

Abstract
Members of the ciliary neurotrophic factor (CNTF)-leukemia inhibitory factor (LIF) gene family play an essential role for survival of developing and postnatal motoneurons. When subunits of the shared receptor complex are inactivated by homologous recombination, the mice die at approximately birth and exhibit reduced numbers of motoneurons in the spinal cord and brainstem nuclei. However, mice in which cntf, lif, or cardiotrophin-1 (ct-1) are inactivated can survive and show less motoneuron cell loss. This suggests cooperative and redundant roles of these ligands. However, their cooperative functions are not well understood. We generated cntf/lif/ct-1 triple-knock-out and combinations of double-knock-out mice to study the individual and combined roles of CNTF, LIF and CT-1 on postnatal motoneuron survival and function. Triple-knock-out mice exhibit increased motoneuron cell loss in the lumbar spinal cord that correlates with muscle weakness during early postnatal development. LIF deficiency leads to pronounced loss of distal axons and motor endplate alterations, whereas CNTF-and/or CT-1-deficient mice do not show significant changes in morphology of these structures. In cntf/lif/ct-1 triple-knock-out mice, various degrees of muscle fiber type grouping are found, indicating that denervation and reinnervation had occurred. We conclude from these findings that CNTF, LIF, and CT-1 have distinct functions for motoneuron survival and function and that LIF plays a more important role for postnatal maintenance of distal axons and motor endplates than CNTF or CT-1.
Keywords: CNTF, LIF, CT-1, motoneuron, skeletal muscle, motor endplates
Introduction
The large variety of neurotrophic molecules that can support motoneuron survival in culture indicates that developing and postnatal motoneurons depend on a cooperation of these molecules that is so far not fully understood. Some of these molecules are only expressed postnatally, indicating that the requirement of motoneurons for these factors changes during development and that postnatal survival and function are also controlled by neurotrophic factors. Members of the ciliary neurotrophic factor (CNTF)-leukemia inhibitory factor (LIF) gene family are potent survival factors for motoneurons in vitro and in vivo (Arakawa et al., 1990; Sendtner et al., 1990; Hughes et al., 1993; Banner and Patterson, 1994; Pennica et al., 1996). At least CNTF plays an essential role for long-term maintenance of axons and paranodal networks, structures that are formed by the Schwann cells and axons near the nodes of Ranvier (Gatzinsky et al., 2003). This factor also protects axons under pathophysiological conditions, i.e., in progressive motoneuronopathy mutant mice (Sendtner et al., 1992a) and in experimental autoimmune encephalomyelitis (Linker et al., 2002).
Additional investigations demonstrate that null mutations in the CNTF gene lead to an earlier onset of disease in patients with sporadic and familial amyotrophic lateral sclerosis and in the human superoxide dismutase-1 G93A mouse model of motoneuron disease (Giess et al., 2002).
Gene-targeting experiments show that CNTF and LIF cooperate in maintaining postnatal survival and functional integrity of motoneurons and that LIF, although expressed only at low levels in peripheral nerves, contributes to compensatory mechanisms that prevent functional deficits in CNTF-deficient mice (Sendtner et al., 1996). This at least partial functional overlap is reflected by the fact that they share gp130 and LIF receptor β (LIFRβ) as signal-transducing receptor components in responsible cells (Davis et al., 1993). Cntf/lif double-knock-out mice do not show any alterations in survival of developing embryonic motoneurons. In contrast, a significant loss of motoneurons is observed in cardiotrophin-1 (CT-1)-deficient mice (Oppenheim et al., 2001). Thus, CT-1 function differs significantly from CNTF and LIF.
We generated cntf/lif/ct-1 triple-knock-out mice to study the individual roles of CNTF, LIF, and CT-1 in postnatal maintenance of motoneurons and whether they act together on motoneuron survival and function. In these mice, significant motoneuron cell loss and functional deficits were found very early during postnatal development. In addition, the size of motoneuron cell bodies was significantly reduced in mice lacking lif gene expression. It became apparent that LIF plays an important role for the maintenance of distal axons, such as in the plantar nerve. Moreover, LIF deficiency was associated with alterations of motor endplates, whereas lack of CNTF and CT-1 did not lead to significant changes. In cntf/lif/ct-1 triple-knock-out mice, various degrees of muscle fiber type grouping were found, indicating that denervation and reinnervation had occurred in these mouse mutants.
Altogether, our results suggest that CNTF, LIF, and CT-1 play distinct roles for motoneuron survival and function and that LIF is more potent than CNTF or CT-1 in compensating deficiency of other members of this family of neurotrophic factors.
Materials and Methods
cntf/lif/ct-1 triple-knock-out, cntf/lif double-knock-out, cntf/ct-1 double-knock-out, and cntf/lif/ct-1 wild-type mice. Mice with homologous recombination of the cntf (Masu et al., 1993), lif (Escary et al., 1993), and ct-1 (Pennica et al., 1996) genes were backcrossed for at least five generations and subsequently at every third generation with C57BL/6 mice (Charles River Wiga, Sulzfeld, Germany). Female mice with homozygous lif gene inactivation do not become pregnant (Stewart et al., 1992). Therefore, we crossbred cntf/lif double-knock-out males with ct-1 single-knock-out females. Resulting triple-heterozygous mice were crossbred, and offspring were analyzed for the individual genotypes by PCR. Mice exhibiting the individual genotypes analyzed in this study were born at a 1.6% ratio. Therefore, mice from the F2 generation were further bred to establish triple-knock-out and corresponding control mouse lines. The number of cntf/lif/ct-1 triple-knock-out mice could be increased to a 50% ratio by mating triple-knock-out males with cntf-/-/lif+/-/ct-1-/- females. Triple-knock-out mice were compared with cntf/lif double-knock-out mice that were generated by crossbreeding cntf/lif double-knock-out males with cntf-/-/lif+/- females, resulting in 50% offspring devoid of CNTF and LIF. As additional control groups, cntf/ct-1 double-knock-out and wild-type mice were generated by crossbreeding mice homozygous for each of these gene loci.
Lumbar spinal cord motoneurons. We determined the number of motoneuron cell bodies in the lumbar spinal cord of newborn and 4-, 12-, 24-, and 48-week-old cntf/lif/ct-1 triple-knock-out and control mice, applying established techniques (Oppenheim et al., 2001). Animals were killed by ether overdose and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 m PBS at pH 7.4. The lumbar spinal cord (L1-L6) was dissected, and 15 μm paraffin sections were prepared. After Nissl staining, motoneurons were counted in every 10th section, and the raw counts were corrected for split nucleoli (Masu et al., 1993). The diameter of nucleoli of at least four mice per group and time point and at least 40 nucleoli per animal were calculated using NIH Image software (Scion, Frederick, MD). The size of motoneuron cell bodies (area in square micrometers) was determined in cross sections of the lumbar spinal cord of 12- and 24-week-old cntf/lif/ct-1 triple-, cntf/lif double-, cntf/ct-1 double-knock-out, and wild-type mice. At least four animals per group and time point and at least 40 motoneurons per animal were analyzed also using the NIH Image program.
Axons in plantar, median, and phrenic nerves. We determined the number of myelinated axons in the plantar nerve of 12-week-old triple-knock-out and control mice. Animals were transcardially perfused with sodium cacodylate buffer containing 4% paraformaldehyde and 2% glutaraldehyde. Plantar and median nerves were carefully removed under a dissection microscope at the level of the ankle joint and wrist, respectively, and postfixed in the same fixative. Tissue preparation (osmification, dehydration, and embedding in Spurr) for electron microscopy was performed as described previously (Carenini et al., 2001). The number of myelinated axons was counted in ultrathin sections. We used a Proscan (Lagerlechfeld, Germany) slow scan camera attached to a Zeiss (Oberkochen, Germany) EM 10 CR and the corresponding software analysis system (Vario Vision Docu; Soft Imaging System, Münster, Germany). To exclude bias, axon counting was performed in all experiments without knowledge of the genotype.
We also determined the number of myelinated axons in the phrenic nerves. Phrenic nerves were removed and postfixed with 2% glutaraldehyde in 0.15 m PBS for 3 d at 4°C. After rinsing in PBS, tissues were incubated with osmium tetroxide (4%) in a buffer containing 4% potassium dichromate and 3.4% NaCl and processed for 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexylcarboxylate (Serva Feinbiochemica, Heidelberg, Germany) embedding according to standard procedures. Semithin sections (1 μm) were prepared and stained with azur-methylenblue. Myelinated axons in the proximal and distal part of the phrenic nerve were counted from photographs taken from nerve sections under an inverse Leica (Nussloch, Germany) light microscope equipped with a digital camera (ActionCam; Agfa, Mortsel, Belgium). We measured axonal circumferences in plantar nerves in three triple-knock-out mice (700-800 randomly chosen axons per mouse) and three wild-type littermates (900-1100 randomly chosen axons per mouse) at the age of 3 months. In the case of phrenic nerves, we scored nerve samples from two triple-knock-out and three wild-type mice.
Myosin ATPase reaction and muscle fiber typing. Gastrocnemic and flexor digitorum brevis (FDB) muscles were dissected from mice deeply anesthetized with a lethal dose of Ketanest/Rompun. The muscles were then freshly frozen in nitrogen-cooled isomethylbutane. ATPase staining was performed on 10-μm-thick cryosections under acidic (pH 4.3 and 4.6) and basic (pH 9.4) conditions. Data presented in this study are from reactions at pH 4.3. Type 1 (slow twitch) muscle fibers are resistant to acidic conditions and show ATPase activity and therefore stain dark. Type 2 (A and B) fibers are not resistant to acidic conditions and do not stain. Type 2C fibers show intermediate (gray color) staining.
Morphometric analysis of muscle fibers. Morphometric analysis of muscle fibers was performed on ATPase-stained 10-μm-thick cryosections of the FDB muscle from 7- to 9-month-old triple-knock-out, cntf/lif double-, ct-1 single-knock-out, and wild-type mice. Sections were viewed and photographed by using a Zeiss Axiophot microscope in conjugation with a CCD digital camera. The cross-sectional areas of type 1, type 2A and 2B, and type 2C fibers showing evidence of having been sectioned normally to their longitudinal axis were determined planimetrically. The fiber profiles were traced with a cursor on a digitizing tablet of the Vario Vision Docu analysis system at a final 45× magnification. An average of 80 type 1, 40 type 2C, and 200 type 2A and 2B optimally transverse-sectioned muscle fibers were evaluated per animal. A total of three mice per group was investigated.
Immunostaining of motor endplates. The FDB muscles from 4-week-old and 6-month-old triple-knock-out and control mice were freshly prepared. Teased muscle fibers were fixed with 2% PFA for 3 h, rinsed in 1× PBS, and blocked overnight in 10% BSA and 0.3% Triton X-100 in 1× PBS. Motor endplates were identified by acetylcholine receptor (AChR) labeling using Alexa Fluor 488-conjugated α-bungarotoxin (5 μg/ml) (Molecular Probes, Eugene, OR) in 1× PBS containing 1% BSA for 30 min. After rinsing in 1× PBS, presynaptic motor nerve terminals were stained overnight with a phospho-neurofilament SMI31 monoclonal antibody (1:500) (Sternberger Monoclonals, Lutherville, MD) in 1× PBS containing 10% BSA and 0.3% Triton X-100. After washing with 1× PBS, muscle fibers were incubated with a secondary antibody (goat antimouse conjugated with cyanine 3, 1:200; Dianova, Hamburg, Germany) in 1× PBS containing 1% BSA for 1.5 h. After rinsing in 1× PBS, muscle fibers were mounted and observed under a Leica confocal microscope. Motor endplate areas were measured using the NIH Image program.
Motor performance test. To evaluate functional motor deficits, cnt/lif/ct-1 triple-knock-out and control mice were subjected to functional motor tests at 4, 12, 24, and 48 weeks of age. The forelimb grip strength (in Newtons) was determined using an automated grip strength meter (Columbus Instruments, Columbus, OH) as described previously (Masu et al., 1993).
Rodent activity wheels (Tecniplast, Hohenpeißenberg, Germany) were used to determine voluntary movement and motor activity over a time period of 7 d. The analysis was done blinded, except for the triple-knock-out mice, which could be identified by their reduced body size.
Statistical analysis. Results are given as mean ± SEM or SD when indicated. Statistical significance of differences was assessed by one-way ANOVA, followed by Bonferroni's post hoc comparison test using Prism software (GraphPad Software, San Diego, CA). The null hypothesis was rejected on the basis of p < 0.05.
Statistical analysis of the number of plantar and median nerve axons was performed using a two-tailed t test, and p values <0.05 were considered significant.
Statistical analysis of size distribution of axonal profiles (Kolmogorov-Smirnov test) was performed using Statistica 6.1 software (StatSoft, Tulsa, OK). As statistical significance, we accepted p < 0.05.
Results
Motoneuron loss in the lumbar spinal cord
Morphological changes associated with functional loss of CNTF, LIF, CT-1, or CNTF/LIF have been described previously (Masu et al., 1993; Banner and Patterson, 1994; Sendtner et al., 1996; Oppenheim et al., 2001). To elucidate how these neurotrophic cytokines act similarly or distinctly on postnatal maintenance of motoneurons, we analyzed cntf/lif/ct-1 triple-knock-out mice compared with cntf/lif double-knock-out, cntf/ct-1 double-knock-out, and wild-type control mice.
Quantification of lumbar spinal cord motoneurons in newborn mice revealed that the number of motoneurons was already substantially reduced in cntf/lif/ct-1 triple-knock-out and cntf/ct-1 double-knock-out mice at birth [2965 ± 47 in triple-knock-out (n = 5) and 3054 ± 39 in cntf/ct-1 double-knock-out (n = 4) compared with 3622 ± 51 (n = 4) in cntf/lif double-knock-out and 3697 ± 87 (n = 4) in wild-type mice] (Fig. 1A). The difference between these groups was statistically significant (p < 0.001) and appeared to be attributable to loss of CT-1 function, as reported previously for ct-1 single-knock-out mice (Oppenheim et al., 2001).
Figure 1.
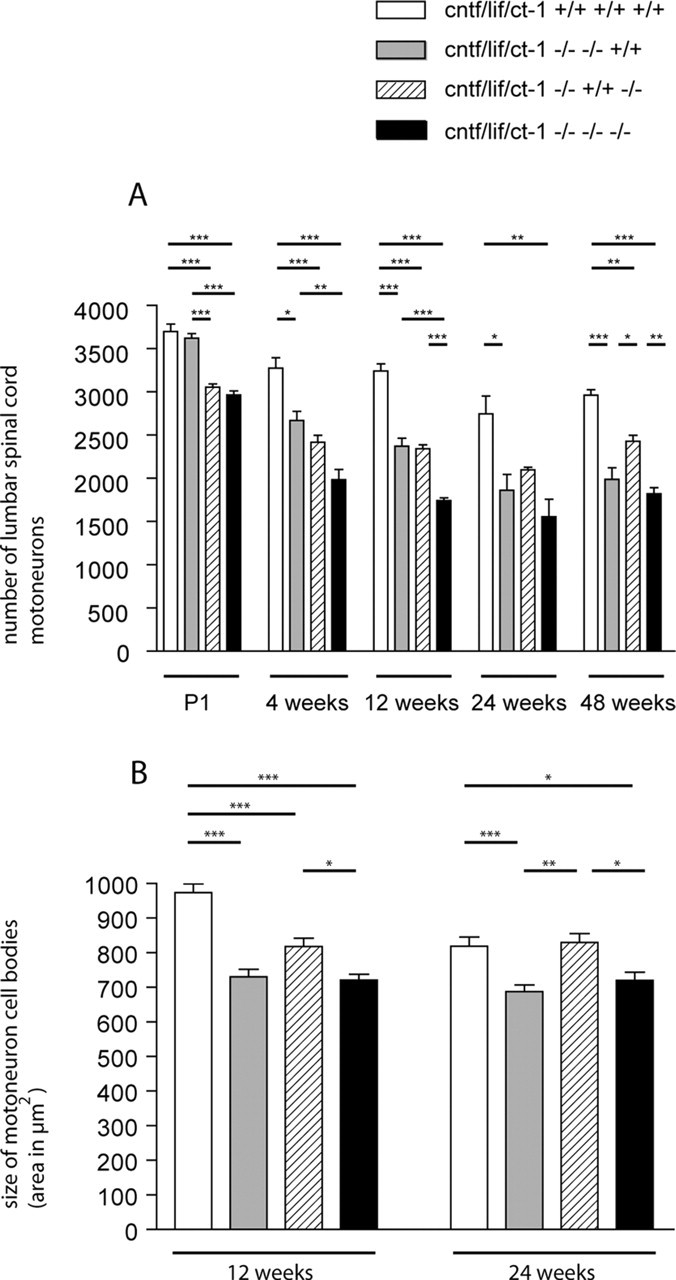
A, Motoneuron counts from lumbar spinal cord. Values shown are mean ± SEM corrected for split nuclei as described previously (Masu et al., 1993). B, Analysis of motoneuron cell body size. Total cell areas from motoneurons with typical Nissl structure and a clearly identifiable nucleolus were determined. The differences between the four groups of mice were tested by one-way ANOVA, followed by Bonferroni's post hoc comparison test. Comparison of individual groups gave the following results: *p < 0.05; **p < 0.01; ***p < 0.001. P1, Postnatal day 1.
In 4-week-old mice, motoneuron numbers were reduced by 39% in cntf/lif/ct-1 triple-knock-out mice (1985 ± 117; n = 4) and by 26% in cntf/ct-1 double-knock-out mice [2419 ± 81 (n = 4) compared with 3276 ± 119 (n = 4) in wild-type mice]. Furthermore, a statistically significant reduction of 19% became apparent in cntf/lif double-knock-out mice at this age (2667 ± 108; n = 3). The differences between these groups and wild-type mice were statistically significant (p < 0.001 for the triple-knock-out and cntf/ct-1 double-knock-out mice and p < 0.05 for the cntf/lif double-knock-out mice). The number of motoneurons did not significantly differ between cntf/lif and cntf/ct-1 double-knock-out mice but differed between cntf/lif double-knock-out and cntf/lif/ct-1 triple-knock-out mice (p < 0.01). There was no additional loss of motoneurons in cntf/lif/ct-1 triple-knock-out and cntf/ct-1 double-knock-out mice in 12-, 24-, and 48-week-old animals. In contrast, progressive loss of motoneurons was observed in cntf/lif double-knock-out mice. The number of motoneurons was reduced from 3622 ± 51 (n = 4) at birth to 2372 ± 94 (n = 3) at postnatal week 12, then to 1860 ± 187 (n = 4) at 24 weeks and to 1987 ± 135 (n = 4) at 48 weeks. At 48 weeks of age, the number of motoneurons was similar in cntf/lif double-knock-out and cntf/lif/ct-1 triple-knock-out mice. The difference of motoneuron loss in cntf/lif/ct-1 triple-knock-out mice at this stage was statistically significant when compared with wild-type (p < 0.001) and cntf/ct-1 double-knock-out (p < 0.01) mice. This indicates that cntf/lif/ct-1 triple-knock-out mice show an earlier loss of motoneurons than cntf/lif double-knock-out mice during postnatal development. However, after 24 weeks of age, motoneuron loss is not significantly different between triple-knock-out and cntf/lif double-knock-out mice.
To exclude the possibility that differences in the size of nucleoli gave rise to errors in determining the motoneuron cell number, we measured the diameter of nucleoli in the triple and double mutants as well as in wild-type control mice. In 12-week-old animals, the diameter of nucleoli in triple-knock-out mice was 3.32 ± 0.12 μm (n = 182) compared with 3.34 ± 0.12 μm in cntf/lif double-knock-out (n = 162), 3.17 ± 0.08 μm in cntf/ct-1 double-knock-out (n = 160), and 3.31 ± 0.09 μm in wild-type (n = 190) mice. The differences between these groups were not statistically significant (p > 0.05). Similarly, no differences were found in 24-week-old mice. At this time point, the diameter of nucleoli was 3.31 ± 0.11 μm in triple-knock-out mice (n = 168) compared with 3.41 ± 0.08 μm in cntf/lif double-knock-out (n = 162), 3.37 ± 0.02 μmin cntf/ct-1 double-knock-out (n = 182), and 3.45 ± 0.08 μm in wild-type (n = 186) mice.
In contrast, we found that, in 12- and 24-week-old triple- and double-knock-out mice, persisting motoneurons showed significant changes in cell body size (Fig. 1B). In particular, LIF deficiency was associated with a pronounced reduction in motoneuron cell body size. In 12-week-old mice, the size of motoneuron cell bodies was reduced by >20% in cntf/lif/ct-1 triple- and cntf/lif double-knock-out mice [721 ± 16.49 μm2 (n = 186) and 730 ± 22.3 μm2 (n = 162)] and by 16% in cntf/ct-1 double-knock-out mice [818 ± 24.45 μm2 (n = 172) compared with 974 ± 24.6 μm2 (n = 192) in wild-type mice]. The differences between these groups and wild-type mice were statistically significant (p < 0.001). Furthermore, a statistically significant difference was also observed between triple- and cntf/ct-1 double-knock-out mice (p < 0.05). In 24-week-old animals, a similar reduction was found. In cntf/lif/ct-1 triple-knock-out mice (n = 175), motoneuron cell body size was 721 ± 23.05 μm2, in cntf/lif double-knock-out mice 688 ± 18.7 μm2 (n = 183), and in cntf/ct-1 double-knock-out mice 829 ± 25.78 μm2 (n = 159) compared with 819 ± 26.53 μm2 in wild-type mice (n = 162). The differences were found statistically significant when triple- and cntf/lif double-knock-out mice were compared with cntf/ct-1 double-knock-out and wild-type mice (Fig. 1B).
However, the size of motoneuron cell bodies was at least 688 μm2 so that they could be clearly separated from other neurons in the ventral horn of the lumbar spinal cord.
Changes in motor performance
To evaluate whether the loss of motoneurons in cntf/lif/ct-1 triple-knock-out mice correlates with reduced motor function, we measured grip strength in single-, double-, and triple-knock-out mice at different time points (Fig. 2A). Triple-knock-out mice showed a reduction in grip strength by ∼42% at 4 weeks of age [0.34 ± 0.03 N (n = 5) compared with 0.59 ± 0.04 N (n = 8) in wild-type mice]. Muscle strength increased when the mice became older but was significantly lower than in wild-type mice at each time point analyzed. At 12 weeks, average grip strength in triple-knock-out mice (n = 9) was 0.57 ± 0.02 N compared with 0.93 ± 0.05 N in wild-type controls (n = 8), 0.74 ± 0.04 N (n = 12) compared with 0.97 ± 0.03 N (n = 13) at 24 weeks, and 0.61 ± 0.04 N (n = 6) compared with 0.94 ± 0.05 N (n = 9) at 48 weeks. The difference between these groups was statistically significant at each time point (p < 0.01).
Figure 2.
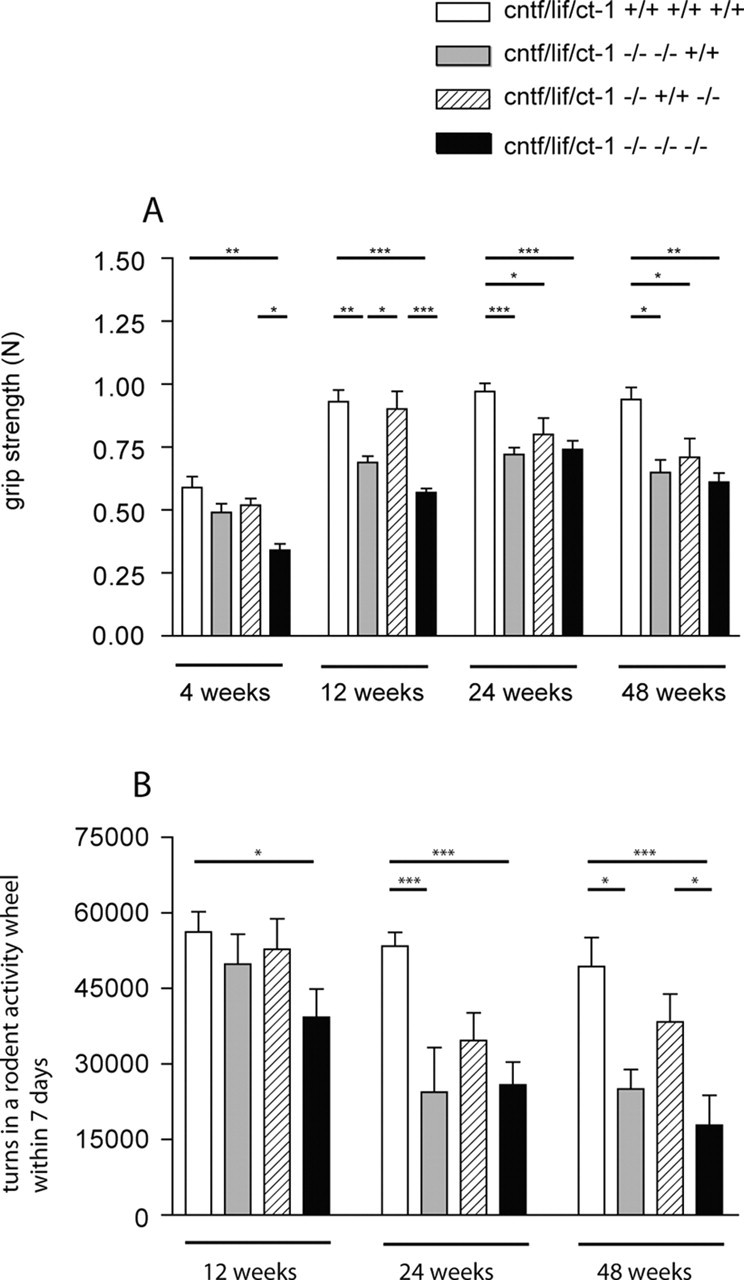
A, Forelimb grip strength was determined in male wild-type control, cntf/lif double-knock-out, cntf/ct-1 double-knock-out, and cntf/lif/ct-1 triple-knock-out mice using an automated grip strength meter as described previously (Masu et al., 1993). Values shown are mean ± SEM from 10 determinations per animal. Statistical significance of the differences between the groups was tested by one-way ANOVA. Comparison of the individual groups by Bonferroni's post hoc comparison test gave the following results: *p < 0.05; **p < 0.01; ***p < 0.001. B, Voluntary movement determined in a rodent activity wheel over a time period of 7 d. Results are given in mean ± SEM. The differences between the groups were tested by one-way ANOVA. Additional analysis by Bonferroni's post hoc comparison test showed that the difference between wild-type and triple-knock-out mice is significant at 12 weeks of age (*p < 0.05) and at 24 and 48 weeks of age (***p < 0.001). Motor function is also significantly reduced in 24-week-old (***p < 0.001) and 48-week-old (*p < 0.05) cntf/lif double-knock-out mice compared with wild-type mice and in 48-week-old triple-knock-out mice compared with cntf/ct-1 double-knock-out mice (*p < 0.05).
Measurement of grip strength in 4-week-old cntf/lif double-knock-out mice revealed a slight but not significant reduction of muscle strength compared with wild-type control mice. At 12-, 24- and 48 weeks, a reduction of >20% was observed in accordance with previous reports (Sendtner et al., 1996). The differences in cntf/lif double-knock-out mice were statistically significant from 12 weeks on when compared with wild-type mice (p < 0.05) but not when compared with cntf/lif/ct-1 triple-knock-out mice. In addition, 12-week-old cntf/lif double-knock-out mice also showed significantly lower grip strength when compared with cntf/ct-1 double-knock-out mice (p < 0.05). In contrast, mice lacking both CNTF and CT-1 did not show impaired motor function before 24 weeks of age. At that time, muscle strength was reduced by ∼18% [0.8 ± 0.07 N(n = 7) compared with 0.97 ± 0.03 N (n = 13) in wild-type mice] and by ∼24% in 48-week-old mice [0.71 ± 0.07 N (n = 7) compared with 0.94 ± 0.05 N (n = 9) in wild-type mice]. The differences between these groups were statistically significant when compared with wild-type mice (p < 0.05).
We also determined voluntary motor activity of cntf/lif/ct-1 triple-knock-out and control mice by counting the number of turns in cages with an activity wheel over a time period of 7 d (Fig. 2B). A significant reduction was observed in cntf/lif/ct-1 triple-knock-out mice already at 12 weeks of age. At this stage, the number of turns was 30% lower (39,290 ± 5631; n = 5) compared with age-matched controls (56,230 ± 3988; n = 12). An additional reduction was observed at 24 (25,830 ± 4591; n = 7) and 48 (17,830 ± 5931; n = 7) weeks compared with wild-type control mice [53,390 ± 2773 (n = 9) and 49,360 ± 5708 (n = 13), respectively]. Cntf/lif double-knock-out mice also showed reduced motor activity in the running wheels at 24 and 48 weeks of age [24,390 ± 8882 (n = 6) and 26,680 ± 4434 (n = 6)]. The differences between cntf/lif double-knock-out and wild-type mice were statistically significant (p < 0.05). These data show that cntf/lif/ct-1 triple-knock-out mice become weak at an earlier time point but that their motor activity does not differ at later developmental periods from that in cntf/lif double-knock-out mice. Moreover, mice lacking both CNTF and CT-1 do not show impaired motor function, indicating that LIF plays a more important role for postnatal maintenance of motor function than CT-1.
Axon loss in the plantar and median nerve
Because cntf/lif/ct-1 triple-knock-out mice showed reduced muscle strength, we investigated the morphology of peripheral nerves and determined the number of myelinated axons in the plantar and median nerves of mutant and control mice. At 12 weeks, the number of myelinated axons in the plantar nerve of triple-knock-out mice (1431 ± 37; n = 3) was significantly lower than that observed in wild-type (1763 ± 22; n = 3) and cntf single-knock-out (1671 ± 105; n = 3) mice. Reduced axon numbers were also found in cntf/lif double-knock-out mice at this age (1487 ± 103; n = 3). The difference was statistically significant when compared with wild-type controls (Fig. 3).
Figure 3.

Numbers of myelinated axons in the plantar nerve of 12-week-old wild-type, cntf single-knock-out, cntf/lif double-knock-out, and cntf/lif/ct-1 triple-knock-out mice were counted in ultrathin sections. Values shown are mean ± SEM. Statistical significance of the difference between the groups was tested by a two-tailed t test. *p < 0.05 was considered significant. **p < 0.01.
We also found a reduced number of myelinated axons in the median nerve in 12-week-old triple-knock-out mice compared with wild-type control mice [1034 ± 21 (n = 3) compared with 1178 ± 93 (n = 3)] (data not shown). However, this difference was smaller than that in plantar nerves. Interestingly, the reduction of myelinated axons (19% in the plantar nerves and 12% in the median nerves) appeared smaller than the loss of motoneurons in the cervical spinal cord of ct-1 single-knock-out mice (Oppenheim et al., 2001), suggesting that a formation of axonal collaterals might compensate for the loss of motoneurons, at least in part.
We could not find any loss of either proximal or distal phrenic nerve axons in triple-knock-out, cntf/lif, and cntf/ct-1 double-knock-out mice at each time point analyzed (data not shown).
To investigate whether axonal sizes have been changed in the plantar nerves of the mutants, we determined the size distribution of axons in the plantar nerves of wild-type and triple-knock-out mice. As a measure for axon size, the circumference of axons was chosen. In both genotypes, a normal distribution of axon sizes was found, and the Kolmogorov-Smirnov test revealed a similar distribution of axon sizes (p > 0.1). Similarly, in the phrenic nerve, the size distribution of axons in wild-type and triple-knock-out mice was comparable using the Kolmogorov-Smirnov test (p > 0.1).
Electron microscopy
Because in neuromuscular disorders, axonopathic changes are often most severe in distal nerves (Martini, 2001), we investigated the plantar nerve, a terminal branch of the sciatic nerve, by electron microscopy. In plantar nerves of wild-type mice, we found myelinated and nonmyelinated axons with ultrastructural characteristics as typically seen in other peripheral nerves (Fig. 4A), such as the sciatic nerve (Peters et al., 1991). No substantial alterations were seen in ct-1 single or cntf/lif double mutants (data not shown). However, subtle but significant changes were seen in plantar nerves of the cntf/lif/ct-1 triple mutants. A typical feature in these mutants was that some nonmyelinated axons appeared as fasciculating, extensively folded processes reminiscent of regrowing sprouts (Fig. 4B,C, arrowheads). Occasionally, we found fiber bundles containing one or two largecaliber axons (Fig. 4D, asterisks). These fiber bundles were collectively enveloped by a thin myelin sheath, possibly induced by the largecaliber axons (Fig. 4D). In some cases, we found large-caliber axons having achieved a 1:1 ratio with a pro-myelinating Schwann cell (Fig. 4B, arrow). This indicates that collaterals had formed in the triple-knock-out mice and that compensatory mechanisms took place even when all three members of the cntf/lif/ct-1 gene family were deficient.
Figure 4.
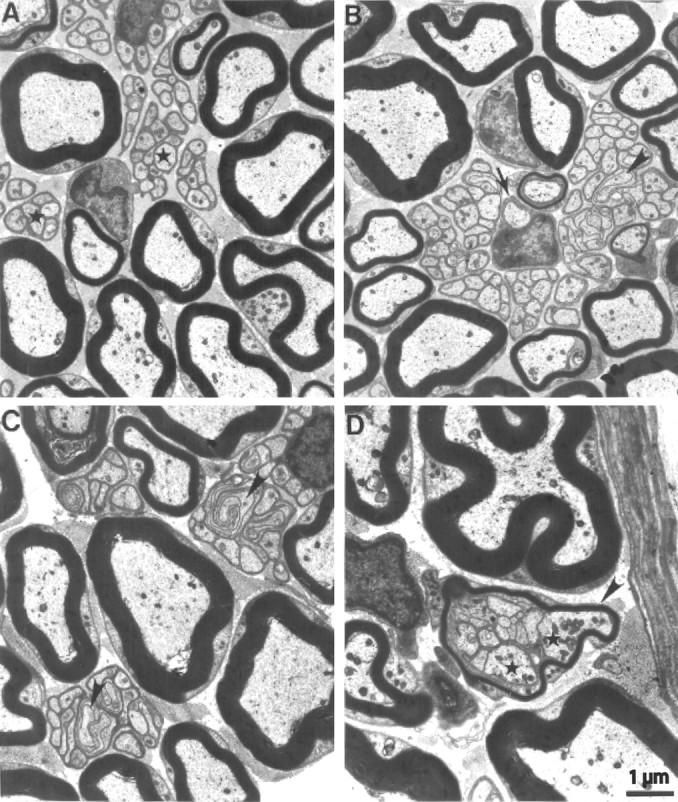
Electron micrographs of cross sections from plantar nerves of 1-year-old wild-type (A) and cntf/lif/ct-1 triple-knock-out (B-D) mice. A, Typical ultrastructural features of wild-type peripheral nerves are myelinated axons of larger-caliber and Remak fibers composed of thin axons (asterisk), which are collectively ensheathed and individually isolated from each other by a nonmyelinating Schwann cell. B, C, Note extensively folded and fasciculating axons (arrowhead) within the Remak bundles. The arrow in B indicates a promyelinated axon. D, Two larger-caliber axons (asterisks) in association with normally sized Remak axons. The bundle is ensheathed by a thin myelin sheath that has putatively been induced by the larger-caliber axons. Scale bar: A-D, 1 μm.
We then investigated the morphology of phrenic nerves of three wild-type and two triple-knock-out mice ∼3 mm proximal to the diaphragm. Neither at the light nor at the electron microscopic level could the nerves be discriminated from each other based on morphological criteria, and there were no hints for axonal degeneration, axonal sprouting, or axon collateralization. In the triple mutants, nonmyelinated axon-Schwann cell units appeared normal and did not contain unusually large axons, fasciculating and extensively folded sprouts, or ectopic myelin sheaths, as observed in the plantar nerves of the triple mutants.
Alterations at motor endplates
We then investigated motor endplate morphology and size in the FDB muscles of 4- and 24-week-old triple-knock-out and control mice. The morphology of endplates appeared grossly normal in all mice investigated. However, quantitative analysis revealed a significant decrease of motor endplate size in mice lacking lif gene expression. In 4-week-old mice, motor endplate areas were reduced by 27% in the lif single-knock-out, by 16% in the cntf/lif double-knock-out, and by 23% in the cntf/lif/ct-1 triple-knock-out mice [159 ± 7.37 μm2 (n = 22), 182 ± 7.35 μm2 (n = 33), and 168 ± 12.33 μm2 (n = 18) compared with 217 ± 8.7 μm2 (n = 28) in wild-type mice]. The differences between these groups and wild-type mice were statistically significant (Fig. 5A). This reduction was even more notable in 24-week-old mice (Fig. 5B,F-H) when compared with wild-type mice (Fig. 5C-E). We found a reduction of 37% in the triple-knock-out (153 ± 6.04 μm2; n = 84), 32% in the cntf/lif double-knock-out (165 ± 11.67 μm2; n = 27), and 24% in the lif single-knock-out (183 ± 13.1 μm2; n = 19) mice when compared with wild-type control mice (241 ± 14.91 μm2; n = 48). The differences between these groups and wild-type mice were statistically significant (p < 0.001 for the triple- and cntf/lif-double-knock-out mice and p < 0.05 for the lif single-knock-out mice).
Figure 5.
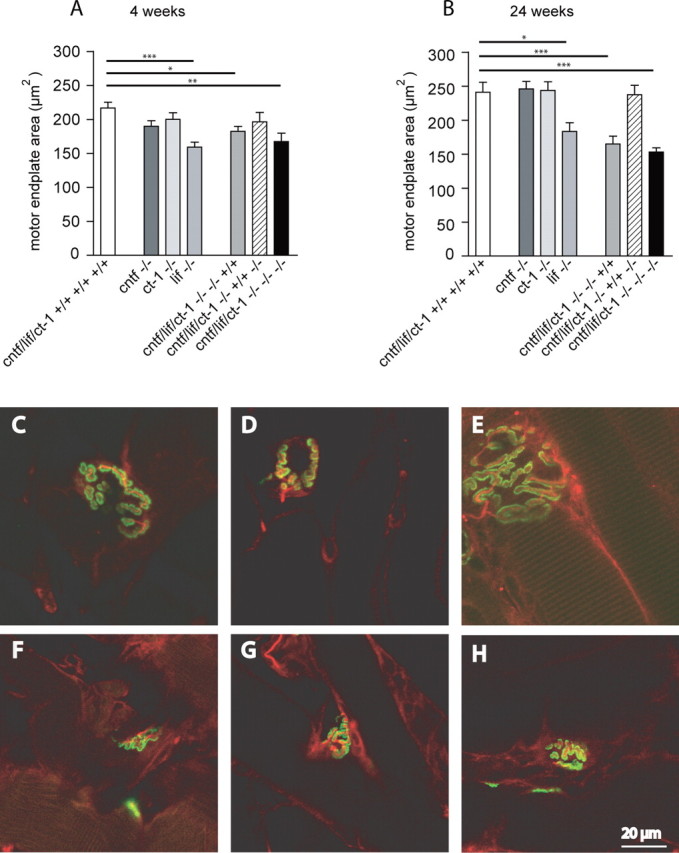
Microscopic analysis of motor endplates. The motor endplate areas were evaluated by confocal microscopy and morphometric analysis as determined by AChR labeling on teased muscle fibers of 4-week-old (A) and 24-week-old (B) control and mutant mice. Values shown are mean ± SEM. Statistical significance of the differences between the groups was tested by one-way ANOVA. Comparison of the individual groups by Bonferroni's post hoc comparison test gave the following results: *p < 0.05; **p < 0.01; ***p < 0.001. A significant reduction of motor endplate area was observed in 4- and 24-week-old mice lacking lif gene expression. C-H, Immunostaining of motor endplates on preparations of teased muscle fibers from 24-week-old wild-type (C-E) and triple-knock-out (F-H) mice. Green, Alexa Fluor 488-conjugated α-bungarotoxin; red, anti-phosphoneurofilament SMI31. Note the significant difference of motor endplate area between wild-type and triple-knock-out mice. Scale bar: C-H, 20 μm.
Fiber type grouping in flexor digitorum brevis and gastrocnemic muscle
We also investigated the ATPase activity in skeletal muscle fibers of two different muscles, the FDB and the gastrocnemic muscle. We chose these muscles because, in many neuromuscular disorders in which degeneration of motoneurons occurs, distal muscles groups are usually more prone to pathological changes than proximal muscles. Using ATPase activity (pH 4.3), we stained the FDB and gastrocnemic muscle of 3-month-old and 7- to 9-month-old wild-type, ct-1 single-knock-out, cntf/lif double-knock-out, and cntf/lif/ct-1 triple-knock-out mice. Type 1 (slow twitch) muscle fibers are resistant to acidic conditions and show ATPase activity and stain dark (Fig. 6A-E, white asterisks). In the FDB muscle, the cross-sectional area of type 1 fibers was not significantly different in 7- to 9-month-old triple-knock-out (506 ± 153 μm2, mean ± SD; n = 234) compared with cntf/lif double-knock-out (401 ± 55 μm2, mean ± SD; n = 119), ct-1 single-knock-out (490 ± 72 μm2, mean ± SD; n = 219), and wild-type (440 ± 67 μm2, mean ± SD; n = 241) mice. The type 2 (A and B) fibers are not resistant to acidic conditions and do not stain (Fig. 6A-E, black asterisks). Type 2C muscle fibers show intermediate (gray color) staining (Fig. 6A-E, arrows).
Figure 6.

Muscle fiber type distribution in the FDB muscle of wild-type (A), ct-1 single-knock-out (B), cntf/lif double-knock-out (C), and triple-knock-out (D, E) mice. Type 1 (slow twitch) muscle fibers are resistant to acidic conditions and show ATPase activity and therefore stain dark (A-E, white asterisks). Type 2A and 2B fibers are not resistant and do not stain (A-E, black asterisks). Type 2C fibers show intermediate (gray color) staining (A-E, arrows). T, Tendon. A, Muscle fibers of a 9-month-old wild-type mouse show the typical chessboard staining pattern (white asterisk, type 1 fiber; black asterisk, type 2A and 2B fibers; arrow, type 2C fiber). B, C, Muscle of 7- to 9-month-old ct-1 single-knock-out (B) and cntf/lif double-knock-out (C) mice. The morphological appearance and the distribution of fiber types is very similar to that found in age-matched wild-type mice (white asterisks, type 1 fibers; black asterisks, type 2A and 2B fibers; arrows, type 2C fibers). Large structures of darker appearance at the top right corner of C are obliquely sectioned type 1 fibers. D, E, Seven- to 9-month-old triple-knock-out mice show various degrees of muscle fiber type grouping (white asterisks, type 1 fibers; black asterisks, type 2A and 2B fibers; arrows, type 2C fibers). ko, Knock-out. Scale bar: A-E, 100 μm.
Both types of muscles showed the typical chessboard staining pattern of wild-type mice. In 3-month-old mutant mice, there was no detectable difference in fiber type distribution compared with age-matched wild-type mice (data not shown). Furthermore, in 7- to 9-month-old ct-1 single (Fig. 6B) and cntf/lif double (Fig. 6C) mutants investigated, the morphological appearance and the distribution of fiber types was very similar to that seen in age-matched wild-type mice (Fig. 6A). Interestingly, the size of type 2C fibers was significantly smaller in cntf/lif double-knock-out (367 ± 33 μm2, mean ± SD; n = 110; p < 0.05) and cntf/lif/ct-1 triple-knock-out (434 ± 150 μm2, mean ± SD; n = 81; p < 0.05) mice compared with wild-type control mice (610 ± 44 μm2, mean ± SD; n = 78). Moreover, we saw various degrees of grouping of muscle fiber types in triple-knock-out mice (Fig. 6D,E).
The gastrocnemic muscles of single- and double-mutant mice did not show any morphological changes or alterations in fiber type distribution at any stage investigated when compared with wild-type mice (data not shown). In the same muscle from 3-month-old cntf/lif/ct-1 triple-mutant mice, more intermediately stained fibers were seen than in wild-type mice. These subtle changes were not detectable in older animals (data not shown).
Discussion
Here we show that CNTF, LIF, and CT-1 exhibit overlapping but also distinct effects on motoneuron survival during development and in the adult. Whereas CNTF and/or LIF deficiency only leads to postnatal loss of motoneurons, lack of CT-1 causes a significant reduction of lumbar spinal motoneurons during prenatal development. In 48-week-old mice, CNTF/LIF deficiency causes higher losses of motoneurons than CNTF/CT-1 deficiency, and lack of LIF in combination with either CNTF or CNTF and CT-1 deficiency appears predominantly responsible for functional motor deficits. This correlates with reduced motor endplate areas, which are detectable in both young (4-week-old) and adult (24-week-old) mice. Interestingly, reduced motor endplate areas are not found in CNTF- or CT-1-deficient mice, and CNTF/CT-1-deficient mice do not show reduction in motor endplate size.
CNTF, LIF, and CT-1 share the signal transducing receptor units gp130 and LIFRβ in the formation of their respective receptor complexes (Stahl and Yancopoulos, 1994). In addition, CNTF binds to a nontransducing α chain (CNTFRα) that acts as a third binding component (Stahl and Yancopoulos, 1994). Genetic deletion of LIFRβ (Li et al., 1995), gp130 (Nakashima et al., 1999), or CNTFRα (DeChiara et al., 1995) results in significant motoneuron loss and early postnatal death, in contrast to deletion of the ligands (Masu et al., 1993; Sendtner et al., 1996; Oppenheim et al., 2001). This suggests that additional factors act through LIFRβ, gp130, and CNTFRα in promoting motoneuron survival and postnatal maintenance. The findings that even in cntf/lif/ct-1 triple-knock-out mice motor function and motoneuron survival are not completely abolished supports this view. Among these potential additional CNTF-like ligands, neuropoietin (Derouet et al., 2004) and the composite cytokine formed by cardiotrophin-like cytokine (CLC) (Senaldi et al., 1999; Shi et al., 1999) and the soluble receptor cytokine-like factor-1 (CLF) (Alexander et al., 1999; Elson et al., 2000) are likely candidates. Both factors have been identified recently as additional ligands for the CNTF receptor complex (Plun-Favreau et al., 2001). CLC and CLF are expressed in embryonic skeletal muscle, and mice that lack CLF show reduced numbers of motoneurons at birth (Forger et al., 2003), suggesting that this factor plays together with CT-1 in the maintenance of developing motoneurons.
Although CNTF, LIF, CT-1, and the other ligands belong to the same family of neuropoietic cytokines, several features differ significantly. First, CT-1 is expressed in skeletal muscle (Pennica et al., 1996), whereas Schwann cells are the major source of CNTF (Stockli et al., 1991; Friedman et al., 1992; Rende et al., 1992) and LIF (Banner and Patterson, 1994; Curtis et al., 1994). Second, the developmental time course of CNTF mRNA expression differs significantly from that of LIF and CT-1. CNTF expression is undetectable in sciatic nerves of newborn rats, only becoming apparent by day 4 and reaching maximum levels in fully differentiated Schwann cells from the end of the third postnatal week on (Stockli et al., 1989; Sendtner et al., 1992b). LIF expression, in contrast, is also detected at earlier stages (Banner and Patterson, 1994). However, only very low levels of LIF mRNA are expressed in intact postnatal nerves, but a rapid and dramatic increase occurs when nerves are lesioned (Banner and Patterson, 1994; Curtis et al., 1994; Sun et al., 1996). LIF acts as an autocrine survival factor for Schwann cells (Dowsing et al., 1999) and may play a role as a regulator of macrophage attraction to the lesioned nerve (Tofaris et al., 2002). Third, LIF can be released from Schwann cells via the classical secretory pathway, in contrast to the CNTF protein, which lacks a conventional leader sequence. Therefore, only a small part of the cytosolic pool of CNTF in differentiated Schwann cells might be available for the motoneurons under physiological conditions. CT-1 and also CLC and CLF, in contrast to CNTF, are produced at relatively high levels in the embryonic limb bud (Pennica et al., 1996; Forger et al., 2003), and thus these factors could act as target-derived neurotrophic factors on responsive motoneurons.
The first effect of LIF identified in neurons was alteration of the neurotransmitter phenotype of sympathetic neurons. In vitro, it induces a switch from noradrenergic to cholinergic transmitter phenotype (Yamamori et al., 1989; Rao et al., 1992) and influences neuropeptide expression, including the induction of vasoactive intestinal peptide (VIP), substance P, and somatostatin (Nawa et al., 1991). However, in vivo, it has been shown that LIF is not required for the target-directed change in neurotransmitter expression from noradrenaline to acetylcholine and VIP that occurs in developing sympathetic neurons innervating sweat glands (Rao et al., 1993; Francis et al., 1997). LIF induces cholinergic function in spinal cord motoneurons and supports survival of embryonic motoneurons and sensory neurons in culture (Martinou et al., 1992). Furthermore, it can induce several neuropeptides through alteration of neuronal gene expression (Patterson and Nawa, 1993) known to be involved in the response of neural tissue to injury (Sun et al., 1996). LIF is retrogradely transported to the cell body of spinal motoneurons and rapidly upregulated when nerve injury occurs (Curtis et al., 1994; Dowsing et al., 2001). LIF also protects facial motoneurons from axotomy-induced cell death (Hughes et al., 1993) and promotes regeneration after nerve injury (Tham et al., 1997; Cafferty et al., 2001). However, lif gene knock-out does not lead to enhanced motoneuron loss but only enhances motoneuron loss when it occurs in combination with CNTF or CNTF/CT-1 deficiency.
In this paper, we show that LIF deficiency leads to significantly reduced motor endplate size, which correlates with reduced muscle strength. In particular, cntf/ct-1 double-knock-out mice, which show motoneuron loss to the same extent as cntf/lif double-knock-out mice, show significantly better grip strength performance at 12 weeks than cntf/lif double mutants. This suggests that the reduced endplate size in LIF-deficient mice contributes to the phenotype, in addition to the loss of motoneurons detectable in the spinal cord of these mutants. It appears interesting that the presence of CNTF from Schwann cells and/or CT-1 from muscle cannot compensate for the deficiency of LIF in controlling motor endplate area. Indeed, the reduction in motor endplate area in lif single-knock-out mice is not higher than the reduction in the cntf/lif/ct-1 triple-knock-out mice, thus pointing to a specific role of LIF.
The development and maturation of the motor endplate is regulated by interactions between presynaptic (nerve terminal and Schwann cells) and postsynaptic (muscle fiber) components (Sanes and Lichtman, 2001). The differentiation of the postsynaptic apparatus, including AChR synthesis and clustering, is promoted and regulated by nerve-derived molecules such as neuregulin and agrin (Gautam et al., 1996; Rosenbaum et al., 1997; Sanes and Lichtman, 2001). In addition, muscle-derived signals, including cell adhesion molecules and neurotrophic factors, control formation of the presynaptic part (Sanes and Lichtman, 1999). LIF has been shown to interfere with maturation of motor units by transiently delaying the onset of synapse withdrawal (Kwon et al., 1995). On the other side, in mice that lack Schwann cells as a result of mutations in neuregulin receptors, motoneurons only transiently form neuromuscular synapses with deranged patterns of innervation (Riethmacher et al., 1997; Lin et al., 2000). This also demonstrates that Schwann cells and trophic factors from Schwann cells are necessary for motor endplate formation.
We also observed muscle fiber type grouping in 7- to 9-month-old cntf/lif/ct-1 triple-knock-out mice. Although the degree of muscle fiber type grouping varies, it seems as if LIF deficiency does not play such a major role here compared with the other ligands. The distribution of fast fibers in the flexor digitorum brevis muscle appears unchanged in cntf/lif double-knock-out mice compared with wild-type mice, and also ct-1 single-knock-out mice do not show major alterations. In contrast, triple-knock-out mice show a high degree of fiber type grouping, which also correlates with significantly reduced voluntary movement, as detected by determining the number of turns in a rodent activity wheel over 1 week. In this case, CT-1 seems to add on the detectable deficits that are observed in cntf/lif double-knock-out mice.
In conclusion, our data suggest that CT-1 is particularly important for developing motoneurons, whereas CNTF and LIF are essential for postnatal maintenance of motoneurons. LIF seems to play a specific role in the control of motor endplate size, and this deficit cannot be compensated by CNTF and/or CT-1. Triple-knock-out mice, compared with CNTF/LIF double and CT-1 single deficient mice, show various degrees of muscle fiber type grouping and a significant reduction in voluntary motor performance, indicating that CT-1 and Schwann cell-derived CNTF and/or LIF play together in the regulation of motoneuron survival and function.
Footnotes
This study was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 581, Teilprojekt B4, Grant 1837/1. We thank Michaela Pfister for genotyping and Karin Urlaub, Jennifer Marcano, and Helga Bruenner for help in histological techniques. We are grateful to Igor Kobsar for his substantial contributions in morphometric analysis and Heinrich Blazyca for skillful technical assistance.
Correspondence should be addressed to Dr. Michael Sendtner, Institute for Clinical Neurobiology, University of Wuerzburg, Josef-Schneider-Strasse 11, D-97080 Wuerzburg, Germany. E-mail: sendtner@mail.uni-wuerzburg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251778-10$15.00/0
References
- Alexander WS, Rakar S, Robb L, Farley A, Willson TA, Zhang JG, Hartley L, Kikuchi Y, Kojima T, Nomura H, Hasegawa M, Maeda M, Fabri L, Jachno K, Nash A, Metcalf D, Nicola NA, Hilton DJ (1999) Suckling defect in mice lacking the soluble haemopoietin receptor NR6. Curr Biol 9: 605-608. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Sendtner M, Thoenen H (1990) Survival effect of ciliary neurotrophic factor (CNTF) on chick embryonic motoneurons in culture: comparison with other neurotrophic factors and cytokines. J Neurosci 10: 3507-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner LR, Patterson PH (1994) Major changes in the expression of the mRNAs for cholinergic differentiation factor/leukemia inhibitory factor and its receptor after injury to adult peripheral nerves and ganglia. Proc Natl Acad Sci USA 91: 7109-7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J, Thompson SW (2001) Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci 21: 7161-7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carenini S, Maurer M, Werner A, Blazyca H, Toyka KV, Schmid CD, Raivich G, Martini R (2001) The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in p0. J Cell Biol 152: 301-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis R, Scherer SS, Somogyi R, Adryan KM, Ip NY, Zhu Y, Lindsay RM, DiStefano PS (1994) Retrograde axonal transport of LIF is increased by peripheral nerve injury: correlation with increased LIF expression in distal nerve. Neuron 12: 191-204. [DOI] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Stahl N, Pan L, Taga T, Kishimoto T, Ip NY, Yancopoulos GD (1993) LIFR beta and gp130 as heterodimerizing signal transducers of the tripartite CNTF receptor. Science 260: 1805-1808. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC, Friedman B, McClain J, Pan L, Stahl N (1995) Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 83: 313-322. [DOI] [PubMed] [Google Scholar]
- Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, Perret D, Diveu C, Guillet C, Preisser L, Dumont A, Barbado M, Morel A, deLapeyriere O, Gascan H, Chevalier S (2004) Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci USA 101: 4827-4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsing BJ, Morrison WA, Nicola NA, Starkey GP, Bucci T, Kilpatrick TJ (1999) Leukemia inhibitory factor is an autocrine survival factor for Schwann cells. J Neurochem 73: 96-104. [DOI] [PubMed] [Google Scholar]
- Dowsing BJ, Romeo R, Morrison WA (2001) Expression of leukemia inhibitory factor in human nerve following injury. J Neurotrauma 18: 1279-1287. [DOI] [PubMed] [Google Scholar]
- Elson GC, Lelievre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, Suard I, de Coignac AB, Delneste Y, Bonnefoy JY, Gauchat JF, Gascan H (2000) CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci 3: 867-872. [DOI] [PubMed] [Google Scholar]
- Escary JL, Perreau J, Dumenil D, Ezine S, Brulet P (1993) Leukaemia inhibitory factor is necessary for maintenance of haematopoietic stem cells and thymocyte stimulation. Nature 363: 361-364. [DOI] [PubMed] [Google Scholar]
- Forger NG, Prevette D, deLapeyriere O, de Bovis B, Wang S, Bartlett P, Oppenheim RW (2003) Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo J Neurosci 23: 8854-8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Asmus SE, Landis SC (1997) CNTF and LIF are not required for the target-directed acquisition of cholinergic and peptidergic properties by sympathetic neurons in vivo. Dev Biol 182: 76-87. [DOI] [PubMed] [Google Scholar]
- Friedman B, Scherer SS, Rudge JS, Helgren M, Morrisey D, McClain J, Wang DY, Wiegand SJ, Furth ME, Lindsay RM, Ip NY (1992) Regulation of ciliary neurotrophic factor expression in myelin-related Schwann cells in vivo. Neuron 9: 295-305. [DOI] [PubMed] [Google Scholar]
- Gatzinsky KP, Holtmann B, Daraie B, Berthold CH, Sendtner M (2003) Early onset of degenerative changes at nodes of Ranvier in alpha-motor axons of Cntf null (-/-) mutant mice. Glia 42: 340-349. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (1996) Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85: 525-535. [DOI] [PubMed] [Google Scholar]
- Giess R, Holtmann B, Braga M, Grimm T, Muller-Myhsok B, Toyka KV, Sendtner M (2002) Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: potential impact of CNTF as a candidate modifier gene. Am J Hum Genet 70: 1277-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RA, Sendtner M, Thoenen H (1993) Members of several gene families influence survival of rat motoneurons in vitro and in vivo. J Neurosci Res 36: 663-671. [DOI] [PubMed] [Google Scholar]
- Kwon YW, Abbondanzo SJ, Stewart CL, Gurney ME (1995) Leukemia inhibitory factor influences the timing of programmed synapses withdrawal from neonatal muscles. J Neurobiol 28: 35-50. [DOI] [PubMed] [Google Scholar]
- Li M, Sendtner M, Smith A (1995) Essential function of LIF receptor in motor neurons. Nature 378: 724-727. [DOI] [PubMed] [Google Scholar]
- Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF (2000) Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA 97: 1299-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Maurer M, Gaupp S, Martini R, Holtmann B, Giess R, Rieckmann P, Lassmann H, Toyka KV, Sendtner M, Gold R (2002) CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med 8: 620-624. [DOI] [PubMed] [Google Scholar]
- Martini R (2001) The effect of myelinating Schwann cells on axons. Muscle Nerve 24: 456-466. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Martinou I, Kato AC (1992) Cholinergic differentiation factor (CDF/LIF) promotes survival of isolated rat embryonic motoneurons in vitro. Neuron 8: 737-744. [DOI] [PubMed] [Google Scholar]
- Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H (1993) Disruption of the CNTF gene results in motor neuron degeneration. Nature 365: 27-32. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Wiese S, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Yoshida K, Kishimoto T, Sendtner M, Taga T (1999) Developmental requirement of gp130 signaling in neuronal survival and astrocyte differentiation. J Neurosci 19: 5429-5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawa H, Nakanishi S, Patterson PH (1991) Recombinant cholinergic differentiation factor (leukemia inhibitory factor) regulates sympathetic neuron phenotype by alterations in the size and amounts of neuropeptide mRNAs. J Neurochem 56: 2147-2150. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Wiese S, Prevette D, Armanini M, Wang S, Houenou LJ, Holtmann B, Gotz R, Pennica D, Sendtner M (2001) Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci 21: 1283-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH, Nawa H (1993) Neuronal differentiation factors/cytokines and synaptic plasticity. Cell [Suppl] 72: 123-137. [DOI] [PubMed] [Google Scholar]
- Pennica D, Arce V, Swanson TA, Vejsada R, Pollock RA, Armanini M, Dudley K, Phillips HS, Rosenthal A, Kato AC, Henderson CE (1996) Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron 17: 63-74. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD (1991) The fine structure of the nervous system, pp 218-232. New York: Oxford UP.
- Plun-Favreau H, Elson G, Chabbert M, Froger J, deLapeyriere O, Lelievre E, Guillet C, Hermann J, Gauchat JF, Gascan H, Chevalier S (2001) The ciliary neurotrophic factor receptor alpha component induces the secretion of and is required for functional responses to cardiotrophin-like cytokine. EMBO J 20: 1692-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Patterson PH, Landis SC (1992) Multiple cholinergic differentiation factors are present in footpad extracts: comparison with known cholinergic factors. Development 116: 731-744. [DOI] [PubMed] [Google Scholar]
- Rao MS, Sun Y, Escary JL, Perreau J, Tresser S, Patterson PH, Zigmond RE, Brulet P, Landis SC (1993) Leukemia inhibitory factor mediates an injury response but not a target-directed developmental transmitter switch in sympathetic neurons. Neuron 11: 1175-1185. [DOI] [PubMed] [Google Scholar]
- Rende M, Muir D, Ruoslahti E, Hagg T, Varon S, Manthorpe M (1992) Immunolocalization of ciliary neuronotrophic factor in adult rat sciatic nerve. Glia 5: 25-32. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C (1997) Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389: 725-730. [DOI] [PubMed] [Google Scholar]
- Rosenbaum C, Karyala S, Marchionni MA, Kim HA, Krasnoselsky AL, Happel B, Isaacs I, Brackenbury R, Ratner N (1997) Schwann cells express NDF and SMDF/n-ARIA mRNAs, secrete neuregulin, and show constitutive activation of erbB3 receptors: evidence for a neuregulin autocrine loop. Exp Neurol 148: 604-615. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389-442. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791-805. [DOI] [PubMed] [Google Scholar]
- Senaldi G, Varnum BC, Sarmiento U, Starnes C, Lile J, Scully S, Guo J, Elliott G, McNinch J, Shaklee CL, Freeman D, Manu F, Simonet WS, Boone T, Chang MS (1999) Novel neurotrophin-1/B cell-stimulating factor-3: a cytokine of the IL-6 family. Proc Natl Acad Sci USA 96: 11458-11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H (1990) Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345: 440-441. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Schmalbruch H, Stockli KA, Carroll P, Kreutzberg GW, Thoenen H (1992a) Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 358: 502-504. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Stockli KA, Thoenen H (1992b) Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J Cell Biol 118: 139-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M, Gotz R, Holtmann B, Escary JL, Masu Y, Carroll P, Wolf E, Brem G, Brulet P, Thoenen H (1996) Cryptic physiological trophic support of motoneurons by LIF revealed by double gene targeting of CNTF and LIF. Curr Biol 6: 686-694. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang W, Yourey PA, Gohari S, Zukauskas D, Zhang J, Ruben S, Alderson RF (1999) Computational EST database analysis identifies a novel member of the neuropoietic cytokine family. Biochem Biophys Res Commun 262: 132-138. [DOI] [PubMed] [Google Scholar]
- Stahl N, Yancopoulos GD (1994) The tripartite CNTF receptor complex: activation and signaling involves components shared with other cytokines. J Neurobiol 25: 1454-1466. [DOI] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, Abbondanzo SJ (1992) Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 359: 76-79. [DOI] [PubMed] [Google Scholar]
- Stockli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Gotz R, Lindholm D, Thoenen H (1989) Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature 342: 920-923. [DOI] [PubMed] [Google Scholar]
- Stockli KA, Lillien LE, Naher-Noe M, Breitfeld G, Hughes RA, Raff MC, Thoenen H, Sendtner M (1991) Regional distribution, developmental changes, and cellular localization of CNTF-mRNA and protein in the rat brain. J Cell Biol 115: 447-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Landis SC, Zigmond RE (1996) Signals triggering the induction of leukemia inhibitory factor in sympathetic superior cervical ganglia and their nerve trunks after axonal injury. Mol Cell Neurosci 7: 152-163. [DOI] [PubMed] [Google Scholar]
- Tham S, Dowsing B, Finkelstein D, Donato R, Cheema SS, Bartlett PF, Morrison WA (1997) Leukemia inhibitory factor enhances the regeneration of transected rat sciatic nerve and the function of reinnervated muscle. J Neurosci Res 47: 208-215. [PubMed] [Google Scholar]
- Tofaris GK, Patterson PH, Jessen KR, Mirsky R (2002) Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci 22: 6696-6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamori T, Fukada K, Aebersold R, Korsching S, Fann MJ, Patterson PH (1989) The cholinergic neuronal differentiation factor from heart cells is identical to leukemia inhibitory factor. Science 246: 1412-1416. [DOI] [PubMed] [Google Scholar]