

Abstract
It has been shown previously that the endogenous opioid system may be involved in the behavioral effects of nicotine. In the present study, the participation of endogenous enkephalins on nicotine responses has been investigated by using preproenkephalin knock-out mice. Acute nicotine-induced hypolocomotion remained unaffected in these mice. In contrast, antinociception elicited in the tail-immersion and hot-plate tests by acute nicotine administration was reduced in mutant animals. The rewarding properties of nicotine were then investigated using the place-conditioning paradigm. Nicotine induced a conditioned place preference in wild-type animals, but this effect was absent in knock-out mice. Accordingly, in vivo microdialysis studies revealed that the enhancement in dopamine extracellular levels in the nucleus accumbens induced by nicotine was also reduced in preproenkephalin-deficient mice. Finally, the somatic expression of the nicotine withdrawal syndrome precipitated in nicotine-dependent mice by mecamylamine was significantly attenuated in mutant animals. In summary, the present results indicate that endogenous opioid peptides derived from preproenkephalin are involved in the antinociceptive and rewarding properties of nicotine and participate in the expression of physical nicotine dependence.
Keywords: nicotine, antinociception, reward, dependence, enkephalin, knock-out mice
Introduction
Nicotine, the main psychoactive component of tobacco, induces its pharmacological effects by acting on nicotinic acetylcholine receptors (nAChRs). These receptors are ligand-gated cation channels composed of five membrane subunits that combine to form a functional receptor (Dani, 2001). Thus far, 12 different subunits (α2-α10 and β2-β4) have been shown to contribute to the formation of nAChRs in the nervous system (Mansvelder et al., 2003). The specific function of the different nAChR subtypes has not been fully understood, and the generation of knock-out mice lacking nAChR subunits has provided a new tool to further clarify the role of these receptors in the CNS (for review, see Cordero-Erausquin et al., 2000). Nicotine modifies the release of many neurotransmitters by acting at presynaptic sites, which is consistent with a modulatory role of nAChRs in the CNS (Wonnacott, 1997).
Animal and clinical studies have suggested the participation of the endogenous opioid system in different behavioral responses to nicotine, mainly concerning its antinociceptive and addictive properties. Thus, the administration of the opioid antagonist naloxone decreases nicotine-induced antinociception in rodents (Tripathi et al., 1982; Zarrindast et al., 1997). In addition, nicotine administration before the μ-opioid antagonist β-funaltrexamine prevents the ability of this antagonist to block morphine antinociception (Davenport et al., 1990), indicating that endogenous opioids released by nicotine administration bind to μ-opioid receptors and protect from β-funaltrexamine effects.
Nicotine has been reported to produce both rewarding and aversive responses by stimulating mesolimbic dopaminergic transmission (Pontieri et al., 1996; Dani and De Biasi, 2001; Laviolette and van der Kooy, 2003), which represents a common substrate for the motivational effects of many drugs of abuse (Koob and Le Moal, 2001). Different clinical studies have indicated an involvement of opioids in the reinforcement of cigarette smoking. Thus, the administration of opioid antagonists such as naltrexone has been shown to reduce cigarette consumption and the satisfaction during ad libitum smoking (Karras and Kane, 1980; Lerner et al., 1998; Wewers et al., 1998; Covey et al., 1999). However, other human studies with opioid antagonists have obtained negative results (Sutherland et al., 1995), and additional trials are needed to identify possible subgroups that may be particularly responsive to opioid antagonism (Modesto-Lowe and Van Kirk, 2002). The participation of opioid mechanisms in nicotine physical dependence has also been reported. Indeed, the opioid antagonist naloxone is able to precipitate a withdrawal syndrome in nicotine-dependent rats (Malin et al., 1993; Adams and Cicero, 1998). Besides, somatic manifestations of withdrawal could be observed in chronic smokers following naloxone challenge (Krishnan-Sarin et al., 1999). The specific involvement of μ-opioid receptors in this functional interaction between nicotine and the opioid system has been recently clarified by using knock-out mice. Thus, the antinociceptive and rewarding effects of nicotine as well as nicotine physical dependence were reduced in mice lacking μ-opioid receptors (Berrendero et al., 2002).
Different endogenous opioid peptides derived from the three protein precursors identified (proopiomelanocortin, proenkephalin and prodynorphin) show high affinity for the μ-opioid receptor. In this study, we evaluated the role of endogenous peptides derived from proenkephalin in acute and chronic nicotine responses by using preproenkephalin-deficient mice (Konig et al., 1996). Locomotor and antinociceptive effects induced by acute nicotine administration were first investigated. The conditioned place-preference paradigm was used to evaluate the rewarding properties of nicotine. In vivo microdialysis studies were performed to determine the effects of nicotine on the extracellular levels of dopamine (DA) in the nucleus accumbens (NAc). Finally, the behavioral expression of mecamylamine-precipitated withdrawal after chronic nicotine treatment was evaluated in these knock-out mice.
Materials and Methods
Animals. The generation of mice lacking the preproenkephalin gene has been described previously (Konig et al., 1996). Mice were housed five per cage in a temperature (21 ± 1°C)- and humidity (55 ± 10%)-controlled room with a 12 h light/dark cycle (light between 8:00 A.M. and 8:00 P.M.). Food and water were available ad libitum. Mice were habituated to their new environment and handled for 1 week before starting the experimental procedure. The mice (8-12 weeks of age) used in this study were on a C57BL/6J genetic background and were obtained from hybrid mutant mice originally created on a 129 SVJ-C57/BL6 background (Konig et al., 1996). Knock-out mice used in these experiments have been crossed for 12 generations to C57BL/6J mice purchased from The Jackson Laboratory (Bar Harbor, ME). Because of this extensive backbreeding, these knock-out mice have virtually no remaining 129Sv genetic background. Furthermore, to avoid a genetic drift, knock-out strains were routinely crossed in the laboratory every third or fourth generation to C57BL/6J mice imported from The Jackson Laboratory.
In each experimental group, mice were matched for age and sex. Animal procedures were conducted in accordance with the guidelines of the European Communities Directive 86/609/EEC regulating animal research and approved by the local ethical committee. The observer was blind to genotype and treatment in all of the experiments.
Drugs. (-)-Nicotine hydrogen tartrate salt [(-)-1-methyl-2(3-pyridyl)pyrrolidine] and mecamylamine hydrochloride (Sigma, Madrid, Spain) were dissolved in physiological saline (0.9%) and administered by subcutaneous route in a volume of 10 ml/kg. All nicotine doses were calculated as nicotine hydrogen tartrate salt. Ketamine hydrochloride (100 mg/kg) (Imalgène 1000; Rhône Mérieux, Lyon, France) and xylazine hydrochloride (20 mg/kg) (Sigma) were mixed and dissolved in ethanol (5%) and distilled water (95%). This anesthetic mixture was administered in an injection volume of 20 ml/kg body weight (i.p.). Tetrodotoxin (TTX) (Sigma) was dissolved in Ringer's solution. All of the other substances used for neurochemical and histological studies were obtained from Sigma.
Locomotor activity. The locomotor responses induced by acute administration of nicotine hydrogen tartrate salt (1, 3, and 6 mg/kg, s.c.) or saline administration were measured by using individual locomotor activity boxes (9 × 20 × 11 cm; Imetronic, Passac, France). The boxes were equipped with one line of photocells placed 2 cm above the floor to evaluate horizontal movements and another line 6 cm above the floor to measure vertical activity (rears). Mice were habituated to the locomotor cages daily (10 min in the morning) on three consecutive days. On day 4, mice were placed in the locomotor cages 5 min after drug injection, and horizontal and vertical locomotor activity were recorded for 10 min in a low-luminosity environment (20-25 lux).
Tail-immersion and hot-plate tests. The tail-immersion test was measured 15 min after acute nicotine hydrogen tartrate salt (1, 3, and 6 mg/kg, s.c.) or saline administration. The water temperature was maintained at 50 ± 0.5°C using a thermo-regulated water circulating pump (Clifton, North Somerset, UK). The time (in seconds) to withdraw the tail from the bath was measured, with a cutoff latency of 5 s to prevent tissue damage.
The hot-plate test was performed 16 min after acute nicotine hydrogen tartrate salt (1, 3, and 6 mg/kg, s.c.) or saline injection. The heated surface of the plate was kept at a temperature of 52 ± 0.1°C (Columbus Instruments, Columbus, OH). The nociceptive threshold evaluated was the jumping response. In absence of jumps, a 240 s cutoff was used to prevent tissue damage.
The data obtained were expressed as percentage of maximum possible effect using the following equation: (MPE %) = (test latency - control latency)/(cutoff time - control latency) × 100.
Conditioned place preference. The rewarding effects of nicotine were evaluated by using the conditioned place-preference paradigm, as described recently (Castañé et al., 2002). The apparatus consisted of two main square conditioning compartments separated by a triangular central division. The movement and location of the mice were monitored by computerized monitoring software (Videotrack; View Point, Lyon, France). During the preconditioning phase, each mouse was placed in the middle of the central division and had free access to both compartments of the conditioning apparatus for 18 min, with the time spent in each compartment recorded. Treatments were counterbalanced between compartments to use an unbiased procedure. No initial place preference or aversion for the different compartments was observed in the experiment. In the conditioning phase, mice were treated during 8 d with alternate injections of nicotine hydrogen tartrate salt (0.25, 0.5, and 1 mg/kg, s.c.) or saline. Mice were confined into the corresponding compartment immediately after injection by using guillotine doors matching the walls for 20 min. Nicotine was administered on days 1, 3, 5, and 7 and saline on days 2, 4, 6, and 8. Control animals received saline every day. The test phase was conducted as in the preconditioning phase (i.e., free access to both compartments for 18 min) and the time spent in each compartment recorded. The time in the central area was proportionally shared and added to the time value of each compartment as reported previously (Maldonado et al., 1997). A score was calculated for each mouse as the difference between the postconditioning and preconditioning time spent in the drug-paired compartment. In an additional experiment, the nicotinic antagonist mecamylamine (1 mg/kg, s.c.) was administered 15 min before nicotine (0.5 mg/kg, s.c.) during the conditioning phase in wild-type animals to verify the mediation of nicotinic acetylcholine receptors in nicotine-rewarding properties.
Surgery and microdialysis procedure. Mice were anesthetized with a ketamine/xylazine mixture (0.2 ml/10 g body weight, i.p.) and placed in a stereotaxic apparatus with a flat skull (Paxinos and Franklin, 1997). A small hole was drilled on the right side of the skull, and the cannula guide (CMA/7; CMA Microdialysis, Stockholm, Sweden) was implanted vertically 1 mm above the NAc (anteroposterior, +1.60 mm; mediolateral, -0.9 mm; dorsoventral, -3.80 mm from bregma) and then fixed to the skull with dental cement. Three days after the surgery, the analytical probe (CMA/7/1 mm; CMA Microdialysis) was inserted into the guide cannula. Two days after probe implantation, animals were habituated to the experimental environment overnight. The following morning, Ringer's solution was pumped through the dialysis probe at a constant rate of 1 μl/min. Samples were taken every 15 min and analyzed by HPLC (see below). Baseline samples were taken during 1.5 h, and subsequently mice were injected with either saline or nicotine hydrogen tartrate salt (0.5, 1, and 3 mg/kg, s.c.). Collection of samples was continued for 3 h after injection. To ensure that the DA content in the dialysate was impulse dependent, baseline samples were taken for 2 h in a separate experiment and then Ringer's solution containing the fast Na+ channel blocker, TTX (5 μm), was perfused for 80 min. Samples were taken for an additional 3 h.
HPLC procedure. Dialysate samples (15 μl) were injected without any purification into an HPLC system that consisted of a pump linked to an automatic injector (Agilent 1100; Agilent Technologies, Palo Alto, CA), a reverse-phase column (Zorbax SB C18, 3.5 μm, 150 × 4.6 mm; Agilent Technologies), and a coulometric detector (Coulochem II; CMA Microdialysis, North Chelmsford, MA) with a 5011 analytical cell to quantify DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA). The first electrode was fixed at -100 mV and the second electrode at +300 mV. The gain of the detector was set at 50 nA. The signal of the second electrode was connected to a HP Chemstation for HPLC (revision A.08.03[847]). The composition of the mobile phase was 50 mm NaH2PO4, 0.1 mm Na2EDTA, 0.65 mm octyl sodium sulfate, and 14% (v/v) methanol, pH 3.5. The flow rate was set at 1 ml/min. The sensitivity of the assay was 1 pg/15 μl for DA, 5 pg/15 μl for DOPAC, and 10 pg/15 μl for HVA. Calibration curves were prepared for DA with standards between 1 and 50 pg/sample, for DOPAC between 20 and 1000 pg/sample, and for HVA between 20 and 1000 pg/sample. Five replicates of a given concentration of DA (15 pg/sample), DOPAC (150 pg/sample), and HVA (150 pg/sample) were analyzed for determination of intra-assay precision and accuracy. Precision, expressed as the percentage relative SD of the estimated concentrations was as follows: 0.95 for DA, 0.98 for DOPAC, and 11.58 for HVA. Accuracy, expressed as the relative error of the calculated concentrations, was 13.12 for DA, 8.67 for DOPAC, and 4.14 for HVA.
One single mean basal level of DA, DOPAC, or HVA was calculated from three baseline samples collected separately, and the values of DA, DOPAC, or HVA after administration of either nicotine hydrogen tartrate salt or saline were normalized to this basal level. DA concentrations after either nicotine hydrogen tartrate salt or saline challenge were converted to area under the curve (AUC) using a standard trapezoid method (Gibaldi and Perrier, 1975). The following equation was used: AUC = [0.5*(B + C1)*d + 0.5*(C1 + C2)*d + 0.5* (C2 + C3)*d +... 0.5*(Cn-1 + Cn)*d) - B*d], where B is the average basal value, C1 to Cn are values during drug challenge, and d is the duration of the sample (15 min in this case).
Histology. At the end of the experiments, animals were killed, and brains were quickly removed and stored at -80°C. To check the position of the probe, brains were cut using a cryostat in 20 μm serial coronal sections, which were then processed with cresyl violet and observed under a microscope. Only those animals that had been implanted correctly were included in the study.
Nicotine dependence and withdrawal. Nicotine dependence was induced by using Alzet osmotic minipumps (model 2001; Alzet, Cupertino, CA) as reported previously (Castañé et al., 2002). These minipumps, implanted subcutaneously under brief ether anesthesia, contained saline or nicotine hydrogen tartrate salt solutions and delivered a constant subcutaneous flow in a rate of 1 μl/h. The concentration of nicotine was adjusted to compensate for differences in the mouse body weight. Thus, the mice received a mean dose of ∼25 mg/kg/d of nicotine hydrogen tartrate salt during 6 d. Nicotine withdrawal syndrome was precipitated 6 d after minipump implantation by injection of the nicotinic receptor antagonist mecamylamine (1 mg/kg, s.c.). The somatic signs of withdrawal were evaluated immediately after mecamylamine injection during 30 min, as reported previously (Castañé et al., 2002). The number of wet dog shakes, front paw tremors, sniffing, and scratches were counted. Body tremor, ptosis, teeth chattering, genital licks, and piloerection were scored 1 for appearance or 0 for nonappearance within each 5 min period. The locomotor activity over 5 min periods was rated 0, 1, or 2 (0 for inactivity, 1 for low activity, and 2 for normal activity). A global withdrawal score was calculated for each animal by giving each individual sign a relative weight: 0.5 for each episode of wet dog shake, front paw tremor, sniffing, and scratching; and 1 for the presence of ptosis, genital lick, tremor, piloerection, and teeth chattering during each 5 min observation period. The relative weight of locomotor activity for each 5 min period was 0 for normal activity, 0.5 for low activity, and 1 for inactivity (Castañé et al., 2002).
Statistical analysis. Acute nicotine effects and global withdrawal scores were compared by using a between-subjects two-way ANOVA (genotype and treatment as factors of variation), followed by one-way ANOVA for individual differences and post hoc comparisons (Dunnett's test) when required. For place-conditioning studies, score values were compared by using a between-subjects two-way ANOVA (genotype and treatment as factors of variation). Time spent in the drug-paired compartment during pretest in the different groups was compared by a one-way ANOVA to ensure use of an unbiased procedure. Individual comparisons of time spent in the drug-paired compartment during preconditioning and test phases were made with a paired two-tailed Student's t test. The microdialysis data were analyzed using three-way ANOVA with treatment (saline and nicotine) and genotype (wild-type and knock-out) as between-subjects factors and time after nicotine injection as within-subjects factor. Basal levels of DA, DOPAC, and HVA were analyzed between genotypes by using a two-tailed Student's t test. AUC values were compared by using a between-subjects two-way ANOVA (genotype and treatment as factors of variation), followed by one-way ANOVA for individual differences and post hoc comparisons (Dunnett's test) when required. Differences were considered significant if the probability of error was <5%.
Results
Nicotine decreased locomotion in wild-type and preproenkephalin-deficient mice
On days 1, 2, and 3, animals were exposed to the locomotor activity boxes to be habituated to the test environment (data not shown), and acute effects of nicotine (1, 3, and 6 mg/kg, s.c.) were evaluated on day 4. Nicotine decreased locomotion in enkephalin knock-out mice and wild-type littermates (Fig. 1). Two-way ANOVA revealed a significant effect of treatment on the horizontal activity (F(3,165) = 70.02; p < 0.0001) but not effect of genotype (F(1,165) = 0.00; NS) nor interaction between treatment and genotype (F(3,165) = 0.74; NS). Subsequent one-way ANOVA (treatment) indicated a significant effect of treatment in wild-type (F(3,86) = 30.32; p < 0.0001) and knock-out mice (F(3,79) = 48.02; p < 0.0001). Post hoc analysis showed a similar decrease of horizontal activity when nicotine was administered in wild-type (3 and 6 mg/kg; p < 0.01) and preproenkephalin-deficient mice (1, 3, and 6 mg/kg; p < 0.01) (Fig. 1A).
Figure 1.
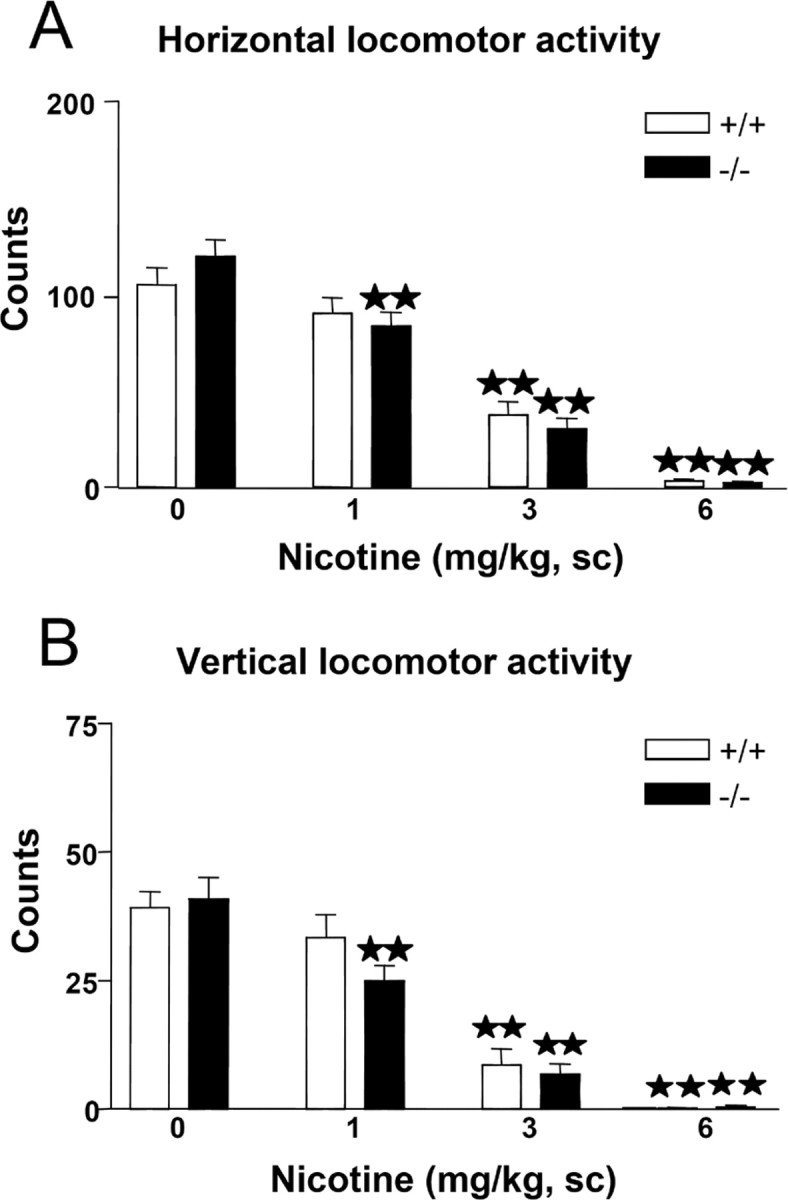
Effects of acute nicotine on locomotion in preproenkephalin-deficient (black bars) and wild-type (white bars) mice. Horizontal (A) and vertical (B) locomotion were measured 5 min after nicotine administration (0, 1, 3, and 6 mg/kg, s.c.). Data are expressed as mean ± SEM of photocell counts during a 10 min period in wild-type (saline, n = 34; nicotine, 1 mg/kg, n = 21; nicotine, 3 mg/kg, n = 20; nicotine, 6 mg/kg, n = 15) and knock-out (saline, n = 33; nicotine, 1 mg/kg, n = 17; nicotine, 3 mg/kg, n = 18; nicotine, 6 mg/kg, n = 15) mice. ★★p < 0.01, when compared with the saline group of the same genotype (Dunnett's test).
Two-way ANOVA also revealed a significant effect of treatment on the vertical activity (F(3,165) = 49.02; p < 0.0001), without effect of genotype (F(1,165) = 0.62; NS) or interaction between these two factors (F(3,165) = 0.71; NS). One-way ANOVA revealed a significant effect of treatment in wild-type (F(3,86) = 28.18; p < 0.0001) and knock-out mice (F(3,79) = 27.51; p < 0.0001). Post hoc comparisons showed a similar reduction of vertical activity when nicotine was administered in wild-type (3 and 6 mg/kg; p < 0.01) and preproenkephalin-deficient mice (1, 3, and 6 mg/kg; p < 0.01) (Fig. 1B).
Nicotine antinociception was reduced in preproenkephalin-deficient mice
Nicotine-induced antinociceptive responses (1, 3, and 6 mg/kg, s.c.) were decreased in preproenkephalin knock-out compared with wild-type mice in the hot-plate and the tail-immersion tests (Fig. 2). In the hot-plate, two-way ANOVA showed a significant effect of treatment (F(3,162) = 91.61; p < 0.0001), genotype (F(1,162) = 12.54; p < 0.001), and interaction between treatment and genotype (F(3,162) = 4.41; p < 0.01). Subsequent one-way ANOVA revealed significant treatment effects in wild-type (F(3,84) = 74.25; p < 0.0001) and knock-out mice (F(3,78) = 25.13; p < 0.0001). Nicotine induced an antinociceptive response at the doses of 3 and 6 mg/kg (p < 0.01) in both genotypes as revealed by post hoc comparisons. Post hoc analysis also showed a reduction of nicotine-induced antinociception in preproenkephalin knock-out mice when compared with wild-type mice at the doses of 3 and 6 mg/kg (p < 0.05) (Fig. 2A).
Figure 2.
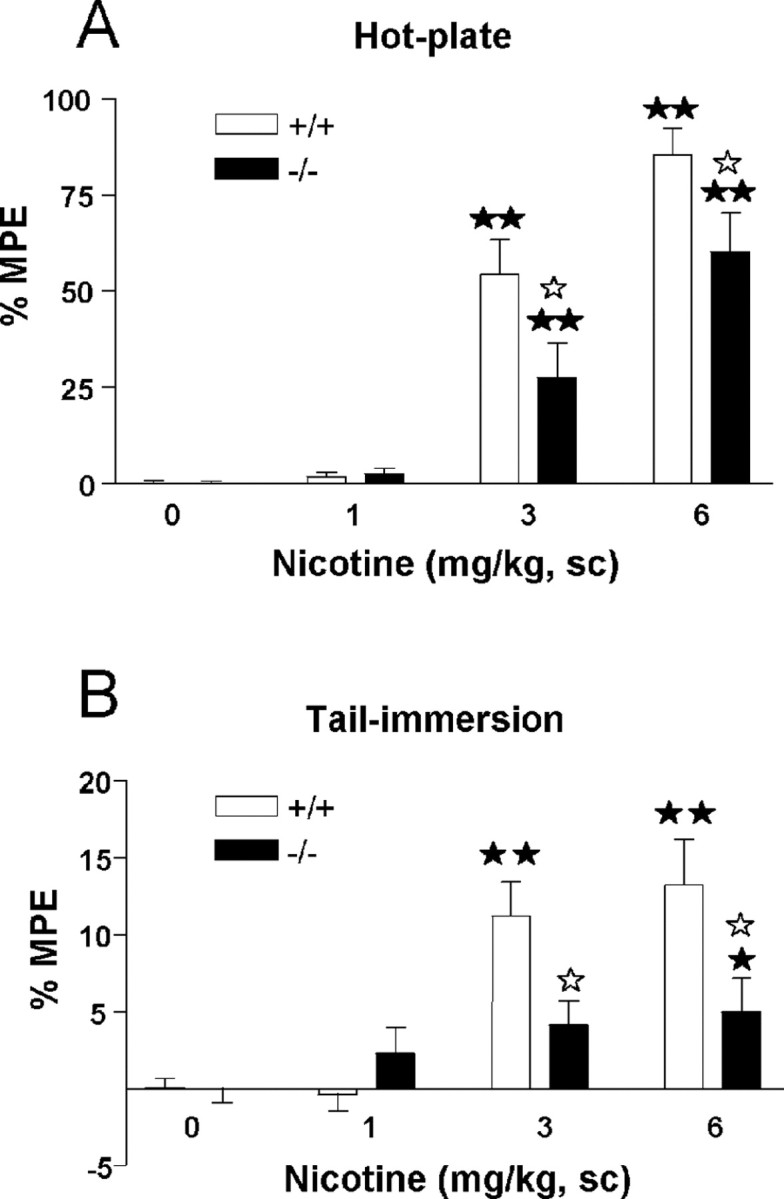
Antinociceptive effects of acute nicotine in preproenkephalin-deficient (black bars) and wild-type (white bars) mice. Antinociceptive responses in the hot-plate (A) and tail-immersion (B) tests were measured at 15 and 16 min, respectively, after nicotine administration (0, 1, 3, and 6 mg/kg, s.c.). Data are expressed as mean ± SEM of percentage of maximum possible effect (MPE) in wild-type (saline, n = 34; nicotine, 1 mg/kg, n = 21; nicotine, 3 mg/kg, n = 19; nicotine, 6 mg/kg, n = 14) and knock-out (saline, n = 33; nicotine, 1 mg/kg, n = 17; nicotine, 3 mg/kg, n = 17; nicotine, 6 mg/kg, n = 15) mice. ★★p < 0.01, when compared with the saline group of the same genotype. ☆p < 0.05, comparison between genotypes (Dunnett's test).
In the tail-immersion test, two-way ANOVA revealed a significant effect of treatment (F(3,162) = 17.75; p < 0.0001) and genotype (F(1,162) = 8.43; p < 0.01) and a significant interaction between treatment and genotype (F(3,162) = 5.85; p < 0.001). Subsequent one-way ANOVA (treatment) indicated a significant effect of treatment in wild-type (F(3,84) = 20.69; p < 0.0001) and knock-out mice (F(3,78) = 2.70; p < 0.05). Post hoc comparisons showed that nicotine induced antinociception in wild-type mice at the doses of 3 and 6 mg/kg (p < 0.01) and knock-out mice at the dose of 6 mg/kg (p < 0.05). Post hoc analysis also revealed a significant difference in the effects induced by 3 and 6 mg/kg nicotine in preproenkephalin knock-out and wild-type mice (p < 0.05) (Fig. 2B).
Nicotine did not produce rewarding responses in the place-preference paradigm in preproenkephalin-deficient mice
One-way ANOVA revealed a similar time spent in the drug-paired compartment during the preconditioning phase in the different groups (F(7,171) = 0.14; NS), ensuring the use of an unbiased procedure (Fig. 3). A significant rewarding effect of nicotine was observed in the place-conditioning paradigm in wild-type but not in mice lacking the preproenkephalin gene. Accordingly, wild-type animals conditioned with a dose of 0.5 mg/kg nicotine (s.c.) spent significantly (p < 0.01) more time in the drug-paired compartment during the testing phase than during the preconditioning phase. In contrast, preproenkephalin-deficient mice receiving such a dose of nicotine spent the same time in the drug-paired compartment during both phases (Fig. 3). The administration of a lower (0.25 mg/kg, s.c.) or a higher (1 mg/kg, s.c.) dose of nicotine did not induce rewarding responses in wild-type and knock-out animals (Fig. 3).
Figure 3.

Rewarding effects of nicotine in preproenkephalin-deficient and wild-type mice on the place-conditioning paradigm. Place conditioning was induced by the administration of saline (Sal) (wild-type mice, n = 39; knock-out mice, n = 37) and 0.25 mg/kg (Nic 0.25) (wild-type mice, n = 15; knock-out mice, n = 14), 0.5 mg/kg (Nic 0.5) (wild-type mice, n = 24; knock-out mice, n = 19), and 1 mg/kg (Nic 1) (wild-type mice, n = 16; knock-out mice, n = 15) of nicotine (s.c.). The figure shows the time spent in the drug-paired compartment during preconditioning (white bars) and test (black bars) phases. All values are expressed as mean ± SEM. ★★p < 0.01, paired two-tailed Student's t test.
Two-way ANOVA calculated for score values of nicotine at the dose of 0.5 mg/kg also indicated a significant main effect of genotype (F(1,115) = 4.47; p < 0.05), without effect of treatment (F(1,115) = 2.75; NS) and a significant interaction between these two factors (F(1,115) = 5.16; p < 0.05). Subsequent one-way ANOVA revealed that nicotine produced a conditioned place preference for the nicotine-assigned compartment in wild-type mice (score values: saline, -26.54 ± 15.89; nicotine, 90.29 ± 29.60) (p < 0.01), whereas no effect was observed in knock-out mice (score values: saline, -21.84 ± 32.30; nicotine, -40.08 ± 40.63) (data not shown).
In an additional experiment, mecamylamine (1 mg/kg) pretreatment blocked the conditioned place preference induced by nicotine (0.5 mg/kg) in wild-type animals, indicating that this effect is mediated by nicotinic acetylcholine receptors. One-way ANOVA revealed a similar time spent in the drug-paired compartment during the preconditioning phase in the different groups (F(3,53) = 0.1072; NS), ensuring the use of an unbiased procedure. A significant rewarding effect of nicotine was only observed in the group receiving saline plus nicotine. Thus, mice treated with saline plus nicotine spent significantly (p < 0.01) more time in the drug-paired compartment during the testing than during the preconditioning phase. In contrast, the other three groups of mice (saline plus saline, mecamylamine plus saline, and mecamylamine plus nicotine) spent the same time in the drug-paired compartment during both phases (Fig. 4). Two-way ANOVA calculated for score values (saline plus saline, -24.04 ± 40.59; mecamylamine plus saline, 41.17 ± 53.02; saline plus nicotine, 144.04 ± 33.00; mecamylamine plus nicotine, 30.35 ± 37.43) indicated no effect of nicotine treatment (F(1,53) = 3.45; NS) or mecamylamine (F(1,53) = 0.33; NS) but a significant interaction between nicotine and mecamylamine treatments (F(1,53) = 4.47; p < 0.05) (data not shown).
Figure 4.

Effects of mecamylamine (1 mg/kg, s.c.) pretreatment on nicotine-induced conditioned place preference (0.5 mg/kg, s.c.) in wild-type animals. The figure shows the time spent in the drug-paired compartment during preconditioning (white bars) and test (black bars) phases. All values are expressed as mean ± SEM. ★★p < 0.01, paired two-tailed Student's t test; n = 13-15 per group.
Nicotine-enhanced extracellular dopamine in the nucleus accumbens is attenuated in preproenkephalin-deficient mice
Basal levels of DA (wild-type, 3.87 ± 0.77; knock-out, 4.78 ± 1.25 pg/15 μl; NS), DOPAC (wild-type, 174.74 ± 40.11; knock-out, 146.69 ± 16.21 pg/15 μl; NS), and HVA (wild-type, 79.20 ± 11.87; knock-out, 88.49 ± 11.96 pg/15 μl; NS) in NAc dialysates were similar in both genotypes. These basal levels are directly related to the physiological dopaminergic transmission because DA, DOPAC, and HVA decreased to undetectable levels 40 min after TTX perfusion and recovered to previous basal levels after TTX was removed.
Nicotine (0.5 mg/kg, s.c.) induced an enhancement of DA extracellular levels in the NAc that was clearly revealed in wild-type mice. A higher dose of nicotine (1 mg/kg, s.c.) did not modify DA efflux in the NAc in both wild-type and mutant mice. Three-way ANOVA revealed a significant main effect of time (F(11,319) = 3.341; p < 0.001) and treatment (F(2,29) = 12.121; p < 0.001), whereas no gene patrimony effect (F(1,29) = 1.284; NS) was observed after nicotine injection. No significant two- or three-way interactions between factors were observed (Fig. 5A). In Figure 5B, AUC values for DA extracellular levels after nicotine (0.5 and 1 mg/kg, s.c.) or saline administration are shown. Two-way ANOVA indicated treatment effect (F(2,29) = 7.293; p < 0.01), no genotype effect (F(1,29) = 1.703; NS), and a significant interaction between these two factors (F(2,29) = 3.172; p < 0.05). Subsequent one-way ANOVA (treatment) revealed a significant effect of nicotine in wild-type mice (F(2,17) = 9.839; p < 0.01) but not in knock-out mice (F(2,12) = 0.535; NS). Post hoc comparisons showed that nicotine induced an enhancement of AUC values for DA extracellular levels in the NAc only in wild-type mice at the dose of 0.5 mg/kg (p < 0.01) when compared with the saline group. A reduction of nicotine effects in AUC values in preproenkephalin mutants when compared with control animals at the dose of 0.5 mg/kg (p < 0.05) was also revealed. These results indicate that the deletion of the preproenkephalin gene impaired the effects of nicotine on the extracellular levels of DA.
Figure 5.
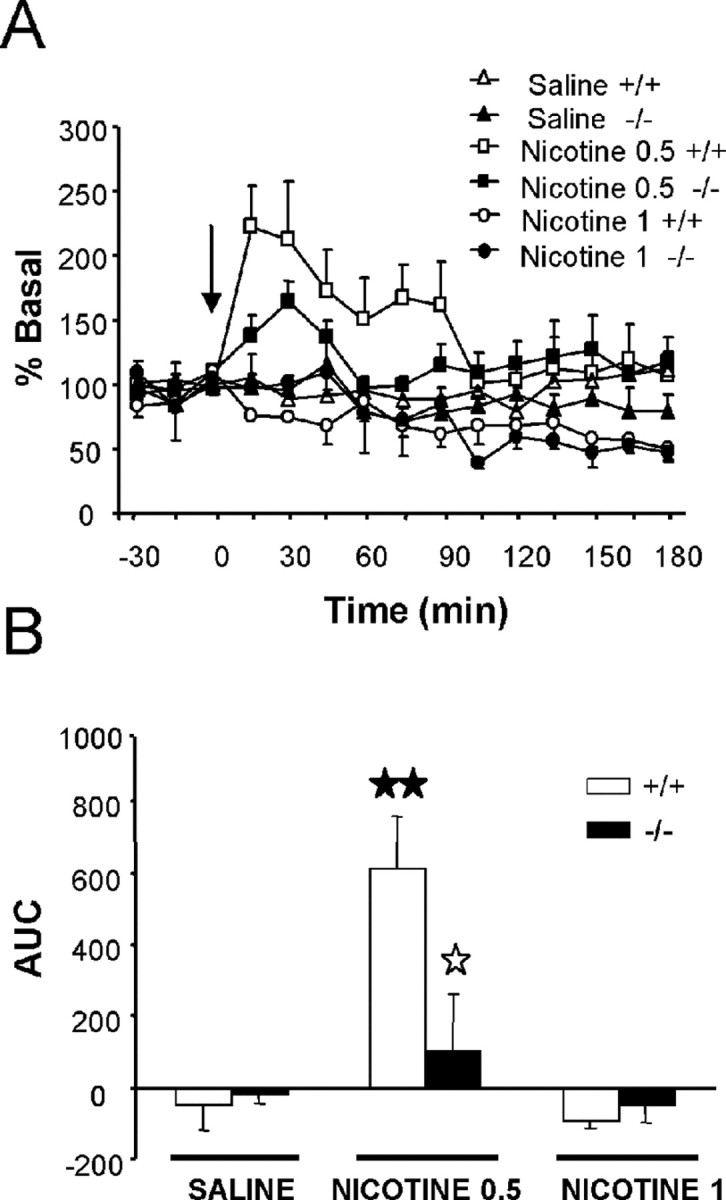
A, Effects of nicotine (0.5 and 1 mg/kg, s.c.) and saline on DA concentrations in dialysates obtained by in vivo microdialysis from the NAc of preproenkephalin-deficient and wild-type mice. The arrow indicates nicotine or saline administration at time 0. Dialysate samples were taken every 15 min during 1.5 h before and 3 h after injection. B, Bar graphs of AUC values for the DA concentration in dialysates after nicotine or saline administration in knock-out (black bars) and wild-type (white bars) mice. All values are expressed as mean ± SEM. ★★p < 0.01, when compared with the saline group of the same genotype; ☆p < 0.05 comparison between genotypes (Dunnett's test).
Nicotine treatment induced a significant increase of DOPAC levels in NAc dialysates in both genotypes, whereas nonsignificant changes were observed in HVA levels. Three-way ANOVA revealed a significant main effect of treatment for DOPAC (F(2,29) = 8.09; p < 0.05) but not for HVA (F(2,21) = 3.00; NS). A significant effect of time was observed for DOPAC (F(11,319) = 7.46; p < 0.01) and HVA (F(11,231) = 2.334; p < 0.05). No effects of gene patrimony were observed for DOPAC and HVA (F(1,29) = 0.235, NS, and F(1,21) = 0.004, NS, respectively). No significant three-way interactions between factors were observed (data not shown).
In an additional experiment, nicotine at the dose of 3 mg/kg (s.c.) induced a decrease of DA extracellular levels in the NAc in wild-type mice (F(12,60) = 4.630; p < 0.01) when compared with basal values. This reduction was significant 165 min (63.46% of reduction regarding the basal value) and 180 min (59.42% of reduction regarding the basal value) (p < 0.05) after nicotine injection (data not shown).
Figure 6 shows a representative coronal section of the mouse brain stained with cresyl violet, illustrating the placement of the microdialysis probe through the NAc. Most probes were located medial to the anterior commissure, between 1.7 and 0.74 mm from bregma, and dorsal to the olfactory tubercule. Mice with the placement of the microdialysis probe outside of the NAc were excluded from the study.
Figure 6.

Representative coronal section (20 μm) of the mouse brain stained with cresyl violet illustrating the placement of the probe in the nucleus accumbens (bregma, +0.98) (A). In B, a diagram showing the representative probe placements between bregma +1.70 and +0.86 mm.
Somatic expression of nicotine withdrawal is attenuated in preproenkephalin-deficient mice
During the behavioral observation performed before mecamylamine administration, no somatic signs of withdrawal were observed in any group of animals. After mecamylamine injection, nicotine withdrawal syndrome was manifested by the presence of a variety of somatic signs in mice receiving chronic nicotine perfusion, as reported previously (Castañé et al., 2002) (Table 1). The intensity of the withdrawal syndrome was decreased in mice lacking the preproenkephalin gene, as shown by two-way ANOVA calculated for global withdrawal scores (treatment: F(1,52) = 19.66, p < 0.0001; genotype: F(1,52) = 5.08, p < 0.05; interaction: F(1,52) = 5.24, p < 0.05) (Fig. 6A). One-way ANOVA revealed significant increases in the global withdrawal scores in wild-type (p < 0.01) and knock-out (p < 0.05) mice receiving chronic nicotine compared with saline-treated controls. A significant decrease of withdrawal was observed in nicotine-treated knock-out mice compared with nicotine-dependent wild-type animals (p < 0.05) (Fig. 7A). The most prominent withdrawal sign attenuated in mutant mice was the number of scratches, as revealed by two-way ANOVA (treatment: F(1,52) = 4.45, p < 0.05; genotype: F(1,52) = 10.95, p < 0.01; interaction: F(1,52) = 4.98, p < 0.05) (Fig. 7B) and subsequent one-way ANOVA (p < 0.05, comparison between nicotine- and saline-treated wild-type mice; p < 0.01, comparison between nicotine-treated wild-type and mutant mice).
Table 1.
Mecamylamine-precipitated nicotine withdrawal in wild-type and preproenkephalin knock-out mice
|
|
Wild-type mice |
Knock-out mice |
||||
|---|---|---|---|---|---|---|
|
|
Saline |
Nicotine |
Saline |
Nicotine |
||
| Locomotor activity | 9.40 ± 0.58 | 7.33 ± 0.67 | 6.92 ± 1.00 | 5.57 ± 0.72 | ||
| Body tremor | 0.00 ± 0.00 | 0.33 ± 0.17 | 0.42 ± 0.25 | 0.29 ± 0.21 | ||
| Genital licks | 1.90 ± 0.62 | 1.22 ± 0.26 | 1.08 ± 0.31 | 1.29 ± 0.24 | ||
| Ptosis | 0.30 ± 0.30 | 1.33 ± 0.41 | 1.58 ± 0.48 | 1.43 ± 0.46 | ||
| Wet dog shakes | 2.40 ± 0.71 | 2.00 ± 0.45 | 1.00 ± 0.32 | 1.43 ± 0.73 | ||
| Teeth chattering | 0.50 ± 0.22 | 2.89 ± 0.55 | 1.25 ± 0.42 | 1.71 ± 0.43 | ||
| Paw tremor | 2.30 ± 0.71 | 4.67 ± 0.84 | 0.92 ± 0.31 | 2.86 ± 0.85 | ||
| Scratches | 5.07 ± 2.45 | 19.79 ± 6.16 | 1.42 ± 0.66 | 1.00 ± 0.60 | ||
| Piloerection | 0.00 ± 0.00 | 0.33 ± 0.22 | 0.08 ± 0.08 | 0.00 ± 0.20 | ||
| GWS |
8.56 ± 1.35 |
21.21 ± 3.06 |
8.62 ± 1.03 |
12.66 ± 1.25 |
||
Data are expressed as mean ± SEM. Two-way ANOVA revealed a significant effect of nicotine treatment for teeth chattering, paw tremor, global withdrawal score (GWS) (p < 0.01), locomotor activity, and scratches (p < 0.05).
Figure 7.

Mecamylamine-precipitated nicotine withdrawal in preproenkephalin-deficient and wild-type mice. Abstinence was precipitated by acute mecamylamine administration (1 mg/kg, s.c.) after 6 d of nicotine perfusion (25 mg/kg/d) by using subcutaneous minipumps. A global withdrawal score was calculated for each animal by giving each individual sign a relative weight (A). A decrease in the number of scratches was the main change observed during nicotine withdrawal in mutant mice (B). Data are expressed as mean ± SEM in wild-type (white bars) and knock-out (black bars) mice (n = 12-15 mice for each group). ★p < 0.05; ★★p < 0.01, when compared with the saline group of the same genotype; ☆p < 0.05; ☆☆p < 0.01, comparison between genotypes (one-way ANOVA).
Discussion
In this study, we show that the effects induced by nicotine on antinociception, reward, and DA extracellular levels in the NAc as well as nicotine withdrawal syndrome precipitated by mecamylamine are reduced in preproenkephalin knock-out mice. These data are in agreement with our recent findings using mice lacking μ-opioid receptors (Berrendero et al., 2002) and identify a family of endogenous opioid peptides that play a major role in these nicotine responses, presumably by activating μ-opioid receptors.
We first analyzed the acute effects induced by nicotine on antinociception and locomotor activity in preproenkephalin knock-out mice. Nicotine has been shown widely to produce antinociception in animal models, and the therapeutic potential of nAChR agonists in pain relief is currently being evaluated (Decker et al., 2004). Nicotine-induced hypolocomotion was unaffected by the enkephalin mutation, suggesting that endogenous enkephalins do not play a relevant role in the acute locomotor effects of nicotine. However, nicotine-induced antinociception in the tail-immersion and hot-plate tests was significantly decreased in preproenkephalin knock-out mice. The participation of opioid mechanisms in the antinociceptive responses elicited by nicotine has been proposed previously by using different pharmacological and molecular approaches. Thus, the nonselective opioid antagonist naloxone blocked the antinociceptive effects of nicotine in some mouse models (Tripathi et al., 1982; Zarrindast et al., 1997). In the same way, the antinociception induced by morphine was enhanced in a dose-dependent manner by nicotine stimulation of supraspinal and spinal nAChRs (Suh et al., 1996a,b). Tolerance to nicotine-induced antinociception after chronic nicotine administration was accompanied by a decrease of met-enkephalin levels and an upregulation of μ-opioid receptors in the striatum (Wewers et al., 1999), suggesting compensation by these receptors for the lower amount of opioid peptides occurred during nicotine tolerance. The specific involvement of μ-opioid receptors in this nicotine-opioid interaction has been demonstrated recently, as revealed by the attenuation of nicotine antinociception in μ-opioid receptor knock-out mice (Berrendero et al., 2002). The present findings suggest that nicotine could enhance the levels of endogenous opioid peptides derived from preproenkephalin, which would participate in the antinociceptive effects induced by nicotine, presumably by stimulating μ-opioid receptors. In agreement, an increase of preproenkephalin mRNA expression in specific brain regions, such as striatum and hippocampus, has been reported after acute nicotine administration (Dhatt et al., 1995; Houdi et al., 1998; Isola et al., 2002). Furthermore, opioid receptors and different subunits for nAChRs involved in nicotine antinociception, such as α4 and β2, overlap in several neuronal regions involved in the control of pain (Mansour et al., 1995; Marubio et al., 1999). Nevertheless, other neurotransmitters apart from opioid peptides also seem to participate in the antinociceptive effects of nicotine (Iwamoto and Marion, 1993; Rogers and Iwamoto, 1993; Cordero-Erausquin and Changeux, 2001).
The rewarding effects of nicotine were investigated in preproenkephalin-deficient mice by using the place-conditioning paradigm. In our experimental conditions, nicotine induced a significant place preference in wild-type but not in preproenkephalin-deficient animals when administered at the dose of 0.5 mg/kg. The administration of higher (1 mg/kg) or lower (0.25 mg/kg) doses of nicotine was ineffective in both genotypes, which excludes a possible shift of the dose-response curve of nicotine in preproenkephalin knock-out mice. In agreement, nicotine-induced conditioned place preference has also been reported previously in rodents (Risinger and Oakes, 1995; Berrendero et al., 2002; Castañé et al., 2002) but only under a narrow range of doses when systemically administered. Indeed, similar ineffective (0.25 and 1 mg/kg) and effective (0.5 mg/kg) doses of nicotine have been reported previously in the mouse place-conditioning paradigm (Risinger and Oakes, 1995; Berrendero et al., 2002; Castañé et al., 2002). Local nicotine microinjection into the ventral tegmental area (VTA) produces conditioned place preference in a wider range of doses (Laviolette and van der Kooy, 2003). Therefore, these results clearly reveal that opioid peptides derived from preproenkephalin are involved in the modulation of nicotine-rewarding properties. Nicotine, like most drugs of abuse, activates the mesocorticolimbic dopaminergic system and increases extracellular DA levels in the NAc (Di Chiara and Imperato, 1988; Pontieri et al., 1996; Zocchi et al., 2003), a feature that has been related to its rewarding properties. Accordingly with our behavioral approach, in vivo microdialysis experiments showed that the enhancement in DA efflux in the NAc induced by nicotine (0.5 mg/kg) in wild-type mice was significantly attenuated in preproenkephalin knock-out animals. The administration of a higher dose of nicotine (1 mg/kg) did not modify DA efflux in the NAc in both wild-type and mutant mice. Two previous microdialysis studies have evaluated the effects of nicotine on mesolimbic dopaminergic activity in mice. A high dose of nicotine (0.8 mg/kg) produced a slight increase in DA levels but only when the probe was located in the shell region of the nucleus accumbens (Zocchi et al., 2003). In the second study, nicotine at the dose of 1 mg/kg increased DA levels in an area shown as the ventral pallidum (Marubio et al., 2003).
The activation of nAChRs located in the VTA has been reported to play a crucial function in the rewarding effects of nicotine. Thus, the local infusion of the nAChR antagonist mecamylamine into the VTA blocks the increase induced by nicotine in the extracellular levels of DA in the NAc (Nisell et al., 1994). This effect is not prevented when mecamylamine is directly infused in the NAc. Thus far, three different types of cells have been reported to express nAChRs in the VTA: DA neurons, GABA neurons, and presynaptic glutamatergic terminals from the medial prefrontal cortex (Mansvelder and McGehee, 2002). Moreover, possible localization of nAChRs in other VTA neurons cannot be excluded at the present moment. Nicotine activation of α7 nAChRs localized on glutamatergic afferents produces an increase in glutamate levels and subsequent stimulation of NMDA receptors found on VTA DA neurons (Schilstrom et al., 1998; Nomikos et al., 2000). Cholinergic and glutamatergic inputs from the pedunculopontine tegmental nucleus (PPTg) also seem to participate in nicotine reward, because PPTg lesions reduce nicotine self-administration (Lança et al., 2000; Picciotto and Corrigall, 2002). Besides these excitatory inputs, DA neurons in the VTA are under inhibitory control, mainly by GABAergic inputs. GABA neurons are also activated by nicotine, but α4 β2 nAChRs on these cells tend to desensitize rapidly during nicotine exposure (Mansvelder et al., 2002). Therefore, the final activation of the mesolimbic dopaminergic activity by nicotine depends on a functional balance between the excitatory and inhibitory inputs on the VTA DA neurons, in addition to the direct nicotine effect on the DA neurons themselves (for reviews, see Mansvelder et al., 2003; Laviolette and van der Kooy, 2004). As mentioned previously, our results indicate that endogenous enkephalins play an important role in mediating the rewarding properties of nicotine. Enkephalinergic interneurons as well as enkephalinergic inputs from the NAc are present in the VTA (Garzón and Pickel, 2002). Therefore, we suggest that nicotine could induce, by a direct mechanism or indirectly through glutamate transmission (Isola et al., 2000), a release of endogenous opioid peptides derived from preproenkephalin. This release would inhibit GABAergic neurotransmission through presynaptic μ-opioid receptor activation (Berrendero et al., 2002) located on the terminals of GABA cells (Bergevin et al., 2002). This opioid-dependent mechanism would be important to remove the inhibitory inputs on VTA DA neurons, finally increasing their firing rate in vivo (Fig. 8). The elucidation of these neurobiological mechanisms is crucial for a better understanding of the differential responsiveness of human nicotine addicts to opioid antagonists (Modesto-Lowe and Van Kirk, 2002). However, other neuroanatomical pathways and neurotransmitters seem to be involved also in the rewarding properties of nicotine (Laviolette and van der Kooy, 2004).
Figure 8.

Diagram showing the possible role of the endogenous enkephalinergic system in the modulation of nicotine positive-rewarding effects. Nicotine exposure could induce the release of endogenous enkephalins in the VTA by a direct mechanism or indirectly through glutamate transmission. The subsequent activation by these opioid peptides of μ-opioid receptors located in GABAergic neurons would remove the inhibitory effect that these GABAergic cells exert on VTA DA neurons, thus contributing to a net increase of DA release in the NAc. Ach, Acetylcholine; ENK, enkephalins; GLUT, glutamate; PFC, prefrontal cortex; PFC, prefrontal cortex; (-) indicates inhibition; (+) indicates stimulation. The arrows indicate the main possible sites of action of nicotine to explain its modulatory effects on opioid transmission.
Nicotine physical dependence was also attenuated in mice lacking the preproenkephalin gene. Common mechanisms underlying opiate and nicotine dependence have been proposed by different studies, although serious controversies have been raised concerning the interpretation of these previous pharmacological results. First, the somatic signs of nicotine withdrawal in rodents resemble those seen in opioid withdrawal (Malin et al., 1993; Hildebrand et al., 1997). In addition, somatic manifestations of nicotine abstinence were precipitated in nicotine-dependent rats by naloxone administration (Malin et al., 1993; Adams and Cicero, 1998; Carboni et al., 2000), although other investigators failed to reproduce this latter effect (Watkins et al., 2000). Also, in humans, naloxone administration has been reported to induce somatic signs of nicotine withdrawal in heavy chronic smokers (Krishnan-Sarin et al., 1999). However, these effects could be a result of, at least in part, direct blockade of nAChRs, because this opioid antagonist was found to bind nAChRs (Tome et al., 2001). Besides these pharmacological data, there are molecular studies showing a possible role of the endogenous opioid system in nicotine abstinence. Thus, an increase in preproenkephalin mRNA levels was found in both striatum and hippocampus of rats during nicotine withdrawal (Houdi et al., 1998) as well as in the striatum of nicotine-abstinent mice (Isola et al., 2002). An additional advance to clarify this controversy was recently reported by using μ-opioid receptor knock-out mice. Indeed, the somatic expression of the nicotine withdrawal syndrome was attenuated by the deletion of μ-opioid receptors (Berrendero et al., 2002). The present findings clearly indicate that endogenous opioid peptides derived from preproenkephalin would be responsible for such an activation of μ-opioid receptors during nicotine dependence. Therefore, this opioid component could play an important role in maintaining smoking behavior under conditions of nicotine deprivation, such as attempted smoking cessation (Malin, 2001). The endogenous opioid system could also participate in the dysphoric effects of nicotine abstinence, because naloxone produces conditioned place aversion in chronically nicotine-treated rodents (Ise et al., 2000; Watkins et al., 2000).
In conclusion, this study shows that opioid peptides derived from preproenkephalin participate in the effects of nicotine on antinociception, reward, the extracellular levels of DA in the NAc, and the somatic expression of nicotine withdrawal. However, most of the behavioral and biochemical effects of nicotine evaluated here were attenuated but not abolished in preproenkephalin knock-out mice, which raises the possibility that other neurotransmitters different from endogenous opioid peptides could also be involved in these nicotine responses. The present results identify a particular family of endogenous opioid peptides involved in the addictive properties of nicotine and provide new information to better understand the mechanisms involved in nicotine addiction, which could help to develop a more rational therapeutic strategy.
Footnotes
This work was supported by National Institute on Drug Abuse-National Institutes of Health Grant 1RO1DA016768, the Human Frontier Science Program Organization (RG0077/2000-B), Generalitat de Catalunya (Distinción de la Generalitat and 2002SGR00193), and Fondo de Investigación Sanitario (FIS) Grants 03/0305, G03/005, and C03/06. P.R. was supported by an FIS investigator contract. V.M. is a postdoctoral fellow supported by Fundación Carolina. F.B. is a researcher supported by the Ramón y Cajal Program of Ministerio de Ciencia y Tecnología.
Correspondence should be addressed to Rafael Maldonado at the above address. E-mail: rafael. maldonado@upf.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251103-10$15.00/0
References
- Adams ML, Cicero TJ (1998) Nitric oxide mediates mecamylamine- and naloxone-precipitated nicotine withdrawal. Eur J Pharmacol 345: R1-R2. [DOI] [PubMed] [Google Scholar]
- Bergevin A, Girardot D, Bourque MJ, Trudeau LE (2002) Presynaptic μ-opioid receptors regulate a late step of the secretory process in rat ventral tegmental area GABAergic neurons. Neuropharmacology 42: 1065-1078. [DOI] [PubMed] [Google Scholar]
- Berrendero F, Kieffer BL, Maldonado R (2002) Attenuation of nicotine-induced antinociception, rewarding effects, and dependence in μ-opioid receptor knock-out mice. J Neurosci 22: 10935-10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni E, Bortone L, Giua C, Di Chiara G (2000) Dissociation of physical abstinence signs from changes in extracellular dopamine in the nucleus accumbens and in the prefrontal cortex of nicotine dependent rats. Drug Alcohol Depend 58: 93-102. [DOI] [PubMed] [Google Scholar]
- Castañé A, Valjent E, Ledent C, Parmentier M, Maldonado R, Valverde O (2002) Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 43: 857-867. [DOI] [PubMed] [Google Scholar]
- Cordero-Erausquin M, Changeux JP (2001) Tonic nicotinic modulation of serotoninergic transmission in the spinal cord. Proc Natl Acad Sci USA 98: 2803-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Erausquin M, Marubio LM, Klink R, Changeux JP (2000) Nicotinic receptor function: new perspectives from knockout mice. Trends Pharmacol Sci 21: 211-217. [DOI] [PubMed] [Google Scholar]
- Covey LS, Glassman AH, Stetner F (1999) Naltrexone effects on short-term and long-term smoking cessation. J Addict Dis 18: 31-40. [DOI] [PubMed] [Google Scholar]
- Dani JA (2001) Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry 49: 166-174. [DOI] [PubMed] [Google Scholar]
- Dani JA, De Biasi M (2001) Cellular mechanisms of nicotine addiction. Pharm Biochem Behav 70: 439-446. [DOI] [PubMed] [Google Scholar]
- Davenport KE, Houdi AA, Van Loon GR (1990) Nicotine protects against μ-opioid receptor antagonism by β-funaltrexamine: evidence for nicotine-induced release of endogenous opioids in brain. Neurosci Lett 113: 40-46. [DOI] [PubMed] [Google Scholar]
- Decker MW, Rueter LE, Bitner RS (2004) Nicotinic acetylcholine receptor agonists: a potential new class of analgesics. Curr Top Med Chem 4: 369-384. [DOI] [PubMed] [Google Scholar]
- Dhatt RK, Gudehithlu KP, Wemlinger TA, Tejwani GA, Neff NH, Hadjiconstantinou M (1995) Preproenkephalin mRNA and methionine-enkephalin content are increased in mouse striatum after treatment with nicotine. J Neurochem 64: 1878-1883. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzón M, Pickel VM (2002) Ultrastructural localization of enkephalin and μ-opioid receptors in the rat ventral tegmental area. Neuroscience 114: 461-474. [DOI] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D (1975) The trapezoid rule. In: Pharmacokinetics (Gibaldi M, Perrier D, eds), pp 293-296. New York: Marcel Dekker.
- Hildebrand BE, Nomikos GG, Bondjers C, Nidell M, Svensson TH (1997) Behavioral manifestations of the nicotine abstinence syndrome in the rat: peripheral versus central mechanisms. Psychopharmacology 129: 348-356. [DOI] [PubMed] [Google Scholar]
- Houdi AA, Dasgupta R, Kindy MS (1998) Effect of nicotine use and withdrawal on brain preproenkephalin A mRNA. Brain Res 799: 257-263. [DOI] [PubMed] [Google Scholar]
- Ise Y, Narita M, Nagase H, Suzuki T (2000) Modulation of opioidergic system on mecamylamine-precipitated nicotine-withdrawal aversion in rats. Psychopharmacology 151: 49-54. [DOI] [PubMed] [Google Scholar]
- Isola R, Duchemin AM, Tejwani GA, Neff NH, Hadjiconstantinou M (2000) Glutamate receptors participate in the nicotine-induced changes of metenkephalin in striatum. Brain Res 878: 72-78. [DOI] [PubMed] [Google Scholar]
- Isola R, Zhang H, Duchemin AM, Tejwani GA, Neff NH, Hadjiconstantinou M (2002) Met-enkephalin and preproenkephalin mRNA changes in the striatum of the nicotine abstinence mouse. Neurosci Lett 325: 67-71. [DOI] [PubMed] [Google Scholar]
- Iwamoto ET, Marion L (1993) Adrenergic, serotonergic and cholinergic components of nicotinic antinociception in rats. J Pharmacol Exp Ther 265: 777-789. [PubMed] [Google Scholar]
- Karras A, Kane JM (1980) Naloxone reduces cigarette smoking. Life Sci 27: 1541-1545. [DOI] [PubMed] [Google Scholar]
- Konig M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A (1996) Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature 383: 535-538. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (2001) Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97-129. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Rosen MI, O'Malley SS (1999) Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56: 663-668. [DOI] [PubMed] [Google Scholar]
- Lança AJ, Adamson KL, Coen KM, Chow BL, Corrigal WA (2000) The pedunculopontine tegmental nucleus and the role of cholinergic neurons in nicotine self-administration in the rat: a correlative neuroanatomical and behavioral study. Neuroscience 96: 735-742. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D (2003) Blockade of mesolimbic dopamine transmission dramatically increases sensitivity to the rewarding effects of nicotine in the ventral tegmental area. Mol Psychiatry 8: 50-59. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D (2004) The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci 5: 55-65. [DOI] [PubMed] [Google Scholar]
- Lerner AG, Oyffe I, Sigal M (1998) Naltrexone-induced reduction of tobacco intake. J Clin Psychiatry 59: 30-31. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E (1997) Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature 388: 586-589. [DOI] [PubMed] [Google Scholar]
- Malin DH (2001) Nicotine dependence. Studies with a laboratory model. Pharmacol Biochem Behav 70: 551-559. [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Carter VA, Cunningham JS, Wilson OB (1993) Naloxone precipitates nicotine abstinence syndrome in the rat. Psychopharmacology 112: 339-342. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci 18: 22-29. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS (2002) Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol 53: 606-617. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS (2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33: 905-919. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, De Rover M, McGehee DS, Brussaard AB (2003) Cholinergic modulation of dopaminergic reward areas: upstream and down-stream targets of nicotine addiction. Eur J Pharmacol 480: 117-123. [DOI] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere M, de Kerchove d'Exaerde A, Huchet M, Damaj MI, Changeux JP (1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398: 805-810. [DOI] [PubMed] [Google Scholar]
- Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, McIntosh JM, Rossi F, Champtiaux N, Zoli M, Changeux JP (2003) Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 17: 1329-1337. [DOI] [PubMed] [Google Scholar]
- Modesto-Lowe V, Van Kirk J (2002) Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmacol 10: 213-227. [DOI] [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH (1994) Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacol Toxicol 75: 348-352. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Schilstrom B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH (2000) Role of α7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res 113: 97-103. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (1997) The mouse brain in stereotaxic coordinates. San Diego: Academic.
- Picciotto MR, Corrigall WA (2002) Neuronal systems underlying behaviors related to nicotine addiction: neural circuits and molecular genetics. J Neurosci 22: 3338-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Orzi F, Di Chiara G (1996) Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382: 255-257. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Oakes RA (1995) Nicotine-induced conditioned place preference and conditioned place aversion in mice. Pharmacol Biochem Behav 51: 457-461. [DOI] [PubMed] [Google Scholar]
- Rogers DT, Iwamoto ET (1993) Multiple spinal mediators in parenteral nicotine-induced antinociception. J Pharmacol Exp Ther 267: 341-349. [PubMed] [Google Scholar]
- Schilstrom B, Nomikos GG, Nisell M, Hertel P, Svensson TH (1998) N-methyl-d-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience 82: 781-789. [DOI] [PubMed] [Google Scholar]
- Suh HW, Song DK, Choi SR, Chung KM, Kim YH (1996a) Nicotine enhances morphine- and β-endorphin-induced antinociception at the supraspinal level in the mouse. Neuropeptides 30: 479-484. [DOI] [PubMed] [Google Scholar]
- Suh HW, Song DK, Lee KJ, Choi SR, Kim YH (1996b) Intrathecally injected nicotine enhances the antinociception induced by morphine but not β-endorphin, d-Pen2,5-enkephalin and U50,488H administered intrathecally in the mouse. Neuropeptides 30: 373-378. [DOI] [PubMed] [Google Scholar]
- Sutherland G, Stapleton JA, Rusell MA, Feyerabend C (1995) Naltrexone, smoking behaviour and cigarette withdrawal. Psychopharmacology 120: 418-425. [DOI] [PubMed] [Google Scholar]
- Tome AR, Izaguirre V, Rosario LM, Cena V, Gonzalez-Garcia C (2001) Naloxone inhibits nicotine-induced receptor current and catecholamine secretion in bovine chromaffin cells. Brain Res 903: 62-65. [DOI] [PubMed] [Google Scholar]
- Tripathi HL, Martin BR, Aceto MD (1982) Nicotine-induced antinociception in rats and mice: correlation with nicotine brain levels. J Pharmacol Exp Ther 221: 91-96. [PubMed] [Google Scholar]
- Watkins SS, Stinus L, Koob GF, Markou A (2000) Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292: 1053-1064. [PubMed] [Google Scholar]
- Wewers ME, Dhatt R, Tejwani GA (1998) Naltrexone administration affects ad libitum smoking behavior. Psychopharmacology 140: 185-190. [DOI] [PubMed] [Google Scholar]
- Wewers ME, Dhatt R, Snively TA, Tejwani GA (1999) The effect of chronic administration of nicotine on antinociception, opioid receptor binding and met-enkephalin levels in rats. Brain Res 822: 107-113. [DOI] [PubMed] [Google Scholar]
- Wonnacott S (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20: 92-98. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Pazouki M, Nassiri-Rad S (1997) Involvement of cholinergic and opioid receptor mechanisms in nicotine-induced antinociception. Pharmacol Toxicol 81: 209-213. [DOI] [PubMed] [Google Scholar]
- Zocchi A, Girlanda E, Varnier G, Sartori I, Zanetti L, Wildish GA, Lennon M, Mugnaini M, Heidbreder CA (2003) Dopamine responsiveness to drugs of abuse: a shell-core investigation in the nucleus accumbens of the mouse. Synapse 50: 293-302. [DOI] [PubMed] [Google Scholar]