

Abstract
A leucine to alanine substitution (L9′A) was introduced in the M2 region of the mouse α4 neuronal nicotinic acetylcholine receptor (nAChR) subunit. Expressed in Xenopus oocytes, α4(L9′A)β2 nAChRs were ≥30-fold more sensitive than wild type (WT) to both ACh and nicotine. We generated knock-in mice with the L9′A mutation and studied their cellular responses, seizure phenotype, and sleep-wake cycle. Seizure studies on α4-mutated animals are relevant to epilepsy research because all known mutations linked to autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) occur in the M2 region of α4or β2 subunits. Thalamic cultures and synaptosomes from L9′A mice were hypersensitive to nicotine-induced ion flux. L9′A mice were ∼15-fold more sensitive to seizures elicited by nicotine injection than their WT littermates. Seizures in L9′A mice differed qualitatively from those in WT: L9′A seizures started earlier, were prevented by nicotine pretreatment, lacked EEG spike-wave discharges, and consisted of fast repetitive movements. Nicotine-induced seizures in L9′A mice were partial, whereas WT seizures were generalized. When L9′A homozygous mice received a 10 mg/kg nicotine injection, there was temporal and phenomenological separation of mutant and WT-like seizures: an initial seizure ∼20 s after injection was clonic and showed no EEG changes. A second seizure began 3-4 min after injection, was tonic-clonic, and had EEG spike-wave activity. No spontaneous seizures were detected in L9′A mice during chronic video/EEG recordings, but their sleep-wake cycle was altered. Our findings show that hypersensitive α4* nicotinic receptors in mice mediate changes in the sleep-wake cycle and nicotine-induced seizures resembling ADNFLE.
Keywords: epilepsy, ADNFLE, nicotinic receptors, sleep disorders, α4β2, nicotine
Introduction
Stimulation of nicotinic acetylcholine receptors (nAChRs) by nicotine triggers a wide range of physiological and behavioral responses, including self-administration, hypothermia, sedation, decreased nociception, increased alertness and locomotion, and at higher doses, violent seizures and death. Questions regarding the specific role of nicotinic receptors are now being addressed: which nicotinic receptor subtypes are responsible for each of the different responses? Where in the brain are the relevant cholinergic pathways? Which responses depend on nicotinic receptor activation, which on desensitization, and which on both?
Nicotinic receptors also figure in several CNS diseases. Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) provides an excellent opportunity to study the link between changes in the structure-function relationships of a specific receptor subtype and the mechanisms underlying a disease. Some cases of ADNFLE are associated with point mutations in the M2 pore-forming region of both α4 and β2 subunits of the nAChR (for review, see Steinlein, 2004), which combine to form the predominant heteromeric nAChR subtype in the brain (Whiting and Lindstrom, 1988; Wada et al., 1989). Similar clinical symptoms (frequent, brief, and sometimes violent partial seizures, starting during childhood and occurring in sleep) are shared by all five ADNFLE alleles isolated from different kindreds (Phillips et al., 1995; Steinlein et al., 1995, 1997; Hirose et al., 1999; De Fusco et al., 2000).
The epileptogenic mechanisms in α4- or β2-associated ADNFLE remain uncertain. Thus far, experimental analysis of ADNFLE mutations has been performed using heterologous expression of nAChRs in Xenopus oocytes and clonal cell lines. In these reports, ADNFLE mutations in the α4 subunit increase ACh affinity (Steinlein et al., 1997), reduce Ca2+ permeability (Kuryatov et al., 1997), decrease Ca2+ potentiation (Figl et al., 1998; Rodrigues-Pinguet et al., 2003), or enhance desensitization (Bertrand et al., 1998; Matsushima et al., 2002). ADNFLE mutations in β2 lead to altered ACh sensitivity (Phillips et al., 2001) and/or prolonged ACh-induced currents (De Fusco et al., 2000). An attractive hypothesis arises from the findings that most or all of the known ADNFLE mutations, when expressed in oocytes, cause an increase in ACh sensitivity (Bertrand et al., 2002), possibly by decreasing Ca2+ dependence (Rodrigues-Pinguet et al., 2003).
We have generated mutated mouse lines that express hypersensitive α4* receptors (Labarca et al., 2001; Fonck et al., 2003; Tapper et al., 2004). These knock-in mice contain mutations at the Leu9′ position in the M2 region of the nAChR α4 subunit; as a result, α4* receptors respond to relatively low agonist concentrations. The Leu9′Ala (L9′A) strain in particular appears to isolate and amplify nicotine responses caused by α4* receptors. At the lowest nicotine doses that we have tested, the L9′A strain is a good model for some aspects of nicotine addiction (Tapper et al., 2004). At higher doses, but still well below the seizure threshold for WT mice, we find that nicotine causes seizures in L9′A mice. The present study examines activation, desensitization, and relative permeability properties of the α4 L9′A mutation when expressed in oocytes. We also studied α4β2 receptor expression and function in forebrain tissue of L9′A and wild-type (WT) mice. We then investigated the seizure phenotype of L9′A mice.
Materials and Methods
L9′A mutation expressed in Xenopus oocytes. Stage V-VI Xenopus oocytes were surgically isolated using a previously published protocol (Quick and Lester, 1994). All surgeries were performed using methods approved by the Institutional Animal Care Committee. Female Xenopus frogs were anesthetized by immersion in 0.2% tricaine methanesulphonate. WT mouse α4 and β2 nicotinic subunits were subcloned into the pCI-Neo vector. The Quikchange kit (Stratagene, La Jolla, CA) was used to construct α4L9′A and α4 Leu9′Ser (L9′S) mutations, which were verified by DNA sequencing. Capped cRNA was synthesized from linearized cDNA using the mMessage mMachine RNA transcription kit (Ambion, Austin, TX). Isolated oocytes were injected with 4 ng of mouse α4 and 6 ng of mouse β2 cRNA in 50 nl of H2O. Before recording, injected oocytes were incubated for 2 d in a solution containing the following (in mm): 96 NaCl, 5 HEPES, 2.5 Na pyruvate, 2 KCl, 1.8 CaCl2, 1 MgCl2, 2.5 μg/ml gentamycin, and 5% horse serum, pH 7.4 (at 18°C). Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO). We used an automated two-electrode voltage-clamp (OpusXpress; Axon Instruments, Union City, CA) to measure the agonist-induced currents in injected oocytes at -60 mV. During recordings, oocytes were continually superfused with a nominally Ca2+-free saline (ND98) containing (in mm) 98 NaCl, 5 HEPES, and 1 MgCl2, pH 7.4, to prevent activation of the endogenous Ca2+-activated Cl- current. Nicotine and ACh were bath applied at a rate of 0.25 ml/s, with a chamber volume of 2 ml. Oocytes with leak currents in ND98 larger than 300 nA were discarded. Agonist concentration-response data were analyzed as described previously (Cohen et al., 1995). EC50 values, IC50 values, and Hill coefficient errors calculated in all oocyte experiments were obtained by fitting these parameters to the mean of normalized concentration-dependence data.
Nicotine-induced desensitization. Similar to Katz and Thesleff (1957), we used periodic ACh test pulses to measure nicotine-induced steady-state desensitization of WT and L9′A mutant receptors expressed in Xenopus oocytes. Receptor desensitization is manifested by a decrease in the amplitude of ACh responses during sustained nicotine exposure. We applied a total of 13 test pulses, 2.5 s in duration at 120 s intervals, to each oocyte using a U-tube microperfusion system (Cohen et al., 1995). Nicotine was bath applied immediately after the third pulse. Five ACh test pulses were delivered during nicotine bath perfusion and five more after. The microperfusate contained 200 nm ACh plus nicotine (equal to the bath-applied nicotine concentration) during nicotine superfusion. In the absence of nicotine, the WT ACh test-pulse response remained constant during the 13 applications. In contrast, the peak L9′A ACh test-pulse response increased linearly by 1.3 ± 0.3% min-1 (n = 4 oocytes), presumably because of increased surface receptor expression. The WT data were recorded 4-6 d after cRNA injections, and L9′A recordings were obtained 1-2 d after cRNA injections. Thus, surface receptor expression apparently stabilized in WT but not in L9′A experiments. To correct for this increase, we divided the peak amplitude of the L9′A test-pulse responses by a factor of (1 + 0.013t), where t is time in minutes since the first test pulse. Steady-state receptor desensitization was estimated using a decreasing exponential fit that included ACh pulses 3 through 8. The first of these six ACh pulses was applied immediately before nicotine, and the last one was applied immediately before switching to nicotine-free solution. The nicotine-induced current was subtracted from the peak test-pulse response to obtain the ACh-induced peak current. Responses during nicotine application were also normalized to the first peak test-pulse response immediately before nicotine bath application. We plotted the six peak test-pulse responses against their time of application and fit the data to the sum of a decreasing exponential and a constant term to obtain the steady-state fraction of undesensitized receptors (SSF). To estimate the half-maximal inhibitory concentration (IC50), we fit the nicotine concentration-response relationship for steady-state desensitization to a sigmoidal inhibitory function:
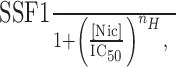 |
where [Nic] is the bath-applied nicotine concentration, and nH is the apparent Hill coefficient for desensitization.
Permeability measurements. We also used the oocyte expression system to measure the permeability of WT and L9′A mutant receptors to Na+ and Ca2+ relative to K+ (PNa/PK, PCa/PK). Permeability estimates were based on shifts in the reversal potential (Er) of the ACh response after external ion substitutions. An ACh-induced current-voltage (I-V) relationship was obtained by ramping the membrane potential from -80 to 50 mV (Na+,K+) or -50 to 30 mV (Ca2+) in the presence and absence of ACh and digitally subtracting the ACh-induced current from the background current. Voltage ramps were 0.5 s in duration and ACh was bath applied. We allowed the ACh response to reach steady state before applying the ramps and used ACh concentrations in the lower half of the ACh concentration-response relationship to minimize desensitization (5 μm ACh for WT, 10 or 100 nm ACh for L9′A). The Er of the ACh-induced current was obtained by fitting the following equation to the I-V relationship between -20 and 20 mV:
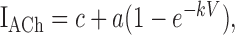 |
where V is the membrane potential in millivolts, IACh is the ACh-induced current in nanoamperes, a is the amplitude of the exponential component, c is a constant, and k is a rate constant. To measure the PNa/PK,we replaced all of the extracellular Na+ (98 mm) with equimolar K+ and measured the shift in the Er of the ACh response. The PNa/PK was calculated using the following equation:
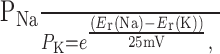 |
where the Er(Na) and Er(K) are the Er values for the ACh response in 98 mm Na+ and K+ solutions, respectively, and 25 mV is the value of RT/F at room temperature (20°C). To estimate PCa/PK, oocytes were injected with 50 nl of a 100 mm BAPTA-K4 solution 2 h before recordings to avoid activating endogenous Ca2+-dependent Cl- currents. We measured the Er for the ACh response in an isosmotic Ca2+ solution containing 65 mm CaCl2 and, assuming that K+ was the sole intracellular permeant cation, calculated PCa/PK from the following equation (Fatt and Ginsborg, 1958):
 |
where [K+]i is the apparent intracellular K+ concentration, [Ca2+]o is the extracellular Ca2+ concentration, and Er(Ca) is the Er for the ACh-induced current in Ca2+.[K+]i in this equation was calculated from the pooled WT and L9′A Er(K), using the Nernst equation, assuming that K+ was the sole intracellular permeant cation. All external solutions contained 1 mm MgCl2 and 5 mm HEPES, pH 7.5, in addition to the cations mentioned above. Extracellular solutions for Na+ and K+ replacement contained no added Ca2+.
Generation of L9′A knock-in mice. This section reproduces the on-line supplemental material in Tapper et al. (2004). L9′A construct design and embryonic stem (ES) cell manipulation was similar to that used in the generation of the L9′S mouse line (Labarca et al., 2001). Previous work generated a 129/SvJ α4 genomic clone containing exon 5 with the L9′S mutation in the vector pKO Scrambler V907 (Labarca et al., 2001). A neomycin resistance cassette with a phosphoglycerate kinase promoter and polyadenylation signal (Neo) flanked by loxP sites was present 163 bp downstream from exon 5 for positive selection. The diphtheria toxin A chain gene with an RNA polymerase II promoter was also added to provide negative selection for random insertion.
A 4.1 kb cassette containing exon 5 was removed from the pKO vector via digestion with SpeI and inserted into pBlueScript (Stratagene) for modification. PCR mutagenesis using the Quikchange Site-directed Mutagenesis kit was used to replace the L9′S mutation with the L9′A mutation (codon changed from TCT to GCT). The presence of the mutation was confirmed by automated sequence analysis. A small 250 bp cassette containing the L9′A mutation was subsequently cut out from pBlueScript with BsrGI and BglII and then unidirectionally ligated into pBlueScript L9′S α4 exon 5. This was done to avoid sequencing the entire 4.1 kb exon 5 cassette that went through PCR amplification. The 4.1 exon 5 cassette was then removed by digestion with SpeI and ligated back into the knock-in construct. Clones in the correct orientation yielded two fragments, 8 and 7 kb in length, during digestion with NsiI (as opposed to 10 and 5 kb fragments in the wrong orientation). The α4L9′A knock-in construct was linearized with NotI and chloroform/phenol extracted in preparation for introduction into mouse ES cells.
CJ7 mouse ES cells were electroporated with 25 μg of linearized knock-in construct. Six rounds of electroporation were performed using 107 ES cells/electroporation. After electroporation, each group of cells was plated on 10 cm dishes containing single layers of mitotically inactivated mouse primary fibroblasts. To select for recombinant clones, G418 (180 μg/ml) was applied to ES cells 24 h after plating. Cell death was apparent 3 d after treatment. After 7 d of G418 treatment, 600 resistant colonies were picked and plated onto 96-well plates, grown for 48 h, trypsinized, and then split onto duplicate plates, one for freezing and the other for screening. Correct recombinant colonies were screened for the presence of the Neo cassette and L9′A mutation by a combination of PCR and automated sequence analysis. DNA for PCR and sequencing was isolated by applying lysis buffer (in mm: 10 Tris, pH 7.5, 10 EDTA, 10 NaCl, 1% SDS, and 1 mg/ml proteinase K) to cells and incubating overnight at 60°C, followed by ethanol precipitation. To confirm the position of the Neo cassette, ES cells were screened via Southern blot. Positive clones were sequenced to verify the presence of the L9′A mutation. Karyotype analysis was used to assess chromosome number. Two positive clones with the highest percentage of proper chromosome number (90%) were selected for the subsequent round of electroporation.
The Neo cassette was excised by electroporating two clones of the Neo-intact L9′A α4 ES cells with cytomegalovirus Cre plasmid, leaving only a single 34 bp loxP. Electroporation was performed as described above. Cells were plated (400 cells per plate) onto 10 cm dishes containing single layers of mitotically inactivated mouse primary fibroblasts. A total of 200 colonies were picked and plated onto 96-well plates, grown for 48 h, trypsinized, and split onto triplicate 96-well plates, one for freezing and two for screening. Clones were initially screened for Neo cassette deletion by G418 (180 μg/ml) treatment. G418-sensitive clones were then further screened using a combination of PCR and automated sequencing. Primers were designed to anneal to a DNA sequence up-stream of the 5′ loxP site and downstream of the 3′ loxP site. All Neo-deleted clones were sequenced to confirm the presence of the L9′A mutation. Karyotype analysis was used to assess chromosome number. Two Neo-deleted L9′A α4 clones were injected into C57BL/6 blastocysts, and chimeras were established.
Chimeras were mated with wild-type animals, and germ-line transmission was established as assessed by PCR analysis of tail DNA and sequence analysis. To obtain a congenic line, L9′A mice have been backcrossed to C57BL/6J for several generations, currently N7.
[Ca2+]i measurement in thalamic cell cultures. Changes in [Ca2+]i were measured in thalamic cell cultures using the ratiometric fura-2 method as described previously (Nashmi et al., 2003). Thalamic tissue obtained from embryonic day 16 embryos was digested with 1 mg/ml papain, and cells were mechanically separated by gentle pipetting. Cells were plated onto 35 mm dishes with glass coverslip bottoms in Neurobasal culture medium containing 2% B27 supplement, 0.5 mm glutamax, and 5% horse serum, all obtained from Invitrogen (Carlsbad, CA). Cell cultures were kept in a temperature-, humidity-, and CO2/O2-controlled incubator. Before imaging, cells were loaded with 1.25 μm fura-2 AM (Invitrogen) in a 0.01% pluronic acid solution for 35 min at room temperature (22 ± 2°C) and in the dark. During experiments, cultures were continually perfused with an extracellular solution (in mm): 150 NaCl, 10 HEPES, 10 glucose, 4 KCl, 2 CaCl2, and 2 MgCl2. Various channel blockers to suppress nonspecific responses were included in the extracellular solution (in μm): 0.5 TTX (voltage-gated Na+ channels), 0.5 atropine (muscarinic receptors), 10 CNQX (AMPA and kainate receptors), 20 bicuculline (GABAA receptors), and 50 AP-5 (NMDA receptors). Ascorbic acid at 1 μm was also added to the extracellular solution to protect neurons from photodamage. Imaging was performed with an inverted fluorescence microscope using a 40× oil-immersed objective (UApo/340, 1.35 numerical aperture; Olympus Optical, Melville, NY). To generate ratiometric estimates of [Ca2+]i, pairs of images were obtained with alternate excitation wavelengths of 340/380 nm and emission wavelength of 510 nm. A U-tube connected to a pipette, placed ∼0.5 mm away from the targeted cell population, was used to deliver nicotine. Nicotinic receptors desensitize at agonist concentrations too low to cause appreciable activation; therefore, between applications, negative pressure was maintained on the U-tube to prevent nicotine from leaking out of the pipette. Nicotine was applied onto cultured cells when solution flow in the U-tube was momentarily (2 s) reversed by stopping outflow with a computer-controlled valve. Images were captured with a 16 bit CCD camera (Photometrics, Tucson, AZ) and analyzed with Slidebook 4.0 imaging software (Intelligent Imaging Innovations, Denver, CO).
Whole-cell patch recordings. To interpret changes in [Ca2+]i measured with ion-sensitive dyes after nicotine or ACh application, one wishes to decide whether the major Ca2+ flux is directly through nAChRs and/or indirectly through voltage-gated Ca2+ channels. This question is usually addressed by using Ca2+ channel blockers. However, concerns arise about the use of these blockers because of previous reports that they also interact with nAChRs (Lopez et al., 1993; Donnelly-Roberts et al., 1995). We therefore studied Ca2+ channel blocker-nAChR interactions. Blockade of nAChR receptors by calcicludine (750 nm) (Stotz et al., 2000), nifedipine (30 μm) (Shen et al., 2000), and ω-conotoxin GVIA (2 μm) (Wang et al., 1992) was examined in voltage-clamped L9′A thalamic cell cultures. Calcicludine and ω-conotoxin were purchased from Alomone Labs (Jerusalem, Israel). Cultures were prepared and maintained as described in the fura-2 experiments. Whole-cell recordings were performed with glass electrodes (2-5 MΩ) filled with an internal solution (in mm): 88 KH2PO4, 4.5 MgCl2, 0.9 EGTA, 9 HEPES, 0.4 CaCl2, 14 creatine phosphate, 4 Mg-ATP, and 0.3 GTP (Tris salt), pH 7.4 with KOH (Nashmi et al., 2003). Channel blockers to suppress nonspecific responses were included in the extracellular solution (in μm): 0.5 TTX (voltage gated Na+ channels), 0.5 atropine (muscarinic receptors), 10 CNQX (AMPA and kainate receptors), 20 bicuculline (GABAA receptors), and 50 AP-5 (NMDA receptors). Recordings were made using an Axopatch 1D (Axon Instruments) amplifier, low-pass filtered at 2-5 kHz, and digitized on-line at 20 kHz (pClamp8; Axon Instruments). The membrane potential was held at -70 mV. The 3 μm ACh test pulses lasting 250 ms were delivered with a two-barrel glass theta tube pulled from borosilicate tubing and connected to a piezoelectric translator (LSS-3100; Burleigh Instruments Fishers, NY). Calcium channel blockers contained in the second barrel of the theta tube were delivered 2 min after an initial pair of ACh test pulses and a few seconds before a third pulse. Recovery from block was assessed after a 10 min wash with extracellular solution.
Nicotine-stimulated 86Rb+ efflux. Nicotine-stimulated 86Rb+ efflux from thalamic and cortical synaptosomes of WT, heterozygous (Het), and homozygous (Hom) mice was measured as described previously (Marks et al., 1999). Cortex and thalamus (excluding habenula) were dissected from adult mice killed by cervical dislocation. Tissue was homogenized by hand in 1 ml of ice-cold 0.32 m sucrose buffered to pH 7.5 with HEPES using a glass/Teflon tissue grinder. After homogenization, the grinder was rinsed three times with 0.5 ml of buffered sucrose solution. A crude synaptosomal pellet was obtained by centrifugation at 20,000 × g for 20 min. After removal of the sucrose, each pellet was resuspended in 0.35 ml of load buffer (in mm: 140 NaCl, 1.5 KCl, 2 CaCl2, 1 MgSO4, 25 HEPES, and 22 glucose) and placed on ice until incubation with 86RbCl. Thalamic and cortical synaptosomes were incubated with 4 μCi 86Rb+ for 30 min in a final volume of 35 μl of load buffer, after which samples were collected by gentle filtration onto a 6-mm-diameter Gelman type A/E glass filter and washed once with 0.5 ml of load buffer. Filters containing the synaptosomes loaded with 86Rb+ were transferred to a polypropylene platform and superfused for 5 min with effluent buffer (in mm): 135 NaCl, 1.5 KCl, 5 CsCl, 2 CaCl2, 1 MgSO4, 25 HEPES, 22 glucose, 50 nm tetrodotoxin, and 0.1% bovine serum albumin. A peristaltic pump applied buffer to the top of the synaptosome containing filter at a rate of 2 ml/min, and a second peristaltic pump set at a faster flow rate of 3 ml/min removed buffer from the bottom of the platform. The greater speed of the second pump prevented pooling of buffer on the filter. Effluent buffer was pumped through a 200 μl Cherenkov cell and into a β-Ram detector (IN/US Systems, Tampa, FL). Radioactivity was measured for 3 min with a 3 s detection window providing 60 data points for each superfusion. Each sample was stimulated by 10 different nicotine concentrations, with a 5 s exposure for each concentration.
Basal 86Rb+ efflux was calculated by nonlinear least-squares curve fit of the samples before and after the nicotine-stimulated peak to a first-order equation: Ct = C0 × e-kt, where Ct is baseline counts at time t,C0 is baseline counts at t = 0, and k is the first-order constant for basal efflux. Nicotine-stimulated efflux was calculated as the difference between the actual sample 86Rb+ counts after nicotine stimulation and the estimated basal efflux. Nicotine-stimulated efflux at each concentration was normalized by dividing by the calculated basal efflux; thus, nicotine-stimulated 86Rb+ efflux is expressed as the response relative to baseline. EC50 values were calculated using the following Hill equation: ENIC = Emax × nicnH/(nicnH + EC50), where ENIC is the efflux measured after stimulation with nicotine concentration nic, and nH is the Hill coefficient.
To evaluate the effects of previous nicotine exposure on nicotine-stimulated 86Rb+ efflux (desensitization), synaptosomes prepared from cortex and thalamus of WT and Hom mice that had been loaded with 86Rb+ were superfused for 10 min with buffer containing nicotine before a 5 s stimulation. WT synaptosomes were treated with one of the following nicotine concentrations (in nm): 0, 1, 3, 10, 30, or 100. Hom samples were treated with one of the following (in nm): 0, 0.1, 0.3, 1, 3, or 10. After treatment with low nicotine concentrations, WT synaptosomes were stimulated with 100 μm nicotine, and Hom samples were stimulated with 10 μm nicotine.
125I-Epibatidine binding. Epibatidine binding was used for quantitating nicotinic receptor levels in various brain regions of WT and L9′A mutant mice as described previously (Whiteaker et al., 2002). Tissue was collected from the thalamus, cerebral cortex, interpeduncular nucleus, medial habenula, and hippocampus and homogenized in ice-cold 0.1× binding buffer (in mm): 14.4 NaCl, 0.2 KCl, 0.2 CaCl2, 1 MgSO4, and 2 HEPES, pH 7.5. Homogenates were washed three times by centrifugation (12,000 - g, 15 min, 4°C) and resuspension into 0.1× binding buffer and stored at -70°C. For saturation binding experiments, 125I-epibatidine concentrations were varied between 3 and 400 pm. The 200 pm 125I-epibatidine (a saturating concentration) was used in inhibition binding experiments. Cytisine inhibition of 125I-epibatidine was measured using drug concentrations of 0.1-3000 nm plus a no-drug control. The amount of membrane protein added was chosen to produce maximum ligand binding to the tissue of ∼1000 Bq/well (<5% of total ligand added, minimizing the effects of ligand depletion) and a minimum of 300 cpm of specific binding. Membranes were incubated for 2 h at 22°C in 30 μl of binding buffer. Nonspecific binding was determined in the presence of 1 mm (-)-nicotine tartrate and fell in the range of 20-50 cpm. Incubations were terminated by filtration onto Gelman GF/F fiber filters using a cell harvester, followed by six washes with ice-cold binding buffer. Bound ligand was measured at 80-85% efficiency using a Packard Cobra gamma counter (PerkinElmer, Boston, MA). Specific binding was calculated for each region from every animal by subtraction of the relevant nonspecific binding value. Saturation binding parameters were calculated by fitting data to the following Hill equation: B = BmaxLn/(Ln + KDn), where B is the binding at the free ligand concentration L, Bmax is the maximum number of binding sites, KD is the equilibrium binding constant, and n is the Hill coefficient. Inhibition of 125I-epibatidine binding was calculated using a two-site fit: B = B1/(1 + (I/IC50-1)) + B2/(1 + (I/IC50-2)), where B was ligand bound at inhibitor concentration I, and B1 and B2 represent 125I-epibatidine binding to sites sensitive to cytisine inhibition with IC50-1 and IC50-2, (cytisine-sensitive and cytisine-resistant sites, respectively).
Behavioral studies. In vivo experiments were performed according to National Institutes of Health and local institutional guidelines for the humane treatment of laboratory animals. Drug-naive mice, 3-5 months old, with similar number of females and males and from N4 through N7 C57BL/6J-backcrossed generations, were used in these experiments. No statistical differences were found between N4 and N7 animals of the same genotype in any of the experiments performed. Nicotine tartrate was dissolved in saline, with a final concentration adjusted to an injection volume of 10 μl/gm body mass and administered subcutaneously. Nicotine doses are expressed as milligrams of base per kilogram of live mouse weight. Acute behavioral responses to nicotine were scored by an experimenter unaware of the mouse's genotype according to a modified behavioral scale (Dobelis et al., 2003): no visible response (0), sedation and loss of locomotion (1), Straub tail and/or shortened gait (2), circling and/or head bobbing (3), severe tremors, wild running, and/or cornering (4), loss of righting response and clonic convulsion (5), clonic-tonic convulsion (6), and tonic hindlimb extension and death (7). Mice with behavioral scores of 5, 6, or 7 were tabulated as having seizures and used to calculate seizure onset averages and SEs (see Fig. 6 B, D). The following compounds were tested for their ability to prevent nicotine-induced seizures in L9′A and WT mice: (1) carbamazepine, a commonly used antiepileptic; (2) dihydro-β-erythroidine (DHβE), a competitive nicotinic antagonist with high affinity for α4/β2 receptors; (3) hexamethonium, a nicotinic antagonist with poor brain-barrier permeability and thus suspected of preferentially targeting nicotinic receptors in the periphery; (4) mecamylamine, a potent wide-spectrum nicotinic antagonist, and (5) nicotine, reported to prevent seizures in an otherwise drug-resistant ADNFLE patient (Willoughby et al., 2003). All pretreatment drugs were dissolved in saline, except for carbamazepine, which was suspended in 1% Tween 80. Dose and delivery schedule of pretreatment drugs are listed in Table 1, except for nicotine, which is shown in Figure 6, C and D. The final concentration of pretreatment drugs and the seizure-inducing nicotine dose were adjusted to an injection volume of 5 μl/gm each so that the total volume of both injections was the same as in single-nicotine injection experiments. To reduce the number of mice killed in seizure-inducing experiments, no Hom mice were tested in the nicotine dose-response pretreatment studies.
Figure 6.

Nicotine-induced seizures and the effect of nicotine pretreatment on seizures. A, B, Seizures in the absence of pretreatment. Nicotine-induced seizures were expressed as percentage of mice that displayed behavioral seizures (A) and time from injection to seizure onset (B). Mice that did not display seizures 10 min after receiving a nicotine injection were considered nonresponsive and correspond to data points next to the 600 s mark. Het and Hom were more sensitive to nicotine than WT. Each data point in B is the mean ± SEM; n = 6. C, D, Effect of nicotine (nic) pretreatment on nicotine-induced seizures in WT and Het mice. C, Percentage of seizing mice (as in A); D, time to seizure onset (as in B). Mice were injected with a nicotine pretreatment dose 10 min before receiving a second test nicotine injection. Nicotine pretreatment (0.1 mg/kg) completely blocked seizures in Het that received a second dose of 0.2, 0.5, or 1 mg/kg nicotine. Seizures in WT mice were not blocked by nicotine pretreatment. Each data point is the mean ± SEM; n = 5.
Table 1.
Ability of various compounds to block nicotine-induced seizures in WT, Het, and Hom mice
|
Pretreatment |
Behavioral response mice with seizures |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug |
Dose (mg/kg) |
Timing (min) |
WT |
Het |
Hom |
||||
| Carbamazepine | 20 | 30 before nic | 4 of 6 | 5 of 6 | 5 of 6 | ||||
| DHβE | 3 | 5 before nic | 5 of 5 | 5 of 5 | 4 of 4 | ||||
| Hexamethonium | 10 | 10 before nic | 5 of 5 | 5 of 5 | 4 of 4 | ||||
| Mecamylamine |
0.5 |
10 before nic |
0 of 5 |
0 of 5 |
0 of 4 |
||||
After an intraperitoneal injection of the pretreatment drug, mice received a seizure-inducing dose of nicotine (nic): 10 mg/kg in WT and 1 mg/kg in Het and Hom. Carbamazepine was suspended in 1% Tween 80. Results are expressed as number of mice that had seizures from total animals tested in each treatment group.
In vivo electrophysiology. Electroencephalographic (EEG) recordings were obtained from freely-moving animals using a low-torque commutator (Dragonfly, Ridgeley, WV). Electrodes were stereotactically implanted in mice anesthetized with a ketamine (100 mg/kg) and xylazine (25 mg/kg) mixture. Field recordings were obtained from (1) stainless steel screws in the skull (head diameter, 1.5 mm; length, 1.8 mm), used for monitoring extended cortical regions, or (2) thin depth electrodes (tungsten, 0.1 mm diameter, ≈200 kΩ), intended to detect voltage changes in deeper brain regions. Depth electrodes were inserted unilaterally through a hole drilled in the cranium. A ground screw electrode was placed in the skull away from the other electrodes. A three-pin microconnector (ground electrode plus two recording electrodes) was cemented to the skull surface with dental acrylic. Electrode placement coordinates in parentheses are expressed in millimeters from bregma, with the first number corresponding to the anteroposterior axis, the second representing midline lateral axis, and, in the case of depth electrodes, the third number representing depth from skull surface. Bilateral screw electrodes were placed in the parietal cortex (-2 ± 1.7), the somatosensory cortex (-1.5 ± 1.7), the primary motor cortex (-0.8 ± 1.1), or the cingulate cortex (0.1 ± 0.3). Depth electrodes were positioned in the hippocampus (-2, 2, 1.5), the ventral thalamus (-1, 1, 3.2), or the insular cortex (0.9, 3.5, 3.6). After surgery, mice were allowed to recover for 5 d. Simultaneous video and EEG recordings started 10 min before and continued for 15 min after a single nicotine injection. Data were acquired with a differential amplifier (Brownlee Precision, San Jose, CA), with a 1 kHz sampling rate and bandpass filtered at 1-200 Hz. EEG data were analyzed with Clampfit 8.2 software (Axon Instruments). Video and electroencephalographic signals were digitally synchronized with a Stellate hardware-software system (Stellate Systems, Montreal, Quebec, Canada). During some EEG recordings, mouse movements were measured with a piezo film sensor mechanotransducer (MSI, Fairfield, NJ) connected to the spring-suspended floor panel. The transducer signal was led to a low-pass Bessel filter (100 Hz, 902 LPF; Frequency Devices, Haverhill, MA) and analyzed with Clampfit 8.2 software.
Chronic EEG recordings. Digitally synchronized video/EEG recordings lasting 24 h were performed to determine whether L9′A mice had spontaneous seizures and/or altered sleep-wake patterns. Screw electrodes were bilaterally implanted above the primary motor cortex (from bregma, anteroposterior, -0.8; lateral, ±1.1 mm). Animals were individually housed with food and water and connected to the recording system through a commutator as described above. Mice were allowed to acclimate in the recording room for 48 h before data acquisition. EEG recordings started at 5:00 P.M., and lights were switched off at 8:00 P.M. and back on at 7:00 A.M. This lights on-off schedule is used by the California Institute of Technology animal facility, where mice are housed from birth. Data were acquired at a sampling rate of 1 kHz, bandpass filtered at 1-100 Hz, and resampled at 200 Hz. The EEG power spectrum was computed for overlapping 2.5 s Hanning-windowed intervals (512 points). The vigilance states of five WT, five Het, and four Hom mice were determined by analyzing EEG power spectral density changes over time (see Fig. 9). Criteria used to identify periods of rapid eye movement (REM) sleep, non-REM (NREM) sleep, or wakefulness were based on previously published reports: REM sleep has a distinct power spectrum peak at 6-7 Hz, NREM sleep has a peak at 2-4 Hz, and wake has an overall low power level, with a peak at 6-8 Hz (smaller than REM) (Franken et al., 1998; Lena et al., 2004). The automated analysis of the power spectra of each mouse was tuned on the basis of 2 h of simultaneous EEG/video recordings. These recordings were manually scored in each of three WT, three Het, and two Hom mice as being awake, sleeping, or having brief movements during sleep; observers were blind to genotype. Algorithms were implemented in Matlab (MathWorks, Natick, MA).
Figure 9.

Chronic EEG recordings in WT, Het, and Hom mice. A, Example of an EEG trace with its corresponding spectrogram from a WT mouse. Power density (in square microvolts per Hertz) changes in the spectrogram are represented by the color intensity of individual pixels: dark coloration means high-power density. The vigilance state of each mouse was determined with an algorithm that analyzed power spectrum changes over time. The blue line above the spectrogram represents the output of the program. B, Brief awakenings were recorded during 13 h with lights on and 11 h with lights off. C, Number of brief awakening events per hour. Het and Hom mice had more brief awakenings than WT (one-way ANOVA, followed by the Tukey's pairwise multiple comparison test, *p < 0.001). Each bar represents the mean and SEM; n = 5 (WT), 5 (Het), and 4 (Hom). D, Wake, NREM sleep, and REM sleep events were quantified in WT, Het, and Hom mice over a 24 h period.
Mouse locomotion. Mouse locomotion was measured over a 24 h period using activity cages with infrared beams (San Diego Instruments, San Diego, CA). A locomotion event was recorded when two contiguous beams were interrupted successively. Locomotion experiments were performed independently from chronic EEG recordings, although housing conditions were the same. Eight mice from each genotype were allowed to acclimate in the recording room for 48 h before data acquisition. The light/dark schedule used was the same as described above. Locomotion activity was also measured in the same group of mice during 24 h of continuous light.
Neuroanatomy. Brain sections obtained from WT and Hom mice (n = 6 per genotype) were Nissl stained for histological analysis (Simmons and Swanson, 1993). Mice were anesthetized with 2-bromo-2-chloro-1,1,1,-trifluoroethane and myocardially perfused with PBS containing 0.1% heparin, followed by 4% paraformaldehyde. Brains were postfixed overnight with 4% paraformaldehyde and immersed for 2 d in a 30% sucrose solution for cryoprotection (4°C). 2-Methylbutane in dry ice was used to rapidly freeze brains, and coronal sections 30 μm thick were cut with a cryostat (Leica, Nussloch, Germany). Alternate sections were mounted onto glass slides and air dried at room temperature for 24 h. Sections were delipidized with successive ethanol washes (70, 95, and 100%), followed by a 1 min immersion in xylene. Sections were then rehydrated with reversed-order ethanol washes, stained with 0.25% thionin, and differentiated with ethanol washes followed by xylene. Finally, glass slides were coverslipped with a 10:1 Permount/xylene mixture, and brain sections were examined under an inverted microscope (Olympus Optical).
Results
The L9′A mutation increases agonist sensitivity in Xenopus oocytes
We designed the L9′A mutation based on two types of previous studies. First, experiments on neuronal and muscle nAChR subtypes expressed in heterologous systems showed that the Leu9′ position is crucial for gating (Revah et al., 1991; Filatov and White, 1995; Labarca et al., 1995). In general, dose-response relationships were shifted to lower agonist concentration by mutations that increase the polarity or hydrophilicity of the 9′ side chain, primarily via increased channel open time (Kearney et al., 1996; Kosolapov et al., 2000). These studies allowed us to predict the approximate degree of hypersensitivity in a receptor mutated at the 9′ position.
The second type of experiment involves the phenotype of knock-in mice bearing such mutations. The homozygous and hemizygous α7 Leu9′Thr mice died within 24 h after birth (Orr-Urtreger et al., 2000), but the heterozygous mice were viable and fertile. The homozygous and heterozygous α4L9′S mice also died within 24 h after birth, and the only viable, fertile mice were L9′S heterozygous (containing Neo) with a reduced mutant allele level of expression (Labarca et al., 2001; Fonck et al., 2003). However, even L9′S-Neo heterozygous mice showed modest deficits of midbrain dopaminergic neurons (Orb et al., 2004).
Therefore, we sought α4 subunits with a more moderate degree of hypersensitivity. We expected to achieve this goal with a 9′ side chain intermediate between the strongly nonpolar WT Leu and the excessively polar Ser or Thr. We replaced the 9′Leu residue with Ala and expressed the mutated neuronal α4 subunit (L9′A) along with WT β2 in oocytes. The L9′A mutation increased the nicotine and ACh sensitivity of the mouse α4β2 nicotinic receptor (Fig. 1). The WT EC50 values for nicotine (7 ± 1 μm) and ACh (48 ± 8 μm) were similar to the values reported previously for human α4β2 nicotinic receptors expressed in human embryonic kidney cells (Buisson and Bertrand, 2001). The L9′A mutation reduced the EC50 for nicotine and ACh by ≥30-fold. The L9′AEC50 for nicotine was 0.23 ± 0.03 μm and for ACh was 0.5 ± 0.1 μm. The ACh EC50 for L9′S was 0.02 ± 0.001 μm. WT and L9′A nicotine Hill coefficients were 1.3 ± 0.1 and 1.1 ± 0.1, respectively. WT, L9′S, and L9′A ACh Hill coefficients were 0.8 ± 0.1, 1.7 ± 0.2, and 1.2 ± 0.1, respectively. Aside from the WT ACh concentration-response relationship, all Hill coefficients were greater than unity and thus do not justify a two-population fit. A WT Hill coefficient of 0.8 for the ACh concentration-response relationship is consistent with previous results for heterologously expressed human WT α4β2 receptors (Buisson and Bertrand, 2001), which suggests receptor heterogeneity. However, fitting the WT ACh data to the sum of two hyperbolic binding functions yielded a low-affinity component that was 93 ± 3% of the total population and displayed an EC50 of 41 ± 5 μm, which resembled the value obtained from the Hill equation fit of 48 ± 8 μm. Consequently, low-affinity receptors formed most of the WT population in our experiments, and the high-affinity WT population was too small for a reliable EC50 estimate. There was no noticeable difference in the health of oocytes expressing α4(L9′A)β2 compared with WT α4β2 receptors.
Figure 1.

Nicotine- and ACh-induced responses of mouse WT α4β2 and α4(L9′A)β2 receptors expressed in Xenopus oocytes. The L9′A mutation increases nicotine and ACh sensitivity of mouse α4β2 receptors. A, Nicotine-induced voltage-clamp currents recorded from oocytes expressing mouse WT or α4(L9′A)β2 receptors. B, WT and L9′A nicotine concentration-response relationships, normalized to the maximal response of each oocyte. Lines are fits to the Hill equation. WT and L9′A nicotine EC50 values are 7 ± 1 and 0.23 ± 0.03 μm, respectively. Hill coefficients are 1.3 ± 0.1 and 1.1 ± 0.1, respectively. Error bars are ± SEM; n = 8-9. C, ACh-induced voltage-clamp currents recorded from oocytes expressing mouse WT or α4(L9′A)β2 receptors. D, ACh concentration-response relationships and fits to the Hill equation. WT, L9′A, and L9′S ACh EC50 values are 48 ± 8, 0.5 ± 0.06, and 0.02 ± 0.001 μm, respectively. Hill coefficients are 0.8 ± 0.1, 1.2 ± 0.1, and 1.7 ± 0.2, respectively. Error bars are ± SEM; n = 5.
Importantly, the α4(L9′A)β2 receptors were, as expected, less hypersensitive than those containing α4 L9′S (Fig. 1D) (rat cDNAs were studied previously; the mouse α4 L9′S cDNAs was constructed and used for the first time in these experiments). On a logarithmic scale, the dose-response relationship for the α4(L9′A)β2 receptor is approximately midway between those for the α4(L9′S)β2 and the WT receptors (Fig. 1D). Because we believe that the lethality of the α7 Leu9′Thr (Orr-Urtreger et al., 2000) and L9′S strains arises partially from endogenously active receptors and/or from activation by choline at concentrations (∼10 μm) existing in CSF (Labarca et al., 2001), we note that α4(L9′A)β2 receptors showed little or no spontaneous activation and little activation (<10% of maximal levels) by 10 μm choline (data not shown).
The L9′A mutation increases nicotine-induced desensitization
We studied the effect of the L9′A mutation on nicotine-induced desensitization using the oocyte expression system. Desensitization was measured by exposing oocytes to a sustained dose of nicotine while eliciting responses to brief ACh pulses (Fig. 2A,B). As reported previously (Jia et al., 2003), recovery from nicotine-induced desensitization was incomplete despite continuous superfusion with nicotine-free saline for 10 min. WT and L9′AIC50 values for steady-state desensitization were 22 ± 5 and 8.2 ± 0.5 nm, respectively (Fig. 2C). The WT IC50 was somewhat less than the previously reported value (61 nm) for rat α4β2 nicotinic receptors expressed in oocytes (Fenster et al., 1997). WT and L9′A receptors showed a similar degree of desensitization when relatively low nicotine concentrations were used (for example, at 10 nm nicotine; Student's t test, p = 0.07), whereas higher nicotine concentrations caused more desensitization in L9′A than in WT (for example, at 100 nm nicotine; Student's t test, p = 0.006) (Fig. 2C). The L9′A mutation reduced the IC50 for steady-state nicotine desensitization approximately threefold compared with a 30-fold reduction in the EC50 for nicotine activation of the receptor (Fig. 2D). Hill coefficients for WT and L9′A desensitization were 0.45 ± 0.06 and 1.13 ± 0.06, respectively. A twofold difference in the Hill coefficient between WT and L9′A desensitization suggests a more homogenous mutant receptor population. The maximum sustained fractional nicotine response [“window” response (Lester, 2004)], measured from the intersection of desensitization and activation concentration-response relationships, occurred at a lower nicotine concentration for L9′A (40-50 nm) than for WT (1-2 μm) receptors (Fig. 2D). Given the uncertainties associated with measurements of small currents and with differently shaped dose-response relationships, it is not possible to compare the size of the windows for WT versus L9′A receptors quantitatively, but such windows clearly do occur for both receptors. Therefore, the L9′A mutation increased steady-state desensitization of the nicotine response over a wide range of nicotine concentrations, and it reduced the nicotine concentration at which the nicotine window response occurred.
Figure 2.

Desensitization of mouse WT α4β2 and α4(L9′A)β2 receptors expressed in Xenopus oocytes. A, WT and L9′A responses to 200 nm ACh pulses. Each oocyte was exposed to 13 ACh pulses, 2.5 s in duration at intervals of 120 s. Immediately after the third ACh pulse, nicotine was bath applied for 10 min (5 pulses). B, Time course of ACh responses at various nicotine concentrations. Data exemplify responses from three or four oocytes tested at each nicotine concentration. L9′A values were corrected to account for changes in the size of the responses over time in the absence of nicotine (see Materials and Methods). No correction was necessary for WT. C, Concentration dependence of desensitization for WT and L9′A. Regression lines are fits to a sigmoidal inhibitory function. IC50 values are 22 ± 5 and 8.2 ± 0.5 nm for WT and L9′A, respectively. Hill coefficients are 0.45 ± 0.06 and 1.13 ± 0.06 for WT and L9′A, respectively. D, Concentration dependence of desensitization and activation (see Fig. 1 B) normalized to maximal response of each oocyte for WT and L9′A.
The L9′A mutation does not affect ion selectivity for Na+, K+, and Ca2+
Previous studies show that mutations in the M2 region can alter the ion selectivity of nicotinic receptors, although mutations at the 2′ position appear to have the greatest effect (Cohen et al., 1992; Wang and Imoto, 1992). To determine whether the L9′A mutation altered ion selectivity of α4β2 receptors for three physiologically relevant cations (Na+, K+, and Ca2+), we substituted K+ or Ca2+ for extracellular Na+ and measured the shift in reversal potential (Er) of the ACh response (see Materials and Methods). The L9′A mutation did not significantly change PNa/PK (Student's t test, p = 0.40) or PCa/PK (Student's t test, p = 0.98) (Fig. 3). WT and L9′A Er values in 98 mm extracellular Na+ were -14 ± 2 mV (n = 5) and -11 ± 1 mV (n = 5), respectively (Fig. 3A). Equimolar K+ substitution shifted WT Er by 8 ± 2 mV (n = 5) and L9′A Er by 7 ± 2 mV (n = 5) (Fig. 3B), giving a PNa/PK for WT of 0.73 ± 0.02 and 0.76 ± 0.03 for L9′A. This shift implies that WT and L9′A channels were 24-27% less permeable to Na+ than K+. Er values in 65 mm Ca+ were equal for WT (8 ± 2 mV; n = 4) and for L9′A (8 ± 3 mV; n = 5) (Fig. 3C). The pooled Er(K) for WT and L9′A was -5 ± 1 mV (n = 10), giving an apparent [K+]i of 119 ± 3 mm (assuming that K+ was the sole intracellular permeant cation). Using this value for [K+]i, WT and L9′A PCa/PK values were 1.5 ± 0.1 (n = 4) and 1.5 ± 0.2 (n = 5), respectively. Thus, WT and mutant channels were 50% more permeable to Ca2+ than K+. Dividing the PCa/PK by PNa/PK gives a PCa/PNa of 2.0 ± 0.1 for WT and 2.0 ± 0.3 for L9′A. These values are similar to those reported previously for rat α4β2 receptors expressed in oocytes (Haghighi and Cooper, 2000).
Figure 3.

Na+ and Ca2+ permeabilities relative to K+ (PNa/PK, PCa/PK) were measured for mouse WT α4β2 and α4(L9′A)β2 receptors expressed in frog oocytes. WT and L9′A ACh-induced I-V relationships in 98 mm K+ (A) and 98 mm Na+ (B) between -20 and 20 mV. The reversal potentials of WT and L9′A I-V relationships (Er values) were -4 and -2 mV in 98 mm K+ and -10 and -11 mV in 98 mm Na+, respectively. C, WT and L9′A ACh-induced I-V relationships in 65 mm Ca2+ between-30 and 30 mV. WT and mutant Er values in 65 mm Ca2+ were 9 and 8 mV, respectively. The I-V relationships were obtained from individual oocytes. D, Relative permeabilities, PNa/PK and PCa/PK, for WT and L9′A receptors. Error bars are means ± SEM; n = 4 oocytes per experiment.
Thalamic cell cultures obtained from L9′A embryos are hypersensitive to nicotine
Thalamic neurons were easily distinguished from glia in culture: cell bodies of neurons were elevated, elongated, and bright, with well defined processes. All cells identifiable anatomically as neurons showed increased 340/380 nm fura-2 fluorescence ratios at the appropriate nicotine concentrations. Thalamic cultured cells derived from Het and Hom embryos were more sensitive to nicotine than WT, as measured by increases in [Ca2+]i (Fig. 4). Het and Hom cultures, but not WT ones, had significant fura-2 responses after bath application of nicotine at concentrations of 0.1 and 1 μm (Fig. 4A-C). In addition, ratiometric measurements showed a 3.5-fold larger fura-2 response in Het and Hom compared with WT cells at saturating nicotine concentrations. There was no significant difference between Het and Hom neurons in their sensitivity to nicotine (ANOVA, p = 0.62) (Fig. 4D). Het and Hom were also more sensitive and had larger maximal responses than WT when ACh was applied to the cells (data not shown). The EC50 values for WT, Het, and Hom thalamic cultures, 7.1 ± 4.3, 0.2 ± 0.1, and 0.6 ± 0.6 μm, respectively, and 3.5-fold greater maximal signals for L9′A cultures, are close to those measured previously for WT and L9′A Het neurons in midbrain cultures (Tapper et al., 2004). Data in Figure 4 correspond to cell groups imaged from a single field of view, from a single Petri dish, and for each genotype (17 WT cells, 22 Het cells, and 16 Hom cells). Similar results were obtained from additional cell cultures derived from embryos from three WT × WT, four Hom × WT, and two Hom × Hom matings.
Figure 4.

Functional characteristics of nAChRs in thalamic primary cell cultures from WT, Het, and Hom L9′A mice. Fura-2 ratiometric [Ca2+]i measurements were made at 2 s intervals, starting 20 s before and ending 80 s after a single application of nicotine in WT (A), Het (B), and Hom (C)-derived cell cultures. Each data point is the average of 17 (WT), 22 (Het), or 16 (Hom) cells. D, Responses to various nicotine concentrations were used to compose a concentration-response relationship. Het- and Hom-derived cell cultures were more sensitive and had larger responses to nicotine compared with WT. Whole-cell patch recordings were obtained from L9′A Hom thalamic cell cultures exposed to calcium channel blockers: 750 nm calcicludine (E), 30 μm nifedipine (F), and 2 μm ω-conotoxin GVIA (G) (n = 2 or 3 neurons per blocker). Receptor activity was elicited with 3 μm ACh test pulses. After a 10 min wash with extracellular solution, recovery from block was assessed with a final 3 μm ACh pulse. All three calcium blockers reversibly decreased ACh responses.
We examined the interaction between calcium channel blockers (calcicludine, nifedipine, and ω-conotoxin) and nAChR expressed in Hom-derived cell cultures with the ultimate goal of determining the source of [Ca2+]i changes after nicotine or ACh exposure. ACh-induced currents were measured in voltage-clamped neurons before and after they were exposed to calcium channel blockers (Fig. 4E-G). Blockers were tested at concentrations reported to achieve maximal block of L-type and N-type calcium channels (Wang et al., 1992; Shen et al., 2000; Stotz et al., 2000). The 30 μm nifedipine and 2 μm ω-conotoxin almost completely abolished the ACh response, whereas 750 nm calcicludine diminished it by 67%. Responses recovered incompletely after a 10 min wash, presumably because some blocker remained and/or rundown effects. Thus, these results reinforce the surprising notion that several calcium channel blockers of widely varying structures also interact directly with nicotinic receptors. Because we lack a specific agent that blocks Ca2+ channels without blocking nAChRs, we desisted from using calcium channel blockers to distinguish between Ca2+ entry directly through nAChRs versus indirectly through voltage-gated calcium channels.
Hypersensitivity of nAChRs in synaptosomes made from L9′A mice
Functional expression of mutated nAChRs in the forebrains of adult L9′A mice was assessed by measuring nicotine-stimulated 86Rb+ efflux in synaptosomes. Once nAChRs are opened by nicotinic ligands, 86Rb+ can exit the cell (or a synaptosome) much in the same way potassium does under physiological conditions. Brief (5 s) exposure to nicotine resulted in a concentration-dependent increase in 86Rb+ efflux for both cortical and thalamic synaptosomes prepared from WT mice (Fig. 5A). Maximal 86Rb+ efflux from thalamic synaptosomes (17 ± 1 relative to baseline) was significantly greater than that from cortical synaptosomes (10 ± 1 relative to baseline). Estimated EC50 values for nicotine also differed between these regions (7 ± 2 μm for cortex and 2.9 ± 0.6 μm for thalamus). However, the L9′A mutation affected nicotine-stimulated 86Rb+ efflux in these two regions similarly, resulting in a gene-dose-dependent reduction in maximal response and a shift to the left (increased sensitivity) of the concentration-effect curves. Maximal 86Rb+ efflux in Hom mice was estimated as 2.1 ± 0.2 for cortex and 4.9 ± 0.1 for thalamus. Estimated EC50 values for nicotine were lower than the EC50 values of WT mice in both brain regions of L9′A Hom mice (0.1 ± 0.1 and 0.18 ± 0.06 μm for cortex and thalamus, respectively). Nicotine-stimulated 86Rb+ efflux for L9′A Het mice was intermediate between that of WT and Hom mice: maximal 86Rb+ efflux was 7.0 ± 1.2 and 12.5 ± 0.8, whereas estimated EC50 values were 2.2 ± 0.7 and 1.0 ± 0.3 μm for cortex and thalamus, respectively. Importantly, despite the reduced maximal nicotine-simulated 86Rb+ efflux observed in Hom, the mutation also increased apparent nicotine affinity, resulting in larger responses for Hom and Het synaptosomes at nicotine concentrations <1 μm.
Figure 5.

Nicotine-stimulated 86Rb+ efflux and 125I-epibatidine binding in WT, Het, and Hom L9′A mice. A, 86Rb+ efflux from synaptosomes made from cortex and thalamus and stimulated with nicotine. Each point is the mean ± SEM; n = 7. Lines are the best fit to the Hill equation. B, Desensitization and activation curves normalized to maximal response for WT and L9′A synaptosomes. Each point is the mean; n = 6 or 7. C, Concentration dependence of 125I-epibatidine binding in the membrane fraction obtained from cortex and thalamus. Each point is the mean ± SEM; n = 4. D, 125I-Epibatidine binding in cortex and thalamus in the presence or absence of cytisine was used to calculate cytisine-sensitive sites and cytisine-resistant sites. Cytisine-sensitive sites are thought to reflect α4β2 receptors. Saturation binding shows fewer epibatidine binding sites in Het and Hom compared with WT in both brain regions examined (one-way ANOVA, followed by Bonferroni's test; §,*p < 0.01). Each bar represents mean and SEM; n = 4.
Desensitization was assessed in cortical and thalamic synaptosomes prepared from WT and Hom mice. We exposed synaptosomes to comparatively low nicotine concentrations and then measured subsequent nicotine-stimulated 86Rb+ efflux (Fig. 5B). Much like the reduction seen in the EC50 for agonist-stimulated 86Rb+ efflux by the L9′A mutation, desensitization also occurred at lower nicotine concentrations in L9′A compared with WT. The IC50 for desensitization of nicotine-stimulated 86Rb+ efflux in WT cortical synaptosomes was 16 ± 6.0 nm, and this value was reduced to 0.93 ± 0.35 nm in Hom. Similarly, the IC50 for desensitization of nicotine-stimulated 86Rb+ efflux in thalamic synaptosomes was reduced from 22 ± 7 nm in WT to 2 ± 0.7 nm in Hom. Thus, in both WT and Hom synaptosomes, the concentration of nicotine required to desensitize a subsequent response to agonist stimulation was at least 100-fold lower than that required for activation. Both the ability to induce desensitization by previous exposure to low concentrations of nicotine and the shift in apparent affinity of nicotine for this process by the L9′A mutation in synaptosomal preparations recall that observed for receptors expressed in Xenopus oocytes (Fig. 2).
125I-Epibatidine binding
To estimate the effect of the L9′A mutation on the expression of nAChRs, we determined the binding of 125I-epibatidine to membranes prepared from cortex and thalamus of WT, Het, and Hom mice (Fig. 5C). Saturation curves for 125I-epibatidine indicated that a gene-dose-dependent reduction in total high-affinity sites occurred as a consequence of the L9′A substitution. Total maximal binding in cortical membranes decreased from 58 ± 8 fmol/mg protein in WT to 33 ± 3 fmol/mg protein in Het (44% decrease) to 19 ± 1 fmol/mg protein in Hom (68% decrease). Similarly, maximal binding in thalamic membranes decreased from 113 ± 4 fmol/mg protein in WT to 77.7 ± 4 fmol/mg protein in Het (31% decrease) to 49 ± 2 fmol/mg protein in Hom (57% decrease). Apparent affinity of 125I-epibatidine for these high-affinity binding sites were also increased slightly by the L9′A mutation (apparent KD values in cortex were 56 ± 3.2, 42 ± 6, and 38 ± 3 pm and in thalamus were 28 ± 2, 20 ± 2, and 15.2 ± 0.6 pm for WT, Het, and Hom mice, respectively).
The data were evaluated for the effects of the L9′A substitution on cytisine-sensitive and cytisine-resistant components of 125I-epibatidine binding (Fig. 5D). Cytisine-sensitive sites, in almost all brain regions, are thought to correspond primarily to α4β2* receptors (Marks et al., 1998). Cytisine inhibition binding data show that most of the reduction in binding occurred from the cytisine-sensitive component. For Hom, the average for cortex and thalamus was ∼56% of WT levels. In the brain regions examined, Het displayed cytisine-sensitive 125I-epibatidine binding that was intermediate between WT and Hom. It should be noted that cytisine-resistant sites comprise a relatively small fraction of the total 125I-epibatidine binding sites in these brain regions (∼4% of the sites in cortex of WT and ∼5.5% of the sites in thalamus of WT). WT, Het, and Hom had similar levels of 125I-epibatidine binding in the presence of cytisine (cytisine-resistant sites; one-way ANOVA, p = 0.12). These sites are thought to correspond to non-α4β2 receptors. Thus, cytisine inhibition data indicate that there was a decrease in the expression of mutated receptors in the Hom and Het, without apparent changes in the expression levels of other nAChR subtypes. Analysis of the effects of the L9′A mutation on cytisine-sensitive and cytisine-resistant sites in interpeduncular nucleus and medial habenula, regions in which the cytisine-resistant sites are highly expressed, also indicated that the effects of the mutation were confined to cytisine-sensitive sites. A previous study determined that cytisine-sensitive, but not cytisine-insensitive, binding was decreased (to 50-60% of WT levels) in the striatum and midbrain of Het L9′A mice (Tapper et al., 2004).
L9′A nicotine-induced seizures and their suppression by nicotine pretreatment
All Het and Hom mice injected with 2 mg/kg nicotine displayed seizures, whereas a dose of 10 mg/kg nicotine was required to elicit seizures in all WT mice (Fig. 6A). No WT mice had seizures when injected with 2 mg/kg nicotine; instead, at this dose, WT displayed sedation and lack of locomotion. ED50 values calculated with a sigmoid fit were 6.3 ± 0.3, 0.3 ± 0.1, and 0.6 ± 0.3 mg/kg for WT, Het, and Hom, respectively. In previous studies of the pharmacokinetics of nicotine and its metabolites, a concentration of ∼1 μm nicotine was detected in the brains of C57BL/6J mice injected with 1 mg/kg nicotine (Petersen et al., 1984). Based on the pharmacokinetic data from the Petersen et al. report, it appears that ED50 values for nicotine-induced seizure effects in L9′A mice produce brain nicotine concentrations (0.3-0.6 μm) near the EC50 values for L9′A receptors in oocytes (Fig. 1B), for thalamic cultures from L9′A mice (Fig. 4D), as well as for 86Rb+ efflux from L9′A synaptosomes (Fig. 5A). Latency to seizure onset in Het and Hom (44 ± 9 and 43 ± 7 s, respectively, with 2 mg/kg nicotine) was much less than in WT (323 ± 49 s with 10 mg/kg nicotine) (one-way ANOVA, followed by the Bonferroni's test, p < 0.001) (Fig. 6B). There was no strong dose dependence of seizure latency in either L9′A mutants and WT. For example, Hom mice that received 0.2, 0.5, or 1 mg/kg nicotine had average latencies of 65 ± 20, 51 ± 5, or 57 ± 8 s, respectively (one-way ANOVA, p = 0.62). Seizure duration was estimated by reviewing video recordings of mice injected with seizure-inducing doses of nicotine. Seizures in L9′A mice injected with 2 mg/kg nicotine lasted 28 ± 6 s (n = 4), whereas in WT injected with 10 mg/kg, seizure duration was 33 ± 12 s (n = 5).
Pretreatment with a comparatively small dose of nicotine (0.1 mg/kg) shifted the Het seizure dose-response relationship to the right (Fig. 6C). Such pretreatment completely blocked 0.2, 0.5, and 1 mg/kg nicotine-induced seizures in Het mice and shifted the ED50 from 0.4 ± 0.1 for saline-preinjected mice to 1.9 ± 0.01 mg/kg for nicotine-preinjected mice. Nicotine-pretreated Het also showed a significant increase in latency to seizure compared with saline-pretreated animals (at 2 mg/kg; Student's t test, p < 0.001) (Fig. 6D). Het mice pretreated with 0.1 mg/kg nicotine, but injected 5 min later (instead of the usual 10 min) with a second nicotine injection, displayed no seizures either. In contrast, seizures in WT were neither blocked nor delayed by nicotine pretreatment (2 mg/kg): ED50 values for the saline- and nicotine-pretreated WT mice were 5.0 ± 0.7 and 5.6 ± 0.2 mg/kg, respectively. Pretreatment of WT mice with 0.1 mg/kg nicotine also failed to prevent seizures or prolong seizure latency. However, pretreatment with 6 mg/kg nicotine slightly reduced WT seizure latency compared with saline-pretreated animals (Student's t test, p = 0.044) (Fig. 6D). Thus, nicotine pretreatment blocked or delayed Het seizures but had no effect or hastened WT seizures. A modest protective effect by nicotine pretreatment against nicotine-induced seizures was reported previously in DBA mice (Miner and Collins, 1988). Discrepancies between the Miner and Collins study and ours could be attributed to differences in the mode of nicotine administration, mouse strain used, or time between nicotine injections.
The antiepileptic drug carbamazepine, injected at a dose of 20 mg/kg and delivered 30 min before a nicotine injection, had a modest protective effect against nicotine-induced seizures in WT, Het, and Hom (Table 1). In previous studies, carbamazepine delivered with the same time and dose successfully blocked seizures and/or diminished their effects in mice treated with pentylenetetrazole (Borowicz et al., 2003) or strychnine (Yamashita et al., 2004). The nicotinic antagonists DHβE and hexamethonium, at the doses and time regimens tested, were unable to suppress or delay nicotine-induced seizures. The DHβE dose (3 mg/kg) was limited by the fact that higher doses cause seizures (Dobelis et al., 2003). Seizures in mice pretreated with carbamazepine, DHβE, or hexamethonium were similar in intensity, latency, and duration to those of saline-pretreated animals. Only one dose and time regimen was tested for each of these drugs; thus, the possibility of successfully blocking seizures at a different dose and/or schedule remains. That neither DHβE nor hexamethonium blocked seizures could be attributed, among other possibilities, to pharmacokinetic factors, including poor ability to cross the blood-brain barrier. Seizures were, however, blocked in all mice pretreated with the nicotinic antagonist mecamylamine (0.5 mg/kg). Damaj et al. (1999) (see also Dobelis et al., 2003) tested various doses of DHβE, hexamethonium, and mecamylamine to assess their ability to block nicotine-induced seizures in various strains of mice. Consistent with our results, these authors found that only mecamylamine could effectively prevent nicotine-induced seizures.
EEG, mechanotransducer, and video recordings
During behavioral seizures, no obvious ictal EEG activity was seen in the traces of Het and Hom mice injected with 2 mg/kg nicotine (Fig. 7). In contrast, EEGs of WT mice having behavioral seizures showed spike-wave discharges after nicotine injection (10 mg/kg). Spike-wave traces are characterized by events at frequencies ranging from 1 to 5 Hz. Occurrences of spike-wave during EEG recordings of seizures are typically interpreted as synchronized firing of neuronal populations in the cerebral cortex.
Figure 7.

Traces from EEG recordings of WT, Het, and Hom mice before and after a single nicotine injection. EEG recordings were obtained from screw electrodes placed on the parietal cortex (above the hippocampus). During behavioral seizures, no changes were seen in the EEG traces of Het and Hom mice injected with 2 mg/kg nicotine. Recordings from WT and Hom (10 mg/kg) were obtained from two pairs of screw electrodes bilaterally implanted into left and right hemispheres. Bilateral and synchronous spike-wave traces were observed in the EEG of WT mice injected with 10 mg/kg nicotine during behavioral seizures. Hom mice injected with 10 mg/kg nicotine had two bouts of convulsions separated by an interictal period of 1-3 min (72 s in the example shown). Bilateral and synchronous spike-wave activity was present in the second convulsive burst of Hom mice injected with 10 mg/kg nicotine. Three WT mice were injected with 2 mg/kg nicotine; five animals were tested in each of the remaining treatment groups.
Interestingly, Hom mice treated with 10 mg/kg nicotine had two successive seizures (n = 5 mice): the first seizure started ∼20 s after nicotine injection, was clonic, and showed no EEG changes (similar to the 2 mg/kg seizures in L9′A Het and Hom described above). The second seizure began 2-4 min after the injection, was tonic-clonic, and had spike-wave discharges (similar to the 10 mg/kg seizures in WT mice described above). The two convulsive episodes in Hom treated with 10 mg/kg were separated by an interictal period that varied with each mouse between 1 and 4 min. Synchronized spike-wave discharges were recorded from both hemispheres of WT and Hom (n = 5 per genotype) with two pairs of screw electrodes bilaterally implanted. Small differences in the amplitude of spike-wave discharges between the left and right hemispheres of WT and Hom mice (10 mg/kg nicotine) were probably caused by variations in the placement of the differential and ground electrodes. Only one Het mouse was injected with 10 mg/kg nicotine, because it died after a quick-onset and violent seizure.
Most EEG recordings were obtained from mice with screw electrodes positioned on their parietal cortex, above the hippocampus (n = 5 per genotype). Other candidate (for ictal activity) brain regions were also examined with either screw or depth electrodes in a smaller set of animals (n = 2 or 3 mice per brain location; for coordinates, see Materials and Methods). No spike-wave discharges were observed in any of the brain regions examined in L9′A mice during seizures with nicotine injections of ≤2 mg/kg. However, modest increases in EEG amplitude and changes in frequency were detected in the traces of two (of three) Hets with depth electrodes in their dorsal hippocampus. For example, after nicotine injection of 2 mg/kg, there was 158% increase in the amplitude (peak to peak) of the EEG signal compared with the amplitude of the preinjection signal in one Het male. A WT injected with 2 mg/kg nicotine (no seizure), also implanted with depth electrodes in the hippocampus, had an increase in EEG amplitude of 63%. EEGs of all WT mice with screw electrodes in parietal cortex, somatosensory cortex, or primary motor cortex, or with depth electrodes in hippocampus or ventral thalamus, had spike-wave activity during nicotine-induced seizures.
Because surprisingly few EEG signals accompanied behaviorally assessed seizures in mutant mice injected with 2 mg/kg nicotine, we sought additional data by making simultaneous electroencephalographic and mechanical measurements, the latter with a mechanotransducer attached to the bottom of the cage (Fig. 8). There was no simple relationship between muscular activity and these mechanotransducer records; nonetheless, in both WT and Hom, the transducer output clearly indicated large movements during behaviorally assessed convulsions. In WT, movements detected by the transducer were accompanied by spike-wave activity. Power spectral analysis of mechanotransducer traces revealed peaks at 6-7 Hz in two Het and two Hom mice while they were having forelimb clonus. It was not always possible to record violent rhythmic movements during seizures with the mechanotransducer because mice sometimes lay on their sides or leaned against the side panels of the cage.
Figure 8.

Traces from simultaneous EEG and mechanotransducer recordings of WT and Hom mice before and after a single nicotine injection. EEG recordings were obtained from screw electrodes placed on the parietal cortex, on each hemisphere. A mechanotransducer connected to the bottom of the cage was used to record the frequency and intensity of the mouse's movements. Photographs were taken during seizures, within the time period of EEG and mechanotransducer traces shown. During seizures, WT displayed a variety of movements that included jumping, rearing, loss of righting response, and limb clonus; examples are shown in the photographic sequence. L9′A mice seizures were characterized by Straub tail, loss of righting response, and violent and repetitive movements of the forelimbs. n = 4 mice per genotype. WT and Hom mice shown in the photographs are N4 littermates.
Animals were also imaged with still and video recordings (Fig. 8) during seizures. WT seizures were characterized by an assortment of movements, with no two mice having the same sequence of movements. Movement during WT seizures included loss of righting response, jumping, rearing, limb clonus, and Straub tail (arching of the tail over the body). Het and Hom seizures displayed less variable behavioral patterns during 2 mg/kg nicotine-induced seizures than the WT group during 10 mg/kg nicotine-induced seizures. Het and Hom seizures were always preceded by Straub tail (Fonck et al., 2003), followed by loss of the righting response, and ending with violent forelimb clonus. The distinction between mutant-like seizures and WT-like seizures was maintained in Hom mice injected with enough nicotine (10 mg/kg) to provoke both seizure types. Thus, Hom mice injected with 10 mg/kg displayed an initial mutant-like seizure (early onset with forelimb clonus) and a second WT-like seizure (onset ∼4 min after injection, with various movements and spike-wave EEGs), with an interictal period of inactivity in between the two seizures. Within treatment groups, male and female mice had similar behavioral and electrophysiological responses.
L9′A mice have more brief awakenings
Spectral analysis of chronic EEG recordings was used to determine the fraction of time mice spent awake, in NREM sleep, and in REM sleep (Fig. 9A). The proportion of time mice spent in each of these states was similar among genotypes (one-way ANOVA, p = 0.16) and consistent with previously reported values for mice with a C57BL/6J background (Fig. 9D) (Franken et al., 1998). Brief movements or twitching events seen during NREM were accompanied by a decrease in power density. These behaviorally distinguishable events were labeled “brief awakenings” following previously published nomenclature (Tobler et al., 1996; Franken et al., 1999) and most likely include and/or are equivalent to “microarousal” episodes as described by Lena and et al. (2004). Brief awakenings were behaviorally distinct from waking episodes because they lasted only a few seconds and mice did not open their eyes, lift their heads, or move around their cages. Our analysis showed that Het and Hom mice had more brief awakenings per hour than WT (Fig. 9B,C). The increment in brief awakenings seen in L9′A compared with WT was evenly distributed over the light and dark phases. The total fraction of time mutant mice spent in these brief awakening was ∼ 0.3%, which does not alter significantly the proportion of time mice spent in the different vigilance states. In addition, Hom mice had 30-41% more sleep-wake transitions (excluding brief awakenings) than Het and WT, respectively (one-way ANOVA, followed by the Tukey's pairwise multiple comparison test, p < 0.01).
Mutant and WT mice also differed in their locomotor activity (Fig. 10). Het and Hom mice moved around their cages twice as much as WT during the lights-on portion of the day (Fig. 10A,C). A similar difference was noted during 24 h constant lights-on experiments (Fig. 10B,D).
Figure 10.

Locomotion of L9′A and WT mice. Locomotion was measured for 24 h in activity cages with infrared beams. Each event was defined as two successive beam breaks. A, Locomotion was recorded during 13 h with lights on and 11 h with lights off. B, Locomotion recorded with lights on for 24 h. C, Average locomotion activity was larger in Het and Hom mice compared with WT during lights-on period as measured in A (each bar represents the mean and SEM; n = 8; one-way ANOVA, followed by the Tukey's pairwise multiple comparison test, *p < 0.05). D, Average locomotion activity was larger in Het and Hom mice compared with WT during a 24 h continuous lights-on experiment (each bar represents the mean and SEM; n = 8; Kruskal-Wallis test, followed by the Tukey's pairwise multiple comparison test, *p < 0.01). The same animals were studied in experiments A and B.
We sought, but did not find, evidence for spontaneous seizures in L9′A mice. No clear ictal activity was identified in chronic EEG recordings of L9′A mice. Likewise, portions of video recordings examined (≥2 h/animal) did not show behaviors that could be construed as convulsive activity.
Neuroanatomy
Nissl-stained brain sections obtained from WT and Hom mice were examined to determine whether expression of the L9′A mutation caused neuroanatomical abnormalities. The morphology of major brain nuclei, including the hippocampus, frontal cortex, midbrain (ventral tegmental area and substantia nigra), striatum, thalamus, and cerebellum, was similar between WT and Hom (images not shown). The absence of gross abnormalities in the brains of L9′A mice is consistent with the neuroanatomical analysis of L9′S mice, whose mutated α4 receptors are even more sensitive to agonists than L9′A ones (Fonck et al., 2003).
Discussion
L9′A mice show a marked increase (∼15-fold) in their sensitivity to nicotine-induced seizures compared with their WT littermates. Het and Hom L9′A mice do not simply have WT-like seizures at abnormally low nicotine concentrations, but rather, nicotine-induced seizures in L9′A differ qualitatively from those of WT. Temporal, pharmacological, electroencephalographic, and behavioral differences between WT and L9′A nicotine seizures strongly suggest the involvement of distinct neuronal circuits, which are likely determined in each case by different activation thresholds of α4-containing nAChRs. Many of the nicotine-induced seizure characteristics displayed by L9′A mice are similar to those experienced spontaneously by ADNFLE patients. Moreover, the L9′A mutation caused changes in receptor function consistent with previous reports of ADNFLE mutations expressed in Xenopus oocytes.
The major conclusion then is that, when one replaces WT α4 subunits in mice by modestly hypersensitive α4 subunits to produce hypersensitive α4* nicotinic receptors (predominantly α4β2), nicotine produces a syndrome with seizures similar to those experienced spontaneously by α4- or β2-linked human ADNFLE. Together with anecdotal evidence that α4- and β2-knock-out mice show neither spontaneous nor enhanced nicotine-induced seizures, these data leave little doubt that human α4- or β2-associated ADNFLE is caused by enhanced α4β2 receptor activity.
Effects of the L9′A mutation on receptor function and expression
Experiments in oocytes show that the α4 L9′A mutation increases the sensitivity of the mouse α4β2 nicotinic receptor to nicotine-induced activation and desensitization. However, it does not affect ion selectivity of the channel for physiologically relevant cations, Na+, K+, and Ca2+. We predicted that L9′A would increase receptor agonist sensitivity based on previous studies showing that the 9′ position is critical for gating and that hydrophilic substitutions increase open-channel time (Labarca et al., 1995; Kearney et al., 1996; Kosolapov et al., 2000). All known ADNFLE-associated mutations in the α4 subunit (S6′F, S10′L, +L18′, and T19′I) also increase agonist sensitivity of α4β2 nAChR (Bertrand et al., 2002; Leniger et al., 2003). Enhanced desensitization was also reported in the ADNFLE S6′F mutation (Weiland et al., 1996; Bertrand et al., 2002).
Our fura-2 and nicotine-stimulated 86Rb+ efflux results indicate that the forebrains of L9′A mice express hypersensitive nAChRs. This hypersensitivity, as evidenced by leftward shifts in nicotine EC50 values, was expected from, and is consistent with, the oocyte expression data. We measured actual receptor expression levels by an appropriately direct method: epibatidine binding in brain tissue homogenates. We found a gene-dose-dependent decrease in such binding in L9′A mice. However, Hom and Het had similar maximum responses for nicotine-induced [Ca2+]i increases in cultured thalamic neurons, 86Rb+ release in synaptosomes, and seizures in mice. These results run contrary to the expectation that Hom would be more sensitive than Het mice.
Other experiments (to be reported elsewhere) show that normally sensitive α4* receptors, still containing the 42 bp loxP site plus associated restriction site in a neighboring intron, are expressed at normal levels in knock-in mice. Hence, the modestly decreased binding level (less than twofold) does not occur because of a molecular genetic artifact associated with construction of the mouse strain. We speculate that the decrement in L9′A receptor expression in vivo arises because of homeostatic mechanisms that respond to hyperactive receptors. If this suggestion is correct, mice are able to compensate functionally for this hyperactivity: they show seizures only after nicotine injections. Humans with α4*- or β2*-linked ADNFLE compensate slightly less completely: they have subtle seizures that escaped clinical description until 1994 (Scheffer et al., 1994).
Nicotine-induced seizures: type, sensitivity, and pharmacology
Comparison of the EEG recordings and behavior between WT and L9′A mice suggests the recruitment of distinct neuronal populations during nicotine-induced seizures. Importantly, nicotine-induced seizures in WT are generalized: they show spike-wave activity in all cortical locations examined, are initiated simultaneously in each hemisphere, and involve violent whole-body movements. Whereas WT mice display spike-wave EEG during seizures (10 mg/kg nicotine), L9′A (2 mg/kg) show no recognizable ictal activity with electrodes positioned at various cortical locations. Previous studies with anesthetized rabbits suggest that nicotine-induced seizures leading to spike-wave discharges originate in the hippocampus (Floris et al., 1964; Stumpf and Gogolak, 1967). We cannot rule out WT seizures with secondary bilateral synchrony in which there is rapid generalization from one or multiple foci, one of which could be the hippocampus. Nicotine-induced seizures in L9′A mice can be characterized as partial because they were electrophysiologically subtle or undetectable in the cortex, and movements were limited to loss of righting response and forelimb clonus. Except for modest increases in the amplitude of the EEGs of two Het mice with depth electrodes in their hippocampus, we were unable to discover the focal origin of L9′A seizures at 2 mg/kg nicotine. Behavioral patterns, homogenous and repetitive in Het and Hom and complex and polymorphic in WT, suggest the involvement of more limited cortical motor control areas during L9′A seizures than in WT ones. Differences in the average seizure latency, 43 s in L9′A but 323 s in WT mice, are also consistent with distinct seizure mechanisms.
Nicotine pretreatment blocked mutant, but not WT, seizures. In oocytes and synaptosomes, nicotine caused more desensitization in L9′A receptors compared with WT ones. Thus, nicotine pretreatment of mice appears to act by desensitizing most L9′A nAChRs and rendering them unresponsive to additional nicotine stimulation (small window activation, on the order of 1%, occurs even at steady-state desensitization). This suggests that nicotine-induced seizures in L9′A mice are initiated by the activation, not desensitization, of mutated hypersensitive α4* nAChRs. A similar mechanism could account for the effects of nicotine on an ADNFLE patient resistant to conventional antiepileptic drugs (including carbamazepine) but successfully treated with a nicotine patch (Willoughby et al., 2003). That nicotine pretreatment did not prevent nicotine-induced seizures in WT mice suggests a different seizure mechanism, perhaps involving desensitization of α4* receptors on inhibitory interneurons, leading to neuronal synchronization and to overexcitation (Mody, 1998; Dobelis et al., 2003).
Although nicotine-induced seizures in L9′A mice are most likely mediated by α4* receptors, it is not clear what role, if any, these receptors play in WT seizures. Because higher nicotine doses are required, we speculate that different nAChR subtypes are activated (and or desensitized) during WT seizures. Indeed, studies on knock-out mice indicate that other nicotinic subunits (α3, α5, and β4) are more important than those containing α4 in mediating nicotine seizures in WT (Kedmi et al., 2004; Salas et al., 2004). Although there are no published reports on the nicotine-induced seizure phenotype of α4 knock-out mice, we recently found that these mice are similar to their WT littermates in their sensitivity to nicotine seizures (our unpublished data). In contrast to α4 knock-out mice, but similar to L9′A mice, mouse lines engineered to express ADNFLE mutations +L264 and S252F are reportedly hypersensitive to nicotine-induced seizures (Klaassen et al., 2004).
Sleep disruptions
Chronic EEG recordings revealed a significant increase in the number of brief awakenings during NREM sleep in L9′A compared with WT mice. This result follows the trend of recent findings that knock-out mice lacking the β2 subunit (the major partner of α4) showed less microarousals and sleep-wake transitions during NREM (Lena et al., 2004). Our results support a role for α4-containing nAChRs in maintaining the structure of NREM sleep. In humans, ADNFLE seizures and transient arousals typically manifest during the same light sleep period (phase 2). Moreover, until a decade ago, ADNFLE patients were misdiagnosed with childhood onset parasomnia, which is a condition that has also been associated with frequent arousals and a consequent fragmentation of NREM sleep (Scheffer et al., 1994; Zucconi and Ferini-Strambi, 2000). These authors suggest that similar mechanisms may underlie excessive transient awakenings during sleep and nocturnal frontal lobe epilepsies and parasomnias. Increased locomotion in L9′A compared with WT mice during lights-on hours may be another indication that sleep-wake patterns are altered as a consequence of expressing gain-of-function α4-containing nAChRs.
Similarities with ADNFLE
In contrast to WT, nicotine-induced seizures in L9′A Het and Hom mice share a number of features with seizures experienced by ADNFLE patients. (1) ADNFLE seizures are typically characterized by repetitive jerky limb movements and asymmetrical posturing (Scheffer et al., 1995) (for review, see Combi et al., 2004). Nicotine seizures in L9′A mice consist of forelimb clonus and loss of righting response (which can also be described as asymmetrical posturing). (2) Nicotine suppressed ADNFLE seizures in the single patient tested to date (Willoughby et al., 2003). Nicotine pretreatment blocked nicotine-induced seizures in L9′A mice. (3) Carbamazepine, which remains the drug of choice in treating ADNFLE, is ineffective in a significant proportion of patients (Oldani et al., 1998). Furthermore, a majority of patients with the S10′L mutation, adjacent to the 9′ position, are resistant to carbamazepine treatment (Ito et al., 2000; Cho et al., 2003). At the dose tested, carbamazepine had a modest effect in preventing nicotine-induced seizures in L9′A mice. (4) Another important parallel between ADNFLE and nicotine seizures in L9′A mice is that ∼70% of ADNFLE patients have nondiagnostic EEGs (Scheffer et al., 1995; Oldani et al., 1998; Rozycka et al., 2003) (for review, see Combi et al., 2004). Nicotine seizures in L9′A were undetected by EEG. Thus, the absence of interictal or ictal EEG activity during chronic recordings of L9′A mice should be considered inconclusive with regards to the occurrence (or lack of) of spontaneous seizures, but many hours of observations also revealed no obvious seizures in L9′A animals. (5) L9′A mice, however, had abnormal NREM sleep with excessive brief awakenings. Many ADNFLE patients also suffer from altered sleep-wake patterns (Oldani et al., 1998; Roberts, 1998).
No existing study satisfactorily explains the pathophysiology of even a monogenic human seizure disorder, much less that of a polygenic disorder. This situation contrasts with the straightforward pathophysiology of monogenic channelopathies in other organs: long QT syndrome, hyperinsulinemia, and cystic fibrosis (Ashcroft, 2000). Our results do, however, serve a more modest purpose: we emphasize the usefulness of iterative experiments comparing the biophysical properties of heterologously expressed receptors with the phenotype of the corresponding knock-in mice. The sequence involved biophysical analysis of the Leu9′Ser mutation, study of the corresponding knock-in mice (Labarca et al., 2001; Fonck et al., 2003), design of the Leu9′Ala mutation, and study of the corresponding knock-in mice. Such a program allows one to target a precise set of receptors, with the consequent activation of specific brain circuits, which in turn trigger unique behavioral responses. We conclude that α4- or β2-linked ADNFLE arises from excessive activation of α4β2 nAChRs.
Footnotes
This work was supported by National Institute of Health Grants NS46464, NS43800, NS11756, DA10156, DA03194, and DA17279 and a National Institutes of Health-National Research Service Award (C.F.). The California Tobacco-Related Disease Research Program and Philip Morris USA/International are gratefully acknowledged. We thank J. Stitzel for the mouse α4 cDNA, Chana Simon for oocyte data analysis, Robert Paz for help with photography, and Sharon Grady, Andrew Tapper, and Johannes Schwarz for insightful discussion.
Correspondence should be addressed to Henry A. Lester, 156-29, California Institute of Technology, Pasadena, CA 91125. E-mail: lester@caltech.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511396-16$15.00/0
References
- Ashcroft FM (2000) Ion channels and disease: channelopathies. San Diego: Academic.
- Bertrand D, Picard F, Le Hellard S, Weiland S, Favre I, Phillips H, Bertrand S, Berkovic SF, Malafosse A, Mulley J (2002) How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia 43 [Suppl 5]: 112-122. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D (1998) Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol 125: 751-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowicz KK, Luszczki JJ, Duda AM, Czuczwar SJ (2003) Effect of topiramate on the anticonvulsant activity of conventional antiepileptic drugs in two models of experimental epilepsy. Epilepsia 44: 640-646. [DOI] [PubMed] [Google Scholar]
- Buisson B, Bertrand D (2001) Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci 21: 1819-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YW, Motamedi GK, Laufenberg I, Sohn SI, Lim JG, Lee H, Yi SD, Lee JH, Kim DK, Reba R, Gaillard WD, Theodore WH, Lesser RP, Steinlein OK (2003) A Korean kindred with autosomal dominant nocturnal frontal lobe epilepsy and mental retardation. Arch Neurol 60: 1625-1632. [DOI] [PubMed] [Google Scholar]
- Cohen BN, Labarca C, Davidson N, Lester HA (1992) Mutations in M2 alter the selectivity of the mouse nicotinic acetylcholine receptor for organic and alkali metal cations. J Gen Physiol 100: 373-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BN, Figl A, Quick MW, Labarca C, Davidson N, Lester HA (1995) Regions of beta 2 and beta 4 responsible for differences between the steady state dose-response relationships of the alpha 3 beta 2 and alpha 3 beta 4 neuronal nicotinic receptors. J Gen Physiol 105: 745-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combi R, Dalpra L, Tenchini ML, Ferini-Strambi L (2004) Autosomal dominant nocturnal frontal lobe epilepsy—a critical overview. J Neurol 251: 923-934. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Glassco W, Dukat M, Martin BR (1999) Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther 291: 1284-1291. [PubMed] [Google Scholar]
- De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, Ballabio A, Wanke E, Casari G (2000) The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet 26: 275-276. [DOI] [PubMed] [Google Scholar]
- Dobelis P, Hutton S, Lu Y, Collins AC (2003) GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J Pharmacol Exp Ther 306: 1159-1166. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Arneric SP, Sullivan JP (1995) Functional modulation of human “ganglionic-like” neuronal nicotinic acetylcholine receptors (nAChRs) by L-type calcium channel antagonists. Biochem Biophys Res Commun 213: 657-662. [DOI] [PubMed] [Google Scholar]
- Fatt P, Ginsborg BL (1958) The ionic requirements for the production of action potentials in crustacean muscle fibres. J Physiol (Lond) 142: 516-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA (1997) Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 17: 5747-5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figl A, Viseshakul N, Shafaee N, Forsayeth J, Cohen BN (1998) Two mutations linked to nocturnal frontal lobe epilepsy cause use-dependent potentiation of the nicotinic ACh response. J Physiol (Lond) 513: 655-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatov GN, White MM (1995) The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol 48: 379-384. [PubMed] [Google Scholar]
- Floris V, Morocutti C, Ayala GF (1964) Effects of nicotine on the cortical, thalamic and hippocampal electrical activity in rabbits. J Cardiovasc Nurs 141: 247-251. [PubMed] [Google Scholar]
- Fonck C, Nashmi R, Deshpande P, Damaj MI, Marks MJ, Riedel A, Schwarz J, Collins AC, Labarca C, Lester HA (2003) Increased sensitivity to agonist-induced seizures, Straub tail, and hippocampal theta rhythm in knock-in mice carrying hypersensitive α4 nicotinic receptors. J Neurosci 23: 2582-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M (1998) Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol 275: R1127-R1137. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M (1999) Genetic determinants of sleep regulation in inbred mice. Sleep 22: 155-169. [PubMed] [Google Scholar]
- Haghighi AP, Cooper E (2000) A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine α3β4 and α4β2 receptors. J Neurosci 20: 529-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S, Iwata H, Akiyoshi H, Kobayashi K, Ito M, Wada K, Kaneko S, Mitsudome A (1999) A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology 53: 1749-1753. [DOI] [PubMed] [Google Scholar]
- Ito M, Kobayashi K, Fujii T, Okuno T, Hirose S, Iwata H, Mitsudome A, Kaneko S (2000) Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia 41: 52-58. [DOI] [PubMed] [Google Scholar]
- Jia L, Flotildes K, Li M, Cohen BN (2003) Nicotine trapping causes the persistent desensitization of alpha4beta2 nicotinic receptors expressed in oocytes. J Neurochem 84: 753-766. [DOI] [PubMed] [Google Scholar]
- Katz B, Thesleff S (1957) A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol (Lond) 138: 63-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PC, Zhang H, Zhong W, Dougherty DA, Lester HA (1996) Determinants of nicotinic receptor gating in natural and unnatural side chain structures at the M2 9′ position. Neuron 17: 1221-1229. [DOI] [PubMed] [Google Scholar]
- Kedmi M, Beaudet AL, Orr-Urtreger A (2004) Mice lacking neuronal nicotinic acetylcholine receptor beta4-subunit and mice lacking both alpha5- and beta4-subunits are highly resistant to nicotine-induced seizures. Physiol Genomics 17: 221-229. [DOI] [PubMed] [Google Scholar]
- Klaassen A, Maguire J, Glykys J, Labarca C, Mody I, Boulter J (2004) Abnormal basal EEG and heightened nicotine-induced seizure response in a mouse model of ADNFLE. Soc Neurosci Abstr 30: 48.14. [Google Scholar]
- Kosolapov AV, Filatov GN, White MM (2000) Acetylcholine receptor gating is influenced by the polarity of amino acids at position 9′ in the M2 domain. J Membr Biol 174: 191-197. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J (1997) Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci 17: 9035-9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA (1995) Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature 376: 514-516. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA (2001) Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci USA 98: 2786-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lena C, Popa D, Grailhe R, Escourrou P, Changeux JP, Adrien J (2004) β2-Containing nicotinic receptors contribute to the organization of sleep and regulate putative micro-arousals in mice. J Neurosci 24: 5711-5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leniger T, Kananura C, Hufnagel A, Bertrand S, Bertrand D, Steinlein OK (2003) A new Chrna4 mutation with low penetrance in nocturnal frontal lobe epilepsy. Epilepsia 44: 981-985. [DOI] [PubMed] [Google Scholar]
- Lester RA (2004) Activation and desensitization of heteromeric neuronal nicotinic receptors: implications for non-synaptic transmission. Bioorg Med Chem Lett 14: 1897-1900. [DOI] [PubMed] [Google Scholar]
- Lopez MG, Fonteriz RI, Gandia L, de la Fuente M, Villarroya M, Garcia-Sancho J, Garcia AG (1993) The nicotinic acetylcholine receptor of the bovine chromaffin cell, a new target for dihydropyridines. Eur J Pharmacol 247: 199-207. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Smith KW, Collins AC (1998) Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther 285: 377-386. [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, Picciotto MR, Changeux JP, Collins AC (1999) Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther 289: 1090-1103. [PubMed] [Google Scholar]
- Matsushima N, Hirose S, Iwata H, Fukuma G, Yonetani M, Nagayama C, Hamanaka W, Matsunaka Y, Ito M, Kaneko S, Mitsudome A, Sugiyama H (2002) Mutation (Ser284Leu) of neuronal nicotinic acetylcholine receptor alpha 4 subunit associated with frontal lobe epilepsy causes faster desensitization of the rat receptor expressed in oocytes. Epilepsy Res 48: 181-186. [DOI] [PubMed] [Google Scholar]
- Miner LL, Collins AC (1988) Effect of nicotine pretreatment on nicotine-induced seizures. Pharmacol Biochem Behav 29: 375-380. [DOI] [PubMed] [Google Scholar]
- Mody I (1998) Interneurons and the ghost of the sea. Nat Neurosci 1: 434-436. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA (2003) Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci 23: 11554-11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldani A, Zucconi M, Asselta R, Modugno M, Bonati MT, Dalpra L, Malcovati M, Tenchini ML, Smirne S, Ferini-Strambi L (1998) Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain 121: 205-223. [DOI] [PubMed] [Google Scholar]
- Orb S, Wieacker J, Labarca C, Fonck C, Lester HA, Schwarz J (2004) Knockin mice with Leu9′Ser α4 nicotinic receptors: substantia nigra dopaminergic neurons are hypersensitive to agonist and lost postnatally. Physiol Genomics 18: 299-307. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Broide RS, Kasten MR, Dang H, Dani JA, Beaudet AL, Patrick JW (2000) Mice homozygous for the L250T mutation in the α7 nicotinic acetylcholine receptor show increased neuronal apoptosis and die within 1 day of birth. J Neurochem 74: 2154-2166. [DOI] [PubMed] [Google Scholar]
- Petersen DR, Norris KJ, Thompson JA (1984) A comparative study of the disposition of nicotine and its metabolites in three inbred strains of mice. Drug Metab Dispos 12: 725-731. [PubMed] [Google Scholar]
- Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Sutherland GR, Mulley JC (1995) Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2. Nat Genet 10: 117-118. [DOI] [PubMed] [Google Scholar]
- Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D, Heron SE, Scheffer IE, Sutherland GR, Berkovic SF, Bertrand D, Mulley JC (2001) CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet 68: 225-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Lester HA (1994) Methods for expression of excitability proteins in Xenopus oocytes. In: Ion channels of excitable cells (Narahashi T, ed), pp 261-279. San Diego: Academic.
- Revah F, Bertrand D, Galzi JL, Devillers-Theiry A, Mulle C (1991) Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353: 846-849. [DOI] [PubMed] [Google Scholar]
- Roberts R (1998) Differential diagnosis of sleep disorders, non-epileptic attacks and epileptic seizures. Curr Opin Neurol 11: 135-139. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Pinguet N, Jia L, Li M, Figl A, Klaassen A, Truong A, Lester HA, Cohen BN (2003) Five ADNFLE mutations reduce the Ca2+ dependence of the α4β2 acetylcholine response. J Physiol (Lond) 550: 11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozycka A, Skorupska E, Kostyrko A, Trzeciak WH (2003) Evidence for S284L mutation of the CHRNA4 in a white family with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia 44: 1113-1117. [DOI] [PubMed] [Google Scholar]
- Salas R, Cook KD, Bassetto L, De Biasi M (2004) The alpha3 and beta4 nicotinic acetylcholine receptor subunits are necessary for nicotine-induced seizures and hypolocomotion in mice. Neuropharmacology 47: 401-407. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann F, Andermann E, Desbiens R, Cendes F, Manson JI (1994) Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet 343: 515-517. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F (1995) Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain 118: 61-73. [DOI] [PubMed] [Google Scholar]
- Shen JB, Jiang B, Pappano AJ (2000) Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J Pharmacol Exp Ther 294: 562-570. [PubMed] [Google Scholar]
- Simmons DM, Swanson LW (1993) The Nissl stain. In: Brain research protocols, pp 1-7. Amsterdam: Elsevier.
- Steinlein OK (2004) Genetic mechanisms that underlie epilepsy. Nat Rev Neurosci 5: 400-408. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11: 201-203. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, Nakken KO, Propping P, Bertrand D (1997) An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet 6: 943-947. [DOI] [PubMed] [Google Scholar]
- Stotz SC, Spaetgens RL, Zamponi GW (2000) Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J Membr Biol 174: 157-165. [DOI] [PubMed] [Google Scholar]
- Stumpf C, Gogolak G (1967) Actions of nicotine in the limbic system. Ann NY Acad Sci 142: 125-128. [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA (2004) Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science 306: 1029-1032. [DOI] [PubMed] [Google Scholar]
- Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC (1996) Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380: 639-642. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW (1989) Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284: 314-335. [DOI] [PubMed] [Google Scholar]
- Wang F, Imoto K (1992) Pore size and negative charge as structural determinants of permeability in the Torpedo nicotinic acetylcholine receptor channel. Proc Biol Sci 250: 11-17. [DOI] [PubMed] [Google Scholar]
- Wang X, Treistman SN, Lemos JR (1992) Two types of high-threshold calcium currents inhibited by omega-conotoxin in nerve terminals of rat neurohypophysis. J Physiol (Lond) 445: 181-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O (1996) An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 398: 91-96. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ (2002) Involvement of the α3 subunit in central nicotinic binding populations. J Neurosci 22: 2522-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ, Lindstrom JM (1988) Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J Neurosci 8: 3395-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby JO, Pope KJ, Eaton V (2003) Nicotine as an antiepileptic agent in ADNFLE: an N-of-one study. Epilepsia 44: 1238-1240. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Ohno K, Amada Y, Hattori H, Ozawa-Funatsu Y, Toya T, Inami H, Shishikura J, Sakamoto S, Okada M, Yamaguchi T (2004) Effects of 2-[N-(4-chlorophenyl)-N-methylamino]-4H-pyrido[3.2-e]-1,3-thiazin-4-one (YM928), an orally active alpha-amino-3-hydroxy-5-methyl-4-isox-azolepropionic acid receptor antagonist, in models of generalized epileptic seizure in mice and rats. J Pharmacol Exp Ther 308: 127-133. [DOI] [PubMed] [Google Scholar]
- Zucconi M, Ferini-Strambi L (2000) NREM parasomnias: arousal disorders and differentiation from nocturnal frontal lobe epilepsy. Clin Neurophysiol 111 [Suppl 2]: S129-S135. [DOI] [PubMed] [Google Scholar]