

Abstract
Anti-GD1a ganglioside antibodies (Abs) are the serological hallmark of the acute motor axonal form of the post-infectious paralysis, Guillain-Barré syndrome. Development of a disease model in mice has been impeded by the weak immunogenicity of gangliosides and the apparent resistance of GD1a-containing neural membranes to anti-GD1a antibody-mediated injury. Here we used mice with altered ganglioside biosynthesis to generate such a model at motor nerve terminals. First, we bypassed immunological tolerance by immunizing GD1a-deficient, β-1,4-N-acetylgalactosaminyl transferase knock-out mice with GD1a ganglioside-mimicking antigens from Campylobacter jejuni and generated high-titer anti-GD1a antisera and complement fixing monoclonal Abs (mAbs). Next, we exposed ex vivo nerve-muscle preparations from GD1a-overexpressing, GD3 synthase knock-out mice to the anti-GD1a mAbs in the presence of a source of complement and investigated morphological and electrophysiological damage. Dense antibody and complement deposits were observed only over presynaptic motor axons, accompanied by severe ultrastructural damage and electrophysiological blockade of motor nerve terminal function. Perisynaptic Schwann cells and postsynaptic membranes were unaffected. In contrast, normal mice were not only unresponsive to immunization with GD1a but also resistant to neural injury during anti-GD1a Ab exposure, demonstrating the central role of membrane antigen density in modulating both immune tolerance to GD1a and axonal susceptibility to anti-GD1a Abmediated injury. Identical paralyzing effects were observed when testing mouse and human anti-GD1a-positive sera. These data indicate that anti-GD1a Abs arise via molecular mimicry and are likely to be clinically relevant in injuring peripheral nerve axonal membranes containing sufficiently high levels of GD1a.
Keywords: neuromuscular, denervation, axon terminal, end plate, MEPP, ganglioside, glycosyltransferase, autoimmunity, neuropathy
Introduction
Guillain-Barré syndrome (GBS) is a post-infectious autoimmune polyneuropathy and the most common cause of acute neuromuscular paralysis worldwide (Hughes and Rees, 1997; Hughes et al., 1999). The acute motor axonal neuropathy (AMAN) variant, in which only motor axons are affected, is characterized by the presence of serum anti-GD1a ganglioside antibodies (Abs) (Lugaresi et al., 1997; Ho et al., 1999; Ogawara et al., 2000; Ilyas et al., 2001). Clinical and pathological evidence indicates multiple locations as potential sites of injury, including the nodes of Ranvier (Griffin et al., 1996), ventral roots and distal nerves (Feasby et al., 1986), and motor nerve terminals (Ho et al., 1997; Kuwabara et al., 2003). The selective injury to motor axons at these sites is presumed to be attributable to Ab binding to axonal membranes relatively enriched in GD1a, although this has never been demonstrated (Ogawa-Goto et al., 1992; De Angelis et al., 2001; Gong et al., 2002).
Anti-GD1a Abs arise in AMAN cases after gastroenteritis caused by Campylobacter jejuni strains that contain GD1a-like antigens on their surface lipo-oligosaccharide (LOS) (Aspinall et al., 1994, 1995). This suggests that molecular mimicry is a mechanism for anti-GD1a Ab induction (Sheikh et al., 1998; Willison and Yuki, 2002).
Gangliosides are sialylated glycosphingolipids involved in development, function, and maintenance of the nervous system (Hansson et al., 1977; Ledeen, 1978; Simons and Ikonen, 1997; Vyas et al., 2002). They are widely distributed and particularly enriched at the neuromuscular junctions (NMJs), along with a diverse range of glycosylated proteins (Martin, 2003). In addition to binding autoantibodies, NMJ gangliosides act aberrantly as target molecules for clostridial neurotoxins (Schiavo et al., 2000; Bullens et al., 2002).
Antibody binding to target membranes results in complement activation with membrane attack complex (MAC) pore formation (Koski et al., 1987; Acosta et al., 1996). Our studies on antidisialosyl ganglioside Ab binding to presynaptic NMJ gangliosides indicate that uncontrolled calcium influx through MAC pores triggers massive neurotransmitter release with electrophysiological failure and calpain-mediated disintegration of the presynaptic axon (Plomp et al., 1999; Bullens et al., 2000; O'Hanlon et al., 2001, 2003; Halstead et al., 2004).
Evidence that anti-GD1a antibodies play a pathogenic role in AMAN is limited (Zhang et al., 2004). The difficulty in generating a mouse model of AMAN is likely attributable to a combination of the poor immunogenicity of gangliosides and the low level of GD1a in mouse motor axons accessible to circulating Abs. Overcoming immunological tolerance has been made possible by using ganglioside-deficient, glycosyl transferase knock-out mice (Lunn et al., 2000; Bowes et al., 2002). However, because these mice no longer contain any target antigen, they are resistant to development of autoimmune disease when immunized with gangliosides, despite having high levels of circulating antibody. Here we overcame these factors in a two-stage immunization protocol. First, we cloned anti-GD1a monoclonal Abs (mAbs) from GD1a-deficient (i.e., immunologically nontolerant) β-1,4-N-acetylgalactosaminyl transferase knock-out (GalNAcT-/-) mice immunized with GD1a-containing C. jejuni LOS. Second, we applied these anti-GD1a mAbs with added complement to distal motor nerve preparations from α-2,8-sialyl transferase II knock-out (GD3s-/-) mice that express abnormally high levels of GD1a (see Fig. 1) (Kawai et al., 2001; Okada et al., 2002) and looked for pathophysiological effects.
Figure 1.

Abbreviated ganglioside biosynthetic pathway. In the GalNAcT-/- mice, disruption of the β-1,4-N-acetylgalactosaminyl transferase gene (Unigene name, Galgt1) results in the absence of all of the complex gangliosides beyond GM3 and GD3 (Takamiya et al., 1996). In the GD3s-/- mice, disruption of the α-2,8-sialyl transferase II gene (Unigene name, Siat8a) results in the absence of b-series gangliosides and a compensatory overexpression of a-series gangliosides, including GD1a (Kawai et al., 2001; Okada et al., 2002). WT mice express varying levels of all gangliosides shown. Nomenclature is according to the IUPAC-IUB Joint Commission on Biochemical Nomenclature (1998) and Svennerholm (1994). CER, Ceramide; ○, galactose; □, N-acetylgalactosamine; •, glucose; ♦, N-acetylneuraminic acid/sialic acid. Carbon-carbon linkages are also shown.
Materials and Methods
Mice
Male GD3s-/- (overexpressing the “a-series” gangliosides) (Okada et al., 2002), GalNAcT-/- (expressing only GM3, GD3, and GT3) (Takamiya et al., 1996), and wild-type (WT) mice were used at 6-8 weeks of age. A simplified scheme of ganglioside biosynthesis with the O-series omitted for clarity is shown in Figure 1. Ganglioside nomenclature is according to IUPAC-IUB Joint Commission on Biochemical Nomenclature (1998) and Svennerholm (1994). All mice were on a C57BL/6-CBA background. Control observations indicate that anti-ganglioside antibodies have the same effects in C57BL/6 mice as in a mixed C57BL/6-CBA background (S. K. Halstead, P. D. Humphreys, and J. A. Goodfellow, unpublished data). All animal experiments were performed according to United Kingdom and Dutch laws and Glasgow and Leiden University guidelines (Glasgow University, government license number PPL60/3096; Leiden University, government license number DEC 01055).
Anti-GD1a-LOS mAbs
mAbs were cloned from GalNAcT-/- mice immunized with C. jejuni HS:19 or HS:4 strains expressing LOS structures identical to GD1a (Aspinall et al., 1994; Bowes et al., 2002). HS:3 was used as an irrelevant C. jejuni isolate devoid of ganglioside-like structures (Aspinall et al., 1995). Fusions were screened against GD1a by ELISA using previously reported protocols (Goodyear et al., 1999). mAbs were characterized for isotype, subclass, and ganglioside-binding profiles (GM1, GM2, GM3, GD1a, GD1b, GD3, GT1b, and GQ1b) using standard ELISA and thin-layer chromatography-based techniques (Goodyear et al., 1999). mAbs were purified from tissue culture supernatants using HiTrap Protein A/G affinity columns (Amersham Biosciences, Bucks, UK), concentrated to 2 mg/ml, and stored at -70°C. Relative levels of mAb reactivity to a panel of gangliosides were determined from ELISA titration curves over antibody concentrations of 10-1 to 10-6 mg/ml and were expressed as the reciprocal of the antibody concentration that gave half-maximal binding (Paterson et al., 1995). The anti-GD1a mAbs cloned from mice immunized with GD1a conjugated to keyhole limpet hemocyanin (GD1a-KLH conjugate) have been reported previously (Lunn et al., 2000; Schnaar et al., 2002).
Antibodies, complement, and immunochemicals
Mouse anti-GD1a antisera were obtained from GalNAcT-/- mice immunized with GD1a-KLH emulsified in complete Freund's adjuvant. GD1a-KLH conjugate was prepared by ozonolysis of GD1a and reductive amination using sodium cyanoborohydride (Helling et al., 1994). Preimmune and hyperimmune sera were tested for anti-GD1a antibody titers by ELISA. Six human anti-GD1a-positive sera (titers from 1:11,000 to 1:23,000) were obtained from acute-phase AMAN patients (subject to local ethical committee approval and guidelines). Mouse and human anti-GD1a sera were depleted of complement by heat inactivation at 56°C for 30 min and diluted 1:2 in Ringer's solution before experiments. Normal human serum (NHS) as a source of heterologous complement was taken from a single healthy donor and stored in multiple aliquots at -70°C to maintain inter-experimental consistency.
The following primary and secondary antibodies were used: rabbit anti-neurofilament (NF) antibody 1211 (Affinity BioReagents, Exeter, UK); mouse IgG1 anti-NF antibody 1217 (Cambridge Bioscience, Cambridge, UK); FITC goat anti-mouse IgG and CY5 or FITC goat antirabbit IgG, and CY5 goat anti-mouse IgG1 (Southern Biotech Associates, Birmingham, AL); FITC-goat anti-human C3c, mouse IgG2a anti-human MAC, and rabbit anti-cow S100 (Dako, Glostrup, Denmark); and FITC goat anti-mouse IgG2a (Bethyl Laboratories, Montgomery, TX).
Immunochemicals and other reagents were as follows: Texas-Redlabeled α-bungarotoxin (TxR-BTx) and Alexa Fluor BTx (Molecular Probes, Eugene, OR; citifluor antifade medium (Citifluor, Canterbury, UK); ethidium homodimer 1 (EthD) (Molecular Probes); and goat serum, Triton X-100, and l-lysine (Sigma-Aldrich, Dorset, UK).
Nerve muscle preparations
Male GalNAcT-/-, GD3s-/-, and WT mice (6-8 weeks old) were killed by CO2 inhalation. Diaphragm or triangularis sterni muscles were dissected out onto a Sylgard-lined base of a dish containing pregassed Ringer's solution (O'Hanlon et al., 2001) at room temperature (RT) (20-22°C). All nerve muscle preparations were exposed to anti-GD1a mAbs/sera (or Ringer's control solution) for 2.5 h at 32°C and then 30 min at 4°C (to accentuate antibody binding) and 10 min at RT unless otherwise stated. Diaphragm was rinsed and then exposed to 40% NHS (or Ringer's control solution) for 1 h at RT. For immunohistology or electron microscopy, a 1 cm square section of tissue was snap frozen on dry ice or fixed in 2.5% paraformaldehyde/2.5% glutaraldehyde, respectively. For analysis of perisynaptic Schwann cell (pSC) integrity, a 1 cm square section was further incubated in EthD for 1 h at RT and then fixed in 0.1% paraformaldehyde, as described previously (Halstead et al., 2004).
Immunohistology
Topical binding studies of anti-GD1a antibodies on diaphragm sections. Diaphragm cryostat sections (8 μm) from GalNAcT-/-, GD3s-/-, and WT mice were placed onto 3-aminopropyltrithoxysilane (APES)-coated slides. Sections were then treated with anti-GD1a mAbs (50 μg/ml in PBS) for 2 h at 4°C with or without 4% NHS for an additional 1 h at RT. Anti-GD1a antibodies or human C3c were detected with FITC goat anti-mouse IgG (1:300) or FITC goat anti-human C3c (1:200;1 h, 4°C). TxR-BTx (1:1000) was included in the secondary antibody solution.
Diaphragm nerve muscle preparations. Eight and 15 μm cryostat sections of snap-frozen diaphragm tissues previously exposed ex vivo to anti-GD1a antibodies/sera and complement (as described above) were placed onto APES-coated slides. Anti-GD1a IgG or human C3c were detected with the FITC goat anti-mouse IgG (1 h, 4°C) or the FITC goat anti-human C3c (1 h, 4°C). TxR-BTx was included in the secondary antibody solution. For detection of NF, sections were first treated with TxR-BTx, fixed in ethanol (20 min, -20°C), and then exposed to rabbit anti-NF 1211 antibody (1:200 in 1% goat serum and 0.1% Triton X-100, overnight, 4°C), followed by FITC goat anti-rabbit IgG (1 h, 4°C). Tissue prepared for analysis of pSC integrity with EthD was sectioned at 20 μm and treated with Alexa Fluor BTx (1:1000; 1 h, RT).
Triangularis sterni nerve muscle preparations. For whole-mount illustrations of anti-GD1a antibody binding, live ex vivo triangularis sterni preparations were incubated with anti-GD1a mAb MOG-35 as above, fixed in 4% paraformaldehyde (20 min, RT), and rinsed in PBS, then 0.1 m glycine, and then PBS (10 min each, 4°C). Tissue was incubated overnight at 4°C in mouse anti-NF 1217 (IgG1) or rabbit anti-cow S100 (both at 1:200; in 1% goat serum, 0.4 m lysine, and 0.5% Triton X-100). Primary antibodies were detected with FITC anti-mouse IgG2b and either CY5 anti-mouse IgG1 or CY5 anti-rabbit IgG (all 1:300), and nicotinic acetylcholine receptors (nAChRs) were detected with TxR-BTx (1:1000; 3.5 h, 4°C).
Electrophysiological studies
Electrophysiological recordings were made from left and right hemidiaphragms as described previously (Plomp et al., 1999). Briefly, intracellular recordings of miniature endplate postsynaptic potentials (MEPPs) were made using standard recording equipment. Randomly within the preparation, muscle fibers were impaled near the NMJ with a 10-20 MΩ glass microelectrode filled with 3 m KCl. MEPP frequency was measured as follows: (1) before addition of anti-GD1a antibody/serum; (2) after their addition (100 μg/ml for MOG-35 anti-GD1a mAb; 1:2 in Ringer's solution for complement-inactivated AMAN serum) for 3 h at 33°C (and 30 min at 4°C in case of AMAN serum); and (3) during subsequent 1 h period of 40% NHS (at 20-22°C). If high MEPP frequency occurred that led to spontaneous fiber twitches (Plomp et al., 1999), μ-conotoxin-GIIIB (1.5 μm; Scientific Marketing, Barnet, UK) was added to block muscle Na+ channels, as well as contractions. In some diaphragm preparations, EPPs were measured under these conditions. Tissue was then washed in Ringer's medium and snap frozen for immunohistology. Signals were digitized and stored for later, off-line analysis. Ten to 25 NMJs were sampled from each experimental condition, and each experimental condition was repeated three times.
Imaging and image analysis
All fluorescence images were acquired using a Bio-Rad (Hemel Hempstead, UK) MRC1024 or Zeiss (Oberkochen, Germany) Pascal confocal microscope. Complete NMJs in whole-mount triangularis sterni or 30 μm diaphragm sections were reconstructed using Voxx threedimensional voxel-based rendering software (Clendenon et al., 2002). Electron micrographs of NMJs were prepared, viewed, and photographed as described previously (O'Hanlon et al., 2001).
For quantitative analysis, the area of the nAChR signal covered by IgG, C3c, or NF is expressed as percentage of nAChR area. Nonparametric data pooled from three to five experimental preparations are presented as the median (horizontal line), 95% confidence intervals (small box), interquartile range (large box), and 1.5× interquartile range (vertical lines). Individual data points lying outside 1.5× interquartile range are not shown in the figures for clarity but were included in all calculations and statistical analysis. Median values of individual mAbs or antisera are indicated in the figures by numbered circles. For topical staining, 436-503 endplates were examined per experimental condition and, for ex vivo preparations, 529-953 endplates. Mann-Whitney mean rank test was used to compare possible statistical differences between groups. *0.05 > p > 0.005; **0.005 > p > 0.001; ***p < 0.001.
Results
Ganglioside and LOS binding patterns of anti-GD1a mAbs
Six IgG monoclonal anti-GD1a antibodies raised in GalNAcT-/- mice immunized with C. jejuni LOS were cloned for this study. MOG-32, MOG-34, and MOG-35 (all IgG2b) were raised against HS:19 LOS. TBG-1 (IgG2b) and TBG-2 and TBG-3 (both IgG3) were raised against HS:4 LOS. All antibodies reacted with the immunizing LOS from the GBS-associated C. jejuni strain HS:19 or HS:4 but not to irrelevant LOS from the uncomplicated enteritis-associated strain HS:3 (data not shown). With respect to ganglioside reactivity, all mAbs bound to GD1a with half-maximal binding values in the 103 range, i.e., at a concentration of ∼1 μg/ml on the midpoint of the titration curve. All six mAbs also bound weakly to GT1b by 1-2 log-fold lower magnitudes. GT1b shares the terminal NeuAc(α2-3)Gal-GalNAc trisaccharide epitope with GD1a. Two mAbs also reacted very weakly with GM1 (TBG-2 and MOG-34) and one with GD1b (MOG-32). Other gangliosides screened (GM2, GM3, GD1b, GD3, and GQ1b) were not bound.
Levels of neural GD1a determine the extent of binding of anti-GD1a-LOS mAbs
We first tested the ability of anti-GD1a-LOS antibodies to bind and fix complement during topical application at the NMJs of diaphragm sections from mice with different levels of expression of GD1a (i.e., GD3s-/-, high GD1a; WT, normal GD1a; GalNAcT-/-, no GD1a). Topical application of anti-GD1a-LOS mAbs and 4% NHS as a source of complement resulted in IgG and complement (measured as the intermediate component, C3c) deposition at NMJs (Fig. 2A). The NMJs from the GD3s-/- mouse showed significantly higher levels of IgG and C3c deposition than WT mice, and the GalNAcT-/- mouse that lacks any GD1a (or GT1b) showed no detectable levels of IgG or C3c. The GalNAcT-/- mouse acts as an ideal negative control because it allows each antibody to act as its own concentration and isotype-matched control and confirms that the specificity of effect is ganglioside dependent rather than being related to nonspecific interactions or cross-reactive glycoconjugate effects. These data indicate that the anti-GD1a-LOS antibodies bind to the NMJ in a GD1a concentration-dependent manner and suggest that the GD3s-/- mouse should be more vulnerable to anti-GD1a antibody-mediated injury than normal WT mice.
Figure 2.

Effects of anti-GD1a-LOS antibodies. Immunohistological analysis of anti-GD1a-LOS antibody binding, complement fixation, and destruction of NF is shown. A, Topical application to diaphragm. “% nAChR area” represents the area of the NMJ stained with TxR-BTx that is covered with fluorescent anti-IgG or anti-C3c signal. These values were calculated as described in Materials and Methods and expressed as the median (horizontal line), interquartile range (large box), and 1.5× the interquartile range (vertical line) of pooled data. IgG (white) or C3c (hatched) levels at NMJs in sections of WT, GD3s-/-, and GalNAcT-/- diaphragm are shown. Highest levels of IgG and C3c are seen in the GD3s-/- mouse. B, Ex vivo GD3s-/- hemidiaphragm preparations. IgG (white), C3c (hatched), and NF (black) levels at NMJs were calculated and expressed as in A. Ringer's solution or anti-GD1a-LOS antibody (100 μg/ml in Ringer's solution) was applied, followed by a source of complement. Anti-GD1a-LOS mAbs bind to the NMJ, fix complement, and cause an NF loss in the GD3s-/- mouse. ***p < 0.001 compared with Ringer's control solution. Median values of individual anti-GD1a-LOS mAbs are plotted as follows: 1, MOG-32; 2, MOG-34; 3, MOG-35; 4, TBG-1; 5, TBG-2; 6, TBG-3.
Anti-GD1a-LOS mAb and complement cause NMJ injury and paralysis ex vivo in GD3s-/- mice
In view of its predicted increase in sensitivity to injury, the GD3s-/- mouse was then used to screen for pathological effects on terminal axons of all six of the anti-GD1a antibodies raised by immunization with GD1a bearing LOS. Axonal injury was monitored by loss of NF immunoreactivity, the main axonal cytoskeletal protein in nerve (O'Hanlon et al., 2001). Exposure of GD3s-/- NMJs ex vivo to anti-GD1a mAbs resulted in IgG binding, C3c deposition as expected from Figure 2A, and concomitant NF loss (Fig. 2B). Thus, all six anti-GD1a mAbs examined induced axonal injury.
To demonstrate the importance of the level of GD1a in rendering the NMJ liable to axonal injury, the effects of one mAb, MOG-35, were examined in WT (normal GD1a), GD3s-/- (high GD1a), and GalNAcT-/- (no GD1a) diaphragm preparations. These data clearly show that GD1a levels influences the magnitude of NF loss as a measure of axonal injury and that this correlates with the level of bound IgG and subsequent C3c deposition (Fig. 3A); thus, the higher levels of IgG and C3c seen at GD3s-/- NMJs translate into a greater injury (low NF signal) than that seen in WT or GalNAcT-/- mice. (Median values for tissue treated with Ringer's solution alone were as follows: IgG, <5%; C3c, <11%; NF, >34% for all genotypes. Therefore, although the anti-GD1a mAb MOG-35 caused a statistically significant loss of NF in the WT compared with GalNAcT-/-, it was a very minor loss that may not be biologically significant.)
Figure 3.

Ex vivo injury by MOG-35 is GD1a dependent. A, The anti-GD1a-monospecific IgG MOG-35 was able to bind to ex vivo NMJs, fix complement, and cause a loss of NF in a GD1a-dependent manner. IgG, C3c, and NF levels are expressed as in Figure 2 B. White, black, and hatched rectangles show IgG, C3c, and NF levels, respectively, for each genotype calculated as described in Materials and Methods. GD3s-/- nerve terminals are more susceptible to antibody binding, complement fixation, and NF loss than WT or GalNAcT-/- nerve terminals. ***p < 0.001. GD3s-/- was compared with WT. WT was compared with GalNAcT-/-. B, MOG-35 causes a massive increase in MEPP frequency in the presence of complement at the ex vivo GD3s-/- NMJ. MEPP recordings before (hatched bars) and after (black bars) MOG-35 addition demonstrate that the mAb alone has no effect on NMJ transmission. Subsequent addition of 40% NHS (white bar) causes a massive increase in asynchronous spontaneous neurotransmitter release at GD3s-/- but not WT NMJs (sample traces shown). IgG, C3c, and NF levels of the preparations that were electrophysiologically tested were equivalent to those shown in A. ***p < 0.001 compared with WT. C, Example of neurotransmission failure at GD3s-/- NMJs induced by MOG-35 and subsequent addition of 40% NHS. The phrenic nerve of a GD3s-/- diaphragm nerve-muscle preparation was stimulated once every 30 s over a 13 min monitoring period, in the presence of μ-conotoxin-GIIIB to prevent muscle action potentials. EPPs were recorded at an NMJ in which MEPP frequency was very high (>100/s, visible at the baseline of traces). EPPs were normalized to -75 mV and superimposed. The EPP decreased with time after complement addition and became blocked. This did not occur at WT or GalNAcT-/- nerve terminals and did not occur without the anti-GD1a mAb. D-H, Ultrastructure of MOG-35- or Ringer's solution-treated GD3s-/- diaphragm preparations. D, F, Control GD3s-/- diaphragm preparations show normal NMJ morphology, including an abundance of synaptic vesicles, electron-dense mitochondria, and healthy pSCs. E, G, H, MOG-35- and complement-treated GD3s-/- diaphragm preparations show grossly abnormal morphological features, including a generally swollen and electron-lucent appearance, depletion of synaptic vesicles, and electron-lucent mitochondria. pSCs retain a normal appearance. Scale bars, 0.25 μm. Ax, Axon terminal.
Electrophysiological examination of MOG-35 plus complement-treated WT, GD3s-/-, and GalNAcT-/- diaphragm preparations showed a massive increase in MEPP frequency at the GD3s-/- mouse NMJs (Fig. 3B), eventually leading to block of synaptic transmission as measured by evoked EPPs (Fig. 3C). WT NMJs were unaffected (Fig. 3B), as were GalNAcT-/- NMJs (data not shown). Subsequent immunohistological analysis of these electrophysiological preparations demonstrated similar IgG, C3c, and NF levels to those in Figure 3A (data not shown).
Previously, we showed that Miller-Fisher syndrome (MFS)-related antidisialosyl antibodies could bind to pSCs, activate complement, and lead to pSC death as monitored by an EthD uptake assay (Halstead et al., 2004). For the anti-GD1a-LOS mAb MOG-35 up to a concentration of 500 μg/ml, there was no significant increase in the proportion of NMJs showing EthD-positive pSCs compared with Ringer's solution-treated preparations (data not shown). This indicates that pSCs are not targeted by anti-GD1a Abs, even in GD1a-enriched GD3s-/- mice.
NMJ tissues were examined for ultrastructural evidence of injury, in accordance with our previous studies (O'Hanlon et al., 2001; Halstead et al., 2004).
Compared with controls (Fig. 3D,F), MOG-35- and complement-treated preparations showed highly abnormal axonal ultrastructure (vesicle depletion, mitochondrial swelling, and electron lucency) at the NMJ (Fig. 3E,G,H). pSCs appeared morphologically normal (Fig. 3E).
Localization of IgG and complement products at MOG-35-exposed NMJs
To determine the immunolocalization of IgG and complement deposits within nerve terminals, serial confocal images of MOG-35-treated diaphragm NMJs were reconstructed. In the preparations from GalNAcT-/- mice that lack GD1a, there was no detectable IgG, C3c, or MAC deposition, and there were normal levels of NF (Fig. 4C,F,I,L). In WT mice, low-level IgG, C3c, and MAC deposits were observed with near-normal NF signal (Fig. 4A,D,G,J). In the GD3s-/- preparations, levels of IgG, C3c, and MAC were very highly aligned with the gutters of the NMJ, and little or no NF signal was present over the NMJ and distal axon (Fig. 4B,E,H,K).
Figure 4.
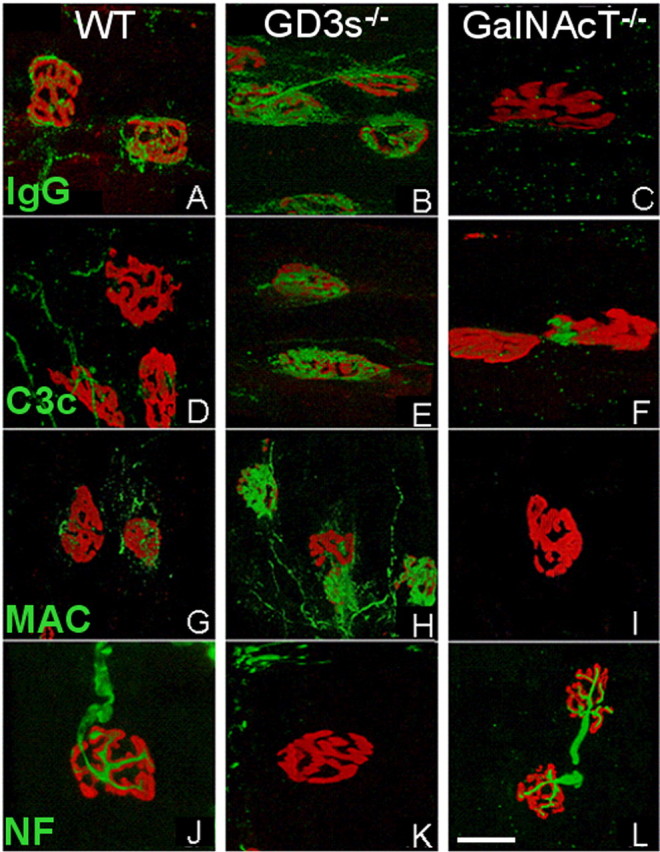
Neurofilament loss is antibody, complement, and GD1a dependent. Reconstructed confocal images of MOG-35- and complement-treated WT, GD3s-/-, and GalNAcT-/- diaphragm NMJs are shown. TxR-BTx staining is shown in red in all panels. Green indicates the following: A-C, mouse IgG; D-F, human C3c; G-I, human MAC; J-L, NF. Images were acquired and reconstructed under identical settings for each genotype. WT NMJs showed low levels of IgG (A), C3c (D), and MAC (G) and high levels of NF (J). GD3s-/- endplates showed high levels of IgG (B), C3c (E), and MAC (H) and low levels of NF (K). GalNAcT-/- NMJs showed essentially no IgG (C), C3c (F), or MAC (I) and high levels of NF (L). Scale bar, 20 μm.
To improve the resolution of MOG-35 localization at the NMJ, we used three-dimensional reconstructions of whole-mount triangularis sterni neuromuscular preparations from GD3s-/- mice (Fig. 5). These studies were conducted in the absence of complement, thereby avoiding tissue injury and allowing NF staining to delineate the axon. MOG-35 immunoreactivity appears exclusively localized to the axolemma, enveloping neurofilament bundles (Fig. 5, blue in A, D, I-K) that sit on the bungarotoxin-labeled gutters beneath and distinct from the pSC (labeled with the Schwann cell marker S100 in blue in E, H, L-N). Parallel preparations treated with Ringer's solution instead of MOG-35 and imaged and reconstructed under the same settings showed no IgG immunoreactivity (data not shown).
Figure 5.
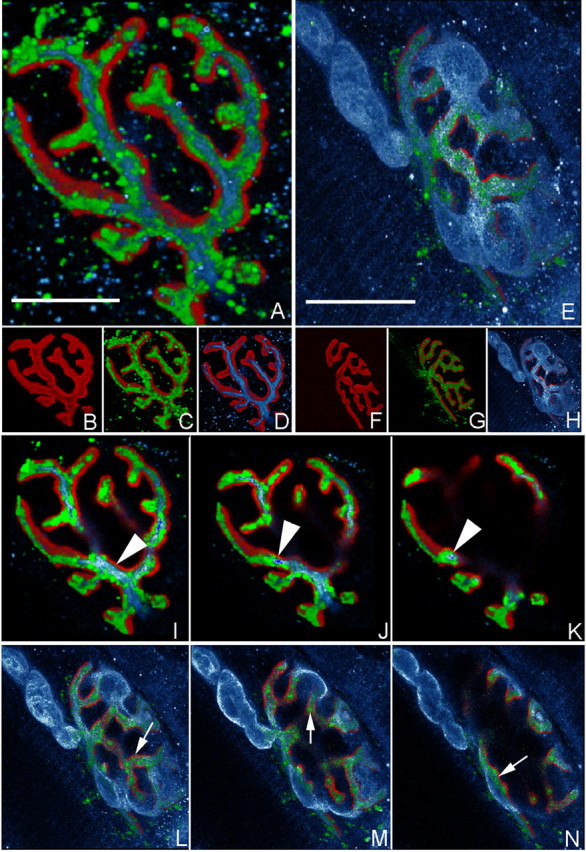
Localization of anti-GD1a-LOS mAb at the NMJ. Immunofluorescent localization of MOG-35 at GD3s-/- triangularis sterni NMJs is shown. Reconstructed confocal images of MOG-35-treated GD3s-/- preparations showing the localization of GD1a (MOG-35 staining), AChRs (TxR-BTx staining), neurofilament (anti-NF antibody staining), and perisynaptic Schwann cells (S100 staining). C, MOG-35 (green) was localized over gutters of the BTx (red) signal (B shows BTx alone). D, NF (blue) shows a similar localization over the BTx signal. A, A merged image of C and D indicated that the MOG-35 signal “wrapped” most of the NF signal. G, Another example of MOG-35 (green) localized over the BTx signal (red) (F shows BTx alone). H, The Schwann cell marker S100 (blue) labeled the pSC and terminal myelinating Schwann cell at this NMJ. E, A merged image of G and H indicated that the majority of MOG-35 staining was “underneath” the S100 signal, whereas the abaxonal pSC membrane was almost unstained.I-K, Serial sections through A. L-N, Serial sections through E. Arrowheads in I-K highlight MOG-35 staining that “wrapped” the NF signal. Arrows in L-N highlight MOG-35 staining that occurred essentially “underneath” the S100 staining. This is consistent with a primarily axonal localization, an adaxonal pSC membrane localization of MOG-35, or both. Scale bars, 10 μm.
Anti-GD1a-positive human and mouse sera bind to terminal motor axons in a GD1a-dependent manner and can cause an NF loss ex vivo
Anti-GD1a-KLH mAbs and anti-GD1a-KLH hyperimmune sera obtained by immunizing GalNAcT-/- mice with GD1a-KLH conjugate had identical pathological effects to anti-GD1a-LOS mAbs at WT, GD3s-/-, and GalNAcT-/- diaphragm NMJs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This demonstrates that anti-GD1a antibodies arising through alternative routes to preceding infection with an LOS-bearing bacterial antigen are similarly pathogenic.
To establish the relevance for human neuropathy of the anti-GD1a mAb data, we investigated the effects of acute-phase anti-GD1a IgG-positive sera from AMAN patients (n = 6; anti-GD1a titers from 1:11,000 to 1:23,000), alongside control sera from multiple sclerosis cases (n = 3; negative for anti-GD1a antibodies). One AMAN sera was also examined in an ex vivo electrophysiological study. In the presence of a source of complement, this latter serum caused a massive increase in MEPP frequency and eventual transmission block in a large proportion of the GD3s-/- NMJs ex vivo, whereas the MEPP frequency increase was very minor at WT NMJs and no transmission block was observed (Fig. 6A). Immunohistological analysis of these preparations (Fig. 6B) revealed that GD3s-/- NMJs had elevated levels of C3c and reduced NF immunoreactivity compared with WT. Human anti-GD1a-specific IgG cannot be identified because it is masked by nonspecific background staining attributable to polyclonal IgG in both the serum and in the NHS used as a source of complement. This same serum sample and five others were then tested, using immunohistological methods, for pathogenic effects ex vivo in the GD3s-/- hemi-diaphragm preparation (Fig. 6C). Four of the six AMAN sera caused a significant loss of NF but spared the pSCs. These data show that human anti-GD1a AMAN serum displays the same pathological potential as mouse anti-GD1a mAbs and antiserum.
Figure 6.

Effects of human AMAN serum. Addition of AMAN serum followed by a source of complement resulted in a massive increase in asynchronous uniquantal ACh release (measured as MEPPs) at GD3s-/- diaphragm NMJs ex vivo. A, Electrophysiological recordings of MEPPs before (hatched bars) and after (black bars) complement-inactivated AMAN serum addition demonstrated that the serum alone had no effect. Subsequent addition of 40% NHS (white bar) caused a massive increase in MEPP frequency (sample traces shown). **p < 0.005 compared with WT. B, Immunohistological investigation of NMJs from these preparations revealed that GD3s-/- (white box) NMJs from AMAN serum-treated preparations had elevated levels of C3c and reduced levels of NF compared with WT (hatched box) NMJs. The median value, the interquartile range, and 1.5× the interquartile range of C3c and NF data are expressed as described in Materials and Methods. ***p < 0.001 compared with WT. C, Six human anti-GD1a-positive AMAN sera were tested for pathogenic effects using immunohistological methods in the GD3s-/- mouse as described in Materials and Methods. Parallel preparations treated with multiple sclerosis (MS) sera or Ringer's solution served as controls. Compared with both multiple sclerosis-(hatched box) or Ringer's solution-(black box) treated preparations, the AMAN sera (white box) led to elevated levels of C3c deposition and decreased levels of NF at the NMJ. The level of complement fixation varied considerably between the AMAN samples, but four of the six caused a very large reduction in NF (median of 0 for samples 1, 3, 4, and 6), whereas the other two caused a minor loss in NF.
Discussion
We found that anti-GD1a antibodies cloned from mice immunized with GD1a-like epitopes on C. jejuni LOS cause acute destructive changes at mouse NMJs ex vivo in a complement- and GD1a-dependent manner. Human anti-GD1a antibody-positive AMAN serum and mouse sera containing polyclonal anti-GD1a antibodies or GD1a mAbs raised from GD1a-KLH immunization all are capable of inducing these effects. Besides illustrating that anti-GD1a antibodies can be generated through the molecular mimicry mechanism, these data demonstrate the capacity of anti-GD1a antibodies to play a direct role in mediating distal motor axonspecific pathology in a model of human disease. This is also the first direct demonstration, to our knowledge, that GD1a is present at motor nerve terminals.
Our data suggest that a defect at the NMJ may contribute to the paralytic clinical features of AMAN, although recognizing this does not preclude injury at other sites along the motor axon (Hafer-Macko et al., 1996; De Angelis et al., 2001; Sheikh et al., 2004). Nevertheless, the absence of a blood-nerve barrier at the motor nerve terminal does render the site especially vulnerable to injury through circulating factors, including autoantibodies and toxins. Several studies have implicated neurotransmission failure as a contributing factor in some GBS patients (Ho et al., 1997; Spaans et al., 2003), including MFS patients (Uncini and Lugaresi, 1999; Wirguin et al., 2002), and in AMAN patients in whom reduced safety factor at the nerve terminal was implicated as an important factor in conduction block (Kuwabara et al., 2003). Although the exact role that NMJ injury may play in the clinical syndromes remains undetermined, this model nevertheless provides an interesting system for studying the acute-phase pathogenic effects of anti-GD1a antibodies and for studying the neuronal and glial response to the degenerative and regenerative phases of autoimmune distal axonal injury.
Remarkably, we found that NMJs from normal mice were resistant to anti-GD1a-mediated injury. Thus, a key strategy was the use of GD3s-/- mice that express higher levels of GD1a than WT mice to act as the tissue target for anti-GD1a Abs. Conversely, because GalNAcT-/- mice do not express GD1a, they are ideal for raising anti-GD1a antibodies because they do not exhibit immunological tolerance (Lunn et al., 2000; Bowes et al., 2002). However, they are not susceptible to developing anti-GD1a-mediated disease because they lack target antigen. We thus manipulated these two mouse strains in a “cross-immunization” paradigm in which mice lacking GD1a (GalNAcT-/-) were used to induce a potent anti-GD1a immune response, and mice expressing high levels of GD1a (GD3s-/-) were used as vehicles to provide an antigen-rich target for the pathophysiological effects of that response. Had we used WT mice throughout this study, we would have encountered both poor humoral immune responses to GD1a and poor levels of tissue injury, thus failing to induce an immunopathological phenotype. This underlines the power of experimentally controlling the level of target antigen, a notion that has been more extremely applied to other models of autoimmune disease precipitated by neoself antigen expression (Cornet et al., 2001). This concept has clear clinical relevance, because, in MFS, the vulnerability of the extraocular nerves to anti-GQ1b antibody-mediated injury is attributed to their higher levels of GQ1b compared with motor nerves innervating the limb (Chiba et al., 1997). The level of expression of GD1a in different regions of the human peripheral nervous system, and the possibility that individuals vary in their ganglioside composition, has not been examined but could provide support for this hypothesis.
Anti-GD1a antibodies did not induce any deleterious effects on pSCs, in contrast to previously studied MFS-associated anti-GQ1b antibodies (Halstead et al., 2004). This underlines the capacity of anti-GD1a antibodies to mediate axon-specific pathology. Electron microscopy and vital dye staining revealed normal pSC integrity and ultrastructure. Moreover, the whole-mount three-dimensional reconstructions of NMJs clearly showed selective axonal anti-GD1a antibody binding. This parallels the selective axonal injury seen in AMAN patients, in which motor axons are damaged whereas Schwann cells are spared (Hafer-Macko et al., 1996; De Angelis et al., 2001).
Not all anti-GD1a antibody-positive AMAN patients show evidence of preceding infection with an LOS-bearing bacterial agent. Viral agents are hypothesized to lead to anti-ganglioside response via presentation of host ganglioside during viral budding from infected cells (Evans and Webb, 1986; Ang et al., 2000). The characteristics and pathological properties of our anti-GD1a antibodies and antisera arising in response to GD1a-KLH immunization were very similar to those seen in response to LOS immunization. This strongly supports the notion that mouse and human anti-GD1a antibodies arising through different mechanisms are similarly capable of inducing highly selective motor axonal injury.
Footnotes
This work was supported by The Wellcome Trust Grants 051930, 060349, and 065261 (H.J.W.), the Guillain-Barré Syndrome Support Group, United Kingdom (J.A.G., H.J.W.), and National Institutes of Health Grant NS42888 (K.S.). We thank Prof. John Griffin for critical comments and discussion.
Correspondence should be addressed to Prof. Hugh J. Willison, Division of Clinical Neurosciences, Department of Neurology, University of Glasgow, Ground Floor Neurology Block, Southern General Hospital, Glasgow G51 4TF, UK. E-mail: h.j.willison@clinmed.gla.ac.uk.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251620-09$15.00/0
References
- Acosta JA, Benzaquen LR, Goldstein DJ, Tosteson MT, Halperin JA (1996) The transient pore formed by homologous terminal complement complexes functions as a bi-directional route for the transport of autocrine and paracrine signals across human cell membranes. J Mol Med 2: 755-765. [PMC free article] [PubMed] [Google Scholar]
- Ang CW, Jacobs BC, Brandenburg AH, Laman JD, van der Meche FG, Osterhaus AD, van Doorn PA (2000) Cross-reactive antibodies against GM2 and CMV-infected fibroblasts in Guillain-Barré syndrome. Neurology 54: 1453-1458. [DOI] [PubMed] [Google Scholar]
- Aspinall GO, Fujimoto S, Mcdonald AG, Pang H, Kurjanczyk LA, Penner JL (1994) Lipopolysaccharides from Campylobacter jejuni associated with Guillain-Barré syndrome patients mimic human gangliosides in structure. Infect Immun 62: 2122-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspinall GO, Lynch CM, Pang H, Shaver RT, Moran AP (1995) Chemical structures of the core region of Campylobacter jejuni O:3 lipopolysaccharide and an associated polysaccharide. Eur J Biochem 231: 570-578. [PubMed] [Google Scholar]
- Bowes T, Wagner ER, Boffey J, Nicholl D, Cochrane L, Benboubetra M, Conner J, Furukawa K, Furukawa K, Willison HJ (2002) Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in Campylobacter jejuni strains associated with Guillain-Barré syndrome. Infect Immun 70: 5008-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullens RWM, O'Hanlon GM, Goodyear CS, Molenaar P, Conner J, Willison HJ, Plomp JJ (2000) Anti-GQ1b antibodies and evoked acetylcholine release at mouse motor endplates. Muscle Nerve 23: 1035-1043. [DOI] [PubMed] [Google Scholar]
- Bullens RWM, O'Hanlon GM, Wagner E, Molenaar PC, Furukawa K, Furukawa K, Plomp JJ, Willison HJ (2002) Complex gangliosides at the neuromuscular junction are membrane receptors for autoantibodies and botulinum neurotoxin but redundant for normal synaptic function. J Neurosci 22: 6876-6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I (1997) Ganglioside composition of the human cranial nerves, with special reference to pathophysiology of Miller Fisher syndrome. Brain Res 745: 32-36. [DOI] [PubMed] [Google Scholar]
- Clendenon JL, Phillips CL, Sandoval RM, Fang S, Dunn KW (2002) Voxx: a PC-based, near real-time volume rendering system for biological microscopy. Am J Physiol Cell Physiol 282: C213-C218. [DOI] [PubMed] [Google Scholar]
- Cornet A, Savidge TC, Cabarrocas J, Deng WL, Colombel JF, Lassmann H, Desreumaux P, Liblau RS (2001) Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn's disease? Proc Natl Acad Sci USA 98: 13306-13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis MV, Di Muzio A, Lupo S, Gambi D, Uncini A, Lugaresi A (2001) Anti-GD1a antibodies from an acute motor axonal neuropathy patient selectively bind to motor nerve fibre nodes of Ranvier. J Neuroimmunol 121: 79-82. [DOI] [PubMed] [Google Scholar]
- Evans NR, Webb HE (1986) Immunoelectron-microscopical labelling of glycolipids in the envelope of a demyelinating brain-derived RNA virus (Semliki Forest) by anti-glycolipid sera. J Neurol Sci 74: 279-287. [DOI] [PubMed] [Google Scholar]
- Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, Zochodne DW (1986) An acute axonal form of Guillain Barre polyneuropathy. Brain 109: 1115-1126. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA (2002) Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain 125: 2491-2506. [DOI] [PubMed] [Google Scholar]
- Goodyear CS, O'Hanlon GM, Plomp JJ, Wagner ER, Morrison I, Veitch J, Cochrane L, Bullens RWM, Molenaar PC, Conner J, Willison HJ (1999) Monoclonal antibodies raised against Guillain Barré syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyse nerve muscle preparations. J Clin Invest 104: 697-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Li CY, Macko C, Ho TW, Hsieh ST, Xue P, Wang FA, Cornblath DR, McKhann GM, Asbury AK (1996) Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol 25: 33-51. [DOI] [PubMed] [Google Scholar]
- Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, McKhann GM, Asbury AK, Griffin JW (1996) Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol 40: 635-644. [DOI] [PubMed] [Google Scholar]
- Halstead SK, O'Hanlon GM, Humphreys PD, Morrison DB, Morgan BP, Todd AJ, Plomp JJ, Willison HJ (2004) Anti-disialoside antibodies kill perisynaptic Schwann cells and damage motor nerve terminals via membrane attack complex in a murine model of neuropathy. Brain 127: 2109-2123. [DOI] [PubMed] [Google Scholar]
- Hansson HA, Holmgren J, Svennerholm L (1977) Ultrastructural localization of cell membrane GM1 ganglioside by cholera toxin. Proc Natl Acad Sci USA 74: 3782-3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helling F, Shang A, Calves M, Zhang S, Ren S, Yu RK, Oettgen HF, Livingston PO (1994) GD3 vaccines for melanoma: superior immunogenicity of keyhole limpet hemocyanin conjugate vaccines. Cancer Res 54: 197-203. [PubMed] [Google Scholar]
- Ho TW, Hsieh S-T, Nachamkin I, Willison HJ, Sheikh K, Kiehlbauch J, Flanigan K, McArthur JC, Cornblath DR, McKhann GM, Griffin JW (1997) Motor nerve terminal degeneration provides a potential mechanism for rapid recovery in acute motor axonal neuropathy after Campylobacter infection. Neurology 48: 717-724. [DOI] [PubMed] [Google Scholar]
- Ho TW, Willison HJ, Nachamkin I, Li CY, Veitch J, Ung H, Wang GR, Liu RC, Cornblath DR, Asbury AK, Griffin JW, McKhann GM (1999) Anti-GD1a antibody is associated with axonal but not demyelinating forms of Guillain-Barré syndrome. Ann Neurol 45: 168-173. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Rees JH (1997) Clinical and epidemiological features of Guillain-Barré syndrome. J Infect Dis 176: S92-S98. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Hadden RD, Gregson NA, Smith KJ (1999) Pathogenesis of Guillain-Barré syndrome. J Neuroimmunol 100: 74-97. [DOI] [PubMed] [Google Scholar]
- Ilyas AA, Chen ZW, Cook SD, Mithen FA, Singhal BS (2001) Immunoglobulin G subclass distribution of autoantibodies to gangliosides in patients with Guillain-Barré syndrome. Res Commun Mol Pathol Pharmacol 109: 115-123. [PubMed] [Google Scholar]
- IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (1998) Nomenclature of glycolipids. Carbohydr Res 312: 167-175. [Google Scholar]
- Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL (2001) Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem 276: 6885-6888. [DOI] [PubMed] [Google Scholar]
- Koski CL, Sanders ME, Swoveland PT, Lawley TJ, Shin ML, Frank MM, Joiner KA (1987) Activation of terminal components of complement in patients with Guillain-Barré syndrome and other demyelinating neuropathies. J Clin Invest 80: 1492-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara S, Bostock H, Ogawara K, Sung J, Kanai K, Mori M, Hattori T, Burke D (2003) The refractory period of transmission is impaired in axonal Guillain-Barré syndrome. Muscle Nerve 28: 683-689. [DOI] [PubMed] [Google Scholar]
- Ledeen RW (1978) Ganglioside structures and distribution: are they localized at the nerve ending? J Supramol Struct 8: 1-17. [DOI] [PubMed] [Google Scholar]
- Lugaresi A, Ragno M, Torrieri F, DiGuglielmo G, Fermani P, Uncini A (1997) Acute motor axonal neuropathy with high titre IgG and IgA anti-GD1a antibodies following Campylobacter enteritis. J Neurol Sci 147: 193-200. [DOI] [PubMed] [Google Scholar]
- Lunn MPT, Johnson LA, Fromholt SE, Itonori S, Huang J, Vyas AA, Hildreth JEK, Griffin JW, Schnaar RL, Sheikh KA (2000) High-affinity antiganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem 75: 404-412. [DOI] [PubMed] [Google Scholar]
- Martin PT (2003) Glycobiology of the neuromuscular junction. J Neurocytol 32: 915-929. [DOI] [PubMed] [Google Scholar]
- Ogawa-Goto K, Funamoto N, Ohta Y, Abe T, Nagashima K (1992) Myelin gangliosides of human peripheral nervous system: an enrichment of GM1 in the motor nerve myelin isolated from cauda equina. J Neurochem 59: 1844-1849. [DOI] [PubMed] [Google Scholar]
- Ogawara K, Kuwabara S, Mori M, Hattori T, Koga M, Yuki N (2000) Axonal Guillain-Barré syndrome: relation to anti-ganglioside antibodies and Campylobacter jejuni infection in Japan. Ann Neurol 48: 624-631. [PubMed] [Google Scholar]
- O'Hanlon GM, Plomp JJ, Chakrabarti M, Morrison I, Wagner ER, Goodyear CS, Yin X, Trapp BD, Conner J, Molenaar PC, Stewart S, Rowan EG, Willison HJ (2001) Anti-GQ1b ganglioside antibodies mediate complement-dependent destruction of the motor nerve terminal. Brain 124: 893-906. [DOI] [PubMed] [Google Scholar]
- O'Hanlon GM, Humphreys PD, Goldman RS, Halstead SK, Bullens RW, Plomp JJ, Ushkaryov Y, Willison HJ (2003) Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain 126: 2497-2509. [DOI] [PubMed] [Google Scholar]
- Okada M, Itoh Mi M, Haraguchi M, Okajima T, Inoue M, Oishi H, Matsuda Y, Iwamoto T, Kawano T, Fukumoto S, Miyazaki H, Furukawa K, Aizawa S, Furukawa K (2002) b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem 277: 1633-1636. [DOI] [PubMed] [Google Scholar]
- Paterson G, Wilson G, Kennedy PGE, Willison HJ (1995) Analysis of anti-GM1 ganglioside IgM antibodies cloned from motor neuropathy patients demonstrates diverse v-region gene usage with extensive somatic mutation. J Immunol 155: 3049-3059. [PubMed] [Google Scholar]
- Plomp JJ, Molenaar PC, O'Hanlon GM, Jacobs BC, Veitch J, Daha MR, van Doorn PA, van der Meché FGA, Vincent A, Morgan BP, Willison HJ (1999) Miller Fisher anti-GQ1b antibodies: α-latrotoxin-like effects on motor end plates. Ann Neurol 45: 189-199. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C (2000) Neurotoxins affecting neuroexocytosis. Physiol Rev 80: 716-766. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Fromholt SE, Gong Y, Vyas AA, Laroy W, Wayman DM, Heffer-Lauc M, Ito H, Ishida H, Kiso M, Griffin JW, Shiekh KA (2002) Immunoglobulin G-class mouse monoclonal antibodies to major brain gangliosides. Anal Biochem 302: 276-284. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Ho TW, Nachamkin I, Li CY, Cornblath DR, Asbury AK, Griffin JW, McKhann GM (1998) Molecular mimicry in Guillain-Barré syndrome. Ann NY Acad Sci 845: 307-321. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G, Gong Y, Schnaar RL, Griffin JW (2004) An antiganglioside antibody-secreting hybridoma induces neuropathy in mice. Ann Neurol 56: 228-239. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569-572. [DOI] [PubMed] [Google Scholar]
- Spaans F, Vredeveld JW, Morre HH, Jacobs BC, De Baets MH (2003) Dysfunction at the motor end-plate and axon membrane in Guillain-Barré syndrome: a single-fiber EMG study. Muscle Nerve 27: 426-434. [DOI] [PubMed] [Google Scholar]
- Svennerholm L (1994) Designation and schematic structure of gangliosides and allied glycosphingolipids. Prog Brain Res 101: XI-XIV. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, Sakae M, Kishikawa M, Shiku H, Aizawa S (1996) Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci USA 93: 10662-10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uncini A, Lugaresi A (1999) Fisher syndrome with tetraparesis and antibody to GQ1b: evidence for motor nerve terminal block. Muscle Nerve 22: 640-644. [DOI] [PubMed] [Google Scholar]
- Vyas AA, Patel HV, Fromholt SE, Heffer-Lauc M, Vyas KA, Dang J, Schachner M, Schnaar RL (2002) Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc Natl Acad Sci USA 99: 8412-8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willison HJ, Yuki N (2002) Peripheral neuropathies and anti-glycolipid antibodies. Brain 2002 125: 2591-2625. [DOI] [PubMed] [Google Scholar]
- Wirguin I, Ifergane G, Almog Y, Lieberman D, Bersudsky M, Herishanu YO (2002) Presynaptic neuromuscular transmission block in Guillain-Barré syndrome associated with anti-GQ1b antibodies. Neuromuscul Disord 12: 292-293. [DOI] [PubMed] [Google Scholar]
- Zhang G, Lopez PH, Li CY, Mehta NR, Griffin JW, Schnaar RL, Sheikh KA (2004) Anti-ganglioside antibody-mediated neuronal cytotoxicity and its protection by intravenous immunoglobulin: implications for immune neuropathies. Brain 127: 1085-1100. [DOI] [PubMed] [Google Scholar]