

Abstract
AMPA-type glutamate receptors play a key role in mediating postsynaptic responses of excitatory neurotransmitters. It is now well accepted that AMPA receptors are also present at the presynapse, where they are thought to modulate neurotransmitter release. However, the mechanisms through which they control synaptic vesicle traffic have remained elusive. We used cultured hippocampal neurons and growth cone particles prepared from fetal rat brain to investigate the functional role of presynaptic AMPA receptors. We show here that stimulation of presynaptic AMPA receptors induces activation of mitogen-activated protein kinase (MAPK) through a nonreceptor tyrosine kinase-dependent and Na+/Ca2+-independent mechanism. This pathway is activated predominantly in axonal growth cones compared with the somatodendritic compartment. After stimulation of presynaptic AMPA receptors, synapsin I is phosphorylated at MAPK-specific sites. These events are paralleled by a prominent increase in evoked synaptic vesicle recycling that is blocked by the specific MAPK inhibitor 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one. Similarly, in synaptosomes isolated from adult brain, AMPA stimulation induces MAPK activation and phosphorylation of synapsin I at MAPK-dependent sites and enhances significantly synaptic vesicle recycling. These results reveal a novel pathway for activation of presynaptic MAPK and suggest a role of this pathway in the regulation of short-term presynaptic plasticity.
Keywords: presynaptic AMPA receptor, MAP kinase, synaptic plasticity, vesicle recycling, growth cone, synaptogenesis
Introduction
AMPA-type glutamate receptors (GluRs) are considered the most important receptors for fast excitatory neurotransmission and are of paramount importance for the postsynaptic modulation of synaptic strength (Malinow and Malenka, 2002). In addition to their well established postsynaptic localization, AMPA receptors (AMPARs) are also present on presynaptic membrane compartments (Fabian-Fine et al., 2000; Satake et al., 2000; Chang and De Camilli, 2001; Lee et al., 2002; Lu et al., 2002; Schenk et al., 2003). In the hippocampus and the cerebral cortex, a major pool of these presynaptic receptors resides on recycling vesicles, so that functional receptors are inserted into the presynaptic membrane in response to neuronal activity (Schenk et al., 2003). Thus, AMPA receptors may participate in an activity-dependent autocrine or paracrine feedback signaling responsible for the modulation of presynaptic function.
At present, little is known about the effects of axonal AMPA/kainate receptor activation on neuronal function. At the mature synapse, their activation was shown to modulate neurotransmitter release with inhibitory or facilitating effects, depending on the system (MacDermott et al., 1999; Satake et al., 2000; Lee et al., 2002). Early in development, they regulate the motility of axonal filopodia (Chang and De Camilli, 2001). Specifically, low kainate concentrations increase filopodia motility, whereas high kainate concentrations have the opposite effect (Tashiro et al., 2003). Thus, an increased local concentration of glutamate at sites of synapse formation may play a role, via an autocrine mechanism, in promoting a transition from filopodia to a mature synapse (Chang and De Camilli, 2001; Tashiro et al., 2003). The molecular mechanisms at the basis of these effects are mostly unknown, and ion influxes through the receptor may only partially account for the observed changes (Lee et al., 2002). Interestingly, several studies have shown that postsynaptic AMPA receptors, in addition to their function as ion channels, also possess metabotropic properties, leading to the activation of mitogen-activated kinase (MAPK) or to the inhibition of adenylate cyclase (Wang and Durkin, 1995; Wang et al., 1997; Hayashi et al., 1999; Perkinton et al., 1999). It is presently not known whether presynaptic AMPA receptors also have metabotropic properties.
MAPKs [extracellular signal-regulated kinase (erk)1,2] are highly conserved enzymes that mediate the actions of cell surface receptors to regulatory targets within the cell (Chang and Karin, 2001). Their activation by phosphorylation is achieved by a signaling cascade of three serially linked protein kinases. Activated MAPKs may either phosphorylate cytosolic proteins or translocate to the nucleus, where they regulate transcription factors (for review, see Chang and Karin, 2001). erk1,2 are highly abundant in the CNS, where they regulate a variety of neuronal processes such as response to growth factors, growth cone dynamics, long-term potentiation, and apoptosis. Presynaptically, MAPK activation is triggered by neurotrophic factors and cytosolic calcium elevations (for review, see Grewal et al., 1999). In this study, we demonstrate that a relevant functional consequence of presynaptic AMPA receptor stimulation consists in the activation of the MAPK signaling pathway. In addition, we show that activated MAPK modulates presynaptic vesicle dynamics by increasing the rate of evoked recycling.
Materials and Methods
Materials
Monoclonal antibodies against synaptobrevin/vesicle-associated membrane protein (VAMP) 2, synaptophysin, and NR1 as well as polyclonal antibodies against vesicular glutamate transporter 1 (vGlut1), vGlut2, and vesicular GABA transporter were from Synaptic System (Goettingen, Germany); antibodies against TI-VAMP were a kind gift from Dr. T. Galli (Institut National de la Santé et de la Recherche Médicale, Paris, France). Monoclonal antibodies against growth-associated protein 43 (GAP43) and activated erk1,2 were purchased from Sigma (Milan, Italy). Phosphospecific polyclonal antibodies against synapsin I were a kind gift from A. Nairn and A. Czernik (Yale University, New Haven, CT). Polyclonal antibodies against GluR1 and GluR2/3 were purchased from Chemicon (Temecula, CA); antibodies against MAP2 were a kind gift from Dr. A. Matus (Friedrich Miescher Institute, Basel, Switzerland). Monoclonal antibodies against tau-1 were from Boehringer Mannheim (Mannheim, Germany). Phalloidin conjugated to FITC or tetramethyl-rhodamine isothiocyanate (TRITC) was purchased from Molecular Probes (Eugene, OR). The secondary antibodies conjugated to FITC, Texas Red, or peroxidase were obtained from Jackson ImmunoResearch (West Grove, PA). AMPA was purchased from Tocris Cookson (Bristol, UK). 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98 059), 4-(8-methyl-9H-1,3-dioxolo [4,5-h][2,3]benzodiazepin-5-yl)benzenamine dihydrochloride (GYKI52466), and pertussis toxin were obtained from Sigma. 3-(4-Chlorophenyl)-1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidine-4-amine (PP2) and methyl-9-(S)-12(R)-epoxy-1H-diindolo[1,2,3-fg:3′2′1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-2,3,9,10,11,12-hexahydro-10-(R)-hydroxy-9-methyl-1-oxo-10-carboxilate (K252a) were purchased from Calbiochem (La Jolla, CA). BDNF was a kind gift from Dr. M. Canossa (University of Bologna, Bologna, Italy).
Hippocampal cell cultures
Primary neuronal cultures were prepared from the hippocampi of 18-d-old fetal rats as described previously (Bartlett and Banker, 1984). The dissociated cells were plated onto glass coverslips coated with poly-l-lysine at densities ranging from 250,000 to 400,000 cells/cm2. After a few hours, the coverslips were transferred to dishes containing a monolayer of cortical glial cells (Bartlett and Banker, 1984). The cells were maintained in MEM (Invitrogen, San Diego, CA) without sera, supplemented with N2 (Invitrogen), 2 mm glutamine, and 1 mg/ml BSA (neuronal medium).
Immunocytochemistry and assays for synaptic vesicle recycling
At 6 d in vitro (DIV), cultured hippocampal neurons were stimulated with 50 μm AMPA in Krebs'-Ringer's solution buffered with HEPES that was devoid of calcium (KRH-Ca) [containing (in mm): 150 NaCl, 5 KCl, 1.2 MgSO4, 10 glucose, 10 HEPES/NaOH, pH 7.4]. In some experiments, sodium was substituted by 150 mm choline (KRH-Ca/Na). Pharmacological inhibitors were added 15-20 min before the stimulation with AMPA. The cultures were fixed and stained as described previously (Verderio et al., 1994). For staining of the activated form of erk1,2, 1 μm vanadate was added to all solutions used after fixation. The images were acquired using a Bio-Rad (Hercules, CA) MRC-1024 confocal microscope equipped with LaserSharp 3.2 software with fixed parameters. Images were analyzed by MetaMorph Imaging Series 6.1 software (Universal Imaging, West Chester, PA). Growth cones were scored as phosphorylated MAPK (P-MAPK)-positive when the fluorescence intensity was at least three times higher compared with growth cones of cultures exposed only to secondary antibodies. Actin was visualized by TRITC- or FITC-conjugated phalloidin (Molecular Probes).
Exo-endocytotic assay. An exo-endocytotic assay to monitor synaptic vesicle recycling was performed using rabbit polyclonal antibodies directed against the intravesicular domain of rat synaptotagmin I (Syt-ecto antibodies) (Matteoli et al., 1992). Where necessary, neurons were preincubated with inhibitors for 15 min at 37°C. Neurons were stimulated with 50 μm AMPA in KRH-Ca for 2 min. After extensive washes, they were kept at 37°C for 2 min. Incubations with the antibody were performed in KRH containing 10 mm KCl as described previously (Verderio et al., 1999). After fixation and staining, cultures were imaged with a Bio-Rad MRC-1024 confocal microscope equipped with LaserSharp 3.2 software.
N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide uptake. Neurons were stimulated with 50 μm AMPA in Ca-free Tyrode's solution for 2 min. In some experiments, cultures were preincubated with 25 μm PD98059. The inhibitor was present during AMPA stimulation and was then removed. After extensive washes, cultures were kept in Ca-free Tyrode's solution at 37°C for 2 min. N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide (FM1-43; 15 μm) was then applied during a stimulation with 300 action potentials (APs) at 10 Hz, followed by washes for 10 min. Then neurons were again stimulated with 900 APs at 10 Hz, and a picture was taken every 10 s. All FM1-43 loading and unloading experiments were done in the presence of 20 μm CNQX. The destaining curve of FM1-43 was obtained using MetaMorph from Universal Imaging. Statistics were performed using SigmaStat (Systat Software, Point Richmond, CA). We first performed a nonparametric Mann-Whitney rank-sum test. It turned out, however, through a Kolmogrov-Smirnov test, that the data follow a Gaussian distribution. Hence, t tests were used throughout (see Results).
RNA interference
For plasmid-based RNA inhibition of GluR1, the following complementary oligonucleotides were annealed and inserted into the HindIII/BglII sites of pSuper vector (Oligoengine, Seattle, WA) (Brummelkamp et al., 2002): 5′-GATCCCCGAACTGGCAGGTAACGGCTTTCAAGAGAAGCCGTTACCTGCCAGTTCTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAGAACTGGCAGGTAACGGCTTCTCTTGAAAGCCGTTACCTGCCAGTTCGGG-3′ (corresponding to nucleotides 503-521 of rat GluR1). GluR2 small interfering RNA (siRNA) was performed as described previously (Passafaro et al., 2003). A sequence (5′-GATCCCCCACTGCAAGCTGTTCTGGATTCAAGAGATCCAGAACAGCTTGCAGTGTTTTTGGAAA-3′) ineffective in GluR2 silencing was also used as a control. Neurons were transfected with calcium phosphate at 2-3 DIV and used for experiments at 6 DIV.
Biochemical techniques
Growth cone particles. Growth cone particles (gcp) were prepared as described previously (Lockerbie et al., 1991; Lohse et al., 1996). Isolated gcp were diluted in 7-8 vol of modified Krebs'-Ringer's solution [containing (in mm): 180 sucrose, 50 NaCl2, 5 KCl, 22 HEPES, pH 7.4, 10 glucose, 1.2 NaH2PO4, 1.2 MgCl] containing antagonists, if needed, and kept on ice for 5 min. For stimulation, gcp were transferred to 37°C for 5-10 min, and then agonists were added for 5-10 min. A fraction of gcp was immediately resuspended in SDS-containing sample buffer for analysis of phosphoproteins. Only gcp, which, when stimulated in parallel, turned out to be responsive to BDNF (50-60% of gcp preparations) were taken into consideration for phosphoprotein analysis after AMPA stimulation. The remaining sample was extracted in 1% Triton X-100 for 10 min on ice and subsequently centrifuged for 1 h at 100,000 × g. Both pellet and supernatant were processed by SDS gel electrophoresis followed by Western blotting.
Synaptosome preparation. The purification of synaptosomes from rat forebrain was performed as described previously (Huttner et al., 1983). Samples with a protein content of 150 μg were pelleted and resuspended in KRH devoid of Ca2+/Na+ [containing (in mm): 150 choline, 5 KCl, 1.2 MgSO4, 10 glucose, 10 HEPES/KOH, pH 7.4] containing various inhibitors. The samples were kept for 10 min on ice and then transferred for 10 min to 37°C. After addition of agonists, they were left for 10 min at 37°C, and then a fraction corresponding to 50 μg of protein was mixed with SDS-containing sample buffer for phosphoprotein analysis. The remaining sample was extracted in 1% Triton X-100 for 30 min on ice and subsequently centrifuged for 1 h at 100,000 × g. Both pellet and supernatant were processed by SDS gel electrophoresis followed by Western blotting.
Phosphoprotein analysis. Equal amounts of growth cone or synaptosomal proteins were loaded on a SDS polyacrylamide gel and transferred electrophoretically to polyvinylidene difluoride membranes. Blots were stained with antibodies specifically recognizing phosphorylated erk1,2 and phosphorylated synapsin I in sites 4,5 (Jovanovic et al., 1996). Immunoreactive bands were visualized with ECL reagent (Pierce, Rockford, IL) and Hyperfilm (Amersham Biosciences, Arlington Heights, IL). For quantitation, all bands were normalized to tubulin and/or total erk1,2, and analysis was performed using NIH Image software.
Immunoisolation of synaptic vesicles. Synaptosomes or growth particles were lysed by hypoosmotic shock (Huttner et al., 1983). Membranes were pelleted by centrifugation at 25,000 × g for 30 min. Immunoisolation of vesicles present in the supernatant was performed with Dyna-beads M-450 rat anti-mouse IgG1 (Dynal, Oslo, Norway) according to the manufacturer's instructions.
Statistical analysis. Results are presented as means ± SE. Data were statistically compared using Student's t test. Differences were considered significant if p < 0.05 (single asterisk), p < 0.01 (double asterisks), p < 0.001 (triple asterisks).
Results
AMPA induces MAPK activation in the axonal growth cone
To investigate how AMPA receptor activation might modulate growth cone dynamics and neurotransmitter release, we first focused on axonal growth cones of cultured hippocampal neurons. In these structures, both AMPA receptors and synaptic vesicles are highly enriched (Pfenninger et al., 1983; Sabo and McAllister, 2003; Schenk et al., 2003) (see Fig. 2A). At 6 DIV, hippocampal neurons were stimulated with 50 μm AMPA for 5 min, fixed, and labeled with antibodies directed against the phosphorylated form of MAPK (Fig. 1A-C) in combination with phalloidin (Fig. 1A-C) or with antibodies directed against the axonal marker tau (data not shown) (Schenk et al., 2003). AMPA stimulation increased the levels of P-MAPK in all neuronal compartments, including the axonal growth cones (Fig. 1B). When AMPA stimulation was performed either in a Krebs'-Ringer's solution devoid of Ca2+ (Fig. 1C,D) or in a Ca2+-free Krebs'-Ringer's solution in which sodium was substituted by choline (data not shown), activation of MAPK occurred prominently at axonal growth cones and distal axons (percentage of P-MAPK-positive growth cones, 31.2 ± 1.8%, n = 148 in control growth cones; 76.8 ± 2.2%, n = 128; p = 1.9 × 10-8 after the application of AMPA), whereas no significant change in MAPK phosphorylation was observed in soma and dendrites (Fig 1B). Stimulation with AMPA under these conditions does not lead to ion influx (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that MAPK is activated in the growth cone through a calcium-independent mechanism. Lower AMPA concentrations (20 μm) also produced an increase of P-MAPK immunoreactivity in growth cones, although less efficiently, whereas 1-10 μm AMPA did not produce a detectable increase of P-MAPK immunoreactivity (data not shown). These results indicate that AMPA-mediated activation of MAPK in axonal growth cones can occur via a mechanism independent from ion influx through the receptor channel and/or depolarization. To further investigate the AMPA-triggered signaling pathway specifically in the axonal growth cone and to prevent the activation of parallel pathways controlled by Ca2+ influx, all subsequent experiments were performed in a Krebs'-Ringer's solution devoid of Ca2+ ions. Activation of MAPK in axonal growth cones, as revealed by an increase in P-MAPK immunoreactivity, was prevented by preincubation of neuronal cultures with either 100 μm GYKI52466, a noncompetitive, selective antagonist of AMPA-type glutamate receptors, or 30 μm PD98059, a selective inhibitor of MAP kinase kinase (MEK), the protein kinase upstream of MAPK (percentage of positive growth cones after AMPA application in the presence of GYKI52466, 33 ± 2.3%, n = 101; percentage of positive growth cones after AMPA application in the presence of PD98059, 18.8 ± 6.4%, n = 119) (Fig. 1D).
Figure 2.

Molecular pathways involved in the AMPA-induced MAPK activation. A-C, Small RNA interference (Rsi) for the GluR2 subunit of AMPA receptor inhibits MAPK activation after application of AMPA. A transfected axon, labeled by GFP and lacking the GluR2,3 subunit of AMPA receptors, does not show activation of MAPK (B). Activation of MAPK is detectable in vector-transfected neurons (A). C, Small RNA interference for the GluR1 subunit of AMPA receptor does not inhibit MAPK activation after application of AMPA. Scale bar, 10 μm. D, At 5 DIV, hippocampal neurons were intoxicated with 150 μg/ml pertussis toxin for 18 h, stimulated with 50 μm AMPA, and double labeled for actin and for activated MAPK. Incubation with pertussis toxin does not prevent activation of MAPK after AMPA stimulation. Scale bar, 12 μm. E, Quantitation of data collected from different experiments performed on 5 and 6 DIV cultures. No significant differences in MAPK activation are detectable between controls (ctrl) and pertussis toxin (PTX)-treated neurons, both under basal conditions and after incubation with 50 μm AMPA. Preincubation with PP2, a specific inhibitor of Src family kinases, but not with K252a, an inhibitor of Trk family kinases, strongly reduces AMPA-stimulated MAPK activation in axonal growth cones. Small RNA interference for the GluR2 subunit, but not for the GluR1 subunit, of AMPA receptors also inhibits MAPK activation after application of AMPA.
Figure 1.

AMPA induces activation of MAPK in axonal growth cones of cultured hippocampal neurons and in growth cone particles. At 5 DIV, hippocampal neurons were incubated for 5 min in Ca2+-free KRH (A), in Ca2+-free KRH containing 50 μm AMPA (C), or in complete KRH containing 50 μm AMPA (B), fixed and double stained with FITC-phalloidin and with an antibody specifically recognizing the phosphorylated form of MAPK. Note the presence of phosphorylated MAPK in the growth cones of neurons stimulated either in the presence or in the absence of extracellular Ca2+ (arrows). Scale bar, 10 μm. D, The AMPA receptor blocker GYKI52466 (GYKI) and the MEK inhibitor PD98059 (PD98) both inhibit AMPA-induced MAPK activation in the growth cones of cultured hippocampal neurons. E, Biochemical characterization of the growth cone particle preparation. The synaptic vesicle markers synaptobrevin (syb), synaptophysin (syp), synaptotagmin (syt), the protein TI-VAMP, and the AMPA receptor subunits GluR2,3 are all heavily enriched in the fraction containing growth cone particles (GCP). LSS, Low-speed supernatant. The enrichment of the growth cone marker GAP43 and of the axonal marker tau in its dephosphorylated form, but not of the NR1 subunit of NMDA receptor, indicates that the majority of growth cone particles are of axonal origin. F, Incubation with 50 μm AMPA stimulates MAPK activation in growth cone particles. No changes in tubulin or total MAPK are detectable. P-MAPK values in the histogram are normalized to tubulin immunoreactivity. G, AMPA-induced activation of MAPK in growth cone particles is prevented by the specific antagonist of AMPA receptors GYKI52466 and by the MEK inhibitor PD98059. ctrl, Control.
To further investigate AMPA-mediated signal transduction, we isolated growth cone particles from fetal rat cortices. These fractions are enriched in pinched-off, resealed, and metabolically active growth cones (Pfenninger et al., 1983; Lockerbie et al., 1991). Accordingly, they are characterized by a prominent enrichment in the growth cone-specific marker GAP43, tau (mainly in its dephosphorylated form) (Lohse et al., 1996), and synaptic vesicle proteins such as synaptobrevin, synaptophysin, and synaptotagmin. They are also enriched in TI-VAMP, a synaptobrevin isoform involved in axonal outgrowth (Coco et al., 1999) (Fig. 1E). Furthermore, vesicles enriched in synaptic vesicle markers, including the vesicular glutamate transporter vGlut2 (Fremeau et al., 2004) and the GluR2,3 subunits of AMPA receptors (Schenk et al., 2003), could be immunoisolated from these particles with anti-synaptotagmin antibodies (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Stimulation of growth cone particles with 50 μm AMPA, followed by SDS-PAGE and Western blotting with an antibody that specifically recognizes the phosphorylated form of MAPK, showed a significant increase in MAPK phosphorylation (P-MAPK level, 140.7 ± 5.1% of control; values normalized to either β-tubulin or total MAPK) (Fig. 1F). AMPA, however, was not as potent as BDNF (100 ng/ml) in activating MAPK in growth cone particles (data not shown) (see Fig. 3B). As in the case of cell cultures, AMPA-induced MAPK phosphorylation in growth cone particles was pharmacologically prevented by the AMPA-specific antagonist GYKI52466 (P-MAPK level, 102 ± 19.05% of control). Furthermore, the MEK inhibitor PD98059 produced a reduction of P-MAPK signal (P-MAPK level, 52.4 ± 24% of control), indicating that the level of P-MAPK observed in controls represents a steady state between dephosphorylation and MEK-dependent phosphorylation of MAPK (Fig. 1G). The observation that activation of MAPK occurs in growth cone particles, a biochemical preparation in which the somatodendritic compartment is mostly excluded, tends to rule out the possibility that activation of somatodendritic AMPARs is sending an anterograde signal to the axon. These data confirm that activation of AMPA receptors on axonal growth cones leads to MAPK phosphorylation and suggest that this signaling pathway may mediate, at least in part, autocrine feedback signaling.
Figure 3.

AMPA-induced MAPK activation leads to phosphorylation of synapsin I. A, Treatment of growth cone particles with 50 μm AMPA increases specifically the Triton X-100 (TX-100) solubility of synapsin I (syn 1), leaving unchanged the solubility of other synaptic vesicle proteins, such as synaptotagmin (stg). B, Both AMPA and BDNF induce activation of MAPK and phosphorylation of synapsin I at MAPK-specific sites (4,5) in growth cone particles. Phosphorylation of synapsin I is paralleled by its increased Triton X-100 solubility. The effects of AMPA are prevented by preincubation of growth cone particles with PD98059 (PD98). C, Synaptosomes from adult brain are incubated with 50 μm AMPA in a Na+/Ca2+-free medium. Note that activation of MAPK precedes phosphorylation of synapsin I at sites 4,5. D, Activation of MAPK in synaptosomes is inhibited by the specific AMPA receptor antagonist GYKI52466 (GYKI), by the MEK inhibitor PD98059, and by the Src tyrosine kinase inhibitor PP2 but not by the Trk family kinase inhibitor K252a. E, AMPA-induced phosphorylation of synapsin I at MAPK-specific sites in synaptosomes is inhibited by the AMPA receptor antagonist GYKI52466, by the MEK inhibitor PD98059, and by the Src tyrosine kinase inhibitor PP2. ctrl, Control.
Molecular pathways of MAPK activation
To test the direct involvement of AMPA receptors in MAPK activation, we inhibited the expression of the GluR1,2,3 subunits of AMPA receptors by siRNA (GluR1,2,3 immunostaining intensity was decreased to <10% of vector-transfected controls; data not shown) (Fig. 2A,B) (Passafaro et al., 2003). All transfected neurons [labeled with green fluorescent protein (GFP)] were double checked for the lack of GluR subunit staining and for MAPK activation (Fig. 2A-C). Silencing of GluR2 (Fig. 2B) and GluR3 (data not shown), but not of GluR1, expression abolished AMPA-induced MAPK activation (Fig. 2C) (percentage of positive growth cones after AMPA application in GluR2 siRNA transfected neurons, 32.8 ± 1.8%, n = 133; percentage of positive growth cones after AMPA application in GluR1 siRNA transfected neurons, 69 ± 3.7%, n = 67) (Fig. 2E). The use of a control construct, ineffective in silencing GluR2 expression, did not abolish AMPA-induced MAPK activation (percentage of positive growth cones after AMPA application in GluR2 control siRNA transfected neurons, 61 ± 5.8%; n = 62). Notably, silencing of either GluR2 (Fig. 2B) or GluR3 (data not shown) resulted in disappearance of immunoreactivity for both subunits from the growth cone and in inhibition of MAPK activation (Fig. 2B,E and data not shown). This effect can probably be explained by the fact that GluR2 plays a critical role in subunit assembly and in the synaptic expression of the receptor complex (Sans et al., 2003). Accordingly, assembling of GluR2 and GluR3 subunits appears to be needed for axonal transport of AMPA receptors to the growth cone (U. Schenk, unpublished observations). These data support a direct involvement of AMPA receptors in the activation of presynaptic MAPK and point to the involvement of the GluR2,3 subunits in this phenomenon.
In other model systems, two pathways have been reported to activate MAPK through AMPA receptor stimulation. The first involves the nonreceptor tyrosine kinase lyn and does not require ion influx through the receptor (Hayashi et al., 1999), whereas the other activates MAPK through a G-protein and requires the ion channel properties of the receptor (Wang and Durkin, 1995; Perkinton et al., 1999). Our observation that activation of MAPK in the growth cone does not depend on ion influx through the receptor (see above) makes unlikely the involvement of a G-protein-mediated process. Further supporting this view, intoxication of cultured hippocampal neurons with 150 ng/ml pertussis toxin for 18 h did not prevent AMPA-induced MAPK activation (Fig. 2D,E), thus excluding the involvement of a Gi/o-protein. To determine whether a tyrosine kinase might be involved in the AMPA-induced MAPK activation, we preincubated cultured hippocampal neurons (Fig. 2D) or growth cone particles (data not shown) for 15 min before AMPA addition with the broad-spectrum tyrosine kinase inhibitor genistein (30 μm) or with the specific inhibitor of Src family tyrosine kinases PP2 (10 μm). Both genistein (data not shown) and PP2 prevented the AMPA-induced MAPK activation both in cultured hippocampal neurons (percentage of positive growth cones after AMPA application in PP2-pretreated cultures, 34.2 ± 5.2%; n = 78) (Fig. 2E) and in growth cone particles (P-MAPK level after PP2 incubation, 84.6 ± 10.2% of control). In contrast, preincubation of cultures with the selective inhibitor of Trk tyrosine kinases, K252a, was ineffective (Fig. 2E). Thus, presynaptic AMPA receptors activate MAPK through a mechanism presumably involving the GluR2,3 subunits of AMPA receptors and a nonreceptor tyrosine kinase.
Effect of AMPA receptor activation at the growth cone and at the mature synapse
We next investigated whether MAPK activation by AMPA regulates key presynaptic effectors or the dynamics of synaptic vesicles. To this aim, we first focused on the synaptic vesicle-associated protein synapsin I, which is the major presynaptic substrate for MAPK (Jovanovic et al., 2000). In isolated growth cone particles, stimulation with AMPA specifically increased the Triton X-100 solubility of synapsin I, without affecting the Triton X-100 solubility of other synaptic vesicle proteins (Fig. 3A). This effect was mimicked by BDNF, a potent activator of MAPK, and was prevented by the MEK inhibitor PD98059 (Fig. 3B) and by GYKI52466 (data not shown). The increased recovery of synapsin I in the Triton X-100-soluble fraction occurred in parallel with MAPK activation and with the phosphorylation of synapsin I at MAPK-specific sites 4 and 5 (Fig. 3B). Thus, the increased Triton X-100 solubility of synapsin I could be a consequence of the MAPK-dependent phosphorylation.
Once it was defined that the activation of presynaptic AMPA receptors in axonal growth cones leads to MAPK activation and to phosphorylation of synapsin I at sites 4,5, we investigated whether the AMPA-induced MAPK activation occurs also at the mature synapse. Synaptosomes were incubated with 50 μm AMPA for 0-20 min in KRH free of Na+ and Ca2+ ions, and the phosphorylation of MAPK and of synapsin I (at the MAPK-dependent sites) was assayed by Western blotting. As in growth cone particles, AMPA stimulation induced the phosphorylation of MAPK in a Na+- and Ca2+-independent manner (P-MAPK level after AMPA stimulation, 161.6 ± 21.2% of control) (Fig. 3C). This process was followed, with a delay of a few minutes, by the MAPK-dependent phosphorylation of synapsin I at sites 4,5 (phosphorylated synapsin I level after AMPA stimulation, 176.4 ± 33.1% of control) (Fig. 3C). Also in synaptosomes, MAPK activation was inhibited by GYKI52466, PD98059, and PP2 but was not affected by K252a (Fig. 3D). Similarly, synapsin I phosphorylation was inhibited by GYKI52466, PD98059, and PP2 (Fig. 3E), thus suggesting, similarly to axonal growth cones, the involvement of a nonreceptor tyrosine kinase in the phenomenon. These data indicate that, not only in axonal growth cones but also at the mature presynaptic terminals, activation of AMPA receptors leads to phosphorylation of MAPK and to regulation of synapsin I. They prompted us to investigate whether AMPA-dependent MAPK activation may regulate presynaptic vesicle dynamics.
The biochemical results obtained on growth cone particles and on synaptosomes were corroborated by the immunocytochemical observation that synapsin I undergoes a relocalization in cultured hippocampal neurons, consequent to AMPA stimulation. Figure 4 shows that synapsin I immunoreactivity, which, in untreated neurons, is clustered in small puncta along the axon and in the growth cones (Fig. 4A), becomes fully dispersed in neurons exposed to 50 μm AMPA (Fig. 4A′). Synapsin I dispersion was specifically observed in growth cones (Fig. 4B) or axonal branches (Fig. 4C) in which MAPK was found to be activated. In growth cones or processes in which MAPK was not activated after AMPA stimulation, synapsin I immunoreactivity maintained its clustered distribution (Fig. 4B,C). Dispersion of synapsin I was reversible (data not shown). Preincubation of cultured hippocampal neurons with 100 μm GYKI52466 (Fig. 4D) or 30 μm PD98059 (Fig. 4E), which prevented the AMPA-induced MAPK activation (Fig. 1) and the increased recovery of synapsin I in the Triton X-100-soluble fraction of growth cone particles (Fig. 3B and data not shown), also prevented synapsin I dispersion (Fig. 4D′,E′). The dispersion of synapsin I immunoreactivity after AMPA stimulation occurred also at presynaptic terminals (Fig. 4F,G). Interestingly, a dispersion of synaptic vesicles paralleled the relocalization of synapsin I immunoreactivity (Fig. 4F). A similar dispersion was also detected when synaptic vesicles were labeled by Syt-ecto antibody, which is internalized in vesicle lumen after fusion with the plasma membrane (Fig. 4H, control; I, AMPA treated), thus suggesting that dispersed vesicles are available for exocytosis. Because the phosphorylation of synapsin I on MAPK-dependent sites does not affect its association with synaptic vesicles (Jovanovic et al., 1996), it is likely that synapsin I remains associated with synaptic vesicles during this relocalization. These results suggest therefore that the dispersion of synapsin I immunoreactivity might result from its dissociation from the actin cytoskeleton, leading to a loss of vesicle clustering.
Figure 4.
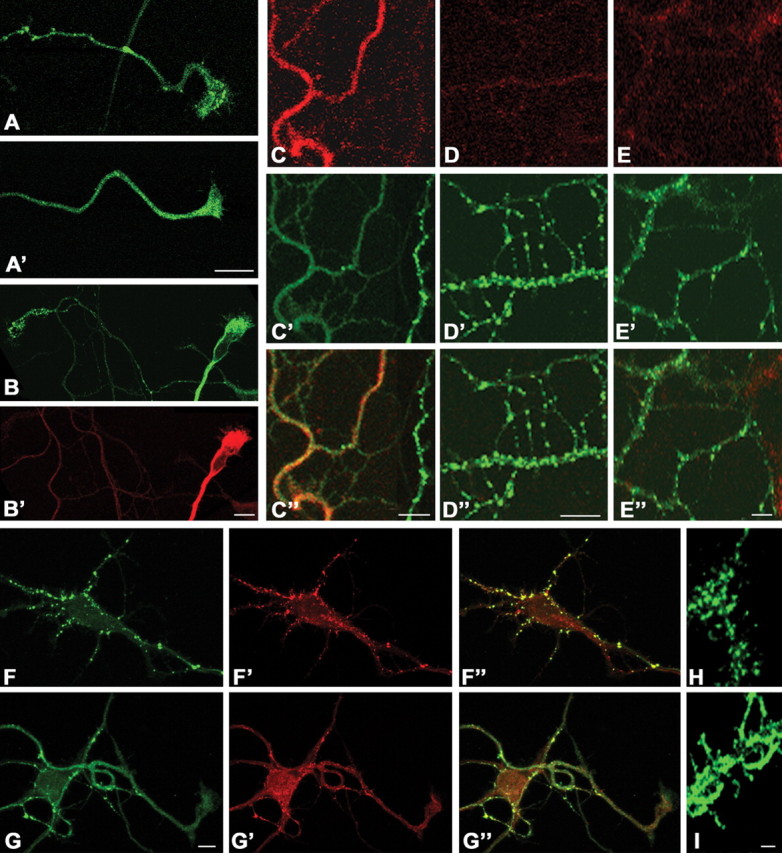
AMPA induces dispersion of synapsin I immunoreactivity in cultured hippocampal neurons. Synapsin I immunoreactivity is mainly found clustered in small puncta along the axon and in the growth cone of untreated neurons. Application of 50 μm AMPA results in dispersion of synapsin I immunoreactivity (A′). B, C, Dispersion of synapsin I immunoreactivity (B′, C′; green) after application of AMPA coincides with MAPK activation both in growth cones (B; red) and in axonal branches (C, red; C″, merged image). Synapsin I dispersion (D′, E′; green) is prevented by GYKI52466 (D′; D″, merged image) or by PD98059 (E′; E″, merged image), which also prevent MAPK activation (D, E; red). F, G, Dispersion of synapsin I immunoreactivity after stimulation with AMPA is paralleled by a dispersion of synaptic vesicles, as evidenced by staining for the synaptic vesicle marker synaptobrevin. F-F ″, Control; G-G″, AMPA treated; F, G, synapsin I (green); F′, G′, synaptobrevin (red); F″, G″, Merged images. H, I, Neuronal processes labeled by internalization of Syt-ecto antibody under control conditions (H) or after AMPA treatment (I). The dispersion of Syt-ecto staining indicates that dispersed vesicles are competent for recycling. Scale bar, 10 μm.
Effect of AMPA receptor activation on synaptic vesicle recycling
Finally, we determined whether AMPA-induced MAPK activation affects synaptic vesicle recycling. At 6-8 DIV, hippocampal neurons were exposed to Ca2+-free Tyrode's solution or Ca2+-free Tyrode's solution containing 50 μm AMPA for 2 min. After extensive washes, cultures were transferred in a Ca2+-containing Tyrode's solution and stimulated for 15 s at 10 Hz in the presence of FM1-43, an amphipatic dye that binds the bilayer and gets internalized into vesicles during recycling (Betz et al., 1992). After extensive washes to remove noninternalized FM1-43, neurons were stimulated for 90 s at 10 Hz, and the kinetics of the destaining of FM1-43 from preloaded puncta was analyzed. To avoid effects caused by “retrograde signaling” from postsynaptic dendritic receptors, we first focused our analysis on the varicosities in isolated axons and growth cone regions. As shown previously (Krueger et al., 2003; Matteoli et al., 2004), these sites are regions of active synaptic vesicle recycling, as indicated by the active internalization of Syt-ecto antibody in MAPK-positive growth cones (Fig. 5D). Figure 5A shows that the kinetics of exocytosis in AMPA-treated varicosities were significantly accelerated compared with that in control puncta (tau value for control, 43.30 ± 1.35, n = 76; AMPA, 21.1 ± 0.89, n = 71; p < 0.001). When cultures were preincubated with 25 μm PD98059 (PD-AMPA) (see Materials and Methods), the effect of AMPA was completely abolished (42.8 ± 1.01; n = 75; p < 0.001), suggesting that MAPK activation is required for AMPA-induced acceleration of exocytosis. We plotted histograms and cumulative populations for the tau of FM1-43 fluorescence decay from individual puncta (Fig. 5B). AMPA treatment increased the number of puncta with smaller tau value and shifted the distribution to higher exocytosis rates (Fig. 5C). This effect was completely abolished by the MEK inhibitor PD98059 (Fig. 5B,C). Furthermore, stimulation of neuronal cultures with 10 μm AMPA did not result in significant acceleration in the kinetics of exocytosis (tau values normalized to control, 1.041 ± 0.09; n = 54). These results indicate that activation of presynaptic AMPA receptors results in an increase of vesicle recycling through a pathway involving MAPK activation.
Figure 5.
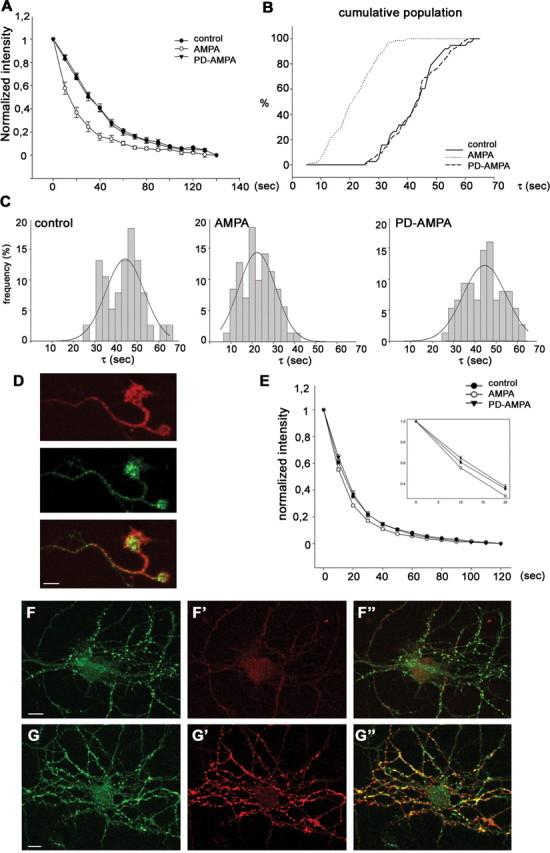
AMPA treatment increases the rate of FM1-43 unloading at axonal growth cone regions and at mature boutons. At 8 DIV, hippocampal neurons were treated with or without AMPA and loaded with FM1-43 using 300 action potentials at 10 Hz. After 10 min of washing, neurons were stimulated to unload FM1-43 (for details, see Materials and Methods). A, Decay of average FM1-43 intensity of the puncta during unloading using 900 action potentials at 10 Hz. Each point represents the average of a total of 71-76 boutons from four to eight different neurons. The average time constant for release of FM1-43 was 43.30 ± 1.35 s (n = 76) in control, 21.1 ± 0.89 s (n = 71) in AMPA-treated (p < 0.001), and 42.8 ± 1.01 s (n = 75) in PD98059 plus AMPA-treated (PD-AMPA) puncta, respectively. B, C, Frequency histograms (B) and cumulative population curve (C) of time constants of puncta unloading for individual puncta from control neurons and neurons treated with AMPA or PD98059 plus AMPA. Note that treatment of AMPA clearly increases the rate of exocytosis, and pretreatment of PD98059 abolishes the stimulatory effect of AMPA on exocytosis. D, At 6 DIV, hippocampal neurons were incubated with Syt-ecto antibody under depolarizing conditions (10 mm KCl); an axonal growth cone displaying MAPK activation (red) actively internalizes Syt-ecto antibody (green). E, Decay of average FM1-43 intensity of presynaptic boutons in 18 DIV cultures during unloading using 900 action potentials at 10 Hz. The average time constant for release of FM1-43 was 20.43 ± 0.75 s (n = 180) in control, 17.02 ± 0.36 s (n = 160) in the AMPA-treated (p < 0.05), and 21.00 ± 0.81 s (n = 160) in PD98059 plus AMPA-treated boutons. Inset, FM1-43 release over the initial 20 s was enlarged to show the differences in release kinetics among experimental groups. F, G, At 8 DIV, hippocampal neurons were incubated with Syt-ecto antibody under depolarizing conditions (F′, G′; red), fixed, and double labeled for synaptobrevin (F, G; green). Note that pretreatment of neurons with 50 μm AMPA results in an increase in the percentage of synapses labeled by Syt-ecto antibody, indicating the occurrence of a higher rate of recycling (F-F″, control; G-G″, AMPA treated; F″, G″, merged images). Scale bar, 10 μm.
A less pronounced, but still significant, increase in the exocytotic rate was obtained at presynaptic boutons of mature hippocampal cultures (18 DIV in culture) after AMPA stimulation (tau value for control, 20.43 ± 0.75, n = 180; AMPA, 17.02 ± 0.36, n = 160, p < 0.05; PD-AMPA, 21.00 ± 0.81, n = 160, p < 0,01) (Fig. 5E). Similar results were obtained in cultures stimulated with AMPA in KRH devoid of Ca2+, followed by exposure to Syt-ecto antibodies under depolarizing conditions in a Ca2+-containing KRH solution. Figure 5F′-G′ shows that a very low level of staining was produced by Syt-ecto antibody in control cultures exposed to 10 mm KCl and then counterstained after fixation with anti-synaptobrevin antibodies. In contrast, most synaptic sites were strongly labeled by Syt-ecto antibody in AMPA-treated cultures. Note that the AMPA-dependent increase in Syt-ecto internalization is paralleled by a loss of clustering of synaptic vesicles that disperse along processes (Fig. 5F,G). Collectively, these results demonstrate that activation of presynaptic AMPA receptors increases synaptic vesicle recycling in growth cones and presynaptic boutons by means of activation of metabotropic signaling cascades involving MAPK and independently of ion influx through the receptor. They also open the possibility that presynaptic AMPA receptors play a role during synaptogenesis as well as in the regulation of synaptic plasticity at mature synapses.
Discussion
Growing evidence points to a role of presynaptic AMPA receptors in the modulation of neurotransmitter release, although the molecular mechanisms at the basis of this phenomenon are still unknown (for review, see Schenk and Matteoli, 2004). In this study, we investigated signaling pathways downstream to AMPA receptors of axon terminals in three model systems: neurons developing in culture before and after synaptogenesis, growth cone particles, and synaptosomes. In all three systems, we have demonstrated that presynaptic AMPA receptors are functionally coupled to MAPK phosphorylation in the axon terminal, and we have shown that this signaling pathway may modulate synaptic vesicle exo-endocytosis.
AMPA induces presynaptic MAPK activation independently from ion influx
Metabotropic properties of AMPA receptors were reported several years ago, when this receptor type was thought to be localized exclusively at postsynaptic density (Wang and Durkin, 1995; Wang et al., 1997; Hayashi et al., 1999; Perkinton et al., 1999). We have now identified AMPA receptors of developing and mature axon terminals as key molecules of a pathway, activated by neuronal activity, which leads to presynaptic MAPK activation. Moreover, we demonstrated that differently from somatodendritic receptors, which appear to activate MAPK predominantly through ion influx, presynaptic AMPA receptors also act via a metabotropic signaling pathway, independently from ion influx. In agreement with our findings, it was reported recently that the AMPA-induced inhibition of neurotransmitter release in cerebellar GABAergic neurons is also independent from ion influx through the receptor (Satake et al., 2004). We also show that AMPA-induced presynaptic MAPK activation is sensitive to the Src kinase family inhibitor PP2. Our findings are convergent with those of Hayashi et al. (1999), who reported MAPK activation by AMPA receptors and a direct association of the GluR2 subunit of AMPA receptor with the nonreceptor tyrosine kinase lyn in the cerebellum. Because lyn is, together with other nonreceptor tyrosine kinases, highly enriched in the growth cone (Helmke and Pfenninger, 1995), it is possible that AMPA might induce MAPK activation through this tyrosine kinase. However, a weak association of the AMPA receptor was also found with other Src family kinases (Hayashi et al., 1999). Thus, other members of this kinase family may be involved in presynaptic AMPA-mediated signaling. Our results are also convergent with the recent genetic demonstration of a key role of intersectin/dynamin associated protein isoform 160 (a ubiquitous presynaptic protein) in the regulation of the development and function of the presynapse (Koh et al., 2004). Intersectin is a large scaffold protein that functionally connects the protein machineries of endocytosis and actin regulation with the Ras-MAPK signaling pathway (Marie et al., 2004).
MAPK-dependent regulation of vesicle dynamics
It is well established that activated MAPK migrates to the nucleus, where it modulates gene expression. Preliminary evidence has indeed indicated that AMPA-activated MAPK migrates to the nucleus, where it leads to cAMP response element-binding protein phosphorylation (Schenk, unpublished observations). However, it appears that activated MAPK also exerts a local action at the cell periphery, possibly through phosphorylation of targets within the growth cone and in the presynaptic terminal. In the present study, we focused on the major presynaptic target of MAPK, synapsin I. We show that activation of presynaptic AMPA receptors leads to MAPK-dependent phosphorylation of synapsin I at sites 4 and 5. Because phosphorylation of sites 4 and 5 reduces the protein affinity for the actin cytoskeleton (Jovanovic et al., 1996; Matsubara et al., 1996), this process could be responsible both for the dispersion of synapsin I immunoreactivity in AMPA-treated cultured neurons and for the increased Triton X-100 solubility of synapsin I in AMPA-stimulated growth cone particles. This possibility is in line with results obtained in Aplysia (Angers et al., 2002; Chin et al., 2002), reporting a dispersion of synapsin I immunoreactivity after MAPK activation.
The dispersion of synapsin I immunoreactivity as a consequence of the activity-dependent phosphorylation of the protein was found to precede evoked synaptic vesicle turnover (Chi et al., 2001, 2003). Moreover, mutations impairing MAPK-dependent synapsin I phosphorylation slowed vesicle turnover and mobilization in response to both high and low stimulation frequencies (Chi et al., 2003). Accordingly, the BDNF-stimulated increase in MAPK-dependent phosphorylation of synapsin I resulted in an increase of basal and evoked neurotransmitter release (Jovanovic et al., 2000). We show here that in parallel to the activation of MAPK and to the phosphorylation of synapsin at 4,5 sites, AMPA also increases the rate of evoked synaptic vesicle recycling, as monitored by the release of the lipophilic dye FM1-43 and by the uptake of Syt-ecto antibodies. This increase was blocked by the MEK inhibitor PD98059, thus demonstrating the critical role of MAPK in mediating these changes. AMPA receptors of nerve terminals could therefore play a major role in the regulation of vesicle turnover by increasing the availability of vesicles for recycling, possibly through MAPK-dependent phosphorylation of synapsin I (for review, see Hilfiker et al., 1999). These changes occur both in developing neurons and at the mature synapse. It is noteworthy, however, that the stimulatory effect of AMPA on synaptic vesicle recycling is less prominent at mature synapses than at varicosities and growth cones of immature neurons. This difference could result from a progressive decrease in the number of presynaptic AMPA receptors as neurons undergo maturation. Such a possibility fits with a higher number of GluR2,3 subunits in vesicles isolated from growth cone particles compared with vesicles isolated from mature synaptosomes (see supplemental Fig. 2, available at www.neurosci.org as supplemental material). Therefore, MAPK modulation via presynaptic AMPA receptors might have a larger impact on immature neurons.
What could be the functional consequence of the activation of this pathway at early stages of neuronal development? During development, glutamate released by the growth cone might rise to the high levels required for MAPK activation through presynaptic AMPA receptors as a result of limitation of the extracellular space around the nascent synapse (Tashiro et al., 2003). At this stage, glutamate may function as an autocrine and paracrine factor that regulates the growth cone and filopodia of the axon from which it is secreted as well as of neighboring axons (Tashiro et al., 2003). Effects mediated by Ca2+ entry may cooperate with activation of the MAPK pathway in the regulation of motility and secretion and in the transition from filopodia to presynaptic structures (Chang and De Camilli, 2001; Goda and Davis, 2003; Tashiro et al., 2003). The persistence of this regulatory pathway after synaptogenesis suggests that AMPA-mediated MAPK phosphorylation, requiring glutamate concentrations that are estimated to be reached in the synaptic cleft at conventionally active or potentiated synapses (Clements, 1996; Diamond and Jahr, 1997; Choi et al., 2000), might also contribute to short-term plasticity phenomena at the mature synapse (Jovanovic et al., 2000; Casey et al., 2002).
Footnotes
This work was supported by the European Community (QLGR3-CT-2000-01343), by Ministero dell'Università e della Ricerca Scientifica e Tecnologica-Progetti di Ricerca di rilevante Interesse Nazionale 2003, by the Fondo per gli Investimenti della Ricerca di Base (RBNE01RHZM), by Fondo Integrativo Speciale per la Ricerca-Consiglio Nazionale delle Ricerche Neurobiotecnologia 2003, and by the Human Frontier Science Program Organization (RGY0027/2001) (M.M.); by the Korean Ministry of Science and Technology (Brain Research Center of the 21st Century Frontier Research Program Grant M103KV010013 03K2201 01320) (S.C.) and by National Institutes of Health Grant NS36251 (P.D.C.). We thank Dr. M. Canossa (University of Bologna, Bologna, Italy) for the gift of BDNF, Dr. T. Galli (Centre National de la Recherche Scientifique, Paris, France) for the gift of anti-TI VAMP antibodies, and Drs. A. Nairn and A. Czernik (Yale University, New Haven, CT; The Rockefeller University, New York, NY) for antibodies against synapsin phosphorylated in sites 4,5.
Correspondence should be addressed to Dr. Michela Matteoli, Department of Medical Pharmacology, Consiglio Nazionale delle Ricerche, Institute of Neuroscience, Cellular and Molecular Pharmacology, University of Milan, via Vanvitelli 32, 20129 Milan, Italy. E-mail: m.matteoli@in.cnr.it.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251654-10$15.00/0
References
- Angers A, Fioravante D, Chin J, Cleary L, Bean A, Byrne J (2002) Serotonin stimulates phosphorylation of Aplysia synapsin and alters its subcellular distribution in sensory neurons. J Neurosci 22: 5412-5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett W, Banker G (1984) An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. II. Synaptic relationships. J Neurosci 4: 1954-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz W, Mao F, Bewick G (1992) Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci 12: 363-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550-553. [DOI] [PubMed] [Google Scholar]
- Casey M, Maguire C, Kelly A, Gooney M, Lynch M (2002) Analysis of the presynaptic signaling mechanisms underlying the inhibition of LTP in rat dentate gyrus by the tyrosine kinase inhibitor, genistein. Hippocampus 12: 377-385. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37-40. [DOI] [PubMed] [Google Scholar]
- Chang S, De Camilli P (2001) Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci 4: 787-793. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan T (2001) Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci 4: 1187-1193. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan T (2003) Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38: 69-78. [DOI] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary L, Eskin A, Byrne J (2002) Transforming growth factor β1 alters synapsin distribution and modulates synaptic depression in Aplysia. J Neurosci 22: RC220(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW (2000) Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.” Nat Neurosci 3: 330-336. [DOI] [PubMed] [Google Scholar]
- Clements JD (1996) Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci 19: 163-171. [DOI] [PubMed] [Google Scholar]
- Coco S, Raposo G, Martinez S, Fontaine JJ, Takamori S, Zahraoui A, Jahn R, Matteoli M, Louvard D, Galli T (1999) Subcellular localization of tetanus neurotoxin-insensitive vesicle-associated membrane protein (VAMP)/VAMP7 in neuronal cells: evidence for a novel membrane compartment. J Neurosci 19: 9803-9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1997) Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17: 4672-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Volknandt W, Fine A, Stewart M (2000) Age-dependent pre- and postsynaptic distribution of AMPA receptors at synapses in CA3 stratum radiatum of hippocampal slice cultures compared with intact brain. Eur J Neurosci 12: 3687-3700. [DOI] [PubMed] [Google Scholar]
- Fremeau Jr RT, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304: 1815-1819. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW (2003) Mechanisms of synapse assembly and disassembly. Neuron 40: 243-264. [DOI] [PubMed] [Google Scholar]
- Grewal S, York R, Stork P (1999) Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol 9: 544-553. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Umemori H, Mishina M, Yamamoto T (1999) The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature 397: 72-76. [DOI] [PubMed] [Google Scholar]
- Helmke S, Pfenninger K (1995) Growth cone enrichment and cytoskeletal association of non-receptor tyrosine kinases. Cell Motil Cytoskeleton 30: 194-207. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone V, Czernik A, Kao H, Augustine G, Greengard P (1999) Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354: 269-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner W, Schiebler W, Greengard P, De CP (1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic J, Benfenati F, Siow Y, Sihra T, Sanghera J, Pelech S, Greengard P, Czernik A (1996) Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA 93: 3679-3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic J, Czernik A, Fienberg A, Greengard P, Sihra T (2000) Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 3: 323-329. [DOI] [PubMed] [Google Scholar]
- Koh TW, Verstreken P, Bellen HJ (2004) Dap160/Intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron 43: 193-205. [DOI] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM (2003) The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron 40: 945-957. [DOI] [PubMed] [Google Scholar]
- Lee C, Bardoni R, Tong C, Engelman H, Joseph D, Magherini P, MacDermott A (2002) Functional expression of AMPA receptors on central terminals of rat dorsal root ganglion neurons and presynaptic inhibition of glutamate release. Neuron 35: 135-146. [DOI] [PubMed] [Google Scholar]
- Lockerbie R, Miller V, Pfenninger K (1991) Regulated plasmalemmal expansion in nerve growth cones. J Cell Biol 112: 1215-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse K, Helmke SM, Wood MR, Quiroga S, de la Houssaye BA, Miller VE, Negre-Aminou P, Pfenninger KH (1996) Axonal origin and purity of growth cones isolated from fetal rat brain. Brain Res Dev Brain Res 96: 83-96. [DOI] [PubMed] [Google Scholar]
- Lu C, Hwang S, Phend K, Rustioni A, Valtschanoff J (2002) Primary afferent terminals in spinal cord express presynaptic AMPA receptors. J Neurosci 22: 9522-9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott A, Role L, Siegelbaum S (1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443-485. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka R (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW (2004) Dap160/Intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43: 207-219. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H (1996) Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem 271: 21108-21113. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P (1992) Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol 117: 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli M, Coco S, Schenk U, Verderio C (2004) Vesicle turnover in developing neurons: how to build a presynaptic terminal. Trends Cell Biol 14: 133-140. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M (2003) Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 424: 677-681. [DOI] [PubMed] [Google Scholar]
- Perkinton M, Sihra T, Williams R (1999) Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci 19: 5861-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger K, Ellis L, Johnson M, Friedman L, Somlo S (1983) Nerve growth cones isolated from fetal rat brain: subcellular fractionation and characterization. Cell 35: 573-584. [DOI] [PubMed] [Google Scholar]
- Sabo S, McAllister A (2003) Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci 6: 1264-1269. [DOI] [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, Wang YX, Chang K, Royle GA, Wang CY, O'Gorman S, Heinemann SF, Wenthold RJ (2003) Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci 23: 9367-9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S, Saitow F, Yamada J, Konishi S (2000) Synaptic activation of AMPA receptors inhibits GABA release from cerebellar interneurons. Nat Neurosci 3: 551-558. [DOI] [PubMed] [Google Scholar]
- Satake S, Saitow F, Rusakov D, Konishi S (2004) AMPA receptor-mediated presynaptic inhibition at cerebellar GABAergic synapses: a characterization of molecular mechanisms. Eur J Neurosci 19: 2464-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk U, Matteoli M (2004) Presynaptic AMPA receptors: more than just ion channels? Biol Cell 96: 257-260. [DOI] [PubMed] [Google Scholar]
- Schenk U, Verderio C, Benfenati F, Matteoli M (2003) Regulated delivery of AMPA receptor subunits to the presynaptic membrane. EMBO J 22: 558-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Dunaevsky A, Blazeski R, Mason C, Yuste R (2003) Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron 38: 773-784. [DOI] [PubMed] [Google Scholar]
- Verderio C, Coco S, Fumagalli G, Matteoli M (1994) Spatial changes in calcium signaling during the establishment of neuronal polarity and synaptogenesis. J Cell Biol 126: 1527-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Coco S, Bacci A, Rossetto O, De CP, Montecucco C, Matteoli M (1999) Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci 19: 6723-6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Durkin J (1995) alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, but not N-methyl-d-aspartate, activates mitogen-activated protein kinase through G-protein beta gamma subunits in rat cortical neurons. J Biol Chem 270: 22783-22787. [DOI] [PubMed] [Google Scholar]
- Wang Y, Small D, Stanimirovic D, Morley P, Durkin J (1997) AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature 389: 502-504. [DOI] [PubMed] [Google Scholar]