

Abstract
Much evidence indicates that fear conditioning involves potentiation of some thalamic inputs to the lateral amygdala (LA). In turn, the LA would excite more neurons in the central nucleus (CE), leading to the generation of fear responses via their brainstem and hypothalamic projections. However, the posterior thalamus not only projects to LA but also to the medial sector of CE (CEm), suggesting that CEm might also be a site of plasticity. To test whether CEm also exhibits activity-dependent synaptic plasticity, we performed whole-cell recordings of CEm neurons in amygdala slices and stimulated thalamic axons coursing through the internal capsule and, as a control, the basolateral (BL) nucleus. High-frequency stimulation of thalamic inputs led to a long-lasting potentiation of thalamic responses, whereas BL-evoked responses remained unchanged. This thalamic long-term potentiation (LTP) developed even when slices were prepared with a cut severing the connections between the LA and CEm but was greatly reduced when an NMDA receptor antagonist was added to the perfusate shortly before and during LTP induction. Yet, intracellular dialysis with the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate did not prevent induction of the thalamic LTP, suggesting that presynaptic NMDA receptors are required for its induction. Consistent with this, the thalamic LTP also developed when the cells were dialyzed with a calcium chelator or kept hyperpolarized during induction. Finally, this thalamic LTP was associated with reduced amounts of paired-pulse facilitation, suggesting that it is expressed presynaptically. These results are consistent with the idea that the CEm plays an active role in fear conditioning.
Keywords: amygdala, long-term potentiation, fear conditioning, PPF, NMDA, GABA, in vitro, learning, memory
Introduction
The lateral nucleus of the amygdala (LA) is thought to be a critical site of plasticity for the acquisition of classically conditioned fear responses (LeDoux, 2000; Blair et al., 2001; Maren, 2001; Walker and Davis, 2002) (but see Cahill et al., 1999). According to the current model of fear learning, convergence of inputs about the conditioned and unconditioned stimuli (CS and US) would increase in the efficacy of thalamic synapses relaying CS information to the LA (LeDoux, 2000; Walker and Davis, 2002). As a result, later presentations of the CS alone would evoke stronger responses in LA neurons (Quirk et al., 1995; Collins and Paré, 2000; Repa et al., 2001). Ultimately, potentiated LA responses would elicit conditioned fear responses by exciting neurons of the central nucleus of the amygdala (CE) that project to brainstem and hypothalamic sites mediating fear responses (Hopkins and Holstege, 1978; Krettek and Price, 1978; Kapp et al., 1979; LeDoux et al., 1988, Bellgowan and Helmstetter, 1996; De Oca et al., 1998; for review, see Davis, 2000).
In the above model, the CE is seen as a passive relay interposed between the LA and downstream effectors. However, accumulating data suggest that the CE may in fact play an active role in fear conditioning. For instance, it was reported that infusing the protein synthesis blocker anisomycin into the CE prevents acquisition of conditioned taste aversion (Bahar et al., 2003). Similarly, injection of an NMDA receptor antagonist in the CE blocks the development of conditioned fear responses (Goosens and Maren, 2003). In keeping with this, NMDA synaptic currents in the CE exhibit a high sensitivity to NR2B-selective antagonists and a slow decay time (Lopez de Armentia and Sah, 2003), which are optimal for associative plasticity. Finally, reversible inactivation of the CE with muscimol during acquisition (Wilensky et al., 2000) or inhibition of protein synthesis in the CE (Wilensky et al., 2001) prevent the acquisition of conditioned fear responses.
Also supporting the idea that the CE may be a site of plasticity in fear conditioning, tracing studies indicate that the medial sector of the CE (CEm) receives inputs from various sensory modalities via the posterior thalamic nucleus (PO) (LeDoux et al., 1985; Turner and Herkenham, 1991; Linke et al., 2000). Indeed, the PO receives auditory inputs from the external, pericentral, and brachial nuclei of the inferior colliculus (Kudo and Niimi, 1980; Kudo et al., 1984; Linke et al., 2000) as well as the dorsal nucleus of the lateral lemniscus (Kudo et al., 1983). In addition, PO receives visual and somatosensory inputs from the superior colliculus and spinal cord (for review, see Jones, 1985).
Together, these results suggest that the CE may also be a site of plasticity in classical fear conditioning. If this were the case, the synapses formed by CE neurons with their sensory afferents should be plastic to allow for behavioral adaptations to change in the significance of sensory stimuli. Thus, in this study, we sought to determine whether thalamic afferents to CEm neurons express activity-dependent synaptic plasticity.
Materials and Methods
Slice preparation. Experiments were conducted on Hartley guinea pigs (200-250 g; 3-5 weeks of age) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with the approval of the Institutional Animal Care and Use Committee of Rutgers University (Newark, NJ). Before decapitation, the animals were deeply anesthetized with intraperitoneal injections of ketamine (80 mg/kg), pentobarbital (60 mg/kg), and xylazine (12 mg/kg). Once extracted from the skull, the brain was placed in an oxygenated artificial CSF (aCSF; 4°C) containing (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.3, 300 mOsm. Coronal sections (400 μm) were prepared with a vibrating microtome and stored for 1 h in an oxygenated chamber at room temperature. One slice was then transferred to a recording chamber perfused with an oxygenated aCSF solution at a rate of 2 ml/min. The temperature of the chamber was gradually increased to 32°C before the recordings began. Because CE neurons are believed to use GABA as a transmitter (Nitecka and Ben-Ari, 1987; McDonald and Augustine, 1993; Paré and Smith, 1993a), and because they have intranuclear axon collaterals (Hall, 1972; McDonald, 1992), the chloride channel blocker picrotoxin (100 μm) was added to the aCSF to prevent CE cells from influencing each other (Lopez de Armentia and Sah, 2004).
Whole-cell recordings. Current- and voltage-clamp recordings were performed with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) under visual control using differential interference contrast and infrared video microscopy. Recording pipettes (4-6 MΩ) were pulled from borosilicate glass capillaries and filled with a solution containing (in mm): 130 K-gluconate, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 10 KCl, 2 MgCl2, 2 ATP-Mg, and 0.2 GTP-tris(hydroxy-methyl)aminomethane. pH was adjusted to 7.2, and osmolarity was adjusted to 280-290 mOsm.
This report only includes neurons with a resting potential negative to -65 mV. Moreover, to be considered in the analyses, these recordings had to remain stable for >1 h with a low (<10 MΩ) and invariant series resistance (<10% variation).
Stimulation sites. Because tracing studies have revealed that thalamic afferents to CEm course through the internal capsule (LeDoux et al., 1985; Turner and Herkenham, 1991), stimulating electrodes were placed there, dorsal to CEm (see Fig. 1 A). Although the CEm has relatively few cortical afferents (for review, see McDonald, 1998) and no striatal or pallidal inputs (Pitkänen, 2000), stimuli delivered in the internal capsule may activate other fiber systems than thalamic axons. Nevertheless, for simplicity, the term thalamic will be used when referring to the afferents that were recruited by these stimulating electrodes.
Figure 1.
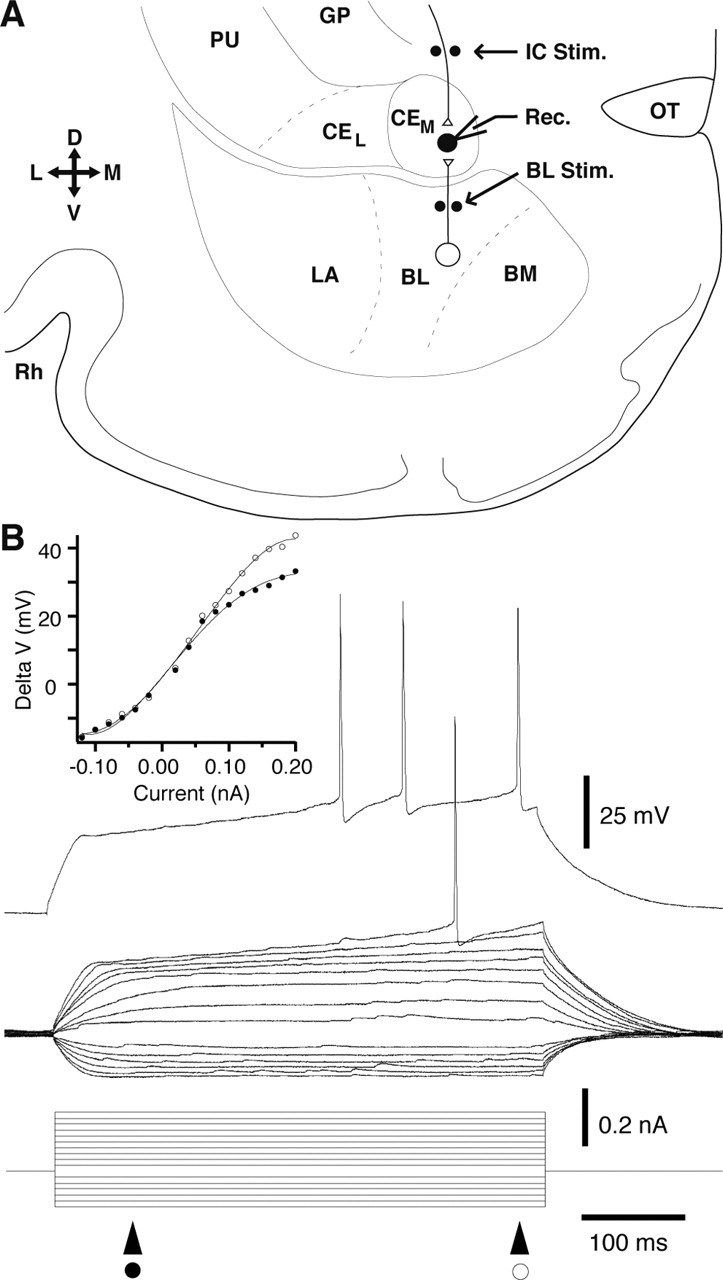
Experimental setup (A) and physiological properties of late-firing CEm neurons (B). A, Scheme showing a coronal slice of the guinea pig amygdala. Black dots indicate position of stimulating electrodes (Stim.) in the internal capsule (IC) and BL nucleus. Pipette shows recording site (Rec.) in CEm. Slice orientation is indicated by the cross on the left where D, V, L, and M indicate dorsal, ventral, lateral, and medial. B, Voltage responses of a representative late-firing cell to a series of current pulses applied from rest (-81 mV). Note delayed firing onset and ramp-like voltage response to depolarizing current pulses that brought the membrane potential positive to -65 mV. The inset on top left shows a plot of voltage response (y-axis) as a function of injected current (x-axis). Voltage measurements were performed at two different times, as indicated by the symbols below the current monitor. BM, Basomedial nucleus; CEL and CEM, lateral and medial sectors of the central nucleus; GP, globus pallidus; PU, putamen; OT, optic tract; Rh, rhinal sulcus.
To test for input specificity of the thalamic high-frequency stimulation (HFS), we also positioned stimulating electrodes ventral to CEm in the basolateral (BL) nucleus (see Fig. 1 A). This nucleus relays information from the LA to the CEm (Krettek and Price, 1978; Smith and Paré, 1994; Paré et al., 1995; Pitkänen et al., 1997). Both thalamic and BL stimulating electrodes consisted of two tungsten rods (80 μm in diameter), separated by 150 μm.
Long-term potentiation induction. The stimulation intensity at each site (100-150 μs pulses of 100-200 μA) was set to generate baseline EPSCs of 0.05-0.15 nA from a holding potential of -80 mV (close to rest in CEm cells; see Results). Electrical stimuli were delivered every 60 s, 10-15 min before, and ≥40 min after long-term potentiation (LTP) induction. To induce LTP, we applied high-frequency stimuli at the thalamic site paired to postsynaptic depolarization in current-clamp mode. HFS consisted of four trains (0.05 Hz) of 10 bursts (3 Hz) of 10 stimuli (100 Hz), in which each stimulus was paired with a brief (2 ms) supra-threshold current pulses. The interval between thalamic stimuli and current pulses ranged between 5 and 7 ms, which allowed postsynaptic spikes to occur at the peak or during the descending phase of the EPSPs. Unless otherwise stated, HFS was applied within 18 min of recording onset to avoid wash-out of intracellular constituents that are necessary for LTP induction (Malinow and Tsien, 1990). Posttetanic potentiation was defined as the difference between control EPSC amplitude and that measured immediately after LTP induction.
Signals were digitized at 10 kHz with a Digidata 1200 interface controlled by the software pClamp 8.1 (Axon Instruments). Analysis was performed off-line with pClamp 9.2 and Igor Pro 4.03 (WaveMetrics, Lake Oswego, OR). All statistical tests were performed on SAS V8 software (SAS Institute, Cary, NC) using a fixed level of significance (p = 0.05). Values are expressed as means ± SE. With the exception of (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801; 1 mm in the intracellular pipette solution) and local pressure applications of glutamate (0.5 mm in aCSF), all drugs were delivered by adding them to the aCSF. Drugs used were picrotoxin (100 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm), and d-2-amino-5-phosphonopentanoic acid (d-APV; 100 μm). All drugs were purchased from Sigma (St. Louis, MO).
Results
Data base
A total of 107 stable whole-cell recordings were obtained in the CEm. Consistent with previous findings in guinea pigs (Martina et al., 1999; Dumont et al., 2002), one physiological cell type prevailed in our sample. Indeed, most CEm cells (78% or 83 cells), displayed a voltage- and time-dependent outward rectification in the depolarizing direction (Fig. 1B). As a result, there was a conspicuous delay between membrane depolarization and firing onset in these cells (hence the designation “late-firing neurons”) (Fig. 1B). On average, late-firing cells had a resting potential of -76.77 ± 0.69 mV, an input resistance of 364 ± 11 MΩ, a spike amplitude of 81.9 ± 1.5 mV, and a spike duration of 1.31 ± 0.07 ms at half amplitude.
The experiments described below were only conducted on late-firing neurons because they are most likely CE output neurons. This view is based on the following facts. Late-firing neurons have the morphological properties of CE projection cells (Martina et al., 1999), as described in previous Golgi studies (for review, see McDonald, 1992). In addition, the vast majority of CEm neurons are retrogradely labeled after large horseradish peroxidase injections in the brainstem (Hopkins and Holstege, 1978). Given that late-firing cells account for most CEm neurons, they must overlap extensively with brainstem-projection cells.
Thalamic and BL stimuli activate distinct sets of inputs to CEm neurons
Both thalamic and BL stimuli evoked EPSCs in almost all tested CEm neurons. To determine whether these EPSCs were mediated by distinct sets of inputs converging onto CEm neurons, we used the occlusion test (n = 44). In this test, the actual and predicted sum of responses evoked by two stimuli is compared. When the response evoked by paired stimuli is smaller than the algebraic sum of individual responses, this constitutes evidence that the two stimulating electrodes activate a partially overlapping set of inputs.
An example of this test is shown in Figure 2A. Stimuli were delivered at each site independently (Fig. 2A, left) or at the same time (Fig. 2A, right). Then, the amplitude of EPSCs evoked by the paired stimuli was compared with the algebraic sum of EPSCs elicited by stimulating each site independently. On average, the response evoked by the paired stimuli was 96.9 ± 3.9% of the summed EPSCs. In fact, the actual and predicted responses were not significantly different from each other (t test, p > 0.05). This suggests that the electrodes located in the internal capsule and BL nucleus activated mostly nonoverlapping sets of inputs converging onto CEm cells.
Figure 2.
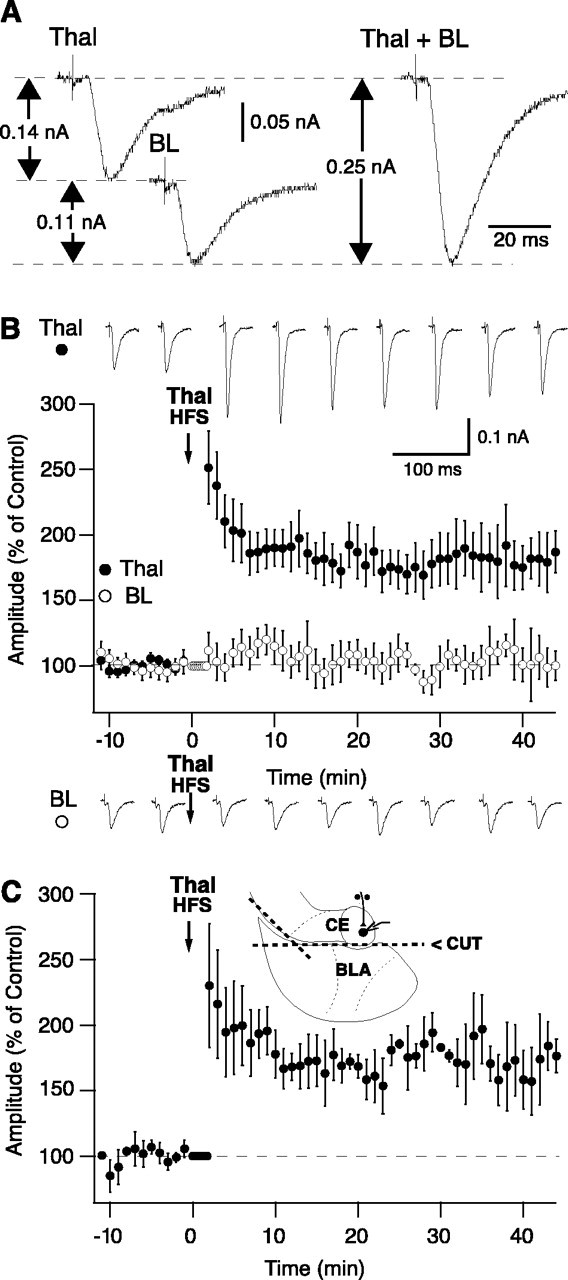
HFS of thalamic afferents induces input-specific LTP. A, Occlusion test. Electrical activation of thalamic (Thal) or BL afferents independently (left) or simultaneously (right; Thal + BL). Amplitude of EPSCs evoked by the paired stimuli (right) was virtually identical to the algebraic sum of EPSCs elicited by stimulating each site independently (dashed lines; left). B, C, Graph plotting the amplitude of EPSCs evoked by activation of thalamic (filled circles) or BL (empty circles) afferents (y-axis) as a function of time (x-axis). Thalamic HFS was applied at the time indicated by the downward arrow. Data were normalized to baseline response amplitudes. Traces above and below the graph in B show examples of responses evoked by activation of thalamic or BL afferents, respectively. The timing of these responses is indicated by the position of their peaks with respect to the x-axis of the graph just below. The data plotted in B were obtained in intact slices, where as the data depicted in C were obtained in slices in which CEm was disconnected from the basolateral complex by means of cuts (inset; dashed lines). Error bars represent SE.
HFS of thalamic axons induces input-specific LTP
LTP experiments began by acquiring a control epoch, 10-15 min in duration, during which thalamic and BL inputs (2 s apart) were activated at a low frequency (one shock per min at each site). This frequency was chosen because pilot experiments had revealed that it produced no time-dependent drift in the amplitude of thalamic and BL-evoked responses. Then, we tested whether repetitive high-frequency trains of thalamic stimuli paired to postsynaptic depolarization produced a long-term enhancement of evoked responses. Note that during HFS of thalamic inputs, no stimuli were delivered in the BL nucleus.
As shown in the population average of Figure 2B (filled circles; n = 45), HFS of thalamic inputs produced a potentiation of evoked responses with no change in the amplitude of EPSCs elicited from the BL nucleus (n = 17) (Fig. 2B, empty circles). Overall, 35 of 45 cells (78%) showed a statistically significant potentiation of thalamic responses after HFS (t test, p < 0.05). This was determined by comparing the amplitude of responses in the control phase versus 30-40 min after HFS. Note that this interval will be used throughout the Results section for statistical comparisons. The HFS-induced enhancement of thalamic EPSCs lasted for as long as the recordings remained stable (up to 130 min) and averaged 180.0 ± 3.7% of control response amplitudes (t test, p < 0.05; n = 45). In contrast, BL-evoked responses were unchanged by thalamic HFS (108.7 ± 4.3% of control; t test, p > 0.05; n = 17), demonstrating that the thalamic LTP was input specific. A representative example of this phenomenon can be seen in the top row of Figure 2B. In this and following figures, the timing of the EPSCs is indicated by the position of their peaks with respect to the x-axis of the adjacent graph. In this cell, HFS of thalamic inputs produced a 69.0 ± 8.3% enhancement of responses from that pathway, whereas BL-evoked EPSCs remained unchanged (Fig. 2B, bottom row). At both thalamic and BL inputs, there was no correlation between control response amplitudes and the effect of thalamic HFS (p > 0.05).
Evidence of a significant potentiation was also obtained when we compared the rising slope of evoked responses. For instance, in the cell of Figure 2B, HFS of thalamic inputs produced a 102.0 ± 8.1% enhancement in the slope of thalamic EPSCs (t test, p < 0.05) but no significant change in the slope of the BL-evoked responses (6.3 ± 5.3%; t test, p > 0.05).
LTP of thalamic inputs to CEm neurons is independent of LA
Because thalamic inputs to LA can undergo activity-dependent LTP (Weisskopf et al., 1999; Bauer et al., 2002; Bissière et al., 2003), it is possible that the LTP we observed is not occurring in the CEm but in the LA. At odds with this possibility, however, HFS of thalamic inputs did not modify response latencies, and their rising phase remained smooth. In the example of Figure 2B, for instance, the latency of thalamic EPSCs was 3.72 ± 0.03 ms before HFS and 3.78 ± 0.05 ms after HFS (t test, p > 0.05).
Nevertheless, to test the possibility that LTP of thalamic inputs in LA was responsible for our results, we repeated the above experiments in slices prepared with cuts that completely isolated the CE from the basolateral complex (Fig. 2C, dashed lines in inset). As shown in Figure 2C, even in the absence of the entire basolateral complex, HFS of thalamic inputs produced a long-lasting enhancement of evoked responses (68.2 ± 14.9%; n = 8) that was statistically indistinguishable from that seen in intact slices (t test, p > 0.05).
LTP induction at thalamic inputs to CEm cells is dependent on NMDA receptor activation
It was shown previously that NMDA receptor activation is required for induction of some forms of LTP in the amygdala (for review, see Blair et al., 2001). For instance, NMDA receptor blockade was reported to prevent LTP of cortical inputs to LA neurons (Huang and Kandel, 1998) and of BLA inputs to intercalated cells (Royer and Paré, 2002, 2003). To test whether LTP induction in CEm neurons is also NMDA dependent, we added APV (100 μm) to the perfusate just before (4 min) and during HFS (n = 13). As shown in Figure 3A, this treatment drastically reduced LTP magnitude (t test, p < 0.05). Indeed, after HFS in the presence of APV, thalamic EPSCs only grew to 121.8 ± 2.5% of baseline response amplitudes (n = 13) (Fig. 3A, triangles) compared with 180.0 ± 3.7% in control conditions (n = 45) (Fig. 3A, circles). Note that this significant difference (t test, p < 0.05) did not result from uncontrolled variables because APV and control cells were interleaved.
Figure 3.
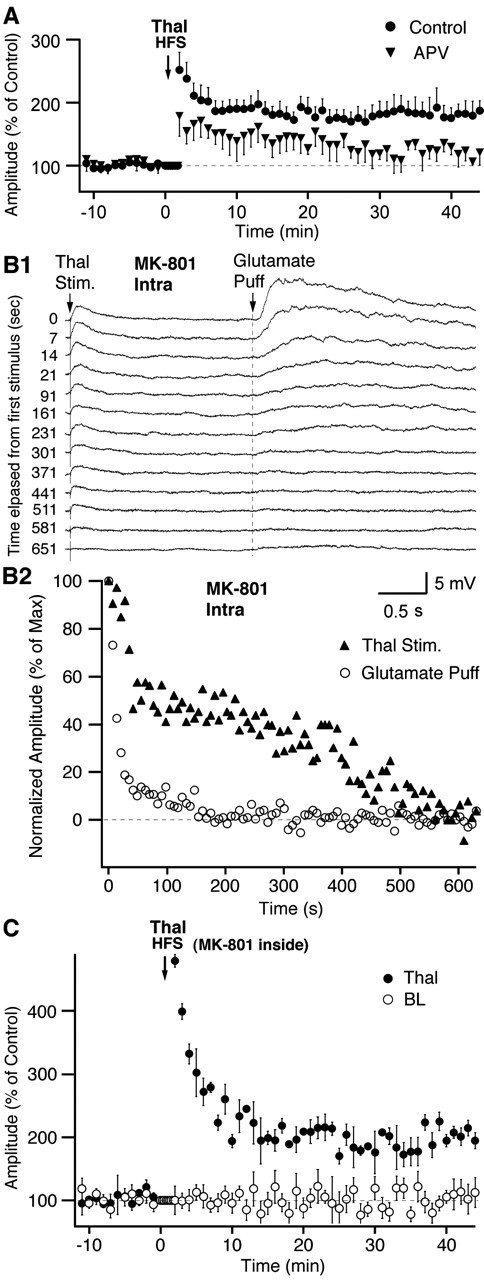
Induction of thalamic LTP is dependent on presynaptic NMDA receptors. A, Graph plotting the amplitude of EPSCs evoked by activation of thalamic afferents (y-axis) as a function of time (x-axis). Circles and triangles show experiments in which HFS was applied in the absence or presence of APV, respectively. B, Time dependence of NMDA receptor block by intracellular MK-801. CNQX (20 μm) and picrotoxin (100 μm) were present in Mg2+-free aCSF. MK-801 (1 mm) was present in pipette solution. B1, NMDA responses evoked by electrical activation of thalamic afferents (left; Thal Stim.) or pressure application of glutamate via a patch pipette positioned close to recorded soma (right; Glutamate Puff). The numbers on left indicate time (in seconds) from first stimulus. B2, Graph plotting NMDA response amplitudes (y-axis) as a function of time (x-axis) for responses evoked by endogenous (triangles) and exogenous (circles) glutamate. Same experiment as in B1. C, Effect of intracellular MK-801 on thalamic LTP. See details in Results. Graph plotting the amplitude of EPSCs evoked by activation of thalamic (filled circles) or BL (empty circles) afferents (y-axis) as a function of time (x-axis). HFS was applied at the time indicated by the downward arrow. Data were normalized to baseline response amplitudes. Error bars represent SE.
A potential caveat in the APV experiments is the possibility that NMDA currents contributed significantly to control response amplitudes or to the expression of potentiated responses. If this were the case, persistence of NMDA block beyond the period of APV application would mislead us into believing that APV interfered with LTP induction when in fact, it interfered with its expression. To test this possibility, we compared the peak amplitude of the thalamic EPSCs in control aCSF versus in the presence of APV from -80 mV, the holding potential at which the LTP experiments were performed. However, because the NMDA contribution to the EPSC peak was negligible, the APV-sensitive component accounted for only 0.68 ± 3.4% of peak EPSC amplitudes (n = 12). Although this result does not exclude the possibility that NMDA currents increase after LTP induction, it indicates that APV did not interfere with our assessment of LTP expression, because it was based on measurements of peak EPSC amplitudes.
LTP of thalamic inputs is induced presynaptically
In the above experiments, it is unclear whether the NMDA receptors that are critical for LTP induction were located presynaptically or postsynaptically, because APV was applied via the aCSF. Thus, we investigated whether the APV effects could be reproduced by blocking only postsynaptic NMDA receptors using intracellular dialysis of the recorded cells with the open NMDA channel blocker MK-801 (1 mm in the pipette solution). Note that intracellular application of MK-801 was used successfully before to block NMDA responses in single cells (Beretta and Jones, 1996; Humeau et al., 2003).
At first, control experiments were performed to test whether MK-801 could completely block NMDA responses within 15 min of recording onset. This is a critical point because prolonged intracellular dialysis often interferes with LTP induction (Malinow and Tsien, 1990). In other words, if MK-801 only blocked NMDA responses after a long delay, it would be unclear whether the failure to induce LTP was caused by the NMDA block or to the loss of intracellular constituents critical to LTP induction.
Thus, we tested the effect of intracellular MK-801 on NMDA responses elicited by electrical stimulation of thalamic afferents (n = 9) (Fig. 3B1, left). However, because MK-801 is an open channel blocker (at least when applied in the extracellular environment), variations in release probability might influence the rate of receptor blockade. Thus, we also tested the effects of MK-801 on NMDA responses evoked by pressure application of glutamate close to the soma (Fig. 3B1, right). Different stimulation frequencies were tested in distinct subsets of cells (every minute or ≥0.1 Hz). NMDA responses were isolated by adding 100 μm picrotoxin and 20 μm CNQX to the perfusate. Because Mg2+ ions are known to inhibit blockade by MK-801 at negative membrane potentials (Huettner and Bean, 1988), the slices were bathed in a Mg2+-free aCSF solution for 10 min before patching the cells.
In these conditions, NMDA responses to peri-somatic glutamate puffs vanished quickly (<3.5 min; n = 4) (Fig. 3B2, circles) compared with 3-10 min for those elicited by electrical stimuli (Fig. 3B2, triangles). The stimulation frequency at thalamic electrodes had little effect on this latency (every minute, n = 5, 5.6 ± 2.1 min; at ≥0.1 Hz, n = 4, 5.3 ± 2.1 min), suggesting that in our conditions, the delay to full NMDA block critically depends on the diffusion time of MK-801 to thalamic synapses at distributed dendritic sites.
Having established that intracellular MK-801 could block NMDA responses in ≤10 min, we examined how MK-801 affects LTP induction. These tests were conducted as above with the following three exceptions: no CNQX was used, the Mg2+ concentration was returned to normal values 4 min before HFS, and the control period lasted 15 min to ensure complete NMDA block.
As shown in Figure 3C, intracellular MK-801 did not prevent LTP induction of thalamic inputs to CEm cells (Fig. 3C, filled circles). The MK-801-resistant enhancement of thalamic EPSCs averaged 192.8 ± 6.2% of baseline response amplitudes (t test, p < 0.05; n = 8). Note that this is not statistically different from the LTP observed in control experiments (180.0 ± 3.7%; t test, p > 0.05). Finally, as was seen in control experiments, the amplitude of BL-evoked responses (Fig. 3C, empty circles) remained unchanged after thalamic HFS (106.4 ± 5.8%; t test, p > 0.05; n = 8).
The sensitivity of the thalamic LTP to extracellular APV coupled to its resistance to intracellular MK-801 suggests that it is induced presynaptically. However, a potential concern with the above experiments is the possibility that the Mg2+ concentration had not returned to control levels before HFS. Although the large posttetanic potentiation (PTP) seen in Figure 3C is consistent with this idea, control experiments suggest that this is not the case. Indeed, we have observed that pharmacologically isolated NMDA responses evoked from rest in 0 Mg2+ disappear completely within 2 min of return to a normal aCSF (A. Popescu, A. Saghyan, and D. Paré, unpublished observations). Moreover, although EPSC amplitudes increased slightly after the removal of extracellular Mg2+ (13.3 ± 4.4%; t test, p < 0.05; n = 16), responses returned to control values within 2-3 min of switching to a normal aCSF. Finally, we found no correlation between PTP and LTP magnitudes in control experiments (r = 0.04; p > 0.05).
Nevertheless, to further probe the possibility that induction of the thalamic LTP is independent of postsynaptic activity, three more tests were performed. We first investigated the effects of the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) at a concentration (10 mm in pipette solution) known to block postsynaptic NMDA-dependent LTP in hippocampal field CA1 (Manabe et al., 1992). As shown in Figure 4, we observed a statistically significant potentiation of thalamic responses after HFS of thalamic inputs (t tests, p < 0.05), suggesting that a postsynaptic rise in the intracellular Ca2+ concentration is not critical for the induction of this form of LTP.
Figure 4.

Induction of thalamic LTP is independent of the postsynaptic calcium concentration. Graph plotting the average (n = 8) amplitude of EPSCs evoked by activation of thalamic (Thal) afferents (y-axis) as a function of time (x-axis) is shown. BAPTA (10 mm) was present in the intracellular solution. Thalamic HFS was applied at the time indicated by the downward arrow. Data were normalized to baseline response amplitudes. Error bars represent SE.
Second, we lengthened the control period. We reasoned that because prolonged intracellular dialysis by the pipette solution abolishes postsynaptically induced forms of NMDA-dependent LTP (Malinow and Tsien, 1990), resistance of the thalamic LTP to such a treatment would indirectly support the notion that it is induced presynaptically. Thus, in five additional CEm neurons, we applied thalamic HFS ≥30 min after gaining whole-cell access. In support of a presynaptic locus of induction, longer pre-HFS periods did not prevent the thalamic LTP (181.8 ± 17.0% of baseline; n = 5).
Third, we voltage clamped CEm neurons at -80 mV during HFS to prevent, or at least reduce, postsynaptic Ca2+ influx via NMDA receptors and voltage-gated Ca2+ channels. Because increases in Ca2+ concentration are known to be critical for the induction of postsynaptically induced LTP (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993), resistance of the thalamic LTP to membrane hyperpolarization would further support the notion that it is induced presynaptically. Thus, to test this idea, we attempted to induce thalamic LTP while voltage clamping the cells at -80 mV during HFS. Note that in these tests, no action currents were observed during HFS. In further support of a presynaptic locus of induction, clamping the cells at -80 mV did not prevent induction of thalamic LTP (175.3 ± 22.5% of baseline; n = 5; t test, p > 0.05).
LTP induction at thalamic inputs is associated with a reduction in paired-pulse facilitation
To address the mechanisms underlying the expression of the thalamic LTP, we studied the effect of HFS on paired-pulse facilitation (PPF). In this analysis (Katz and Miledi, 1968), two stimuli of equal intensity are applied in brief succession. When the interval between the two shocks is sufficiently brief, the second one elicits a larger response that is believed to result from residual Ca2+ in the presynaptic terminals (for review, see Zucker and Regehr, 2002). The amount of PPF is thought to be inversely proportional to transmitter release probability, because manipulations that increase release probability decrease PPF and conversely (Creager et al., 1980; Manabe et al., 1993).
Thus, we used the same protocol as above with the exception that control and postinduction thalamic stimuli were applied in pairs. An interstimulus interval of 50 ms was chosen to minimize response contamination via multisynaptic pathways. Below, the term PPF will be used when referring to the ratio of the second to the first EPSC (expressed in percentage).
In control conditions, paired thalamic stimuli elicited a clear PPF (162.3 ± 3.8%; n = 19) that was not correlated to the amplitude of the first EPSC (r = 0.24; p > 0.05). As was seen with single stimuli (Fig. 2B), thalamic HFS induced a response potentiation (EPSC1, 185.1 ± 7.6% of control) that was associated with a significant decrease in PPF (-18.1 ± 3.0%; paired t test, p < 0.05; n = 19) (Fig. 5A). Examples of responses to paired thalamic stimuli are shown in the top part of Figure 5A (control, left; post-HFS, right). In further support of the idea of a presynaptic locus of expression, the PPF change and LTP magnitude were significantly correlated (r =-0.51; p < 0.05) (Fig. 5B).
Figure 5.
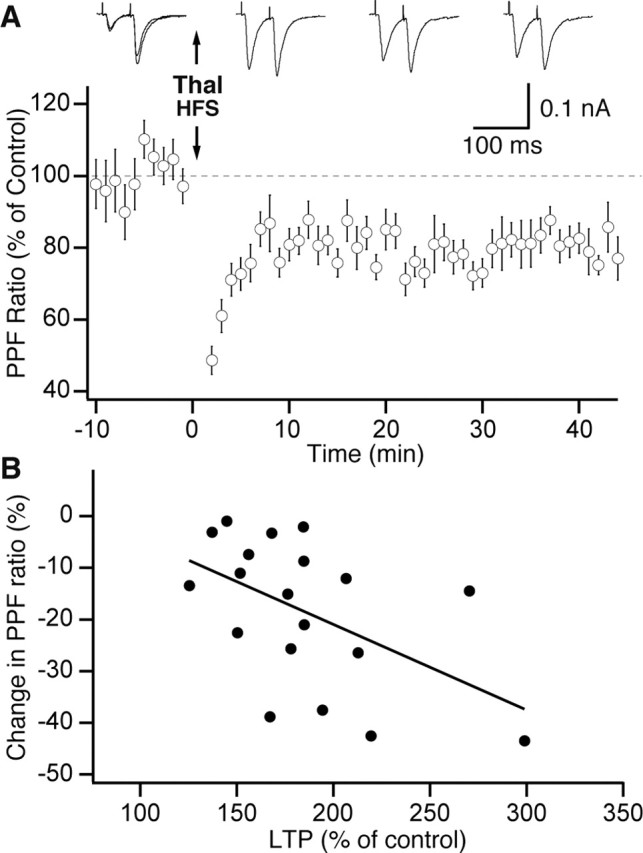
HFS of thalamic (Thal) inputs causes a decrease in PPF. A, Graph plotting the PPF ratio (y-axis) as a function of time (x-axis). Data were normalized to control values (average of 19 cells). HFS was applied at the time indicated by the downward arrow. Traces above graph show a case in which HFS of thalamic inputs produced a marked decrease in PPF. The timing of the responses shown on top is indicated by the position of the peaks of the first EPSC with respect to the x-axis of the graph just below. B, Graph plotting the change in PPF ratio (y-axis) as a function of the amount of LTP (x-axis) in our sample (n = 19). Error bars represent SE.
Discussion
This study was undertaken to test whether glutamatergic afferents to CEm neurons can exhibit activity-dependent synaptic plasticity. The importance of this issue comes from recent data indicating that the CEm might not be a passive relay in classical fear conditioning, but a critical site of plasticity (Paré et al., 2004). In keeping with this possibility, our results suggest that thalamic inputs to CEm neurons exhibit an unusual NMDA-dependent form of LTP that is induced and expressed presynaptically.
Identity of stimulated afferents
As in previous in vitro studies of synaptic plasticity in the amygdala (Blair et al., 2001), the identity of the synapses that were activated by stimulating the internal capsule is uncertain. However, thalamic inputs represent the main glutamatergic afferent to course through the internal capsule and end in the CEm. First, the CEm receives light cortical inputs compared with the CEl and the basolateral complex (McDonald, 1998). Second, although it is possible that stimulation of the internal capsule backfired some BL axons, our occlusion tests suggest that they did not target CEm cells. Third, tract-tracing studies do indicate that the internal capsule contains an important contingent of axons that originates in the PO thalamic complex and end in the CEm. Thus, although it is clear that our electrical stimuli activated a number of afferent systems, thalamic axons likely constituted the main one.
Thalamic inputs to CEm neurons exhibit activity dependent LTP
In the present study, we observed that HFS of thalamic inputs to CEm neurons leads to a long-term enhancement of the efficacy of these synapses. This potentiation was input specific because BL-evoked responses remained stable after thalamic HFS. Moreover, it did not depend on plastic events in the LA because it was observed in slices prepared with cuts isolating the CEm from the basolateral complex. Finally, the induction of this LTP was dependent on NMDA receptor activation, because application of APV shortly before and during HFS produced an ∼75% reduction in LTP magnitude.
Surprisingly, several observations suggest that this LTP is induced and expressed presynaptically. Indeed, it developed normally after blockade of postsynaptic NMDA receptors with intracellular MK-801 or when the cells were dialyzed with BAPTA. Moreover, it persisted when the control period was lengthened to ≥30 min or when the cells were kept hyperpolarized during HFS. Finally, induction of this thalamic LTP was associated with decreased amounts of PPF, a phenomenon commonly believed to reflect synaptic release probability (Creager et al., 1980; Manabe et al., 1993). In keeping with this, we found a significant correlation between the amount of LTP and the change in PPF. However, we cannot completely exclude the possibility that a network effect is at play. Demonstration of an NMDA-induced presynaptic facilitation of release in the presence of extracellular tetrodotoxin and intracellular MK-801 will be required to do so.
Although many forms of LTP in the amygdala were reported to depend, at least in part, on NMDA receptors for their induction, in most cases, the critical NMDA receptors were inferred to be located postsynaptically. This conclusion was based on the fact that postsynaptic manipulations such as membrane hyperpolarization and/or Ca2+ chelation interfered with LTP induction. For instance, this was the case of the potentiation of cortical inputs to LA cells (Huang and Kandel, 1998; Tsvetkov et al., 2002, 2004) and of BL inputs to intercalated neurons (Royer and Paré, 2002, 2003).
However, there is at least one precedent in the amygdala for a form of LTP that requires presynaptic NMDA receptor activation for its induction. Indeed, Humeau et al. (2003) reported that poisson-like trains of cortical and thalamic shocks produce LTP of cortical but not thalamic inputs to principal LA neurons. Activation of either pathway in isolation had no effect, except after pharmacological inhibition of glutamate uptake. As was observed here, this LTP was APV sensitive but resisted intracellular-MK-801 and was independent of postsynaptic activity. These properties were interpreted as evidence that glutamate released by thalamic axons activated presynaptic NMDA receptors located on cortical axon terminals.
Although the concept of a presynaptically induced NMDA-dependent LTP might seem unorthodox, it has much experimental support (MacDermott et al., 1999). First, presynaptic NMDA receptors are not unusual. They have been observed on primary afferent terminals in the spinal cord and gracile nucleus (Lu et al., 2003), on GABAergic axon terminals in the basal forebrain, thalamus, hypothalamus, and some brainstem nuclei (Paquet and Smith, 2000), as well as on unidentified excitatory terminals in the cortex (Aoki et al., 1994) and amygdala (Farb et al., 1995). Consistent with our PPF results, presynaptic NMDA receptors have been reported to enhance spontaneous transmitter release in the entorhinal and visual cortices (Beretta and Jones, 1996; Woodhall et al., 2001; Sjöström et al., 2003). Moreover, presynaptic NMDA receptors were reported to mediate enduring forms of transmitter release modulations (Liu et al., 1997; Glitsch and Marty, 1999; Sjöström et al., 2003). Thus, there are numerous precedents in the literature for presynaptic NMDA receptors producing long-term alterations in synaptic efficacy.
Our results suggest that significant activation of presynaptic NMDA receptors only occurs during bouts of high-frequency activity. This is supported by the fact that low-frequency test stimuli applied in the absence of extracellular Mg2+ did not induce LTP (Fig. 3C). A possible explanation for this observation is that presynaptic NMDA receptors are located outside the synaptic cleft. As a result, they would only be activated when presynaptic activity is high, allowing for glutamate levels outside the synaptic cleft to rise beyond a critical level for LTP induction. Consistent with this possibility, a previous study (Humeau et al., 2003) reported that inhibition of glutamate reuptake lessens the presynaptic requirement for induction of the presynaptically induced NMDA-dependent LTP in the lateral nucleus of the amygdala. Another possibility is that basal levels of presynaptic NMDA receptors on the plasma membrane are low but that high rates of presynaptic activity lead to a translocation of NMDA receptors. In support of this idea, two ultrastructural studies reported that presynaptic NMDA receptors are concentrated in dense core vesicles in basal conditions (Zhang et al., 1998; Paquet and Smith, 2000).
Implication of CEm plasticity for the intraamygdaloid substrates of pavlovian fear conditioning
That thalamic inputs to CEm express LTP does not prove that this nucleus is a site of plasticity in fear conditioning. However, it does make this idea seem more plausible. This possibility was considered (Pascoe and Kapp, 1985) but later abandoned as more and more data implicated the LA as the critical storage site (Campeau et al., 1992; Quirk et al., 1995; Muller et al., 1997; for review, see LeDoux, 2000). Although this data remains strong, it has become evident that the LA lacks direct links with the sector of CE that projects to brainstem sites mediating fear responses: the CEm (Hopkins and Holstege, 1978; Krettek and Price, 1978; Smith and Paré, 1994; Pitkänen et al., 1995) (but see Koo et al., 2004). This realization, combined with evidence that the CEm receives sensory inputs from the thalamus, plus reports that muscimol (Wilensky et al., 2000) or APV (Goosens and Maren, 2003) injections in CE block acquisition of conditioned fear responses, indicates that CE may also be a site of plasticity in fear conditioning. The present study strongly supports this idea.
However, because the LA does not project to the CEm, the question becomes: how do we reconcile the fact that interfering with LA (Muller et al., 1997) or CE activity (Wilensky et al., 2000; Goosens and Maren, 2003) during training prevents acquisition of conditioned fear responses. Recently, it was proposed that the key resides in the intercalated cell masses (Paré et al., 2004).
Intercalated cell masses are clusters of GABAergic neurons sandwiched between the basolateral amygdala (BLA) and the CE (McDonald and Augustine 1993; Nitecka and Ben-Ari 1987; Paré and Smith 1993a). They receive glutamatergic inputs from BLA and cause feedforward inhibition in the CE (Paré and Smith 1993b; Royer et al., 1999). It was observed that there is a lateromedial correspondence between the position of intercalated neurons, where they project in the central nucleus, and where they derive their inputs from the BLA (Royer et al., 1999). Importantly, there are unidirectional connections between intercalated cell clusters, directed lateromedially (Royer et al., 2000). Consequently, activation of LA depolarizes intercalated cells located at the same lateromedial level, causing an inhibition of more medially located intercalated neurons and the disinhibition of CEm neurons (Royer et al., 1999). The final consequence is a facilitation of CEm output by LA activation.
Thus, we submit that potentiation of CS responsiveness in the LA is critical to the acquisition of conditioned fear responses because it causes a disinhibition of brainstem-projecting CEm cells by way of intercalated neurons (Paré et al., 2004). In parallel, thalamic inputs to CEm neurons would undergo activity-dependent potentiation. Thus, the acquisition of conditioned fear responses may well depend on distributed storage in the amygdala. It is also likely that other brain regions, on the input and/or output sides, keep a trace of the CS-US association (Weinberger, 1995; Sanders and Fanselow, 2003).
Footnotes
This work was supported by Grant IBN-0208712 from the National Science Foundation and Grant RO1 MH-066856-01 from the National Institutes of Health.
Correspondence should be addressed to Denis Paré, Center for Molecular and Behavioral Neuroscience, Rutgers, The State University of New Jersey, 197 University Avenue, Newark, NJ 07102. E-mail: pare@axon.rutgers.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251847-09$15.00/0
References
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM (1994) Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci 14: 5202-5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar A, Samuel A, Hazvi S, Dudai Y (2003) The amygdalar circuit that acquires taste aversion memory differs from the circuit that extinguishes it. Eur J Neurosci 17: 1527-1530. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE (2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22: 5239-5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellgowan PS, Helmstetter FJ (1996) Neural systems for the expression of hypoalgesia during nonassociative fear. Behav Neurosci 110: 727-736. [DOI] [PubMed] [Google Scholar]
- Beretta N, Jones RS (1996) Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75: 339-344. [DOI] [PubMed] [Google Scholar]
- Bissière S, Humeau Y, Lüthi A (2003) Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci 6: 587-592. [DOI] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE (2001) Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8: 229-242. [DOI] [PubMed] [Google Scholar]
- Bliss T, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Cahill L, Weinberger NM, Roozendaal B, McGaugh JL (1999) Is the amygdala a locus of “conditioned fear”? Some questions and caveats. Neuron 23: 227-228. [DOI] [PubMed] [Google Scholar]
- Campeau S, Miserendino M, Davis M (1992) Intra-amygdala infusion of the N-methyl-d-aspartate receptor antagonist AP5 blocks acquisition but not expression of fear-potentiated startle to an auditory conditioned stimulus. Behav Neurosci 106: 569-574. [DOI] [PubMed] [Google Scholar]
- Collins DR, Paré D (2000) Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS(+) and CS(-). Learn Mem 7: 97-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager R, Dunwiddie T, Lynch G (1980) Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 299: 409-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M (2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: The amygdala: a functional analysis (Aggleton JP, ed), pp 213-287. Oxford: Oxford UP.
- De Oca BM, De Cola JP, Maren S, Fanselow MS (1998) Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci 18: 3426-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont ÉC, Martina M, Samson RD, Drolet G, Paré D (2002) Physiological properties of central amygdala neurons: species differences. Eur J Neurosci 15: 545-552. [DOI] [PubMed] [Google Scholar]
- Farb CR, Aoki C, LeDoux JE (1995) Differential localization of NMDA and AMPA receptor subunits in the lateral and basal nuclei of the amygdala: a light and electron microscopic study. J Comp Neurol 362: 86-108. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A (1999) Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci 19: 511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S (2003) Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci 117: 738-750. [DOI] [PubMed] [Google Scholar]
- Hall E (1972) The amygdala of the cat: a Golgi study. Z Zellforsch Mikrosk Anat 134: 439-458. [DOI] [PubMed] [Google Scholar]
- Hopkins DA, Holstege G (1978) Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res 32: 529-547. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1998) Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21: 169-178. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP (1988) Block of N-methyl-d-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA 95: 1307-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A (2003) Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426: 841-845. [DOI] [PubMed] [Google Scholar]
- Jones EG (1985) The thalamus. New York: Plenum.
- Kapp BS, Frysinger RC, Gallagher M, Haselton JR (1979) Amygdala central nucleus lesions: effects on heart rate conditioning in the rabbit. Physiol Behav 23: 1109-1117. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R (1968) The role of calcium in neuromuscular facilitation. J Physiol (Lond) 195: 481-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Han JS, Kim JJ (2004) Selective neurotoxic lesions of basolateral and central nuclei of the amygdala produce differential effects on fear conditioning. J Neurosci 24: 7654-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krettek JE, Price JL (1978) A description of the amygdaloid complex in the rat and cat with observations on intra-amygdaloid axonal connections. J Comp Neurol 178: 255-280. [DOI] [PubMed] [Google Scholar]
- Kudo M, Niimi K (1980) Ascending projections of the inferior colliculus in the cat: an autoradiographic study. J Comp Neurol 191: 545-556. [DOI] [PubMed] [Google Scholar]
- Kudo M, Itoh K, Kawamura S, Mizuno N (1983) Direct projections to the pretectum and the midbrain reticular formation from auditory relay nuclei in the lower brainstem of the cat. Brain Res 288: 13-19. [DOI] [PubMed] [Google Scholar]
- Kudo M, Tashiro T, Higo S, Matsuyama T, Kawamura S (1984) Ascending projections from the nucleus of the brachium of the inferior colliculus in the cat. Exp Brain Res 54: 203-211. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155-184. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Ruggiero DA, Reis DJ (1985) Projections to the subcortical forebrain from anatomically defined regions of the medial geniculate body in the rat. J Comp Neurol 242: 182-213. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ (1988) Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8: 2517-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke R, Braune G, Schwegler H (2000) Differential projection of the posterior paralaminar thalamic nuclei to the amygdaloid complex in the rat. Exp Brain Res 134: 520-532. [DOI] [PubMed] [Google Scholar]
- Liu HT, Mantyh PW, Basbaum AI (1997) NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature 386: 721-724. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P (2003) Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci 23: 6876-6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P (2004) Firing properties and connectivity of neurons in the rat lateral central nucleus of the amygdala. J Neurophysiol 92: 1285-1294. [DOI] [PubMed] [Google Scholar]
- Lu CR, Hwang SJ, Phend KD, Rustioni A, Valtschanoff JG (2003) Primary afferent terminals that express presynaptic NR1 in rats are mainly from myelinated, mechanosensitive fibers. J Comp Neurol 460: 191-202. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA (1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443-485. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1993) NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16: 521-527. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW (1990) Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346: 177-180. [DOI] [PubMed] [Google Scholar]
- Manabe T, Renner P, Nicoll RA (1992) Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature 355: 50-55. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA (1993) Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol 70: 1451-1459. [DOI] [PubMed] [Google Scholar]
- Maren S (2001) Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24: 897-931. [DOI] [PubMed] [Google Scholar]
- Martina M, Royer S, Paré D (1999) Physiological properties of central medial and central lateral amygdala neurons. J Neurophysiol 82: 1843-1854. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1992) Cell types and intrinsic connections of the amygdala. In: The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction (Aggleton JP, ed), pp 67-96. New York: Wiley.
- McDonald AJ (1998) Cortical pathways to the mammalian amygdala. Prog Neurobiol 55: 257-332. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Augustine JR (1993) Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52: 281-294. [DOI] [PubMed] [Google Scholar]
- Muller J, Corodimas KP, Fridel Z, LeDoux JE (1997) Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci 111: 683-691. [DOI] [PubMed] [Google Scholar]
- Nitecka L, Ben-Ari Y (1987) Distribution of GABA-like immunoreactivity in the rat amygdaloid complex. J Comp Neurol 266: 45-55. [DOI] [PubMed] [Google Scholar]
- Paquet M, Smith Y (2000) Presynaptic NMDA receptor subunit immunoreactivity in GABAergic terminals in rat brain. J Comp Neurol 423: 330-347. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y (1993a) Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience 57: 1061-1076. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y (1993b) The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience 57: 1077-1090. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y, Paré JF (1995) Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: phaseolus vulgarisleucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience 69: 567-583. [DOI] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, LeDoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1-9. [DOI] [PubMed] [Google Scholar]
- Pascoe JP, Kapp BS (1985) Electrophysiological characteristics of amygdaloid central nucleus neurons during Pavlovian fear conditioning in the rabbit. Behav Brain Res 16: 117-133. [DOI] [PubMed] [Google Scholar]
- Pitkänen A (2000) Connectivity of the rat amygdaloid complex. In: The amygdala: a functional analysis (Aggleton JP, ed), pp 31-115. Oxford: Oxford UP.
- Pitkänen A, Stefanacci L, Farb CR, Go GG, LeDoux JE, Amaral DG (1995) Intrinsic connections of the rat amygdaloid complex: projections originating in the lateral nucleus. J Comp Neurol 356: 288-310. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE (1997) Organization of intraamygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20: 517-523. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Repa JC, LeDoux JE (1995) Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15: 1029-1039. [DOI] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE (2001) Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci 4: 724-731. [DOI] [PubMed] [Google Scholar]
- Royer S, Paré D (2002) Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115: 455-462. [DOI] [PubMed] [Google Scholar]
- Royer S, Paré D (2003) Conservation of total synaptic weights via inverse homovs. heterosynaptic LTD and LTP. Nature 422: 518-522. [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (1999) An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19: 10575-10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (2000) Polarized synaptic interactions between intercalated neurons of the amygdala. J Neurophysiol 83: 3509-3518. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Fanselow MS (2003) Opioid receptor blockade in subregions of the periaqueductal gray produce distinct effects on the acquisition and expression of context fear. Soc Neurosci Abstr 29: 624.16. [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641-654. [DOI] [PubMed] [Google Scholar]
- Smith Y, Paré D (1994) Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with post-embedding GABA and glutamate immunocytochemistry. J Comp Neurol 342: 232-248. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon Jr W, Benes FM, Kandel ER, Bolshakov VY (2002) Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 34: 289-300. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Shin RM, Bolshakov VY (2004) Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron 41: 139-151. [DOI] [PubMed] [Google Scholar]
- Turner BH, Herkenham M (1991) Thalamoamygdaloid projections in the rat: a test of the amygdala's role in sensory processing. J Comp Neurol 313: 295-325. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M (2002) The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav 71: 379-392. [DOI] [PubMed] [Google Scholar]
- Weinberger NM (1995) Dynamic regulation of receptive fields and maps in the adult sensory cortex. Annu Rev Neurosci 18: 129-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE (1999) L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci 19: 10512-10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE (2000) Functional inactivation of amygdala nuclei during acquisition of Pavlovian fear conditioning. Soc Neurosci Abstr 26: 465.9. [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE (2001) Does the central nucleus of the amygdala contribute to the consolidation of auditory fear conditioning? Soc Neurosci Abstr 27: 187.9. [Google Scholar]
- Woodhall G, Evans DI, Cunningham MO, Jones RS (2001) NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J Neurophysiol 86: 1644-1651. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao L, Arvidsson U, Elde R, Hökfelt T (1998) Localization and regulation of the δ-opioid receptor in dorsal root ganglia and spinal cord of the rat and monkey: evidence for association with the membrane of large dense-core vesicles. Neuroscience 82: 1225-1242. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]