs in a subpopulation of D1 receptors containing medium-size striatal neurons and requires the stimulation of both D1 and NMDA receptors (Valjent et al., 2000, 2005). Thus, ERK activation in these neurons behaves as a functional coincidence detector of dopamine and glutamate signals, and DARPP-32, a protein phosphatase-1 inhibitor, is necessary for the cross talk between the two types of receptors (Valjent et al., 2005). Once activated, ERK is translocated rapidly to the nucleus. This localization supports its role in gene transcription. Accordingly, in most eukaryotic cells ERK plays a dual role in gene expression programs by phosphorylating transcription factors and promoting chromatin modification (Yang et al., 2003).
CREB has long been proposed as an important transcription factor implicated in adaptations to drugs of abuse (for review, see Chao and Nestler, 2004; Nestler, 2004). Administration of drugs of abuse, including cocaine, stimulates the cAMP pathway and causes CREB activation in the striatum. Although CREB is a well known protein kinase A (PKA) substrate, pharmacological evidence indicates that the ERK pathway is essential for its phosphorylation in striatal neurons both in vitro in response to dopamine D1 or D2 dopamine agonists or glutamate (Vanhoutte et al., 1999; Yan et al., 1999; Zanassi et al., 2001) and in vivo after electrical stimulation of the corticostriatal pathway (Sgambato et al., 1998). Here we show that the ERK pathway is necessary for cocaine-induced phosphorylation of CREB in the striatum. Because CREB Ser133 phosphorylation was prevented in MSK1-KO mice, MSK1 appears to be the major intermediate kinase phosphorylating CREB after activation of the ERK pathway. This is an important result, because another kinase, RSK2, was found to be the main ERK-activated kinase responsible for CREB phosphorylation in some cell types (Xing et al., 1996; De Cesare et al., 1998). Our data obtained in living mice are in agreement with recent evidence implicating MSK1 as the major CREB kinase downstream from ERK in fibroblasts (Wiggin et al., 2002) and in neurotrophin-treated cortical neurons in culture (Simon et al., 2004).
In MSK1-KO mice, cocaine-induced expression of c-Fos and dynorphin, but not of Egr-1, was prevented. These restricted effects contrast with the total inhibition of both c-Fos and Egr-1 produced by pharmacological blockade of ERK activation (Valjent et al., 2000; Derkinderen et al., 2003). Although the preprodynorphin gene is known to be a target for CREB, we provide here the first evidence that the ERK pathway controls its expression after acute cocaine administration. Thus, ERK exerts pleio-tropic effects on gene expression by controlling the phosphorylation state of CREB via MSK1, as evidenced here, but also by other mechanisms. For instance, ERK phosphorylates the ternary complex factor Elk-1, which binds the serum response element present in the promoter of specific genes, including c-fos and Egr-1 (Treisman, 1995; Vanhoutte and Caboche, 2002). Activation of such factors by ERK appears to be sufficient for the transcriptional regulation of a set of genes independently of MSK1, for example Egr-1, and may account for the rescued transcription of c-fos in response to a high dose of cocaine.
Modulation of chromatin structure has been implicated in mammalian associative learning and long-term memory formation (Alarcon et al., 2004; Levenson et al., 2004). In the nucleus, DNA is packed tightly into chromatin, a DNA protein complex, comprising a group of highly basic proteins, the histones. Unmodified histones are linked tightly to DNA, preventing interactions of other proteins, including RNA polymerase II, which is required for transcription (Fischle et al., 2003). Thus for transcription to occur, the native structure of chromatin must be disrupted by mechanisms that include histone acetylation and phosphorylation and the action of ATP-dependent remodeling complexes. The phosphorylation of histone H3 Ser10 has been correlated with transcriptional activation of several IEGs, including c-fos (Mahadevan et al., 1991; Clayton et al., 2000; Clayton and Mahadevan, 2003). In response to mitogenic stimuli, H3 Ser10 phosphorylation involves the ERK cascade, and both RSKs and MSKs have been proposed to be histone H3 kinases in vitro (De Cesare et al., 1998; Sassone-Corsi et al., 1999; Thomson et al., 1999). In MSK1/MSK2-deficient fibroblasts H3 phosphorylation, but not acetylation, was reduced severely in response to mitogens (Soloaga et al., 2003). In the present work we show that MSK1 is a critical kinase for H3 in striatal neurons in response to cocaine in vivo, an event that is totally dependent, like ERK activation (Valjent et al., 2000), on D1 and NMDA receptor stimulation. No modification of global levels of H3 acetylation was found in response to cocaine in either wild-type or MSK1-KO mice, supporting an independence between H3 phosphorylation and acetylation in vivo.
The results reported here show that some molecular effects of cocaine are blocked in MSK1-KO, but not others. As expected, the alterations were more restricted than those observed in mice treated with SL327, a MEK inhibitor that prevents the activation of ERK, because MSK1 is only one of ERK multiple targets. The selective blockade of some of the signaling pathways activated by cocaine in MSK1-KO mice provides an excellent model to examine the mechanisms of the behavioral alterations that depend on the ERK pathway. Although ERK inhibition by SL327 partially decreased cocaine-induced hyperlocomotion (Valjent et al., 2000), we did not observe such an alteration in MSK1-KO mice. This indicates that the MSK1 role may be restricted to delayed effects, as might be expected for a protein kinase regulating transcription. ERK is critical for long-lasting effects of cocaine because its inhibition totally blocks both locomotor sensitization (Valjent et al., 2005) and CPP (Valjent et al., 2000). Interestingly, MSK1-KO mice displayed a decreased locomotor sensitization that contrasted with an apparent increased sensitivity to cocaine in the place preference paradigm, indicated by a leftward shift of the CPP dose-response curve. Cocaine-induced locomotor sensitization is thought to arise from neuronal plasticity at several levels, including in dopamine neurons and in the striatum (Pierce and Kalivas, 1997). Our results show that MSK1 is an essential player in the establishment of locomotor sensitization. Additional experiments are now necessary to identify the genes specifically regulated by MSK1 that are necessary for this long-term alteration of behavioral responses to cocaine.
CPP is also a long-term behavioral alteration thought to reflect neuronal plasticity, albeit presumably at different sites. We found that in MSK1-KO mice CPP was not diminished but was even increased at the lowest dose of cocaine used. The critical role of MSK1 in the phosphorylation of CREB provides a possible explanation for this observation, because CREB has been reported to play a negative role in CPP (Carlezon et al., 1998). This has been attributed to the role of CREB in the regulation of dynorphin (Carlezon et al., 1998), a peptide that decreases dopamine release by activating κ-opioid receptors on dopamine terminals (Spanagel et al., 1992). Because stimulation of κ-opioid receptors has aversive effects in both humans and rats (Shippenberg and Rea, 1997), the induction of dynorphin by CREB may be responsible for an aversive component in response to drugs. In support of this hypothesis we observed a lack of induction of dynorphin in MSK1-KO mice. More importantly, our result strongly indicate that phosphorylation of CREB and expression of c-Fos are not required for the induction of CPP.
In conclusion, this study shows that a single cocaine injection activates MSK1 in the striatum downstream from ERK. MSK1 plays a selective role in some molecular effects of cocaine, including phosphorylation of CREB and histone H3 and induction of c-Fos and dynorphin. Study of behavioral responses of MSK1-KO mice revealed a specific inhibition of cocaine-induced locomotor sensitization, whereas CPP for cocaine was enhanced slightly. Thus our results allow for the parsing of signaling pathways underlying specific behavioral responses to cocaine and may help to design strategies to interfere specifically with selective effects of drugs of abuse.
Footnotes
Work at Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7102, was supported in part by grants from Mission Interministérielle de Lutte contre la Drogue et la Toxicomanie (MILDT) and Ministère délégué à l'Enseignement supérieur et à la Recherche [Action Concertée Incitative (ACI): Développement et Biologie Intégrative]. Work at Institut National de la Santé et de la Recherche Médicale, Unité 536, was supported by grants from MILDT, ACI, Fondation Schlumberger pour l'Enseignement et la Recherche (FSER), and Bettencourt-Schueller Foundation. S.A. was funded by the British Medical Research Council, Astra-Zeneca, Boehringer-Ingelheim, GlaxoSmith-Kline, Merck and Company, Merck Germany, and Pfizer. K.B.-C. was supported by a fellowship from the Jérôme Lejeune Foundation. E.V. was supported by FSER and Foundation for Medical Research. We thank Dr. J. M. Trzaskos for the gift of SL327, P. Cohen and Dr. P. Vanhoutte for a critical reading of this manuscript, and Philippe Bernard for his help with mouse breeding.
Correspondence should be addressed to Jocelyne Caboche, Université Pierre et Marie Curie, Unité Mixte de Recherche 7102, 9 quai Saint Bernard, 75005 Paris, France. E-mail: jocelyne.caboche@snv.jussieu.fr.
K. Brami-Cherrier's present address: Université Pierre et Marie Curie, Institut National de la Santé et de la Recherche Médicale, Unité 536, 17 rue du Fer à Moulin, 75005 Paris, France.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511444-11$15.00/0
K.B.-C. and E.V. contributed equally to this work.
References
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A (2004) Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947-959. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Cohen P (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett 482: 44-48. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD (1998) The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1: 602-609. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515-532. [DOI] [PubMed] [Google Scholar]
- Berke JD, Paletzki RF, Aronson GJ, Hyman SE, Gerfen CR (1998) A complex program of striatal gene expression induced by dopaminergic stimulation. J Neurosci 18: 5301-5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon Jr WA, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ (1998) Regulation of cocaine reward by CREB. Science 282: 2272-2275. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ (2004) Molecular neurobiology of drug addiction. Annu Rev Med 55: 113-132. [DOI] [PubMed] [Google Scholar]
- Clayton AL, Mahadevan LC (2003) MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett 546: 51-58. [DOI] [PubMed] [Google Scholar]
- Clayton AL, Rose S, Barratt MJ, Mahadevan LC (2000) Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J 19: 3714-3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol JC, Studler JM, Schonn JS, Girault JA, Hervé D (2001) Gαolf is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem 76: 1585-1588. [DOI] [PubMed] [Google Scholar]
- Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P (2003) Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 116: 4905-4914. [DOI] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17: 4426-4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P (1998) Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci USA 95: 12202-12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA (2003) Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci 23: 2371-2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172-183. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004) Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279: 40545-40559. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Mahadevan LC, Willis AC, Barratt MJ (1991) Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 65: 775-783. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R (2002) Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34: 807-820. [DOI] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS (2005) MSK1 activity is controlled by multiple phosphorylation sites. Biochem J 387: 507-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2004) Molecular mechanisms of drug addiction. Neuropharmacology 47 [Suppl 1]: 24-32. [DOI] [PubMed] [Google Scholar]
- Nowak SJ, Corces VG (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 20: 214-220. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW (1997) A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Brain Res Rev 25: 192-216. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285: 886-891. [DOI] [PubMed] [Google Scholar]
- Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J (1998) Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci 18: 8814-8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Rea W (1997) Sensitization to the behavioral effects of cocaine: modulation by dynorphin and κ-opioid receptor agonists. Pharmacol Biochem Behav 57: 449-455. [DOI] [PubMed] [Google Scholar]
- Simon J, Arthur C, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S (2004) Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci 24: 4324-4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS (2003) MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 22: 2788-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS (1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA 89: 2046-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311-317. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signaling and synaptic plasticity. Nat Rev Neurosci 5: 173-183. [DOI] [PubMed] [Google Scholar]
- Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J 18: 4779-4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R (1995) Journey to the surface of the cell: Fos regulation and the SRE. EMBO J 14: 4905-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ (2004) Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci 24: 5603-5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J (2000) Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci 20: 8701-8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Caboche J, Vanhoutte P (2001a) Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Mol Neurobiol 23: 83-99. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J (2001b) Δ9-Tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur J Neurosci 14: 342-352. [DOI] [PubMed] [Google Scholar]
- Valjent E, Hervé D, Caboche J, Girault JA (2003) Possible role of the extracellular signal-regulated kinase (ERK) in reward-controlled learning and addiction. Curr Neuropharmacol 1: 165-174. [Google Scholar]
- Valjent E, Pages C, Hervé D, Girault JA, Caboche J (2004) Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci 19: 1826-1836. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault JA (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102: 491-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Caboche J (2002) Elk-1: an important regulator of immediate early gene expression in the brain. In: Handbook of chemical neuroanatomy, Vol 19 (Kaczmarek L, Robertson HA, eds), pp 287-307, Amsterdam: Elsevier. [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J (1999) Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol 19: 136-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS (2002) MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol 22: 2871-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10: 439-448. [DOI] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 18: 1946-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Feng J, Fienberg AA, Greengard P (1999) D2 dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci USA 96: 11607-11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD, Whitmarsh AJ (2003) Transcriptional regulation by the MAP kinase signaling cascades. Gene 320: 3-21. [DOI] [PubMed] [Google Scholar]
- Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S (2001) cAMP-dependent protein kinase induces cAMP response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem 276: 11487-11495. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lou D, Jiao H, Zhang D, Wang X, Xia Y, Zhang J, Xu M (2004) Cocaine-induced intracellular signaling and gene expression are oppositely regulated by the dopamine D1 and D3 receptors. J Neurosci 24: 3344-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]