

Abstract
The efficiency of neural circuits is enhanced not only by increasing synaptic strength but also by increasing intrinsic excitability. In contrast to the detailed analysis of long-term potentiation (LTP), less attention has been given to activity-dependent changes in the intrinsic neuronal excitability. By stimulating hippocampal CA1 pyramidal neurons with synaptic inputs correlating with postsynaptic neuronal spikes, we elicited an LTP of intrinsic excitability (LTP-IE) concurring with synaptic LTP. LTP-IE was manifested as a decrease in the action potential threshold that was attributable to a hyperpolarized shift in the activation curve of voltage-gated sodium channels (VGSCs) rather than activity-dependent changes in synaptic inputs or A-type K+ channels. Cell-attached patch recording of VGSC activities indicated such an activity-dependent change in VGSCs. Induction of LTP-IE was blocked by the NMDA receptor antagonist APV, intracellular BAPTA, the CaM kinase inhibitors KN-62 and autocamtide-2-related inhibitory peptide, and the protein synthesis inhibitors emetine and anisomycin. The results suggest that induction of LTP-IE shares a similar signaling pathway with the late phase of synaptic LTP and requires activation of the NMDA glutamate receptor subtype, Ca2+ influx, activity of CaM kinase II, and function of the protein synthesis. This new form of hippocampal neuronal plasticity could be a cellular correlate of learning and memory besides synaptic LTP.
Keywords: long-term potentiation, intrinsic excitability, Na+ channel, NMDA receptors, CaM kinases, protein synthesis
Introduction
The intrinsic excitability of neurons has a crucial impact on the function of neural networks and is modified during the learning task in a variety of species (Alkon et al., 1985; Cleary et al., 1998; Antonov et al., 2001; Burrell et al., 2001; Aizenman et al., 2003). In contrast to the detailed analysis of synaptic long-term potentiation (LTP), a cellular model for learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Choi et al., 2000), less attention has been given to activity-dependent changes in the intrinsic excitability of neurons. In CA1 hippocampal pyramidal neurons, after induction of LTP, postsynaptic spike firing increases more than would be expected from synaptic LTP, a phenomenon referred to as EPSP-to-spike (E-S) potentiation (Bliss and Lomo, 1973; Andersen et al., 1980). E-S potentiation has been reported to depend on disinhibition (Abraham et al., 1987; Chavez-Noriega et al., 1990), but later studies have suggested a change in the intrinsic excitability (Wathey et al., 1992; Pugliese et al., 1994; Jester et al., 1995; Wang et al., 2003). More recent studies suggest that full expression of E-S potentiation requires both GABAA receptor-mediated disinhibition and intrinsic potentiation (Daoudal et al., 2002; Staff and Spruston, 2003). The detail mechanisms underlying this form of activity-dependent plasticity are not fully understood. Two recent studies in cerebellar slices reported that high-frequency stimuli were capable of enhancing intrinsic excitability by increasing input resistance and decreasing action potential (AP) threshold (Aizenman and Linden, 2000; Armano et al., 2000). In low-density hippocampal cultures, increased intrinsic excitability of presynaptic neurons has been detected by delivering correlated stimulation (CS) to postsynaptic neurons, which was related to protein kinase C (PKC)-mediated changes in voltage-dependent activation and inactivation of sodium channels (Ganguly et al., 2000). A recent study in cortical layer V pyramidal neurons reported that high-frequency APs induced a decrease in the AP threshold that involved the protein kinase A (PKA) pathway (Cudmore and Turrigiano, 2004). LTP of intrinsic excitability (LTP-IE) also was reported to be expressed as a reduction of afterhyperpolarization (AHP) (Ireland and Abraham, 2002; Oh et al., 2003; Sourdet et al., 2003), an increase in input resistance (Armano et al., 2000), and an enhanced local excitability of dendrites through a hyperpolarized shift in the inactivation curve of A-type K+ channels (Frick et al., 2004). The activity-dependent changes in the firing threshold may work together with the activity-dependent inhibition of A-type K+ channels and reduction of AHP to modify neuronal input-output property during learning and memory. Here, we report that correlation of synaptic stimuli with postsynaptic APs induced both synaptic LTP and a decreased AP threshold (LTP-IE) in CA1 postsynaptic pyramidal neurons in hippocampal slices. LTP-IE involved a hyperpolarized shift in the activation and inactivation curves of voltage-gated sodium channels (VGSCs). Moreover, we found that induction of LTP-IE required activation of the NMDA receptor (NMDAR), activity of the calcium/calmodulin-dependent protein kinase (CaMK) pathway, and function of protein synthesis.
Materials and Methods
Slice preparation. Brain slices were prepared as described previously (Kang et al., 1998; Jiang et al., 2001). Briefly, 14- to 20-d-old (P14-P20) Sprague Dawley rats were anesthetized with pentobarbitone sodium (55 mg/kg) and decapitated. Brains were removed rapidly and glued with the anterior surfaces down. Transverse brain slices of 300 μm were cut with a vibratome (Technical Products International, St. Louis, MO) in a cutting solution containing (in mm) 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 10 glucose, 26 NaHCO3, and 230 sucrose. Slices containing the hippocampus were incubated in the slice solution gassed with 5% CO2 and 95% O2 for 1-7 h and then transferred to a recording chamber (1.5 ml) that was perfused with the slice solution gassed with 5% CO2 and 95% O2 at room temperature (23-24°C) for recording. The standard slice solution contained (in mm) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 10 glucose and 26 NaHCO3, pH 7.4 when gassed with 95% O2 and 5% CO2.
Whole-cell patch-clamp recording. Cells were visualized with a 63× water immersion lens on an Olympus BX51 upright microscope (Olympus, New Hyde Park, NY) equipped with differential interference contrast (DIC) optics. Patch electrodes with a resistance of 4-7 MΩ for somatic recordings and 7-10 MΩ for dendritic recordings were pulled from KG-33 glass capillaries (inner diameter, 1.0 mm; outer diameter, 1.5 mm; Garner Glass, Claremont, CA) using a P-97 electrode puller (Sutter Instruments, Novato, CA). Cells with the seal resistance <5 GΩ and a holding current more than -200 pA were rejected. Pyramidal neurons were patched in either the voltage-clamp or the current-clamp configuration (Hamill et al., 1981). The pipette solution for whole-cell recordings contained (in mm) 123 K-gluconate, 10 KCl, 1 MgCl2, 10 HEPES, 1 EGTA, 0.1 CaCl2, 1 K2ATP, 0.2 Na4GTP, and 4 glucose, pH adjusted to 7.2 with KOH. The pipette solution for cell-attached patches contained (in mm) 140 NaCl, 20 TEA-Cl, 5 4-AP, and 10 HEPES, pH adjusted to 7.3 with NaOH. To perform single pipette experiments, we used an Axopatch 200B amplifier (Axon Instruments, Union City, CA), and to perform dual recordings, we used the Axopatch 200B for single-channel recordings and Multiclamp 700A for whole-cell recordings. Cases in which the series resistance changed by >10% of control were rejected. The AP threshold was measured as the potential of the AP start point. The value of membrane potentials was not adjusted by the pipette liquid junction potential that was 13.7 ± 0.1 mV in our experimental conditions according to the methods reported previously (Neher, 1992).
Cell-attached patch recording of VGSC activities in dual recordings. Dual recordings with one whole-cell and one cell-attached patch were both performed on the soma of a pyramidal neuron (see Fig. 4 A). The pipette solution for cell-attached patches contained (in mm) 140 NaCl, 20 TEA-Cl, 5 4-AP, and 10 HEPES, pH adjusted to 7.3 with NaOH. The membrane potential was held at -64 mV by the whole-cell pipette. The CS protocol was delivered through the whole-cell pipette, and single-channel activities were examined every 5 min by delivering depolarizing protocol through the cell-attached patch pipette before and after the CS. Activation property was tested by voltage steps that ranged from -84 to -4 mV in 5 mV steps with a duration of 50 ms and intervals of 10 s. The inactivation protocol was a set of prepulses from -104 to -34 mV in 10 mV steps, followed by a depolarization step that depolarized the patch membrane to -34 mV. The peak amplitude of channel activities from three traces were averaged for each step to obtain activation and inactivation curves. An on-line subtraction program and capacitance compensation were used for cell-attached recordings to reduce the capacitance currents induced by depolarization steps. Capacitance transients in cell-attached patch recordings were further subtracted by off-line analysis using Clampfit version 8.2.
Figure 4.

A hyperpolarized shift in the activation curve of VGSCs during induction of LTP-IE. A, Whole-cell voltage-clamp recordings from a representative cell showing the voltage-dependent activation of the Na+ current (INa, arrows) during control (Con) and 100 min after CS (CS). After CS, the depolarization step required to activate the INa became -60 mV. B, Summary of changes in the voltage-dependent activation of the INa (ΔThNa) from time control (open bar; n = 10 cells) and CS (filled bar; **p < 0.01; paired t test; n = 9 cells) experiments. C, A representative patch showing activities of fast and transient VGSC activities at different steps (left numbers in millivolts) before (Con) and 1 h after CS (CS). The bottom lines indicate the depolarization steps (in millivolts) delivered to the cell-attached patch. The straight line in each trace indicates baseline (closed state). D, A DIC image showing a whole-cell pipette (arrowhead) and a cell-attached pipette (arrow) on the soma of a pyramidal neuron. Scale bar, 10 μm. E, The I-V curve for unitary currents of control VGSCs (n = 8 patches). F, The activation curve of VGSCs during control (open circle) and after CS (filled circle). Normalized conductance was obtained through dividing the chord conductance by the maximal conductance. The data were fitted by the Boltzman equation. The numbers are V1/2 during control (right) and after CS (left). V1/2 is the voltage at which the chord conductance of VGSCs is at half of the maximum.
CS. To evoke presynaptic glutamate release, a bipolar tungsten electrode was placed in the stratum radiatum 100-200 μm from the soma of recorded pyramidal neurons to deliver extracellular stimulation to the Schaffer collateral pathway. Stimuli were produced by a Master eight-pulse generator (A.M.P.I., Jerusalem, Israel) and passed through an ISOFlex isolator (A.M.P.I.). The duration of each pulse was 0.1 ms with the intensity of 100-400 μA. Four trains of 100 stimuli (20 Hz, 5 s) (see Fig. 1 B) were delivered with a 5 min intertrain interval. Current injection-evoked postsynaptic APs within 10 ms of the onset of each individual EPSP (see Fig. 1 B) (current: 0.4-0.8 nA, 2 ms) were coapplied to induce synaptic plasticity and intrinsic plasticity.
Figure 1.

CS induced both synaptic LTP and neuronal LTP-IE. A, The intracellular Ca2+/EGTA ratio affects the stability of the AP threshold. The AP threshold changed as a function of recording time when a 0 mm Ca2+/0 mm EGTA (filled circle) or a 0 mm Ca2+/10 mm EGTA (filled square) pipette solution was used but was relatively stable when a 0.1 mm Ca2+/1 mm EGTA (open circle) or 1 mm Ca2+/10 mm EGTA (open diamond) pipette solution was used (n = 5 cells for each group). B, Protocol of CS. Top trace, A recording of synaptic EPSPs and APs evoked by 5 s of CS. An enlarged trace from the indicated period shows four EPSCs and APs. The two bottom lines correspond to the synaptic stimulation (Syn) and current injection (Cur) protocols. C, Whole-cell voltage-clamp (top traces) and current-clamp (bottom traces) recordings from a representative pyramidal neuron showing CS-induced synaptic LTP (top, EPSC) and gradually developed LTP-IE (bottom, arrows). Bottom lines, Injecting currents (in picoamperes). D, The time course of synaptic LTP (top) and LTP-IE (bottom).
Calcium image. A customized two-photon laser-scanning Olympus BX61WI microscope with a 60× objective lens was used to detect Ca2+ signals. A Mai/Tai laser (Solid-State Laser, Mountain View, CA) tuned to 810 nm was used for excitation. Both red and green fluorescence were detected simultaneously by two photonmultiplier tubes. Image acquisition was controlled by Olympus software Fluoview FV300 (Olympus America, Melville, NY). In the transfluorescence pathway, a 565 nm dichroic mirror was used to separate green and red fluorescence. HQ525/50 and HQ605/50 filters were placed in the “green” and “red” pathways, respectively, to eliminate transmitted or reflected excitation light (Chroma Technology, Rockingham, VT). Neurons were loaded with dyes through the patch pipette for 20 min before image acquisition. Alexa Fluor-594 was used to outline the neuronal structures, and Alexa Fluor-4 was used to detect Ca2+ signals. To measure Ca2+ dynamics, the fluorescence was collected by scanning at 4 Hz in a surface-scanning mode (XYT). Fluorescence was averaged from the specified structures to obtain F(t). Baseline fluorescence (F0) was the average of four images during control, and ΔF/F was calculated as (ΔF/F) (t) = (F(t) - F0)/F0.
Data analysis. Recording signals were filtered through an eight-pole Bessel low-pass filter with a 1 or 2 kHz cutoff frequency and sampled by pClamp version 8.2 software (Axon Instruments) with an interval of 50 μs. Data were further analyzed by Clampfit version 8.2 (Axon Instruments). Unitary currents for current-voltage (I-V) relationship curves were measured by two horizontal cursers. Analyzed data were further processed with Origin 5.1 (Microcal Software, Northampton, MA) and CorelDraw 9.0 (Corel, Ottawa, Ontario, Canada) programs. Statistical data are presented as mean ± SEM if not indicated otherwise.
Results
Whole-cell patch-clamp recordings were performed in CA1 pyramidal neurons from acutely isolated hippocampal slices. To study intrinsic excitability, stable recordings of the AP threshold are necessary. We found that recording from pyramidal neurons with a Ca2+-free pipette solution (Fig. 1A, filled circle and filled square) caused a decrease in the AP threshold, which was accompanied by a hyperpolarized resting membrane potential (RMP) (in 0 mm Ca2+/10 mm EGTA at 0 min, -64.4 ± 0.8 mV; at 15 min, -78.3 ± 1.8 mV; p < 0.01; paired t test; n = 5 cells). In contrast, recording with an EGTA-buffered Ca2+ solution (0.1 mm Ca2+/1 mm EGTA or 1 mm Ca2+/10 mm EGTA) maintained a relatively stable AP threshold (Fig. 1A, open circle and open diamond). Therefore, we used a pipette solution containing 0.1 mm Ca2+ and 1 mm EGTA in all following experiments. The CS used in this study was composed of synaptic stimuli (Fig. 1B, Syn) (four 20 Hz trains of 5 s duration with an intertrain interval of 5 min) and current injection-evoked postsynaptic APs within 10 ms of the onset of each individual EPSP (Fig. 1B, Cur) (0.4-0.8 nA, 2 ms) (Markram et al., 1997; Bi and Poo, 1998; Debanne et al., 1998).
LTP is accompanied by a decrease in the AP threshold
Under the control conditions, the AP threshold in hippocampal CA1 pyramidal neurons is in a range of -38 to -51 mV. CS induced a decrease (>4 mV) in the AP threshold in 92% (46 of 50) of tested pyramidal neurons (Fig. 1C, bottom, arrows). The time course of the changes (Fig. 1D, bottom) showed that the decrease in the postsynaptic AP threshold took 100-120 min to attain its maximum. The decreased AP threshold lasted for as long as we could keep recordings in these cells (the longest recording in our study was 6 h; data not shown). At the same time, CS induced a persistent increase in the synaptic strength, indicating the onset of synaptic LTP (Fig. 1C,D, EPSC). Pooled data showed that the postsynaptic AP threshold was significantly lower after CS (Fig. 2A, bottom, filled circle) (n = 12 cells) than in the control group (Fig. 2A, bottom, open circle) (n = 13 cells; p < 0.01; two-way ANOVA). Enhancement of synaptic strength (Fig. 2A, top, filled circle) occurred immediately after CS, whereas the decrease in the AP threshold progressed gradually (Fig. 2A, bottom, filled circle). CS-induced synaptic LTP showed a slow progressive increase with time (Figs. 1C, 2A), which is similar to the observation reported by Magee and Johnston (1997), and may be attributable to a slow enhanced NMDAR function (D'Angelo et al., 1999; Watt et al., 2004). In contrast to the effect of the Ca2+-free pipette solution, which induced a shift in the postsynaptic AP threshold (Fig. 1A, filled circle and filled square) and RMP, CS did not change the RMP significantly (control, -63.1 ± 0.3 mV; after CS, -63.9 ± 0.2 mV; p = 0.35; paired t test; n = 5 cells).
Figure 2.

Association of presynaptic inputs and postsynaptic AP is required for the induction of LTP and LTP-IE. A, Statistical data showing changes in synaptic EPSCs (top) and postsynaptic AP threshold (bottom) induced by CS (filled circle; n = 12 cells), synaptic stimulation alone (open square; n = 6 cells), post synaptic APs alone (open diamond; n = 5 cells), and time control (open circle; n = 13 cells). The amplitude of the EPSCs was normalized according to the amplitude of EPSCs during the control period, and the mean changes in the postsynaptic AP threshold (ΔThAP) were plotted against time. B, Left, The averaged amplitude of AHP before (Con) and after CS (**p < 0.01; paired t test; n = 10 cells) and in the time control group (T-Con; **p < 0.01; t test; n = 10 cells for each). Right, The firing frequency evoked by injecting a 60 pA current with 1 s duration. The firing frequency was significantly higher after CS (filled bar) than during control (open bar; p < 0.01; paired t test; n = 11). C, The somatic (open circle) and dendritic (filled circle) input resistance plotted against time. Dual recording with two pipettes in the soma and proximal dendrite was used. The somatic I-V curve was obtained by measuring somatic membrane potentials with dendritic current injection steps, and the dendritic I-V curve was obtained by measuring dendritic membrane potentials with somatic current injection steps. The slope input resistance was calculated from the linear region of the I-V curve. D, Recording traces of APs evoked by synaptic stimulation (arrows) during control conditions (Con) and after induction of LTP-IE (CS). Inset, Summary of changes in the threshold of synaptically evoked APs in the time control (open bar) and CS (filled bar) groups. **p < 0.01 compared with control; t test; n = 5 cells for each group.
To establish whether the occurrence of LTP-IE and synaptic LTP requires correlation of synaptic inputs and postsynaptic firing of APs, we stimulated pyramidal neurons either by synaptic stimulation alone (20 Hz) or by injecting currents to generate APs alone. We found that neither synaptic stimulation (Fig. 2A, bottom, open square) (n = 6 cells; p = 0.67 compared with control; two-way ANOVA) nor postsynaptic firing of APs (Fig. 2A, bottom, open diamond) (p = 0.80 compared with control; two-way ANOVA; n = 5 cells for each group) induced significant changes in the postsynaptic AP threshold. In addition, synaptic strength was not enhanced significantly by synaptic stimuli alone or by APs alone (Fig. 2A, top) (synaptic stimulation, 106.0 ± 27%; AP, 96.7 ± 26%; p = 0.70 and 0.31 for synaptic stimulation and AP, respectively; paired t test). The results suggest that both LTP-IE and synaptic LTP require the cooperation of synaptic inputs and postsynaptic firing of APs when 20 Hz synaptic stimuli were used.
CS also induced a reduction of AHP similar to that reported previously (Saar et al., 1998; Sourdet et al., 2003). AHPs were measured from the baseline to the peak of hyperpolarization valley at end of the current injection that generated seven spikes per trace. Six consecutive traces with 20 s intervals were used to get the average amplitude of AHP. The amplitude of AHP decreased significantly 100 min after CS (Fig. 2B, left, filled bar) (-1.8 ± 0.2 mV) compared with either AHP during control (Fig. 2B, left, Con) (p < 0.01; paired t test; n = 10 cells) or time control (Fig. 2B, left, T-Con) (p < 0.01; t test). As a result of the decreased firing threshold and reduced AHP, the number of spikes induced by injecting a 50 pA current was increased after LTP-IE induction (Fig. 2B, right, filled bar) (9.8 ± 1.4) compared with control (Fig. 2B, right, open bar) (3.8 ± 0.4; p < 0.001; paired t test; n = 11 cells). The input resistance was reported to increase after theta-burst stimulation (Armano et al., 2000). Activity-dependent changes in the input resistance may differ at the dendrite from at the soma. To examine changes in the input resistance in the soma and dendrite during induction of LTP-IE, we used pair whole-cell recordings with one pipette in the soma and one pipette in the proximal dendrite. To exclude the effects of increased access resistance during long-time recording, we measured the slope input resistance from the linear region of the I-V relationship for current-clamp recordings from one pipette with current steps injected through the other pipette. The input resistance at both the soma and dendrite increased during induction of LTP-IE (Fig. 2C). The increase in the input resistance at the dendrite (Fig. 2C, filled circle) was larger than that at the soma (Fig. 2C, open circle) (p < 0.01; two-way ANOVA; n = 5 cells), suggesting that CS induces larger changes in dendritic channels than somatic channels. The results are in accordance with the previously reported activity-dependent increase in the input resistance (Armano et al., 2000). Activity-dependent increase in the input resistance will work together with the decreased AP threshold to facilitate the AP activation. To test whether synaptic inputs can trigger APs with the decreased AP threshold during LTP-IE, we elicited postsynaptic APs through synaptic activation (300-400 μA). The CS-induced decrease in the threshold of synaptically evoked APs (Fig. 2D, CS) (-8.5 ± 0.9 mV; n = 5 cells) was not significantly different from that of APs evoked by somatic current injections (Fig. 2A) (p = 0.90; paired t test; n = 5 cells).
The decrease in the AP threshold is not attributable to increased excitatory inputs, reduced inhibitory inputs, and inhibition of A-type K+ channels
Induction of synaptic LTP may increase spontaneous EPSPs that can influence the intrinsic excitability by depolarizing the membrane toward the AP threshold. To test whether glutamatergic synaptic inputs contribute to LTP-IE, we perfused slices with the Ca2+ channel blocker Cd2+ (0.2 mm) and the ionotropic glutamate receptor antagonists amino-phosphonopentanoic acid (APV; 50 μm) and cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm) after the induction of LTP-IE. The decrease in the AP threshold induced by CS (Fig. 3A,B, CS) remained after perfusion with Cd2+, APV, and CNQX (Fig. 3A,B, APV/CNQX/Cd), suggesting that excitatory inputs do not contribute to the expression of LTP-IE. To test whether inhibitory inputs were involved in induction and expression of LTP-IE (Abraham et al., 1987; Chavez-Noriega et al., 1990; Breakwell et al., 1996), we used the GABAA and GABAB receptor antagonists bicuculline and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl) ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845A). In the presence of bicuculline (30 μm) and CGP 55845A (3 μm), CS still induced LTP-IE (Fig. 3C, filled bar) (p = 0.77; t test; n = 5 cells for each group), suggesting that activity-dependent disinhibition does not contribute significantly to CS-induced LTP-IE. To examine whether previously reported activity-dependent inhibition of A-type K+ channels (Frick et al., 2004) contributes to the expression of LTP-IE, we did two experiments. First, we added the A-type K+ channel inhibitor 4-AP (10 mm) in the pipette solution to inhibit A-type K+ channels intracellularly. Control recording was performed for 20 min to allow the diffusion of 4-AP into the cell. 4-AP prolonged the half width of APs from 2.8 ± 0.2 to 5.3 ± 0.8 ms (p < 0.05; paired t test; n = 6 cells), indicating that intracellular 10 mm 4-AP indeed inhibited A-type K+ channels. Then the same CS protocols were applied. In the presence of internal 4-AP, CS still induced LTP-IE (Fig. 3D, filled circle) that was not different from the absence of 4-AP (Fig. 3D, open circle) (100 min after CS; p = 0.26; t test; n = 5 cells for each group). Next, we perfused slices with the slice solution containing 4-AP (5 mm) and examined changes in the AP threshold. The APs were evoked by current injection through the whole-cell recording pipette in the soma. 4-AP alone caused a small but significant decrease in the AP threshold (Fig. 3F, 4-AP alone) (Con, -46.9 ± 0.5 mV; 4-AP, -49.9 ± 0.8 mV; p < 0.01; paired t test; n = 10 cells). However, when eliminating the presynaptic effects of 4-AP by adding APV (50 μm), CNQX (20 μm), and Cd2+ (0.2 mm), 4-AP did not significantly alter the AP threshold, although an increase in bursting of APs attributable to inhibition of A-type K+ channels was observed (Fig. 3E,F, APV/CNQX/Cd) (ΔThAP, 0.7 ± 0.3; p = 0.12; paired t test; n = 5 cells). Furthermore, 4-AP did not significantly alter the AP threshold in the presence of APV/CNQX (Fig. 3F, APV/CNQX) or APV/CNQX/bicuculline (Fig. 3F, APV/CNQX/Bicu), suggesting that synaptic inputs are the major contributor for 4-AP-induced decrease in the AP threshold. The results suggest that the decrease in the AP threshold is not attributable to the activity-dependent inhibition of A-type K+ channels.
Figure 3.

Synaptic inputs and A-type K+ channels did not contribute to the CS-induced decrease in the AP threshold. A, Blockade of synaptic activities did not attenuate expression of LTP-IE. Cells were stimulated by CS first, and then the slice solution containing APV (50 μm), CNQX (20 μm), and Cd2+ (0.2 mm) was perfused (APV/CNQX/Cd). B, The mean AP threshold during control (Con), after CS, and after perfusion of APV/CNQX/Cd. The AP threshold in the presence of APV, CNQX, and Cd2+ (APV/CNQX/Cd) was not significantly different from that in the absence of blockers (CS; p = 0.31; paired t test; n = 5 cells). C, LTP-IE was induced by CS in the absence (CS) or presence of the GABAA and GABAB receptor antagonists bicuculline (30 μm) and CGP 55845A (3 μm) (CS/Bicu/CGP). The CS-induced decrease in the AP threshold in the presence of bicuculline and CGP 55845A (gray bar) was not significantly different from that in the absence of the blockers (filled bar; p = 0.77; paired t test; n = 5 cells). D, Induction of LTP-IE in the absence (open circle) and presence (filled circle) of intracellular 4-AP. E, Inhibition of A-type K+ channels by 4-AP (5 mm) did not induce changes in the AP threshold in the presence of the synaptic transmission blockers APV (50 μm), CNQX (20 μm), and Cd2+ (0.2 mm). F, The mean AP threshold during control (open bar) and application of 4-AP in the absence (4-AP alone) or presence of APV/CNQX/Cd, APV/CNQX, or APV/CNQX/bicuculline (APV/CNQX/Bicu). The AP threshold during perfusion of 4-AP was not significantly different from control in the presence of synaptic transmission blockers (p = 0.09, 0.10, and 0.71 for B, C, and D, respectively; paired t test; n = 5, 7, and 9 cells) but significantly decreased in the absence of the blockers (**p < 0.01; paired t test; n = 10 cells).
CS-induced a hyperpolarized shift in the activation and inactivation curves of VGSCs
APs are generated by the activation of VGSCs. Voltage dependence of gating is a major factor in determining the AP threshold (Moore and Westerfield, 1983). To investigate whether LTP-IE is attributable to a change in the voltage-dependent activation of VGSCs, we first examined the activation of voltage-dependent Na+ currents in the voltage-clamp configuration before and after induction of LTP-IE. The VGSC-mediated current (INa), which was sensitive to TTX (1 μm), could be detected when the membrane was depolarized to approximately -45 mV during control (Fig. 4A, Con, INa). After CS, the depolarization required for activating the INa decreased to -60 mV (Fig. 4A, CS, INa). Pooled data showed that CS induced a negative shift in the activation of the INa (Fig. 4B, filled bar) (-9.2 ± 0.3 mV) that was significantly different from the time control group (Fig. 4B, open bar) (-2.1 ± 0.5 mV; p < 0.01; unpaired t test). The CS-induced shift in the activation of the INa was not significantly different from the CS-induced decrease in the AP threshold (p = 0.72; paired t test; n = 6 cells), implying that the change in the activation property of the INa might account for LTP-IE.
Whole-cell recording of Na+ currents has limitations because of the problem of spatial clamping. To further test the change in the activation property of VGSCs during LTP-IE, we used dual patch-clamp techniques, with one pipette in the cell-attached voltage-clamp configuration and one in the whole-cell current-clamp configuration (Fig. 4D) (Hamill et al., 1981; Kang et al., 2000). Cell-attached patch recording was used to detect VGSC channel activities, and whole-cell recording was used to deliver the CS protocol, detect LTP-IE, and control membrane potential. To standardize the results, we held the membrane at -64 mV, the mean value of RMP in these cells. VGSC channel activities, depending on depolarization steps, were detected in 42 of 45 patches. Thirty-eight of 42 patches contained fast and transient VGSCs (Fig. 4C, Con), and four patches contained persistent Na+ channels. In dual recording experiments, we successfully performed recordings in 42 of 85 cells. Both cell-attached patch and whole-cell recordings kept in good conditions were attained for >40 min in 21 cells. In these 21 cells, six cell-attached patches that contained multiple fast transient VGSCs and showed a decreased threshold of channel activities after CS stimulation were selected for analyzing the CS-induced changes in activation and inactivation. In a representative patch from these six patches, channel activities started at -44 mV during control (Fig. 4C, Con). Channel openings were slower and less synchronized around threshold voltage (Fig. 4C, Con) (-44 and -39 mV) and became transient and fast at more depolarized steps (Fig. 4C, Con) (-29 mV), which is in accordance with previous reported properties of VGSCs by other groups (Magee and Johnston, 1995). In the same patch 60 min after CS, channel activities started at -54 mV (Fig. 4C, CS), and synchronized openings also shifted to -39 mV. When TTX (2 μm) was added to the cell-attached pipette solution, the fast and transient openings were not detected either before or after CS (15 patches; data not shown), suggesting that the channel is a TTX-sensitive VGSC. The I-V relationship (I-V curve) was obtained by measuring the amplitude of unitary currents chosen from all 38 patches. The channel activity at just above the threshold potential (Fig. 4A) (Con, -39 and -44 mV; CS, -49 and -54 mV) was less synchronized. Single-channel openings could be sometimes observed at these voltage steps, especially for patches that contained a few channels as reported previously by other groups (Magee and Johnston, 1995; Colbert and Johnston, 1996). The amplitude of unitary currents was measured by cursors manually. The I-V curve for unitary currents of control VGSCs (Fig. 4E) showed a unitary conductance of 16.3 ± 0.6 pS and a reversal potential (Vrev) of 52.3 ± 3.6 mV (n = 8 patches). The activation curve for VGSCs was obtained by normalizing chord conductance at each depolarization step by the maximal conductance from the six patches that contained multiple VGSCs (maximal amplitude, >10 pA) and showed a decreased opening threshold. Chord conductance was calculated from the averaged peak amplitude and the driving force (Vm - Vrev). The activation curve for the control VGSCs showed a typical voltage-dependent activation (Fig. 4F, open circle) with a V1/2 (voltage at which the chord conductance of VGSCs is an half of the maximum) of -29.5 ± 1.2 mV similar to the value reported previously by the other groups (Magee and Johnston, 1995). The activation curve for the VGSCs after CS (Fig. 4F, filled circle) showed a shift to the hyperpolarized direction. The V1/2 of the activation curve after CS was -37.6 ± 1.0 mV and was significantly different from that for the control channels (p < 0.01; paired t test; n = 6 patches). The results suggest that LTP-IE is attributable to a CS-induced hyperpolarized shift in the activation curve of VGSCs. Inactivation of channel activities before and after CS was tested by delivering a prepulse protocol to cell-attached patches (Fig. 5A, bottom lines). Both control VGSCs and CS-induced VGSCs were inactivated in a voltage-dependent manner (Fig. 5A). The inactivation curve was obtained by plotting the normalized ensemble average current against the voltage. CS also induced a hyperpolarized shift in the inactivation curve for the VGSCs (Fig. 5B, filled circle), and the V1/2 of the inactivation curve for control channels was -57.2 mV and shifted to -69.3 mV after CS (p < 0.05; paired t test; n = 6 patches). The results suggest that during induction of LTP-IE, both the activation and inactivation curves of VGSCs shift in a hyperpolarized direction. The CS-induced changes in the steady-state inactivation of VGSCs may cause more VGSCs to be inactivated at rest and thereby attenuate the AP amplitude. Therefore, we carefully examined the AP amplitude in the time control and CS-stimulation groups. The amplitude of APs was measured from the starting point (threshold) to the peak of APs that were evoked by the threshold current step. The average amplitude of APs from five consecutive traces (20 s intervals) was used as one time point in each cell, and the mean of AP amplitude from five cells is plotted as a function of time. In control conditions (Fig. 5C, open circle), the amplitude of APs decreased in 20 min after forming the whole-cell recording configuration and maintained relatively constant after 20 min. The CS protocols were applied 20 min after breaking the patch membrane. In the CS group (Fig. 5C, filled circle), a progressing decrease 40 min after CS was observed. The AP amplitude 100 min after CS was significantly different from the AP amplitude in the time-control group (p < 0.01; t test; n = 5 cells for each group). This decrease in the AP amplitude during LTP-IE is consistent with the hyperpolarized shift in the steady-state inactivation of VGSCs.
Figure 5.

Changes in the inactivation curve of VGSCs during induction of LTP-IE. A, Voltage-dependent inactivation of VGSCs before (Con) and after CS tested by a prepulse protocol (bottom lines). The number at the left of each trace indicates the prepulse steps in millivolts. Prepulse steps from -104 to -44 mV in 10 mV steps (bottom lines) were given through cell-attached patch pipettes. Patches were held at -84 mV by adding +20 mV to the cell-attached patch pipette. The depolarization step after prepulses was -34 mV. Each trace was an ensemble average trace from six patches for each prepulse step. B, The inactivation curve of VGSCs before (open circle) and after (filled circle) CS. The inactivation curve was obtained by plotting normalized amplitude of ensemble trace (divided by the maximal amplitude) against prepulse steps. C, The mean amplitude of APs in the time control (open circle) and LTP-IE (filled circle) groups. The AP amplitude in the LTP-IE group was not significantly different from the time control before CS (p = 0.89; t test; n = 5 cells for each group) but significantly decreased 100 min after CS (p < 0.05; t test).
Induction of LTP-IE requires activation of NMDARs
It is accepted that the induction of synaptic LTP in CA1 pyramidal neurons is mediated by activation of NMDARs (Collingridge et al., 1983; Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). We tested whether activation of NMDARs is necessary for induction of LTP-IE. APV (50 μm) was perfused 5 min before CS and completely blocked LTP-IE (Fig. 6A,B, CS/APV). No significant difference in the postsynaptic AP threshold was found between the CS/APV (Fig. 6B, bottom, open square) and control (Fig. 6B, bottom, open circle) groups (p = 0.73; two-way ANOVA; n = 5 cells for each group). Consistent with previous reports (Collingridge et al., 1983; Malenka, 1991; Malenka and Nicoll, 1993), APV also prevented the induction of synaptic LTP (Fig. 6A,B, CS/APV) (p = 0.41 compared with control; two-way ANOVA). These results imply that the induction mechanism of LTP-IE shares similarities with that of synaptic LTP. However, APV did not block the CS-induced reduction of AHP (Fig. 6B, inset, CS, 2), suggesting that the reduction in AHP is induced through an NMDA-independent pathway. To test the role of metabotropic glutamate receptors (mGluRs) in the induction of LTP-IE, we used the group I/II mGluR antagonist α-methyl-4-carboxyphenylglycine (MCPG; 1 mm) and the group III mGluR antagonist (RS)-a-methylserine-O-phosphate (MSOP; 100 μm). In the presence of MCPG and MSOP, CS still induced a decrease in the AP threshold (Fig. 6B, filled square) (p = 0.83; 100 min after CS) and a reduction of AHP (Fig. 6B, inset, CS, 3) (p < 0.01 compared with control; paired t test; n = 7 cells). However, CS-induced reduction in AHP in the presence of MCPG/MSOP (Fig. 6B, inset, CS, 3) was significantly smaller than that in the absence of the blockers (Fig. 6B, inset, CS, 1) (p < 0.01; t test). The results suggest that activation of mGluR receptors is not involved in the CS-induced decrease in the AP threshold but contributes partially to the CS-induced reduction of AHP.
Figure 6.

Activation of NMDARs and Ca2+ influx are required for induction of LTP-IE. A, EPSCs (top traces) and APs (bottom traces) from a representative cell showing that APV (50 μm) blocked both the increase in EPSCs and decrease in the postsynaptic AP threshold (arrow) induced by CS. B, Pooled data showing that, in the presence of APV, CS-induced changes in EPSCs (top) and in the postsynaptic AP threshold (bottom; open square; n = 5 cells) were not different from those in the time control group (open circle; n = 6 cells). Using dual whole-cell recording, BAPTA (10 mm) in the dendritic pipette blocked the CS-induced decrease in the AP threshold in somatic recording (bottom; open diamond; n = 6 cells). In the presence of MCPG (1 mm) and MSOP (100 μm), CS still induced LTP-IE (filled square). Inset, The mean amplitude of AHP during control (open bar) and after CS (filled bar) in the absence (1) or presence of APV (50 μm) (2) or MCPG (1 mm)/MSOP (100 μm) (3). In the presence of MCPG/MSOP, CS-induced reduction of AHP was significantly smaller than the absence the blockers (Fig. 2 B) (*p < 0.01; t test; n = 7) but still significantly different from control AHP (*p < 0.01; paired t test).
NMDAR-mediated Ca2+ influx plays a key role in induction of synaptic LTP (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). However, because reducing intracellular Ca2+ by the calcium chelator decreased the AP threshold by itself (Fig. 1A, filled square), use of the single whole-cell pipette with BAPTA to chelate intracellular Ca2+ is not reliable. Therefore, we used dual whole-cell recordings with the somatic pipette containing 0.1 mm Ca2+/1 mm EGTA to stabilize the firing threshold and the dendritic pipette containing 10 mm BAPTA to chelate Ca2+ signals in dendrites. In the presence of BAPTA in the dendrite, CS did not induce the decrease in the AP threshold (Fig. 6B, bottom, open diamond) (p = 0.31 compared with time control; two-way ANOVA). The results suggest that Ca2+ signals in dendrites are required for induction of LTP-IE. To further study CS-induced Ca2+ signals in local dendrites, we used two-photon microscopy to measure Ca2+ signals in spines and local dendrites induced by stimuli similar as those used in Figure 2A. Alexa Fluor-4 (100 μm) was used to monitor the Ca2+ signals, and Alexa Fluor-594 (100 μm) was used to outline the dendritic structure of pyramidal neurons. Stimuli were delivered in a random order. Synaptic stimulation (20 Hz, 5 s) induced a transient Ca2+ increase in spines (Fig. 7A,B, II, 1 and 2), parent dendritic shafts (Fig. 7A,B, II, 3), and dendritic branch points (Fig. 7A,B, II, 4). Postsynaptic APs induced Ca2+ signals that were longer and larger than the Ca2+ signals induced by synaptic stimulation in the three structures (Fig. 7A,B, IV). The AP-induced Ca2+ signals in spines were in accordance with previous studies (Sabatini et al., 2002). CS induced Ca2+ signals that lasted even longer than those evoked by postsynaptic APs (Fig. 7A,B, VI and VII). Pooled data showed that CS-induced Ca2+ signals (Fig. 7C, red) lasted significantly longer than those evoked by synaptic stimulation (Fig. 7C, black) or by postsynaptic APs (Fig. 7C, cyan) in the three structures. The results suggest that large and prolonged Ca2+ signals in dendrites and spines may be required for induction of LTP-IE.
Figure 7.
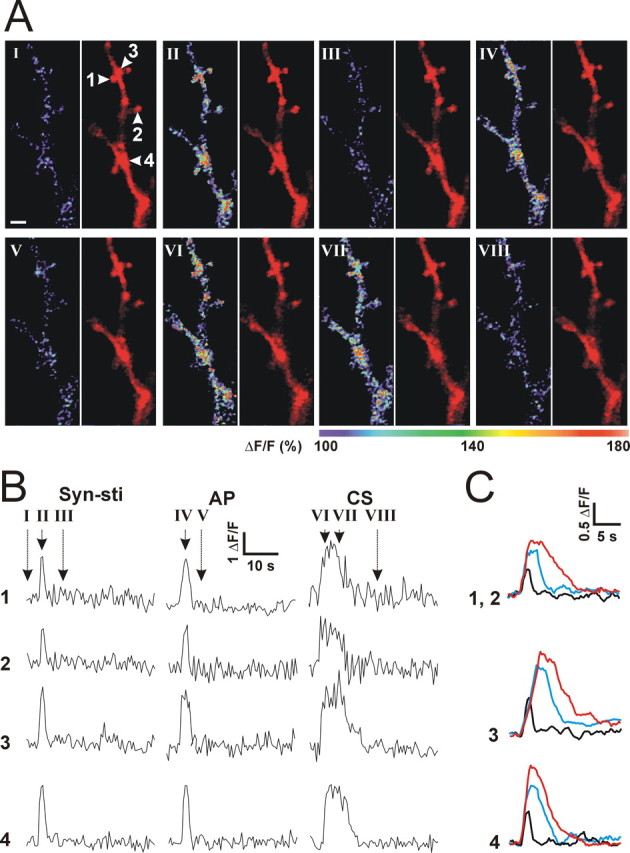
CS-induced Ca2+ signals in local dendrites. A, Images obtained with a two-photon microscopy showing Ca2+ signals in spines (1, 2), parent dendritic shafts of the spine (3), and dendritic branch points (4) during control (I), after synaptic stimulation (II), after APs (IV), and after CS (VI and VII). The left image is a Fluor-4 fluorescent signal showing Ca2+ levels, and the right image is an Alexa Fluor-594 fluorescent signal showing the morphology of dendrites and spines. Images were recorded at the time indicated in B. Scale bar, 2 μm. B, Ca2+ levels (ΔF/F) in response to synaptic stimulation (Syn-sti), postsynaptic APs, and CS from the dendritic structures (1-4) indicated in I. C, Averaged traces for Ca2+ levels induced by synaptic stimulation (black), APs (cyan), and CS (red) in spines (1 and 2; n = 7 cells), parent dendritic shafts of spines (3; n = 4 cells), and dendritic branch points (4; n = 4 cells).
Induction of LTP-IE requires activities of the CaMK pathway and function of protein synthesis
The CaMK pathway is required for induction of synaptic LTP (Malinow et al., 1989; Braun and Schulman, 1995; Lisman et al., 1997). To examine whether the CaMK pathway also is required for induction of LTP-IE, we pretreated the slices with the CaMK inhibitor KN-62 (10 μm) for 30 min and added KN-62 (10 μm) to the pipette solution. LTP-IE induced by CS was blocked by KN-62 (Fig. 8A,B, CS/KN-62) (p = 0.47 compared with control; two-way ANOVA). As expected, synaptic LTP also was blocked by KN-62 (Fig. 8B, top) (p = 0.30 compared with control; two-way ANOVA). The results suggest that activation of CaMK is required for induction of both LTP-IE and synaptic LTP. Because KN-62 inhibits both CaMKII and CaMKVI (Enslen et al., 1994), to further test whether activation of CaMK II is required for induction of LTP-IE, we used the specific CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP) (Ishida et al., 1995). AIP (5 μm) was dissolved in the pipette solution for whole-cell recording, and CS was applied 20 min after the whole-cell configuration was formed. CS-induced LTP-IE was fully blocked by AIP (Fig. 8B, bottom, open diamond) (p = 0.85 compared with time control; two-way ANOVA; n = 5 cells). Synaptic LTP was also blocked by AIP (Fig. 8B, top, open diamond) (p = 0.30 compared with time control; two-way ANOVA). The results suggest that induction of both synaptic LTP and LTP-IE requires CaMKII. The AP amplitude was not changed significantly by either KN-62 or AIP (Fig. 8B, inset, AP) (p = 0.41 and 0.58 for KN-62 and AIP, respectively; t test; n = 8 cells for each group), indicating KN-62 and AIP did not directly affect activation of VGSCs. The reduction of AHP was not blocked by either KN-62 or AIP (Fig. 8B, inset, AHP), suggesting that CaMKs are not required for the CS-induced reduction of AHP.
Figure 8.

Activity of CaMK II is required for the induction of LTP-IE. A, Representative recordings of EPSCs (top traces) and APs (bottom traces) during control and 100 min after CS in the presence of KN-62. B, Pooled data showing the CS-induced changes in EPSCs (top) and AP threshold (bottom) in the presence of KN-62 (open square; n = 6 cells) or AIP (open diamond; n = 5 cells) compared with data from time control cells (open circle). Inset, The mean AP amplitude in the presence of KN-62 (gray bar) or AIP (filled bar) compared with control cells (open bar) and the mean AHP amplitude (AHP) before (open bar) and after CS in the presence of KN-62 (gray bar) or AIP (filled bar). *p < 0.05; paired t test.
The slow time course of LTP-IE induction (Figs. 1D, 2A) implies that LTP-IE may require protein synthesis, similar to the late phase of LTP (L-LTP) (Frey et al., 1993). To test whether protein synthesis is involved in LTP-IE, we pretreated brain slices with the protein synthesis inhibitor emetine (5 μm) for 30 min and added emetine (5 μm) to the pipette solution. Emetine completely blocked LTP-IE (Fig. 9A,B, bottom, open diamond) (p = 0.74 compared with time control; two-way ANOVA; n = 5 cells for each group) but only attenuated the L-LTP (Fig. 9A,B, top, CS 100 min), leaving the early phase of LTP unaffected (Fig. 9A,B, top, CS 30 min). In the presence of emetine, EPSCs at 30 min after CS (182 ± 7%) were significantly different from control (Con, 97 ± 6%; p < 0.01; unpaired t test), whereas EPSCs at 100 min after CS (94 ± 8%) were not significantly different from control (p = 0.58; unpaired t test). To confirm the involvement of protein synthesis in induction of LTP-IE, we used another protein synthesis inhibitor, anisomycin (5 μm), to pretreat slices for 30 min and also added anisomycin (5 μm) to the pipette solution. Anisomycin had a similar effect as emetine and blocked synaptic L-LTP and LTP-IE (Fig. 9B, open square). The results suggest that both LTP-IE and L-LTP involve the synthesis of proteins. The AP amplitude was not changed significantly by emetine and anisomycin (Fig. 9B, inset, AP) (p = 0.48 and 0.57 for emetine and anisomycin, respectively; t test; n = 8 and 5 cells for emetine and anisomycin, respectively), indicating emetine and anisomycin did not directly affect activity of VGSCs. The reduction of AHP was not blocked by either inhibitor (Fig. 9B, inset, AHP), suggesting that protein synthesis is not involved in the CS-induced reduction of AHP.
Figure 9.

Function of protein synthesis is required for induction of LTP-IE. A, Representative recordings of EPSCs (top traces) and APs (bottom traces) during control and 30 and 100 min after CS in the presence of emetine (5 μm). B, Pooled data showing the CS-induced changes in EPSPs (top) and AP threshold (bottom) in the presence of emetine (open diamond; n = 5 cells) or anisomycin (5 μm; open square; n = 5 cells) compared with data from time control cells (open circle). Inset, The mean AP amplitude in the presence of emetine (gray bar) and anisomycin (filled bar) compared with control cells (open bar) and the mean AHP amplitude before (open bar) and after CS in the presence of emetine (gray bar) or anisomycin (filled bar). *p < 0.05; paired t test.
Discussion
Characteristics of CS-induced LTP-IE
In this study, we report that correlation of presynaptic activities with APs is capable of inducing an activity-dependent LTP-IE that is primarily expressed as a progressive decrease in the AP threshold (Figs. 1C, 2A) in hippocampal CA1 pyramidal neurons. The decrease in the AP threshold makes neurons more sensitive to membrane depolarization and easier to generate APs (Fig. 1B). This decrease in the AP threshold works together with the increase in the input resistance (Fig. 2C) to enhance the excitability of postsynaptic neurons. This type of LTP-IE serves, at least partially, as the mechanism for E-S potentiation, which is consistent with previous studies reported by other groups (Chavez-Noriega et al., 1990; Jester et al., 1995). The activity-dependent increase in the intrinsic excitability is concurring with synaptic LTP to increase the efficiency of active neurons for information relay. The slow and gradual induction of LTP-IE comparing with quick induction of synaptic LTP (Fig. 2A) may reflect the slow expression of LTP-IE.
A hyperpolarized shift in the activation and inactivation curves of VGSCs during LTP-IE
Results from several experiments in this study support that CS-induced changes in VGSCs are the mechanism for expression of LTP-IE. First, the decrease in the AP threshold is attributable to changes in intrinsic properties rather than increased excitatory synaptic inputs because blockade of synaptic activity by APV, CNQX, and Cd2+ after induction of LTP-IE did not recover the AP threshold (Fig. 3A,B). Furthermore, application of GABAA and GABAB receptor blockers affected neither induction nor expression of LTP-IE (Fig. 3C), suggesting that activity-dependent depression of inhibitory transmission is not involved in the CS-induced decrease in the AP threshold. Second, activity-dependent downregulation of A-type K channels does not contribute to the decease in the AP threshold, because blockade of A-type K channels with 4-AP in the presence of the synaptic transmission blockers does not change the AP threshold (Fig. 3E,F). Our results indicate that only in the absence of synaptic transmission blockers does 4-AP cause a small but significant decrease in the AP threshold (Fig. 3E, 4-AP alone), which is mainly attributable to increased excitatory inputs. The effects of 4-AP here are different from the observation reported by Nedergaard (1999). The discrepancy may be attributable to use of either different recording techniques (whole-cell patch-clamp vs intracellular recording) or different neurons (hippocampal CA1 pyramidal neurons vs substantial nigra neurons). Third, dual recording with one whole-cell and one cell-attached patch provided direct evidence for the expression mechanism for LTP-IE. A hyperpolarized shift in the activation curve of VGSCs during induction of LTP-IE (Fig. 4) indicates that the decrease in the threshold of APs and INa is attributable to such alteration of VGSCs. The hyperpolarized shift in the activation curve of VGSCs represents both more channel activities and more synchronized openings of VGSCs at given membrane potential above the threshold potential (Fig. 4A). CS also induced a negative shift and decreased slope of the steady-state inactivation curve of VGSCs (Fig. 5B). These changes in the inactivation curve may cause less VGSC channels ready to open in response to depolarization during LTP-IE than during control conditions. The observation that a slight but significant reduction in the amplitude of APs (Fig. 5C) during LTP-IE compared with the time control is consistent with such changes in the inactivation curve of VGSCs.
The CS-induced modification of VGSCs found in this study is different from the change in presynaptic VGSCs previously reported by Poo and colleagues (Ganguly et al., 2000) in the following several views: (1) the change in our study occurs in postsynaptic pyramidal neurons in slices rather than in cultured presynaptic neurons, although the stimulation protocol is similar; (2) the CS-induced changes in the AP threshold in presynaptic neurons depend on activity of the PKC pathway, whereas the decrease in the AP threshold in postsynaptic pyramidal neurons depends on activity of CaMKs and protein synthesis; and (3) Ganguly et al. (2000) reported that CS induced a depolarized shift in the inactivation curve of presynaptic VGSCs, whereas we found that CS also induced a hyperpolarized shift in the inactivation curve of postsynaptic VGSCs. The parallel shifts in the activation and inactivation property of VGSCs and requirement of protein synthesis imply new synthesized VGSC channels, because the VGSC channel is expressed with coupled activation and inactivation determinant residues (Catterall, 2000). However, we cannot rule out modulation of the existing channel by new synthesized enzymes.
Induction of LTP-IE requires the NMDAR, CaMK pathway, and protein synthesis
The decreased AP threshold has been reported to be induced by CS (Ganguly et al., 2000), tetanic synaptic stimulation (Chavez-Noriega et al., 1990; Jester et al., 1995; Staff and Spruston, 2003), or postsynaptic depolarization (Aizenman and Linden, 2000; Armano et al., 2000; Cudmore and Turrigiano, 2004). Both NMDAR-dependent (Aizenman and Linden, 2000; Armano et al., 2000; Ganguly et al., 2000) and NMDAR-independent (Cudmore and Turrigiano, 2004) induction of this type of intrinsic plasticity has been reported. Our results suggest that the induction mechanism for this type of LTP-IE shares the similar pathways with synaptic LTP. First, the induction of both synaptic LTP and LTP-IE requires the correlation of synaptic activities and postsynaptic APs in our experimental conditions (Fig. 2A). Second, the requirement of NMDARs and Ca2+ signals suggests that Ca2+ influx through NMDARs is a key factor for induction of both synaptic LTP and LTP-IE. Third, CaMKs are required for induction of both synaptic LTP and LTP-IE. Fourth, protein synthesis is involved in induction of both late phase synaptic L-LTP and LTP-IE. Other kinases that have been suggested to be involved in induction of synaptic LTP, such as PKA (Abel et al., 1997), PKC (Colley and Routtenberg, 1993), and mitogen-activated protein kinase (English and Sweatt, 1996, 1997), should be tested in future studies.
Cooperation of activity-dependent intrinsic plasticity
Activity-dependent intrinsic plasticity has been suggested to be the other side of the engram for learning and memory (Aizenman and Linden, 2000; Armano et al., 2000; Ganguly et al., 2000; Zhang and Linden, 2003; Cudmore and Turrigiano, 2004; Frick et al., 2004). The correlation of decrease in the AP threshold with learning-related behavior changes has been established by several groups (Schreurs et al., 1997, 1998; Burrell et al., 2001). LTP-IE expressed as a hyperpolarized shift in the activation property of VGSCs in this study adds another piece of evidence to support this important concept. Activity-dependent plasticity of intrinsic excitability in postsynaptic neurons could target on modulating Na+, K+, and Ca2+ channels. Modulation of K+ channels has been previously reported and expressed as inhibition of the A-type K+ channel (Frick et al., 2004) and inhibition of AHP (Saar et al., 1998, 2001; Ireland and Abraham, 2002; Melyan et al., 2002, 2004; Sourdet et al., 2003). In our study, CS also induced a reduction in AHP (Fig. 2B). However, CS-induced reduction of AHP is only partially dependent on the mGluR (Fig. 6B, inset, 3) that is different from the observation of 10 Hz burst stimulation-induced attenuation of AHP in neocortical neurons (Ireland and Abraham, 2002; Sourdet et al., 2003). Thus, the mGluR-independent part of CS-induced reduction of AHP may involve other cellular mechanisms, such as activation of KA receptors (Melyan et al., 2002, 2004) and cholinergic-muscarinic receptors (Saar et al., 1998, 2001). Consistent with NMDAR independency, blockade of CaMK and protein synthesis did not inhibit CS-induced reduction of AHP, further suggesting the decreased AP threshold and attenuated AHP are different forms of intrinsic plasticity through separate cellular mechanisms.
The activity-dependent modulation of Na+ channels expressed as the decreased AP threshold found in this study may work together with the modulation of the input resistance, the A-type K+ channel, and AHP to modify neuronal input-output property. The increase in input resistance will cause a larger depolarization with a given synaptic current, and the larger increase in input resistance at dendrites than at the soma found in this study (Fig. 2C) will specifically facilitate dendritic depolarization evoked by synaptic activation of ionotropic receptors. Inhibition of the dendritic A-type K+ channels will make EPSPs easier to depolarize membrane to the firing threshold and facilitate propagation of EPSPs. The attenuated AHP will reduce the interspike interval and increase neuronal bandpass in the high-frequency range. The decreased firing threshold will lower the size requirement for EPSPs to evoke APs and enhance output for all synapses in the neuron. The form of LTP-IE in this study may serve as a cellular mechanism for learning and memory in certain learning tasks. This idea is supported by a study in which an increase in the voltage-gated Na+ current was observed in optical tectal neurons of Xenopus tadpoles with persistent visual stimulation (Aizenman et al., 2003).
In this study, we demonstrated that synaptic activities cooperated with postsynaptic APs induce two forms of plasticity, synaptic LTP and neuronal LTP-IE, through the same signaling pathway, NMDARs/Ca2+/CaMK/protein synthesis. Under physiological conditions, subthreshold synaptic inputs may not induce LTP-IE because postsynaptic firing is required. However, when synaptic strength is enhanced by previous synaptic activity, such as after LTP or short-term potentiation, or when activity at multiple synapses is synchronized, postsynaptic firing may occur and cooperate with synaptic inputs to induce LTP-IE. Because synaptic LTP consists of an increase in synaptic strength and LTP-IE is expressed as a decrease in the AP threshold, these two forms of plasticity would work cooperatively at the same circuit. In addition, LTP-IE also may serve as the mechanism for some pathological brain disorders with increased excitability, such as epileptic seizures.
Footnotes
This work was supported by National Institutes of Health Grants NS 37349 and NS 39997.
Correspondence should be addressed to Dr. Jian Kang, Department of Cell Biology and Anatomy, New York Medical College, Basic Science Building, Room 220, Valhalla, NY 10595. E-mail: jian_kang@NYMC.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251750-11$15.00/0
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615-626. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Gustafsson B, Wigstrom H (1987) Long-term potentiation involves enhanced synaptic excitation relative to synaptic inhibition in guinea-pig hippocampus. J Physiol (Lond) 394: 367-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman CD, Linden DJ (2000) Rapid, synaptically driven increases in the intrinsic excitability of cerebellar deep nuclear neurons. Nat Neurosci 3: 109-111. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Akerman CJ, Jensen KR, Cline HT (2003) Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron 39: 831-842. [DOI] [PubMed] [Google Scholar]
- Alkon DL, Sakakibara M, Forman R, Harrigan J, Lederhendler I, Farley J (1985) Reduction of two voltage-dependent K+ currents mediates retention of a learned association. Behav Neural Biol 44: 278-300. [DOI] [PubMed] [Google Scholar]
- Andersen P, Sundberg SH, Sveen O, Swann JW, Wigstrom H (1980) Possible mechanisms for long-lasting potentiation of synaptic transmission in hippocampal slices from guinea-pigs. J Physiol (Lond) 302: 463-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov I, Antonova I, Kandel ER, Hawkins RD (2001) The contribution of activity-dependent synaptic plasticity to classical conditioning in Aplysia J Neurosci 21: 6413-6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armano S, Rossi P, Taglietti V, D'Angelo E (2000) Long-term potentiation of intrinsic excitability at the mossy fibergranule cell synapse of rat cerebellum. J Neurosci 20: 5208-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM (1998) Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci 18: 10464-10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 232: 331-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AP, Schulman H (1995) The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol 57: 417-445. [DOI] [PubMed] [Google Scholar]
- Breakwell NA, Rowan MJ, Anwyl R (1996) Metabotropic glutamate receptor dependent EPSP and EPSP-spike potentiation in area CA1 of the submerged rat hippocampal slice. J Neurophysiol 76: 3126-3135. [DOI] [PubMed] [Google Scholar]
- Burrell BD, Sahley CL, Muller KJ (2001) Non-associative learning and serotonin induce similar bi-directional changes in excitability of a neuron critical for learning in the medicinal leech. J Neurosci 21: 1401-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13-25. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Halliwell JV, Bliss TV (1990) A decrease in firing threshold observed after induction of the EPSP-spike (E-S) component of long-term potentiation in rat hippocampal slices. Exp Brain Res 79: 633-641. [DOI] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW (2000) Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.” Nat Neurosci 3: 330-336. [DOI] [PubMed] [Google Scholar]
- Cleary LJ, Lee WL, Byrne JH (1998) Cellular correlates of long-term sensitization in Aplysia J Neurosci 18: 5988-5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert CM, Johnston D (1996) Axonal action-potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. J Neurosci 16: 6676-6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley PA, Routtenberg A (1993) Long-term potentiation as synaptic dialogue. Brain Res Brain Res Rev 18: 115-122. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H (1983) Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol (Lond) 334: 33-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore RH, Turrigiano GG (2004) Long-term potentiation of intrinsic excitability in LV visual cortical neurons. J Neurophysiol 92: 341-348. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Rossi P, Armano S, Taglietti V (1999) Evidence for NMDA and mGlu receptor-dependent long-term potentiation of mossy fiber-granule cell transmission in rat cerebellum. J Neurophysiol 81: 277-287. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Hanada Y, Debanne D (2002) Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc Natl Acad Sci USA 99: 14512-14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM (1998) Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol (Lond) 507: 237-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1996) Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem 271: 24329-24332. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272: 19103-19106. [DOI] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR (1994) Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem 269: 15520-15527. [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER (1993) Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260: 1661-1664. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D (2004) LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 7: 126-135. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Kiss L, Poo M (2000) Enhancement of presynaptic neuronal excitability by correlated presynaptic and postsynaptic spiking. Nat Neurosci 3: 1018-1026. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391: 85-100. [DOI] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC (2002) Group I mGluRs increase excitability of hippocampal CA1 pyramidal neurons by a PLC-independent mechanism. J Neurophysiol 88: 107-116. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H (1995) A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun 212: 806-812. [DOI] [PubMed] [Google Scholar]
- Jester JM, Campbell LW, Sejnowski TJ (1995) Associative EPSP-spike potentiation induced by pairing orthodromic and antidromic stimulation in rat hippocampal slices. J Physiol (Lond) 484: 689-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Xu J, Nedergaard M, Kang J (2001) A kainate receptor increases the efficacy of GABAergic synapses. Neuron 30: 503-513. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1: 683-692. [DOI] [PubMed] [Google Scholar]
- Kang J, Huguenard JR, Prince DA (2000) Voltage-gated potassium channels activated during action potentials in layer V neocortical pyramidal neurons. J Neurophysiol 83: 70-80. [DOI] [PubMed] [Google Scholar]
- Lisman J, Malenka RC, Nicoll RA, Malinow R (1997) Learning mechanisms: the case for CaM-KII. Science 276: 2001-2002. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1995) Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol (Lond) 487: 67-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275: 209-213. [DOI] [PubMed] [Google Scholar]
- Malenka RC (1991) Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6: 53-60. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1993) NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16: 521-527. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999) Long-term potentiation—a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862-866. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B (1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275: 213-215. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B (2002) Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34: 107-114. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Lancaster B, Wheal HV (2004) Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci 24: 4530-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JW, Westerfield M (1983) Action potential propagation and threshold parameters in inhomogeneous regions of squid axons. J Physiol (Lond) 336: 285-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard S (1999) Regulation of action potential size and excitability in substantia nigra compacta neurons: sensitivity to 4-aminopyridine. J Neurophysiol 82: 2903-2913. [DOI] [PubMed] [Google Scholar]
- Neher E (1992) Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207: 123-131. [DOI] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF (2003) Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol 90: 2171-2179. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Ballerini L, Passani MB, Corradetti R (1994) EPSP-spike potentiation during primed burst-induced long-term potentiation in the CA1 region of rat hippocampal slices. Neuroscience 62: 1021-1032. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E (1998) Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci 10: 1518-1523. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E (2001) Long-lasting cholinergic modulation underlies rule learning in rats. J Neurosci 21: 1385-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K (2002) The life cycle of Ca2+ ions in dendritic spines. Neuron 33: 439-452. [DOI] [PubMed] [Google Scholar]
- Schreurs BG, Tomsic D, Gusev PA, Alkon DL (1997) Dendritic excitability microzones and occluded long-term depression after classical conditioning of the rabbit's nictitating membrane response. J Neurophysiol 77: 86-92. [DOI] [PubMed] [Google Scholar]
- Schreurs BG, Gusev PA, Tomsic D, Alkon DL, Shi T (1998) Intracellular correlates of acquisition and long-term memory of classical conditioning in Purkinje cell dendrites in slices of rabbit cerebellar lobule HVI. J Neurosci 18: 5498-5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourdet V, Russier M, Daoudal G, Ankri N, Debanne D (2003) Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J Neurosci 23: 10238-10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Spruston N (2003) Intracellular correlate of EPSP-spike potentiation in CA1 pyramidal neurons is controlled by GABAergic modulation. Hippocampus 13: 801-805. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xu NL, Wu CP, Duan S, Poo MM (2003) Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37: 463-472. [DOI] [PubMed] [Google Scholar]
- Wathey JC, Lytton WW, Jester JM, Sejnowski TJ (1992) Computer simulations of EPSP-spike (E-S) potentiation in hippocampal CA1 pyramidal cells. J Neurosci 12: 607-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG (2004) A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci 7: 518-524. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4: 885-900. [DOI] [PubMed] [Google Scholar]