

Abstract
Although protein tyrosine phosphatases (PTPs) are expressed abundantly in the brain, their roles in synaptic plasticity have not been well elucidated. In this study, we have examined the physiological functions of Ptprz, which is a receptor-type PTP expressed predominantly in the brain as a chondroitin sulfate proteoglycan. We have examined phenotypes of mutant mice deficient in Ptprz using electrophysiological, pharmacological, and behavioral approaches. Mutant mice exhibit enhanced long-term potentiation (LTP) in the CA1 region of hippocampal slices and impaired spatial learning abilities in an age-dependent manner: young adult (<10 weeks old) mutant mice show normal LTP and learning abilities in the Morris water maze task, whereas adult (>13 weeks old) mutant mice exhibit enhanced LTP and impairment in the task. The enhanced LTP is specifically canceled out by pharmacological inhibition of Rho-associated kinase (ROCK), a major downstream effector of Rho. These findings suggest that the lack of Ptprz leads to aberrant activation of ROCK and resultantly to enhanced LTP in the slice and learning impairments in the animal.
Keywords: tyrosine, phosphatase, synapse, memory, hippocampus, LTP
Introduction
Long-term potentiation (LTP) in the hippocampus is thought to be a cellular basis for learning and memory (Lynch, 2004). The induction of LTP is modified by various regulatory molecules including protein kinases and phosphatases (Soderling and Derkach, 2000). Among them, Src family tyrosine kinases have been reported to be involved in the modification of excitatory synaptic transmission and its plasticity (Salter and Kalia, 2004): LTP is inhibited by tyrosine kinase inhibitors (O'Dell et al., 1991), and knock-out mice lacking tyrosine kinases such as Fyn show impaired LTP and abnormal learning ability (Grant et al., 1992). Furthermore, properties of the NMDA receptor, which is essential for the induction of LTP in the hippocampal CA1 region (Nicoll and Malenka, 1995), are dynamically regulated by Src family tyrosine kinases (Wang and Salter, 1994; Manabe et al., 2000; Nakazawa et al., 2001; Salter and Kalia, 2004). On the other hand, mice deficient in some protein tyrosine phosphatases (PTPs) show different phenotypes in LTP and learning (Uetani et al., 2000; Lei et al., 2002; Petrone et al., 2003). Thus, the regulation of synaptic plasticity through PTPs is rather complex, and the precise roles of PTPs still remain to be elucidated.
Ptprz (also known as PTPζ or RPTPβ) is a receptor-type PTP that is expressed abundantly as a chondroitin sulfate proteoglycan in the brain (Nishiwaki et al., 1998; Peles et al., 1998). The extracellular region of Ptprz consists of a carbonic anhydrase-like domain, a fibronectin-type III-like domain, and a serine-glycine-rich region, which is considered to be the chondroitin sulfate attachment region. Three splice isoforms exist: the two transmembrane forms, Ptprz-A and Ptprz-B, and the secreted form, Ptprz-S (also known as 6B4 proteoglycan or phosphacan) (Nishiwaki et al., 1998). It is known that the extracellular domain of Ptprz binds various cell adhesion molecules [Nr-CAM, L1/Ng-CAM, F3/contactin, neural cell adhesion molecule (NCAM), and TAG1/axonin-1], growth factors [pleiotrophin (PTN)/heparin-binding growth-associated molecule (HB-GAM), midkine, and fibroblast growth factor-2], and extracellular matrix molecules (tenascin-C and tenascin-R) (Maeda et al., 1996, 1999; Maeda and Noda, 1998; Milev et al., 1998; Peles et al., 1998). Ptprz is expressed from the early developmental stage to adulthood in neurons as well as in astrocytes (Nishiwaki et al., 1998; Shintani et al., 1998). This suggests that this gene plays variegated roles in brain development and brain function. However, knock-out mice lacking Ptprz exhibit no obvious anatomical abnormalities in the CNS (Shintani et al., 1998; Harroch et al., 2000).
In the mature brain, the expression of Ptprz persists in some brain regions including the hippocampus and cerebellum (Snyder et al., 1996; Nishiwaki et al., 1998; Shintani et al., 1998), where the structure and function of synapses are modified continually throughout life. Several molecules (NCAM, tenascin-C, tenascin-R, F3/contactin, and PTN) interacting with the extracellular domain of Ptprz are shown to be implicated in synaptic plasticity in the hippocampus (Muller et al., 1996; Amet et al., 2001; Saghatelyan et al., 2001; Evers et al., 2002; Murai et al., 2002; Pavlov et al., 2002). In addition, Ptprz interacts intracellularly with the postsynaptic density-95 (PSD-95)/synapse-associated protein-90 family proteins (Kawachi et al., 1999), which are the synaptic scaffolding proteins involved in synaptic plasticity. These findings suggest the possibility that Ptprz plays critical roles in synaptic plasticity in the adult brain. In this study, we examined phenotypes of mutant mice deficient in Ptprz using electrophysiological, pharmacological, and behavioral approaches.
Materials and Methods
Mice. Generation of mutant mice lacking Ptprz was described previously (Shintani et al., 1998). In the mutant mice, all Ptprz isoforms are missing. The mice used for experiments were backcrossed to the inbred C57BL/6 strain for 10 generations. Male mice were used in all experiments. All experiments were performed according to the guidelines of the Animal Care and Experimentation Committee of Kobe University, the University of Tokyo, and the National Institute for Basic Biology.
Electrophysiology. Transverse hippocampal slices (400 μm thickness) were prepared from 5- to 10-week-old mice or from 13- to 31-week-old mice and placed in an interface-type holding chamber for at least 1 h. A single slice was then transferred to the recording chamber and submerged beneath a continuously perfusing medium that had been saturated with 95% O2 and 5% CO2. The composition of the medium was (in mm) 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11 glucose. All of the perfusing solutions contained 100 μm picrotoxin to block GABAA receptor-mediated inhibitory synaptic responses. The field potential recordings were made with a glass electrode (3 m NaCl) placed in the stratum radiatum. The whole-cell pipette solution contained (in mm) 122.5 Cs gluconate, 17.5 CsCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 Mg-ATP, and 0.3 Na3-GTP, pH 7.2; 290-300 mOsm. In the whole-cell recordings, the values of the membrane potential were compensated for the liquid junction potential at the electrode tip. When NMDA receptor-mediated EPSCs were recorded, 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX; 10 μm) was present to block AMPA receptor-mediated EPSCs. For evoking synaptic responses, a bipolar tungsten stimulating electrode was placed in the stratum radiatum, and Schaffer collateral/commissural fibers were stimulated at 0.1 Hz. An Axopatch 1D amplifier (Axon Instruments, Union City, CA) was used, and the signal was filtered at 1-5 kHz, digitized at 10 kHz, and stored on a personal computer. The stimulus strength was adjusted so that it gave rise to AMPA receptor-mediated EPSPs of the slope value between 0.10 and 0.15 mV/ms. For the analysis of EPSPs, we measured their early rising phase to avoid contamination of voltage-dependent components as much as possible. Each data point represents the averaged slope value for 1 min that was normalized to the baseline slope value. For experiments in which input-output relationships were examined, a low concentration of CNQX (1 μm) was present to block AMPA receptor-mediated synaptic responses partially, because fiber volleys were usually much smaller than EPSPs, and then 10 μm CNQX was applied to isolate fiber volley components. These experiments were done in the presence of d-2-amino-5-phosphonovaleric acid (d-APV; 25 μm) to block NMDA receptor-mediated synaptic responses. When paired-pulse facilitation and posttetanic potentiation were examined, d-APV (25 or 50 μm and 50 μm, respectively) was present. For the experiments of LTP saturation, a set of trains of high-frequency stimulation (100 Hz, 1 s; four times at 10 s intervals) was repeatedly applied at intervals of ∼15 min until there was no more potentiation; thus, the number of sets of the trains applied was variable among the slices examined. All experiments were performed at 25°C. The data are expressed as mean ± SEM. Student's t test or ANOVA was used to determine whether there was a significant difference (p < 0.05) in the mean. The majority of electrophysiological experiments were performed in a blind manner, and the results were essentially identical to those of the nonblind experiments; thus, all of the data were pooled. Picrotoxin was purchased from Wako (Osaka, Japan), d-APV and CNQX were purchased from Tocris Cookson (Avonmouth, UK), and Y-27632 was purchased from Calbiochem (Darmstadt, Germany).
Behavioral analysis. The system for the Morris water maze task consists of a round water tank (110 cm in diameter, filled with ∼24°C water), a transparent platform (6 cm in diameter), and a computer-based video tracking apparatus (AXIS90; Neuroscience, Osaka, Japan). White curtains suspended from rails attached to the laboratory ceiling were drawn around the pool. In the visible-platform task, mice were trained with a randomly located platform in the pool (two trials per day for 6 consecutive days; the maximum trial duration was 60 s). One week after the visible-platform task, the mice were then trained on the hidden-platform task to find the transparent platform that was submerged 1 cm below the surface of the water at a fixed location in the pool (two trials per day for 14 consecutive days; the maximum trial duration was 120 s). In both the visible-platform and hidden-platform tasks, the time required to reach the platform (escape latency) was recorded, and mice were allowed to spend 30 s on the platform at the end of each trial. On the day after the hidden-platform task, a transfer test was performed. Mice were allowed to swim for 60 s in the pool without the platform. The movement of each mouse was monitored by the computer-based video tracking system, and the time spent in each of four quadrants of the pool and the number of crossings of the platform position were recorded. Data obtained from the visible-platform and hidden-platform tasks were analyzed by two-way repeated-measures ANOVA. When a significant interaction was found (p < 0.05), a post hoc comparison using the Fisher PLSD test was performed between the genotypes. Data from the transfer tests were analyzed by unpaired Student's t test. All statistical analyses were performed with StatView software (version 5.0J; SAS Institute, Cary, NC).
Northern blot analysis. Total RNA was isolated from mouse hippocampi using TRIzol (Invitrogen, San Diego, CA) according to the manufacturer's instructions. Northern blot analysis using total RNA was performed as described previously (Suzuki et al., 2000). Templates used for the preparation of probes were as follows: rat Ptprg cDNA (nucleotide residues 1-1588; GenBank accession number AY177703) and rat Ptpra cDNA (nucleotide residues 1-427; GenBank accession number U57500).
Results
Enhanced LTP in the CA1 region of the hippocampus in adult Ptprz-deficient mice
To investigate synaptic plasticity in Ptprz-deficient mice, first we analyzed LTP in the CA1 region of hippocampal slices. After obtaining stable baseline EPSPs with the field potential extracellular recording technique, high-frequency stimulation (100 Hz, 1 s) was applied to afferent fibers, and LTP of similar magnitudes was induced in young adult (5-10 weeks old; designated as “young adult mice” in this study) wild-type and mutant mice (Fig. 1A,B). However, the same conditioning stimulation gave rise to a significantly larger LTP (50-60 min after the tetanus; p < 0.02) in adult (13-31 weeks old; designated as “adult mice”) mutant mice than in adult wild-type mice (Fig. 1C,D), whereas there was no significant difference in the earlier phase (11-20 min after the tetanus; p > 0.2). We also examined the saturation level of LTP by applying tetanic stimulation repeatedly (Fig. 1E,F) and found that it was significantly higher (p < 0.03) in adult mutant mice than in wild-type adult mice (Fig. 1F).
Figure 1.
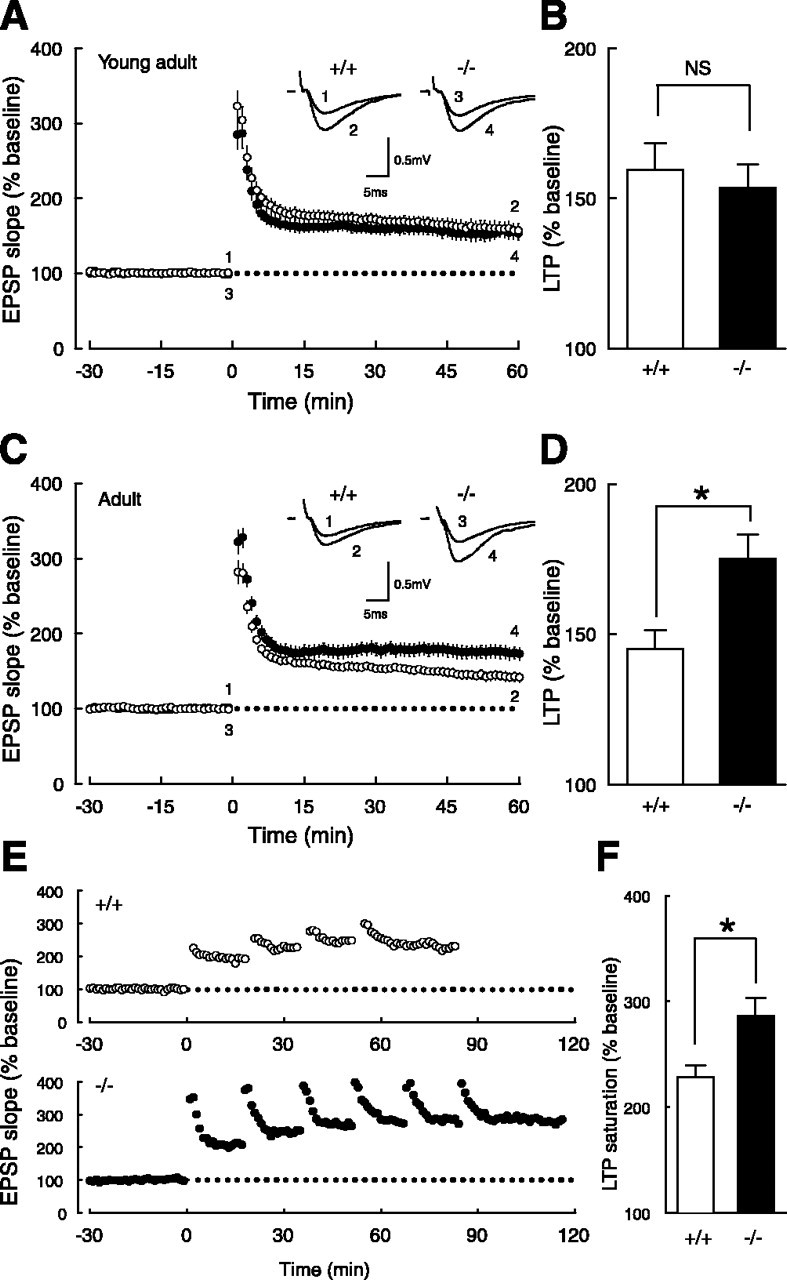
Age-dependent enhancement of LTP in Ptprz-deficient mice. A, The time course of LTP in young adult wild-type (○) and mutant (•) mice. Initial EPSP slopes were measured, and the values were normalized in each experiment to the averaged slope value measured during the control period (-30 to 0 min). Tetanic stimulation (100 Hz, 1 s) was applied at 0 min. Sample traces in the inset represent EPSPs (average of 10 consecutive responses) of wild-type (+/+) and mutant (-/-) mice recorded at the times indicated by the numbers. B, Summary of LTP experiments in young adult mice. There was no significant difference between wild-type (159.4 ± 9.0% of baseline; n = 13) and mutant (153.5 ± 7.9% of baseline; n = 13) mice. Potentiation ratios were calculated by dividing the averaged EPSP slope values from 50 to 60 min by those from -30 to 0 min. NS, Not significant. C, The time course of LTP in adult wild-type (○) and mutant (•) mice. D, Summary of LTP experiments in adult mice. There was a significant difference (*p < 0.02) between wild-type (145.0 ± 6.3% of baseline; n = 14) and mutant (175.0 ± 8.3% of baseline; n = 18) mice, whereas there was no significant difference (p > 0.2) in the earlier phase between wild-type (163.1 ± 4.6% of baseline; n = 14) and mutant (176.1. ± 8.4% of baseline; n = 18) mice. E, Representative examples of LTP saturation in adult wild-type (+/+) and mutant (-/-) mice. High-frequency stimulation was applied repeatedly until there was no more potentiation (see Materials and Methods). F, Summary of LTP saturation experiments. There was a significant difference (*p < 0.03) between adult wild-type (228.5 ± 11.4% of baseline; n = 9) and mutant (285.8 ± 17.5% of baseline; n = 10) mice.
To clarify the mechanism involved in the enhanced LTP in adult mutant mice, we examined the basic properties of synaptic transmission. The input-output relationship of AMPA receptor-mediated EPSPs was indistinguishable between the genotypes in young adult (Fig. 2A) and adult (Fig. 2B) mice. We next examined the possible involvement of presynaptic factors in the enhanced LTP in adult mutant mice. Paired-pulse facilitation (50, 100, and 200 ms intervals), which is dependent on the presynaptic release probability, was indistinguishable between the two genotypes in young adult (Fig. 2C) and adult (Fig. 2D) mice. Furthermore, posttetanic potentiation, a purely presynaptic phenomenon, induced by 100 Hz, 1 s conditioning stimulation in the presence of d-APV (50 μm), an NMDA receptor antagonist, was also unaltered in both young adult (Fig. 2E) and adult (Fig. 2F) mice. This experiment also indicates that the enhanced LTP in adult mutant mice was not brought about by the addition of any NMDA receptor-independent potentiation, because d-APV completely blocked the enduring phase of synaptic potentiation.
Figure 2.
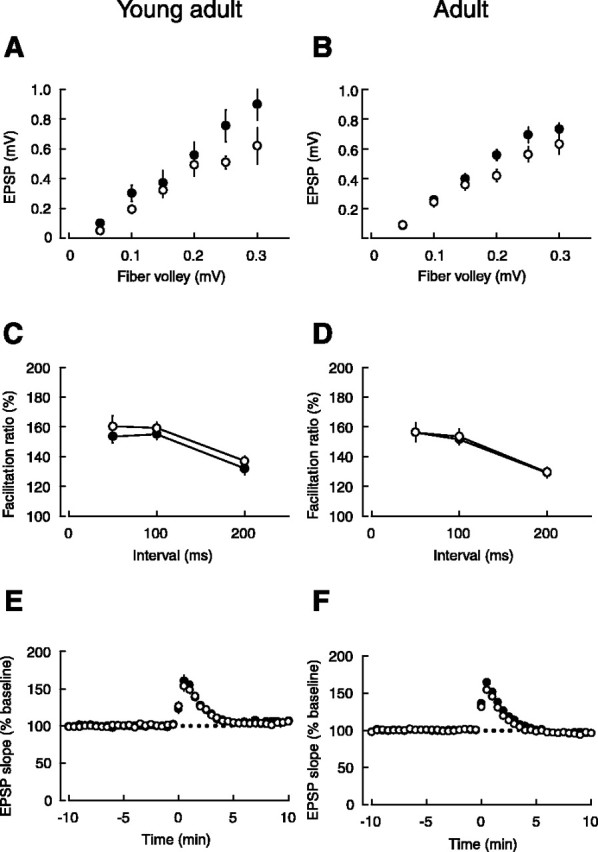
Normal basic properties of synaptic transmission in Ptprz-deficient mice. A, The input-output relationships of AMPA receptor-mediated EPSPs in young adult wild-type (○; n = 11) and mutant (•; n = 11) mice, which were not significantly different between the two genotypes. The data were first sorted by the range of fiber volley amplitudes, and then EPSP amplitudes were averaged within each range. B, The input-output relationships of AMPA receptor-mediated EPSPs in adult wild-type (○; n = 16) and mutant (•; n = 17) mice, which were not significantly different between the two genotypes. C, Paired-pulse facilitation induced by stimulating afferent fibers twice at intervals of 50, 100, or 200 ms was not statistically different between young adult wild-type (○; n = 11) and mutant (•; n = 9) mice. D, Paired-pulse facilitation was not statistically different between adult wild-type (○; n = 17) and mutant (•; n = 24) mice. E, Posttetanic potentiation was not statistically different between young adult wild-type (○; n = 11) and mutant (•; n = 11) mice. F, Posttetanic potentiation was not statistically different between adult wild-type (○; n = 14) and mutant (•; n = 22) mice.
Because the activity of NMDA receptors is upregulated by tyrosine phosphorylation (Wang and Salter, 1994; Manabe et al., 2000; Nakazawa et al., 2001; Salter and Kalia, 2004) and therefore NMDA receptor-mediated synaptic responses may be downregulated by PTPs, it is possible that the enhanced LTP in adult mutant mice is caused by the modification of NMDA receptor functions. Therefore, we next examined properties of NMDA receptors in mutant mice. The ratio of NMDA receptor-mediated EPSC amplitudes/AMPA receptor-mediated EPSC amplitudes was not different between the two genotypes in young adult (Fig. 3A,B) and adult (Fig. 3C,D) mice. Because the input-output relationship of AMPA receptor-mediated EPSPs was indistinguishable between the genotypes (Fig. 2A,B), this result indicates that the input-output relationship of NMDA receptor-mediated EPSPs was also normal in mutant mice. The current-voltage curve of NMDA receptor-mediated EPSCs was also unaltered in mutant mice both in young adult (Fig. 3E) and adult (Fig. 3F) mice, suggesting that the Mg2+ block of NMDA receptor channels was intact in mutant mice. Furthermore, the summation of NMDA receptor synaptic currents during tetanic stimulation (100 Hz, 100 ms) was indistinguishable between the genotypes in adult mice (Fig. 3G,H). These results demonstrated that the enhancement of LTP in adult mutant mice is not caused by direct modification of NMDA receptor properties but is associated with some intracellular signaling downstream from NMDA receptor activation.
Figure 3.

Unaltered properties of the NMDA receptor in Ptprz-deficient mice. A, Sample traces of NMDA EPSCs (upward traces; recorded at +40 mV) and AMPA EPSCs (downward traces; recorded at -90 mV) in young adult wild-type (+/+) and mutant (-/-) mice. B, The ratio of amplitudes of the NMDA EPSC to those of the AMPA EPSC was not significantly different (p > 0.8) between young adult wild-type (79.2 ± 15.7%; n = 6) and mutant (76.3 ± 8.2%; n = 8) mice. The ratio was calculated for each cell, and the values were then averaged for all cells. C, Sample traces of NMDA EPSCs and AMPA EPSCs in adult wild-type (+/+) and mutant (-/-) mice. D, The ratio of the NMDA EPSC to those of the AMPA EPSC was not significantly different (p > 0.06) between adult wild-type (77.4 ± 6.2%; n = 16) and mutant (60.3 ± 5.9%; n = 15) mice. E, The current-voltage relationships of NMDA receptor-mediated EPSCs in young adult wild-type (○; n = 6) and mutant (•; n = 8) mice. Current amplitudes, which were not different between the two genotypes, were normalized to the value obtained at +40 mV for easier comparison. F, The current-voltage relationships of NMDA receptor-mediated EPSCs in adult wild-type (○; n = 15) and mutant (•; n = 14) mice. G, Sample traces showing the summation of NMDA EPSCs during repetitive stimulation (100 Hz, 100 ms) in adult wild-type (+/+) and mutant (-/-) mice. The baseline EPSC evoked by a single pulse (smaller trace; average of 10 consecutive responses) is superimposed on the synaptic response evoked by 10 pulses (larger trace). H, The ratio of the amplitude of the 10th NMDA EPSC during the repetitive stimulation to that of the baseline NMDA EPSC evoked by a single pulse was not significantly different between adult wild-type (518.5 ± 44.1% of baseline; n = 7) and mutant (531.9 ± 69.6% of baseline; n = 6) mice.
The intracellular signaling pathway regulated by Ptprz
Recently, we identified p190 RhoGAP, a GTPase-activating protein that switches off Rho GTPases, as one of the substrates of Ptprz (Fukada et al., 2005). Rho GTPases are known to regulate neuronal morphogenesis, including cell migration, and axonal growth and guidance (Luo, 2000), through the downstream kinase ROCK activities (Nakayama et al., 2000). Thus, we investigated possible involvement of the ROCK pathway in the enhanced LTP in adult mutant mice. We first confirmed that the magnitude of LTP in adult mutant mice was larger (p < 0.05) than that in adult wild-type mice (Fig. 4A,B). However, in the presence of the ROCK inhibitor Y-27632 (10 μm), LTP in adult wild-type mice was not significantly affected, but LTP in adult mutant mice was reduced to yield no statistically significant difference between the two genotypes (Fig. 4C,D), suggesting that the enhancement of LTP is mediated by activation of the Rho/ROCK pathway.
Figure 4.

Involvement of the ROCK pathway in LTP enhancement in adult Ptprz-deficient mice. A, The time course of LTP in adult wild-type (○) and mutant (•) mice in control conditions. Tetanic stimulation (100 Hz, 1s) was applied at 0 min. Sample traces in the inset represent EPSPs (average of 10 consecutive responses) of wild-type (+/+) and mutant (-/-) mice recorded at the times indicated by the numbers. B, Summary of LTP experiments in control conditions. There was a significant difference (*p < 0.05) between wild-type (145.0 ± 5.9% of baseline; n = 8) and mutant (164.7 ± 5.7% of baseline; n = 9) mice. C, The time course of LTP in adult wild-type (○) and mutant (•) mice in the presence of the ROCK inhibitor Y-27632, which was applied from -30 to 0 min. D, Summary of LTP experiments in the presence of Y-27632. There was no significant difference between wild-type (143.5 ± 7.4% of baseline; n = 9) and mutant (155.2 ± 3.6% of baseline; n = 8) mice. NS, Not significant.
Age-dependent memory impairments in Ptprz-deficient mice
We concomitantly addressed the learning ability of Ptprz-deficient mice in the Morris water maze task (Morris, 1984). At the age of 8 weeks (young adult mice), in both the visible-platform and hidden-platform tasks, the performance of wild-type and mutant mice was improved during training (visible: F(5,150) = 27.04, p < 0.0001; hidden: F(13,390) = 15.42, p < 0.0001), and there was no difference between the two genotypes (visible: F(1,30) = 1.8 × 10-2, p = 0.8920; hidden: F(1,30) = 0.30, p < 0.5872) (Fig. 5A,C). In the transfer test, there were no differences between the two genotypes in either the time spent in the trained quadrant (p > 0.11) or the number of crossings of the trained platform position (p > 0.74) (Fig. 5E,G).
Figure 5.

Age-dependent memory impairment in Ptprz-deficient mice. A-D, Wild-type (○) and mutant (•) mice were subjected to the Morris water maze task at the age of 8 weeks (young adult mice; n = 16 each) (A, C) or at the age of 20 weeks (adult mice; n = 21 each) (B, D). Escape latencies in the visible-platform task (A, B) and the hidden-platform task (C, D) are shown. The data points represent the mean ± SEM of two trials per day. E-H, The time spent (E, F) and the number of platform crossings (G, H) in each of four quadrants of the pool in the transfer test. Values are expressed as the mean ± SEM. *p < 0.05; Student's t test. T, Target; the quadrant where the platform was constantly located in the hidden-platform task; L, the quadrant left of the target; R, the quadrant right of the target; O, the quadrant opposite of the target; □, wild-type mice; ▪, mutant mice.
At the age of 20 weeks (adult mice), there was no difference in the ability to perform the visible-platform task between the genotypes (genotypes: F(1,40) = 0.19, p = 0.6637; trained blocks: F(5,200) = 53.35, p < 0.0001) (Fig. 5B). In the hidden-platform task, however, the overall escape latencies of mutant mice were significantly longer than those of wild-type mice (genotypes: F(1,40) = 4.88, p = 0.0330; trained blocks: F(13,520) = 9.40, p < 0.0001): a significant increase was observed at the seventh training block (p < 0.01; post hoc Fisher PLSD test) (Fig. 5D). In the transfer tests, adult mutant mice showed a significant decrease in the time spent in the trained quadrant (p < 0.05) (Fig. 5F), and, although it was not statistically significant, there was a tendency of decrease in the number of crossings of the trained platform position (p > 0.12) (Fig. 5H). We found no differences in the swimming speed between the two genotypes: young adult wild-type mice, 21.9 ± 1.2 cm/s (n = 16); young adult mutant mice, 23.2 ± 0.8 cm/s (n = 16); adult wild-type mice, 20.1 ± 1.0 cm/s (n = 21); adult mutant mice, 21.0 ± 0.9 cm/s (n = 21). Thus, mutant mice exhibited a slower rate of spatial learning specifically at the age of 20 weeks, but not at the age of 8 weeks, which was in accordance with the age-dependent impairment of LTP (Fig. 1).
No expressional alteration of Ptprg and Ptpra in Ptprz-deficient mice
Ptprz and Ptprg (PTPα) resemble structurally and constitute the type V subfamily of RPTPs with these two members (Andersen et al., 2001). High-level expression of Ptprz and Ptprg was observed in the adult hippocampal formation (Barnea et al., 1993; Shintani et al., 1998). To address the possibility that Ptprg expression is upregulated in the mutant mice to compensate the deficiency in Ptprz, we examined the mRNA level of Ptprg in the hippocampus by Northern blot analysis. We also examined the expression of Ptpra (PTPα), which is another RPTP with a highly glycosylated extracellular domain (Daum et al., 1994). It is known that both interact with NCAM, contactin (Peles et al., 1995; Zeng et al., 1999), and a Helicobacter pylori vacuolating cytotoxin, VacA (Fujikawa et al., 2003; Yahiro et al., 2003). As shown in Figure 6, the expression of Ptprg and Ptpra was not affected either in young adult or adult Ptprz-deficient mice. Thus, Ptprg and Ptpra, functionally relevant PTP candidates to Ptprz, were not upregulated to compensate the Ptprz deficiency.
Figure 6.
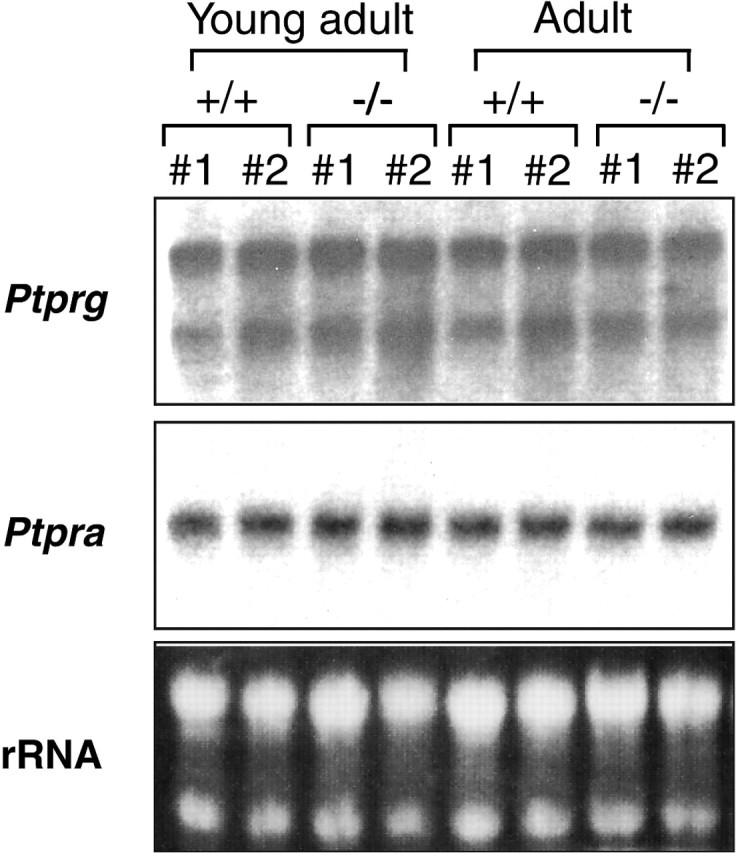
Developmental expression of Ptprg and Ptpra in Ptprz-deficient mice. Total RNA prepared from hippocampi was loaded in each lane (10 μg). The cDNA fragment of rat Ptprg or Ptpra was 32P labeled and used as a probe. Staining with ethidium bromide for ribosomal RNA (rRNA) is shown as a loading control. The expression of Ptprg and Ptpra was similar between wild-type and Ptprz-deficient mice both at the young adult and adult stages.
Discussion
The roles of PTPs in synaptic plasticity have not been elucidated, although several papers have appeared examining the role of PTPs in synaptic transmission in the CNS. The activation of Ptpra, which binds to the PSD-95/Discs large/zona occludens-1 (PDZ) domain of PSD-95, enhances NMDA receptor-mediated synaptic responses, and the blockade of endogenous Ptpra results in impairment of the induction of hippocampal LTP (Lei et al., 2002). Mutant mice lacking Ptpra also exhibit impaired LTP (Petrone et al., 2003). In this study, we found that Ptprz regulates synaptic plasticity without affecting basal synaptic transmission, and the regulation is age dependent. Furthermore, the ability of spatial learning is also modulated by Ptprz in an age-dependent manner. The phenotype appeared only when the mice reached 10 weeks of age. This result supports the view that the LTP phenotype is not related to the developmental abnormality because young Ptprz-deficient mice show normal LTP. Interestingly, the age-dependent enhancement of LTP is canceled out when the Rho/ROCK pathway is pharmacologically blocked.
We recently showed that Ptprz interacts with PSD-95 in the postsynaptic density including the spine of hippocampal pyramidal cells, which contains the NMDA receptor complex (Kawachi et al., 1999). This finding intimates that the enhanced LTP in adult mutant mice may be caused by the functional modification of NMDA receptors through tyrosine phosphorylation (Salter and Kalia, 2004). However, we failed to identify any change in the properties of NMDA receptors in this study (Fig. 3), which is in accordance with our unpublished observations that the level of NMDA receptor tyrosine phosphorylation in mutant mice is unaltered. Thus, facilitation of NMDA receptor activities is unlikely to be associated with the enhanced LTP in adult mutant mice. Instead, the modification of intracellular signaling mechanisms downstream of NMDA receptor activation is likely to be responsible for the enhancement.
We previously observed that Ptprz is implicated in neuronal migration, neurite extension, and cell attachment (Maeda et al., 1996; Maeda and Noda, 1998; Fujikawa et al., 2003). Recently, using our yeast substrate-trapping system (Kawachi et al., 2001), we identified p190 RhoGAP, a negative regulator of Rho GTPases, as a candidate of substrates for Ptprz (Fukada et al., 2005). p190 RhoGAP is demonstrated to play an important role in the memory formation in adult animals (Lamprecht et al., 2002). In this study, we showed that the enhanced component of LTP in adult mutant mice is cancelled out by the inhibitor for ROCK, a downstream effector of Rho GTPase. It is thus possible that Ptprz regulates LTP induction through regulation of p190 RhoGAP activity. The Rho/ROCK pathway is known to play an important role in the organization of the actin cytoskeleton in various cellular phenomena including neurite extension, cell migration, cell adhesion, and cell morphology (Luo, 2000). Because actin reorganization has been implicated in LTP in the adult nervous system (Krucker et al., 2000; Fukazawa et al., 2003), Ptprz likely modulates LTP through the regulation of actin organization by the Rho/ROCK pathway leading to the morphological modification of dendritic spines.
In the brain, p190 RhoGAP is tyrosine phosphorylated by Src family kinases (Brouns et al., 2001). Therefore, it is conceivable that the tyrosine phosphorylation level of p190 RhoGAP is determined by the balance between the activities of Src kinases and Ptprz. Providing that p190 RhoGAP activity is negatively mediated by Src, as suggested by some reports (Billuart et al., 2001; Lamprecht et al., 2002), p190 RhoGAP in Ptprz-deficient mice might be hyperphosphorylated, and the activity of Rho GTPase is augmented. In contrast to Ptprz-deficient mice, the targeted disruption of Fyn, a member of the Src kinase family, causes deficits in the induction of LTP (Grant et al., 1992). This phenotype might be ascribed to the reduction of Rho activity. Interestingly, Fyn-deficient mice also show an age-dependent impairment of LTP (Kojima et al., 1997): the LTP phenotype is observed only after 10 weeks after birth, which coincides with the period when the enhanced LTP is detected in Ptprz-deficient mice. These observations suggest that Ptprz and Fyn exert their functions, at least in part, in the same context in the induction of LTP. At present, the mechanism underlying the age dependencies observed in Ptprz-deficient mice is not clear. Additional studies are necessary to understand the age-dependent changes in the hippocampal plasticity.
In many kinds of knock-out mice, LTP impairment often leads to abnormal learning abilities in behavioral tests (Crawley and Paylor, 1997), and, consistently, we have reported that learning ability is facilitated when hippocampal LTP is enhanced (Manabe et al., 1998). However, in the present study, we found that LTP is enhanced in adult mutant mice, but their learning ability is impaired rather than upregulated. Although the underlying mechanism is unclear at present, there are some papers reporting similar phenotypes of mutant mice lacking functional molecules. In PSD-95-deficient mice, LTP in the hippocampus was enhanced, whereas these mutant mice exhibited impaired spatial learning (Migaud et al., 1998). Hippocampal slices from the mice lacking PTN (HB-GAM), a ligand for Ptprz, also showed enhanced LTP, although the learning ability was impaired (Amet et al., 2001; Pavlov et al., 2002). The mice overexpressing PTN demonstrated opposite electrophysiological and behavioral phenotypes (Pavlov et al., 2002). These results suggest that Ptprz negatively regulates the induction of LTP in corporation with PTN. In contrast, mutant mice deficient in Ptprd (PTPδ) exhibit learning impairment in the Morris water maze task, whereas hippocampal LTP is unexpectedly enhanced (Uetani et al., 2000); however, paired-pulse facilitation was enhanced in Ptprd-deficient mice but not in our mutant mice. Thus, some presynaptic change can be involved in LTP enhancement in Ptprd-deficient mice, and therefore precise mechanisms for abnormal LTP could be essentially different.
We have examined the effect of prolonged low-frequency stimulation (data not shown), but in mice in adulthood, it is generally difficult to induce LTD, and this was also the case in this study. Thus, the deficiency in hippocampal LTD may not be a major cause of the abnormal behavior. We also found that the LTP saturation level in adult mutant mice was significantly higher than in adult wild-type mice. Therefore, it is likely that the abnormal hippocampal LTP mechanisms are involved in the abnormal behavior observed in adult mutant mice, although we cannot exclude possible contribution of abnormal synaptic plasticity in other brain regions. Although LTP has been regarded as a cellular model of memory (Lynch, 2004), it should be more critical in understanding behavioral phenotypes how synaptic transmission is modified by a specific signaling pathway affected by mutations, because more LTP might not necessarily be beneficial.
Footnotes
This work was supported by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, Culture, and Technology of Japan (T.M., M.N.); by the Center for Brain Medical Science, 21st Century Center of Excellence Program, University of Tokyo (T.M.); by the Uehara Memorial Foundation (T.M.), Terumo Life Science Foundation (T.M.), Sumitomo Foundation (T.M.), Naito Foundation (T.M.), Takeda Foundation (T.M.), Novartis Foundation (M.N.), Yamada Science Foundation (M.N.), and Research Institute of Science and Technology for Society (K.N., T.M.) and Core Research for Evolutional Science and Technology (T.M., M.N.), Japan Science and Technology Agency. We thank M. Mizoguchi, M. Ishida, and K. Yamada for technical assistance in behavioral experiments and T. Nakazawa for valuable advice.
Correspondence should be addressed to either of the following: Dr. Toshiya Manabe, Division of Neuronal Network, Department of Basic Medical Sciences, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan, E-mail: tmanabe-tky@umin.ac.jp; or Dr. Masaharu Noda, Division of Molecular Neurobiology, National Institute for Basic Biology, 5-1 Higashiyama, Myodaiji-cho, Okazaki, Aichi 444-8787, Japan, E-mail: madon@nibb.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251081-08$15.00/0
K.N., A.F., and S.K. contributed equally to this work.
References
- Amet LE, Lauri SE, Hienola A, Croll SD, Lu Y, Levorse JM, Prabhakaran B, Taira T, Rauvala H, Vogt TF (2001) Enhanced hippocampal long-term potentiation in mice lacking heparin-binding growth-associated molecule. Mol Cell Neurosci 17: 1014-1024. [DOI] [PubMed] [Google Scholar]
- Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol 21: 7117-7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnea G, Silvennoinen O, Shaanan B, Honegger AM, Canoll PD, D'Eustachio P, Morse B, Levy JB, Laforgia S, Huebner K, Musacchio JM, Sap J, Schlessinger J (1993) Identification of a carbonic anhydrase-like domain in the extracellular region of RPTP γ defines a new subfamily of receptor tyrosine phosphatases. Mol Cell Biol 13: 1497-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Winter CG, Maresh A, Zhao X, Luo L (2001) Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell 107: 195-207. [DOI] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Settleman J (2001) p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol 3: 361-367. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Paylor R (1997) A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav 31: 197-211. [DOI] [PubMed] [Google Scholar]
- Daum G, Regenass S, Sap J, Schlessinger J, Fischer EH (1994) Multiple forms of the human tyrosine phosphatase RPTPα. J Biol Chem 269: 10524-10528. [PubMed] [Google Scholar]
- Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bosl MR, Morellini F, Bartsch U, Dityatev A, Schachner M (2002) Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J Neurosci 22: 7177-7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M, Shintani T, Wada A, Aoyama N, Hirayama T, Fukamachi H, Noda M (2003) Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori Nat Genet 33: 375-381. [DOI] [PubMed] [Google Scholar]
- Fukada M, Kawachi H, Fujikawa A, Noda M (2005) Yeast substrate-trapping system for isolating substrates of porotein tyrosine phosphatases: isolation of substrates for protein tyrosine phosphatase receptor type z. Methods 35: 54-63. [DOI] [PubMed] [Google Scholar]
- Fukazawa, Y Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo Neuron 38: 447-460. [DOI] [PubMed] [Google Scholar]
- Grant SGN, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER (1992) Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258: 1903-1910. [DOI] [PubMed] [Google Scholar]
- Harroch S, Palmeri M, Rosenbluth J, Custer A, Okigaki M, Shrager P, Blum M, Buxbaum JD, Schlessinger J (2000) No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase ζ. Mol Cell Biol 20: 7706-7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawachi H, Tamura H, Watakabe I, Shintani T, Maeda N, Noda M (1999) Protein tyrosine phosphatase ζ/RPTPβ interacts with PSD-95/SAP90 family. Mol Brain Res 72: 47-54. [DOI] [PubMed] [Google Scholar]
- Kawachi H, Fujikawa A, Maeda N, Noda M (2001) Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphataseζ/β by the yeast substrate-trapping system. Proc Natl Acad Sci USA 98: 6593-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER (1997) Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA 94: 4761-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S (2000) Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA 97: 6856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, LeDoux JE (2002) Fear memory formation involves p190 RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron 36: 727-738. [DOI] [PubMed] [Google Scholar]
- Lei G, Xue S, Chery N, Liu Q, Xu J, Kwan CL, Fu YP, Lu YM, Liu M, Harder KW, Yu XM (2002) Gain control of N-methyl-d-aspartate receptor activity by receptor-like protein tyrosine phosphatase α. EMBO J 21: 2977-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L (2000) Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 1: 173-180. [DOI] [PubMed] [Google Scholar]
- Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84: 87-136. [DOI] [PubMed] [Google Scholar]
- Maeda N, Noda M (1998) Involvement of receptor-like protein tyrosine phosphatase ζ/RPTPβ and its ligand pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) in neuronal migration. J Cell Biol 142: 203-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Nishiwaki T, Shintani T, Hamanaka M, Noda M (1996) 6B4 proteoglycan/phosphcan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTP β, binds pleiotrophin/heparin-binding growth associated molecule (HB-GAM). J Biol Chem 271: 21446-21452. [DOI] [PubMed] [Google Scholar]
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M (1999) A receptor-like protein-tyrosine phosphatase PTPζ/RPTPβ binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPζ. J Biol Chem 274: 12474-12479. [DOI] [PubMed] [Google Scholar]
- Manabe T, Noda Y, Mamiya T, Katagiri H, Houtani T, Nishi M, Noda T, Takahashi T, Sugimoto T, Nabeshima T, Takeshima H (1998) Facilitation of long-term potentiation and memory in mice lacking nociceptin receptors. Nature 394: 577-581. [DOI] [PubMed] [Google Scholar]
- Manabe T, Aiba A, Yamada A, Ichise T, Sakagami H, Kondo H, Katsuki M (2000) Regulation of long-term potentiation by H-Ras through NMDA receptor phosphorylation. J Neurosci 20: 2504-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433-439. [DOI] [PubMed] [Google Scholar]
- Milev P, Chiba A, Haring M, Rauvala H, Schachner M, Ranscht B, Margolis RK, Margolis RU (1998) High affinity binding and overlapping localization of neurocan and phosphacan/protein-tyrosine phosphatase-ζ/β with tenascin-R, amphoterin, and the heparin-binding growth-associated molecule. J Biol Chem 273: 6998-7005. [DOI] [PubMed] [Google Scholar]
- Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47-60. [DOI] [PubMed] [Google Scholar]
- Muller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V, Rougon G, Kiss JZ (1996) PSA-NCAM is required for activity-induced synaptic plasticity. Neuron 17: 413-422. [DOI] [PubMed] [Google Scholar]
- Murai KK, Misner D, Ranscht B (2002) Contactin supports synaptic plasticity associated with hippocampal long-term depression but not potentiation. Curr Biol 12: 181-190. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L (2000) Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci 20: 5329-5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T (2001) Characterization of Fyn-mediated phosphorylation sites on GluRϵ2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J Biol Chem 276: 693-699. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC (1995) Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 377: 115-118. [DOI] [PubMed] [Google Scholar]
- Nishiwaki T, Maeda N, Noda M (1998) Characterization and developmental regulation of proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ isoforms. J Biochem 123: 458-467. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER, Grant SGN (1991) Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature 353: 558-560. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Voikar V, Kaksonen M, Lauri SE, Hienola A, Taira T, Rauvala H (2002) Role of heparin-binding growth-associated molecule (HB-GAM) in hippocampal LTP and spatial learning revealed by studies on overexpressing and knockout mice. Mol Cell Neurosci 20: 330-342. [DOI] [PubMed] [Google Scholar]
- Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, Clary DO, Schilling J, Barnea G, Plowman GD, Grumet M, Schlessinger J (1995) The carbonic anhydrase domain of receptor tyrosine phosphatase β is a functional ligand for the axonal cell recognition molecule contactin. Cell 82: 251-260. [DOI] [PubMed] [Google Scholar]
- Peles E, Schlesinger S, Grumet M (1998) Multi-ligand interactions with receptor-like protein tyrosine phosphatase β: implications for intercellular signaling. Trends Biochem Sci 23: 121-124. [DOI] [PubMed] [Google Scholar]
- Petrone A, Battaglia F, Wang C, Dusa A, Su J, Zagzag D, Bianchi R, Casaccia-Bonnefil P, Arancio O, Sap J (2003) Receptor protein tyrosine phosphatase α is essential for hippocampal neuronal migration and long-term potentiation. EMBO J 22: 4121-4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelyan AK, Dityatev A, Schmidt S, Schuster T, Bartsch U, Schachner M (2001) Reduced perisomatic inhibition, increased excitatory transmission, and impaired long-term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin-R. Mol Cell Neurosci 17: 226-240. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5: 317-328. [DOI] [PubMed] [Google Scholar]
- Shintani T, Watanabe E, Maeda N, Noda M (1998) Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ: analysis of mice in which the PTPζ /RPTP β gene was replaced with the LacZ gene. Neurosci Lett 247: 135-138. [DOI] [PubMed] [Google Scholar]
- Snyder SE, Li J, Schauwecker PE, McNeill TH, Salton SR (1996) Comparizon of RPTPζ/β, phosphacan, and trkB mRNA expression in the developing and adult rat nervous system and induction of RPTPζ/β and phosphacan mRNA following brain injury. Mol Brain Res 40: 79-96. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA (2000) Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23: 75-80. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Shintani T, Sakuta H, Kato A, Ohkawara T, Osumi N, Noda M (2000) Identification of RALDH-3, a novel retinaldehyde dehydrogenase, expressed in the ventral region of the retina. Mech Dev 98: 37-50. [DOI] [PubMed] [Google Scholar]
- Uetani N, Kato K, Ogura H, Mizuno K, Kawano K, Mikoshiba K, Yakura H, Asano M, Iwakura Y (2000) Impaired learning with enhanced hippocampal long-term potentiation in PTPδ-deficient mice. EMBO J 19: 2775-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Salter MW (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369: 233-235. [DOI] [PubMed] [Google Scholar]
- Yahiro K, Wada A, Nakayama M, Kimura T, Ogushi K, Niidome T, Aoyagi H, Yoshino K, Yonezawa K, Moss J, Hirayama T (2003) Protein-tyrosine phosphatase α, RPTP α, is a Helicobacter pylori VacA receptor. J Biol Chem 278: 19183-19189. [DOI] [PubMed] [Google Scholar]
- Zeng L, D'Alessandri L, Kalousek MB, Vaughan L, Pallen CJ (1999) Protein tyrosine phosphatase α (PTPα) and contactin form a novel neuronal receptor complex linked to the intracellular tyrosine kinase fyn. J Cell Biol 147: 707-713. [DOI] [PMC free article] [PubMed] [Google Scholar]