

Abstract
Rapid trafficking of glutamate receptors contributes importantly to synaptic plasticity, but whether similar trafficking extends to other ionotropic receptors is unknown. Nicotinic acetylcholine receptors containing α7 subunits are widely expressed in the nervous system and allow calcium influx. Because of this, α7-containing receptors regulate diverse events, depending on the signaling pathways available. We find that the receptors codistribute with target soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) postsynaptically and that nicotinic stimulation rapidly induces SNARE-dependent vesicular endocytosis accompanied by receptor internalization. At the same time, a SNARE-dependent process recruits receptors to the cell surface from internal pools. Overall, the trafficking does not markedly change the number of surface receptors or their combined whole-cell response to nicotine. SNARE-dependent trafficking is needed, however, for the receptors to remain capable of activating the transcription factor cAMP response element-binding protein and attendant gene expression when repeatedly challenged. Thus, trafficking appears to be essential for maintaining functional coupling between α7-receptor responses and downstream signaling.
Keywords: nicotinic, receptor, synapse, trafficking, ciliary, postsynaptic, spine, SNARE
Introduction
Rapid trafficking of glutamate receptors in the postsynaptic neuron is an important vehicle for synaptic plasticity. Receptor trafficking can generate long-term potentiation by quickly increasing the number of postsynaptic AMPA receptors. Similarly, long-term depression can result from decreases in AMPA receptors brought on by rapid trafficking. Receptor transport in these cases can occur between surface and intracellular pools or between synaptic and extrasynaptic pools on the cell surface. Elaborate intracellular and transmembrane machinery including components such as stargazin, postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1 (PDZ)-containing proteins, and soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor (SNARE)/NSF-dependent processes promote the trafficking (Malinow and Malenka, 2002; Song and Huganir, 2002; Bredt and Nicoll, 2003; Tomita et al., 2003).
Like glutamate receptors, nicotinic acetylcholine receptors (nAChRs) are cation-selective channels capable of mediating excitatory transmission. One of the most abundant and widely expressed is a species composed of α7 subunits (Broide and Leslie, 1999; Berg and Conroy, 2002). Such receptors (α7-nAChRs) reach the highest levels in early postnatal life (Adams et al., 2002; Falk et al., 2003), possibly providing early forms of excitation and help with maturation in the nervous system. The α7-nAChR combines features of several glutamate receptor subtypes. It rapidly desensitizes like many AMPA receptors and has a high relative permeability to calcium, like NMDA receptors (Bertrand et al., 1993; Seguela et al., 1993). This latter feature enables α7-nAChRs to influence numerous calcium-dependent events, including gene expression (Chang and Berg, 2001; Hu et al., 2002), as do NMDA receptors (Greenberg and Ziff, 2001; Vanhoutte and Bading, 2003). Also like NMDA receptors, α7-nAChRs and related subtypes codistribute with postsynaptic PDZ-containing proteins of the PSD-95/synapse-associated protein 90 family (Conroy et al., 2003; Parker et al., 2004).
In the CNS, α7-nAChRs are found both presynaptically and postsynaptically at a variety of synapses (McGehee et al., 1995; Gray et al., 1996; Alkondon et al., 1997, 1998; Frazier et al., 1998; Hefft et al., 1999; Jones et al., 1999; Fabian-Fine et al., 2001; Hatton and Yang, 2002; Levy and Aoki, 2002). Studies on hippocampal neurons in culture indicate the receptors are initially localized, in part, on filopodial-like extensions and on spine-like structures (Kawai et al., 2002). High levels of α7-nAChRs are found on chick ciliary ganglion (CG) neurons, where they are concentrated on somatic spines and mediate synaptically driven calcium-dependent regulation of signaling and gene expression (Liu and Berg, 1999; Shoop et al., 1999, 2001; Chang and Berg, 2001). These findings raise the question of whether α7-nAChRs are subject to the kinds of trafficking found for glutamate receptors.
We show here that α7-nAChRs on somatic spines undergo rapid activity-driven, calcium-dependent trafficking that requires functional SNAREs. The trafficking produces vesicular endocytosis and internalization of surface α7-nAChRs. Coordinated with the internalization is a recruitment of internal α7-nAChRs to the cell surface. Although the trafficking does not produce obvious changes in the number of surface receptors or their whole-cell response, it is required for sustained coupling with downstream signaling pathways. Without it, the receptors lose their ability to activate the transcription factor cAMP response element-binding protein (CREB) when reexposed to agonist. These results suggest a novel postsynaptic role for SNARE-dependent trafficking in neurons.
Materials and Methods
Cell cultures. Dissociated embryonic day 8 (E8) CG neurons were grown in culture for 6-8 d on glass coverslips coated with poly-d-lysine, fibronectin, and lysed fibroblasts as described previously (Nishi and Berg, 1981; Zhang et al., 1994). Dissociated E14 CG neurons were prepared and maintained for 1-4 h on glass coverslips lacking the lysed fibroblasts. For binding experiments, cells were plated in plastic dishes coated with poly-d-lysine. Drug treatments initiated before nicotine exposure were as follows, unless indicated otherwise: d-tubocurarine (dTC), methyllycaconitine (MLA), KN93, and sucrose for 30 min; thapsigargin for 15 min; brefeldin A (BFA) and N-ethylmaleimide (NEM) for 1 h; botulinum toxins C1, D, and the heat-inactivated versions for 2 h.
Transfections. CG neurons in cell culture for 1 h were transfected as described previously (Conroy et al., 2003). The constructs encoded either green fluorescent protein (GFP:pEGFP-N1; Clontech, Palo Alto, CA) or GFP fused to α7-nAChR on the receptor C terminus (α7-nAChR-GFP). The α7-nAChR sequence was obtained by PCR from a chick α7-nAChR cDNA/pCDNA3.1 construct using 5′-AAACTCGAGATGGGCCTCCGGGCGCTGATG and 3′-AAAGGATCCCGAGCAAAATCTTTAGAGACAG as the primers. PCR products of the appropriate size were cloned into the pEGFP-N1 vector and sequenced. Usually 1-2% of the neurons were successfully transfected, displaying significant levels of the protein 1 week later in culture.
Fluorescence imaging. Most fluorescence imaging was performed on freshly dissociated E14 CG neurons using techniques and reagents described in detail previously (Shoop et al., 1999; Kawai et al., 2002; Conroy et al., 2003). The following primary antibodies and toxins were used: Alexa488-αBungarotoxin (AlexaαBgt; 200 nm; Molecular Probes, Eugene, OR) and rhodamine-αBgt for α7-nAChRs; anti-SNAP-25 monoclonal antibody (mAb) SP12 and anti-syntaxin 1 mAb HPC1 (1:250 dilutions; Santa Cruz Biotechnology, Santa Cruz, CA); anti-SV2 mAb (purified from a hybridoma line; a gift from Kathleen M. Buckley, Harvard Medical School, Boston, MA); mAb 35 for heteromeric α3-containing nicotinic receptors (α3*-nAChRs); rhodamine-phalloidin for filamentous actin (F-actin; 1:500 dilution; Molecular Probes); and rabbit anti-horseradish peroxidase (HRP) cyanine 3 (Cy3) antibody (1: 250; Jackson ImmunoResearch, West Grove, PA). Secondary antibodies were used as described previously (Conroy et al., 2003).
For dye-uptake experiments, E14 neurons were stimulated at room temperature for the indicated time with 10 μm nicotine in the presence of either 5 μm FM4-64 or 10 mg/ml HRP in Ringer's solution containing (in mm) 150 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES, pH 7.4, unless indicated otherwise. The cells were then washed three times with Ringer's solution, stained with AlexaαBgt in calcium-free Ringer's solution for 5 min, and washed three more times. The cells were then viewed immediately in the case of FM4-64 staining. For HRP staining, the cells were viewed after fixing with 2% (w/v) paraformaldehyde (PFA) for 5-10 min, then washed three times with PBS, incubated with anti-HRP Cy3 antibody for 1 h at room temperature in PBS solution containing 0.25% Triton X-100 and 5% donkey serum, and then washed three times with PBS. Randomly selected cells were scored positive for colocalization of FM4-64 and AlexaαBgt staining if they had at least three clear sites at which the two stains codistributed. Cells were viewed through a 63×, 1.4 numerical aperture oil objective on a Axiovert microscope (Zeiss, Oberkochen, Germany) equipped with a CCD camera and digital imaging using Slidebook deconvolution software (Intelligent Imaging Innovations, Santa Monica, CA). Reconstructed images were generated from z-axis stacks of 0.3-μm-thick deconvolved optical sections (usually eight sections per stack). Control and experimental images were always taken with the same exposure settings and displayed with the same dynamic range of pixel intensities to facilitate direct comparison.
For experiments in which receptor internalization was to be examined, E8 neurons in culture were transfected with a construct encoding α7-nAChR-GFP and 6 d later were stained with anti-GFP mAb JL-8 (Clontech). After rinsing three times, cells were stimulated with 10 μm nicotine for 10 min at room temperature, washed three more times, and then stripped for 5 min on ice in 0.2 m acetic acid plus 0.5 m sodium chloride. After washing and fixing with 2% PFA for 10 min, the cells were then stained for 1 h with donkey Cy3-conjugated secondary antibody in PBS containing 5% (v/v) normal donkey serum and 0.25% (v/v) Triton X-100. After rinsing three more times with PBS, the cells were viewed with fluorescence microscopy. For experiments examining possible internalization of α3*-nAChRs, freshly dissociated E14 neurons were incubated 10 min with mAb 35 (1:500 dilution) (Conroy et al., 2003), rinsed three times, and treated as described above.
For experiments in which receptor appearance on the cell surface was to be examined, freshly dissociated E14 neurons were stimulated with 10 μm nicotine for 1 min at room temperature, rinsed three times in Ringer's solution and then incubated for 3 min with 4 μm αBgt in Ringer's solution before being rinsed an additional three times, and incubated with 200 nm AlexaαBgt in Ringer's solution for 20 min. After one to two quick rinses in Ringer's solution, the cells were fixed with 2% PFA for 10 min, rinsed three times with PBS, and viewed with fluorescence microscopy. In some cases, the cells were costained with rhodamine-conjugated phalloidin for 1 h at room temperature in PBS containing 5% (v/v) normal donkey serum and 0.25% (v/v) Triton X-100, rinsed, and then viewed with fluorescence microscopy.
Quantification of fluorescence staining was performed with ImagePro software. Threshold conditions for automatic selection of clusters or puncta were that they contain at least 20 contiguous pixels (0.2 μm2) and that their average pixel intensity, after subtracting nonspecific background, be at least a fixed percentage of the maximum value obtained for clusters previously selected visually in the experiment. Thresholds of 10 and 15% of maximum were chosen for α7-nAChR and syntaxin 1 clusters, respectively, in colabeling experiments; thresholds of 16 and 24% of maximum were chosen for α7-nAChR and FM4-64 puncta, respectively, in dye-uptake experiments. For experiments quantifying α7-nAChR reappearance on the surface, threshold was taken to be 2% of the maximum seen for α7-nAChR clusters in untreated control cultures. Thresholds were chosen to exceed by at least threefold nonspecific background, which varied with the probe and conditions.
Electron microscopy. Freshly dissociated E14 CG neurons were incubated with 10 μm nicotine at room temperature for 5-10 min in the presence of 10 mg/ml HRP in Ringer's solution (in mm: 150 NaCl, 3 KCl, 5 CaCl2, 2 MgCl2, 17 glucose, and 10 HEPES, pH 7.2), rinsed 5 min in Ringer's solution and then in PBS, and fixed in 2% (v/v) glutaraldehyde/2% formaldehyde (Electron Microscopy Sciences, Fort Washington, PA). After staining with Vector VIP (Vector Laboratories, Burlingame, CA) for 15 min according to the manufacturer's instructions, the cells were dehydrated in an ethanol series and then embedded in Embed 812 resin (Electron Microscopy Sciences). Sections (70 nm) were cut on a Reichert ultracut E and examined on a JEOL transmission electron microscope.
125I-αBgt binding. To quantify surface α7-nAChRs, cells were incubated for 20 min at 37°C with 10 nm 125I-αBgt (263 Ci/mmol; Amersham Biosciences, Piscataway, NJ). Nonspecific labeling was determined by including 1 μm unlabeled αBgt (Biotoxins, St. Cloud, FL). After rinsing three times with PBS, the cells were scraped in 1N NaOH and taken for gamma counting. To determine the proportion of α7-nAChRs present in intracellular pools, cells were first incubated with and without 200 nm unlabeled αBgt for 30 min to block surface receptors, then rinsed in PBS and permeabilized with 0.1% (w/v) saponin for 30 min, followed by labeling with 125I-αBgt for 2 h all on ice, washing, scraping, and gamma counting. Controls comparing surface binding of 125I-αBgt with and without a subsequent saponin treatment indicated that ∼40% of the specific binding was lost by the detergent treatment; this reduced efficiency was assumed to apply equally to surface and internal pools of receptors.
Receptor function assays. Patch-clamp recording in voltage-clamp mode was used to measure whole-cell current responses induced in CG neurons by rapid application of nicotine from a large bore pipette as described previously (Zhang et al., 1994). Long-term activation of CREB was determined by immunostaining for pCREB 20 min after stimulation with nicotine in the presence of 0.2 mm cadmium to block voltage-gated calcium channels (VGCCs). Cells were scored positive in an all-or-none manner if they displayed specific nuclear pCREB staining (Chang and Berg, 2001). Both for pCREB scoring here and for immunofluorescence staining above, cells were selected at random by inspecting adjacent fields of view across the diameter of the coverslip and choosing for analysis the first 60-100 neurons encountered. Results are presented as “percentage of long-term pCREB,” meaning the proportion of neurons that displayed nuclear staining for pCREB at least 20 min after the stimulus had been removed; expression of pCREB for this duration in CG neurons has previously been shown to correlate with changes in gene expression (Chang and Berg, 2001). Statistical significance was assessed by two-way ANOVA.
Materials. White leghorn chick embryos were obtained locally and maintained at 37°C in a humidified incubator. FM4-64 was purchased from Molecular Probes, HRP was purchased from Jackson ImmunoResearch, and BFA and the botulinum toxins C1 and D were purchased from Calbiochem (San Diego, CA). All other reagents were purchased from Sigma (St. Louis, MO), unless indicated otherwise.
Results
Postsynaptic target SNAREs colocalize with α7-nAChR clusters
Large numbers of α7-nAChRs are concentrated on somatic spines arranged in clumps or mats on chick ciliary neurons (Shoop et al., 1999). Staining freshly dissociated E14 CG neurons with AlexaαBgt to label α7-nAChRs showed the expected large clusters of receptors. Costaining the cells with mAbs either to SNAP-25 (Fig. 1A-C) or to syntaxin 1 (Fig. 1D-F) revealed that target SNAREs (tSNAREs) codistribute with the clusters. No significant staining was seen for SV2, indicating that presynaptic compartments were essentially absent (Fig. 1G-I); ultrastructural analysis previously yielded the same conclusion (Shoop et al., 1999). The tSNARE staining was specific because it was not seen when the primary antibody was replaced with nonimmune IgG (Fig. 1J-L). Quantifying the codistribution indicated that 77 ± 5% of the syntaxin 1 clusters overlapped with α7-nAChR clusters, whereas 80 ± 5% of α7-nAChR clusters overlapped with syntaxin 1 (mean ± SEM; n = 60 cells from four experiments). Of the total area occupied by syntaxin 1 clusters, 72 ± 4% overlaid the α7-nAChR cluster area, whereas 59 ± 3% of the α7-nAChR cluster area overlaid syntaxin 1. For reference, 16 ± 1% of the cell perimeter was occupied by α7-nAChR clusters (n = 10 cells).
Figure 1.
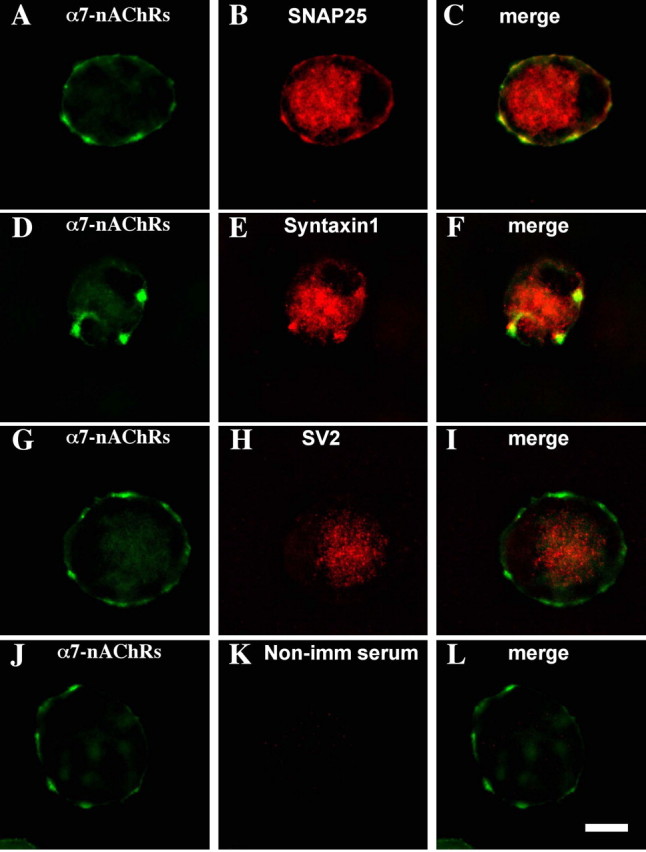
Codistribution of tSNAREs with α7-nAChR clusters on CG neurons. Freshly dissociated E14 CG neurons were costained with AlexaαBgt for α7-nAChRs (A, D, G, J) and antibodies for SNAP-25 (B), syntaxin 1 (E), SV2 (H), or nonimmune serum as a negative control (K), and the corresponding pairs of fluorescence images were merged (C, F, I, L). Both SNAP-25 and syntaxin 1 codistribute with α7-nAChRs. Similar results were obtained in three additional experiments. Scale bar, 10 μm.
tSNAREs do not appear to interact directly with α7-nAChRs because coimmunoprecipitation experiments similar to those performed previously with nicotinic receptors and PDZ-containing proteins (Conroy et al., 2003) failed to show association of either syntaxin 1 or SNAP-25 with α7-nAChRs. Instead, the codistribution appears to depend on lipid rafts. Treating freshly dissociated E14 CG neurons with methyl-β-cyclodextrin to extract cholesterol and disperse lipid rafts dispersed not only the α7-nAChR clusters as reported previously (Brusés et al., 2001) but also the tSNARE coclusters (data not shown).
Nicotinic stimulation drives SNARE-dependent postsynaptic endocytosis
SNARE proteins are known to mediate both vesicular exocytosis and endocytosis (Pelham, 1999). We tested for endocytosis by incubating E14 CG neurons with the dye FM4-64, rinsing, and examining with fluorescence microscopy. In the absence of nicotine, only occasional FM4-64-stained puncta were seen, and they rarely codistributed with α7-nAChR clusters (Fig. 2A-C). After a 5 min incubation with 10 μm nicotine, however, nearly half of the cells showed marked FM4-64 labeling codistributing with α7-nAChR clusters (Fig. 2D-F). Similar results were obtained with HRP in place of the dye. Fixing, permeabilizing, and immunostaining for intracellular HRP showed little uptake in controls (Fig. 2G-I) but significant HRP uptake after nicotinic stimulation, with the uptake being mostly confined to areas defined by α7-nAChR clusters (Fig. 2J-L).
Figure 2.
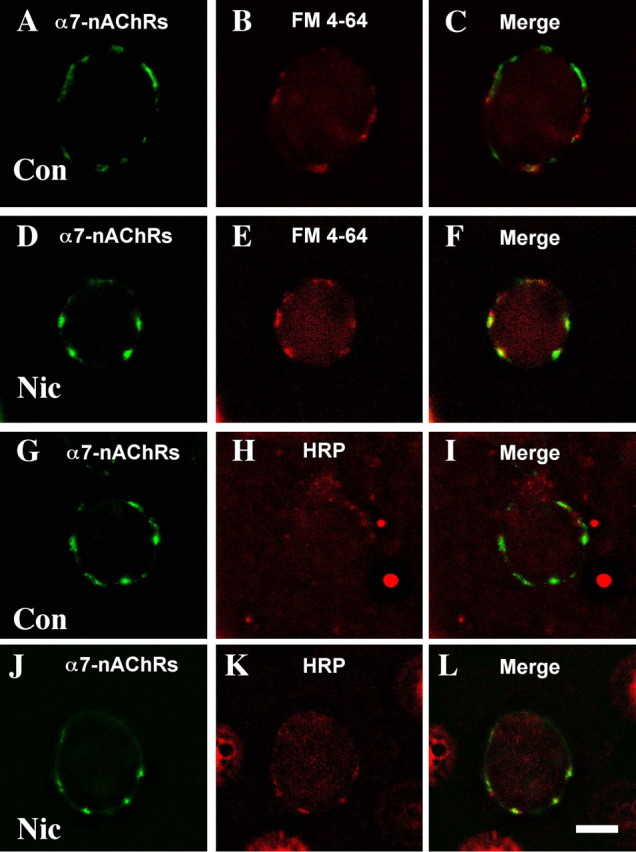
Nicotine-driven uptake into postsynaptic compartments. E14 CG neurons were stimulated with nicotine (10 μm, 5 min) in the presence of either FM4-64 or HRP, then rinsed, labeled with AlexaαBgt for α7-nAChRs (A, D, G, J), and then either viewed immediately in the case of FM4-64 (B, E) or fixed, permeabilized, and reacted with anti-HRP antibody followed by fluorescent secondary antibody in the case of HRP (H, K) before viewing. In the absence of nicotine treatment (Con), little FM4-64 (A-C) or HRP (G-I) stain was retained at α7-nAChR clusters. After nicotine treatment (Nic), both FM4-64 (D-F) and HRP (J-L) staining were prominent at the receptor clusters. Similar results were obtained in three additional experiments. Scale bar, 10 μm.
Quantification showed that in the control population only 4 ± 1% of the area occupied by α7-nAChR surface clusters codistributed with FM4-64 staining. The codistribution increased dramatically to 58 ± 3% after nicotinic stimulation (mean ± SEM; n = 60 cells per condition from four experiments) for those cells showing dye uptake (about half of the population). The overall increase included a sevenfold increase in area stained by FM4-64 after stimulation and a twofold increase in the fractional area of FM4-64 staining codistributing with α7-nAChR clusters. Nicotinic stimulation did not change the portion of the perimeter occupied by α7-nAChR clusters (14 ± 1vs13 ± 1% for control vs nicotine-stimulated cells; n = 10 cells). Thus, nicotinic stimulation dramatically increased the total amount of FM4-64 uptake, and much of the uptake was localized at sites demarcated by α7-nAChR clusters.
On ciliary neurons, α7-nAChR clusters correspond to spine mats enriched in the receptors (Shoop et al., 1999, 2002). Electron microscopy confirmed that nicotinic stimulation drove HRP uptake and demonstrated that the sequestered HRP could be detected in small vesicles dispersed throughout spines 15 min later (Fig. 3A). Examining 65 such neurons revealed, on average, >30 HRP-labeled vesicles per spine. The mean size of the stained vesicles was 20 ± 6 nm (±SEM; n = 110). None was found in the cell body. Few stained vesicles were present in spines of HRP-treated cells lacking the nicotine exposure (Fig. 3B). Quantifying the number of stained vesicles per unit area of spine yielded 29 vesicles per square micrometer for nicotine-stimulated neurons versus 0.5 vesicles per micrometer squared for unstimulated neurons (n = 3 experiments).
Figure 3.
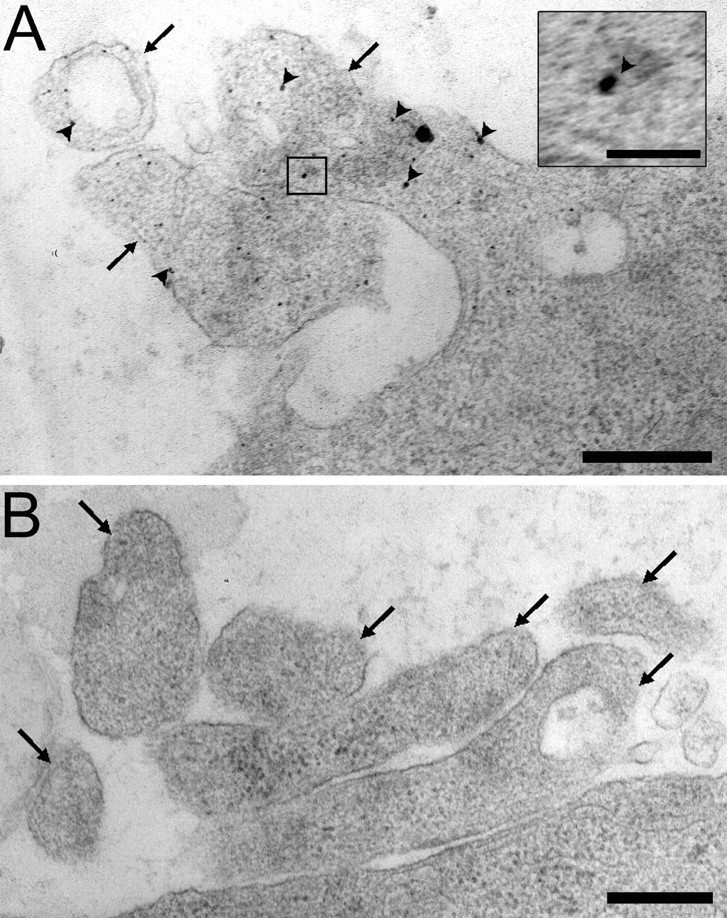
Electron micrographs of nicotine-induced HRP-filled vesicles present in postsynaptic spines. A, E14 CG neurons were stimulated with 10 μm nicotine for 5 min in the presence of HRP and were then rinsed, fixed, stained for HRP activity, and prepared for electron microscopy. Prominent HRP-stained vesicles (arrowheads) can be seen in the cross section of somatic spines (arrows). The inset shows an enlargement of one such labeled vesicle. B, Omitting the nicotine prevented accumulation of HRP-stained vesicles in the spines. Scale bars: A, B, 500 nm; inset, 100 nm.
The vesicular endocytosis visualized by FM4-64 uptake depended on α7-nAChR activation. It was blocked not only by the general nicotinic antagonist dTC but also by the specific α7-nAChR blocker MLA (Fig. 4). Replacing the nicotine with 90 mm KCl, which depolarizes the cells, did not induce uptake (data not shown). Previous studies have shown that CG α7-nAChRs influence downstream events in part by causing calcium influx, calcium release from internal stores, and activation of calcium/calmodulin-dependent protein kinase II (CamKII) (Liu and Berg, 1999; Chang and Berg, 2001). Similar mechanisms appear to be involved in vesicular endocytosis induced by α7-nAChRs. Removal of extracellular calcium, blockade of calcium release from internal stores by pretreatment with thapsigargin, and inhibition of CamKII by KN93 each blocked the ability of nicotine to induce FM4-64 uptake (Fig. 4). Nicotine was unable to stimulate dye uptake in the cold (on ice), indicating that the process was temperature dependent. Neither sucrose (400 mm) used to block clathrin-mediated endocytosis (Gicquiaux et al., 2002) nor cyclosporin used to block calcineurin prevented the nicotine-induced uptake (data not shown).
Figure 4.

Proportion of cells displaying nicotine-induced FM4-64 staining of α7-nAChR clusters. Cells were subjected to the indicated treatments and then processed for FM4-64 uptake as described in Figure 3. AlexaαBgt was used to label α7-nAChRs. Control, No nicotine; Nic-treated, 10 μm nicotine for 5 min; dTC, 100 μm; MLA, 10 nm; Ca-free, no extracellular calcium; Thaps, 1 μm thapsigargin; KN93, 10 μm; Bot C1, 66 nm botulinum toxin C1; Bot D, 110 nm botulinum toxin D; Inact C1, 66 nm inactive Bot C1; Inact D, 110 nm inactive Bot D; NEM, 1 mm. Values represent the mean ± SEM of results from six or more cultures from three or more experiments per condition.
Other treatments confirmed the role of SNAREs in the endocytosis. Treating the neurons with botulinum toxins C1 and D to cleave tSNAREs and vesicle SNAREs (vSNAREs), respectively, each prevented the nicotine-induced uptake of FM4-64 (Fig. 4). Inactivating the toxins by heat (95°C for 60 min) rendered them ineffective in the assay. NSF, which often works in concert with vSNAREs and tSNAREs to promote vesicular release and endocytosis (Sudhof and Scheller, 2001), also appeared to be involved. Treating the neurons with low concentrations of NEM blocked the uptake (Fig. 4). NEM has previously been used to identify a role for NSF in vesicular transport (Block et al., 1988).
SNARE-dependent endocytosis internalizes α7-nAChRs
The abundance of α7-nAChRs on somatic spines on ciliary neurons and their proximity to tSNAREs suggested that SNAREs may mediate α7-nAChR trafficking. Because receptor activation was required to drive SNARE-dependent endocytosis, it was not possible to visualize receptor internalization by prelabeling with fluorescent αBgt derivatives that block receptor function. Instead, CG neurons maintained in cell culture were transfected with a construct encoding a fusion protein: α7-nAChR coupled with GFP via the C terminus. Because the C terminus of α7-nAChR is extracellular, accumulation of α7-nAChR-GFP on the cell surface should be accessible with anti-GFP antibodies on living cells. Indeed, cells positive for GFP (expressing the transfected construct) also displayed punctate immunostaining for GFP on the cell surface, indicating the presence of α7-nAChR-GFP clusters (Fig. 5A,B). Similar clusters of native α7-nAChRs can be detected with AlexaαBgt on the neurons (data not shown). No surface immunostaining was seen for cells transfected with GFP alone as a negative control (Fig. 5C,D), demonstrating that the surface staining obtained with the fusion protein represented receptor in the plasma membrane.
Figure 5.
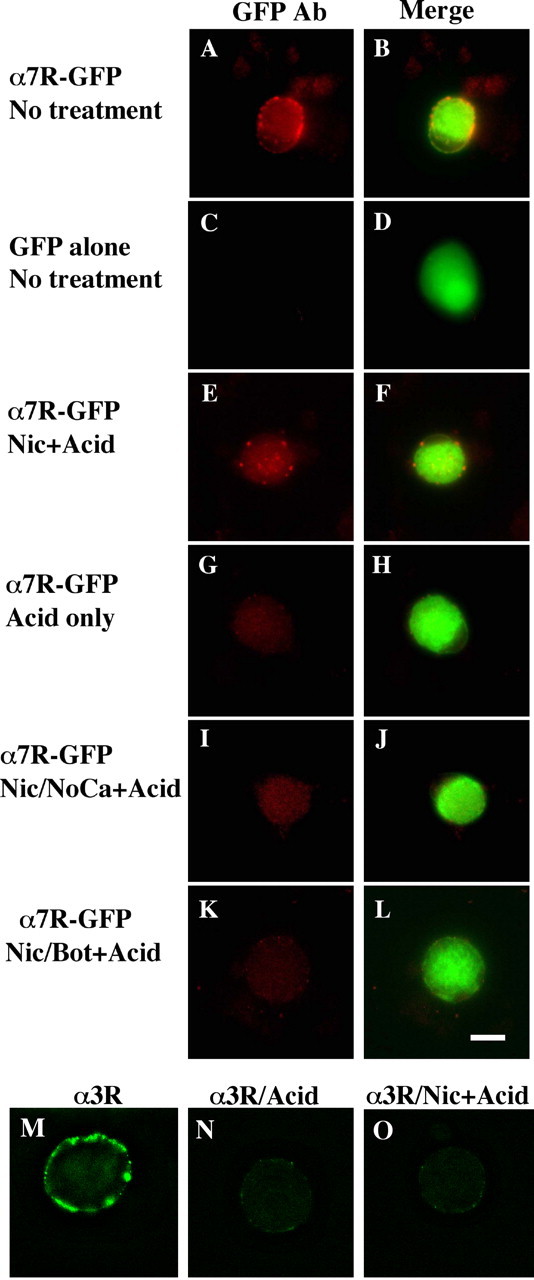
Nicotine-induced internalization of surface α7-nAChRs in a SNARE-dependent manner. E8 CG neurons were grown in culture for 6-7 d after being transfected with constructs encoding either the fusion protein α7-nAChR-GFP (α7R-GFP) (A, B, E-L) or GFP alone (C, D). The cells were then treated as indicated below and either imaged directly (A-D) or acid-stripped and then processed for imaging (E-L). The left panel of each pair shows immunostaining with anti-GFP antibody in red, and the right panel shows the image merged with the green GFP fluorescence to yield yellow. Treatments are as in Figure 4: A-D, no treatment; E, F, nicotine plus acid stripping (+Acid); G, H, negative control, no nicotine plus acid; I, J, nicotine plus acid without extracellular calcium; K, L, nicotine plus acid after Bot C1. The α7-nAChR-GFP fusion protein appears in the surface membrane where it can be stained with anti-GFP antibody; nicotine drives internalization of the receptor (revealed by protection of bound anti-GFP antibody against acid stripping) in a calcium-dependent manner that is blocked by botulinum C1. In other cultures, α3*-nAChRs on the cell surface were labeled with mAb 35, and after rinsing, the cells were imaged after fixation, permeabilization, and labeling with fluorescent secondary antibody (M); or acid-washed first to remove mAb 35 and then fixed, permeabilized, and labeled (N); or nicotine treated to induce internalization, acid-washed, and then fixed, permeabilized, and labeled (O). In contrast to α7-nAChRs, the nicotine treatment did not protect mAb 35 labeling against acid-wash, indicating that no detectable α3*-nAChR/antibody complex had been internalized. Similar results were obtained in two additional experiments. Scale bar, 10 μm.
This enabled the critical experiment of labeling surface α7-nAChR-GFPs with anti-GFP antibodies, rinsing, treating with nicotine to induce endocytosis, and then acid-stripping to remove antibody bound to receptor remaining on the surface. Antibody retained after the acid-strip represented internalized material. Significant amounts of α7-nAChR-GFP were internalized by the nicotine treatment, and the internalized receptors were located in clusters near the cell surface (Fig. 5E,F). In the absence of nicotinic stimulation, the receptors remained on the surface as evidenced by the fact that acid-stripping removed bound antibody and left very little detectable fluorescent clusters on or near the cell perimeter (Fig. 5G,H). As in the case of dye uptake, little internalization of receptor was detected in the absence of extracellular calcium (Fig. 5I,J) or after treatment with botulinum toxin (Fig. 5K,L). The experiment was quite reproducible (six experiments; ≥60 cells examined per condition) with all transfected cells displaying the same phenotype. The results indicate nicotine-induced SNARE-dependent internalization of α7-nAChRs.
The internalization did not extend to heteromeric α3*-nAChRs expressed by the neurons. Such receptors can be visualized by binding mAb 35 and fluorescent secondary antibody (Conroy et al., 2003). The protocol involved labeling first with mAb 35, which does not block receptor function, then challenging with nicotine, rinsing, allowing internalization, acid-stripping, fixing, permeabilizing, and staining with fluorescent secondary antibody. No internalization was seen, and the level of residual surface staining was equivalent to that found for cells lacking the nicotine exposure (Fig. 5M-O). Nicotine-driven SNARE-dependent endocytosis apparently removes α7-nAChRs preferentially.
Nicotinic stimulation recruits intracellular α7-nAChRs to the surface via SNAREs
In addition to inducing α7-nAChR internalization, nicotinic stimulation of CG neurons also triggered a rapid recruitment of internal α7-nAChRs to the cell surface. This was demonstrated using a multi-step protocol in which freshly dissociated E14 neurons were first stimulated with nicotine for 1 min and then rinsed quickly and incubated with saturating amounts of unlabeled αBgt (4 μm for 3 min) to rapidly block all α7-nAChRs remaining on the surface. After rinsing away unbound αBgt, the neurons were incubated with AlexaαBgt for 15 min to detect new α7-nAChRs appearing on the cell surface during that time period. Rinsing, fixing, and imaging the cells revealed distinctive clusters of labeled α7-nAChRs (Fig. 6A). The labeling was specific because the clusters were not seen if nicotinic stimulation was omitted, if dTC was used to compete the nicotine, or if αBgt was included along with the AlexaαBgt during the 15 min labeling period (Fig. 6B-D). The new receptor clusters appeared to localize at spine mats as defined by costaining for the spine constituent F-actin (Fig. 6E-G), as done previously (Shoop et al., 1999).
Figure 6.
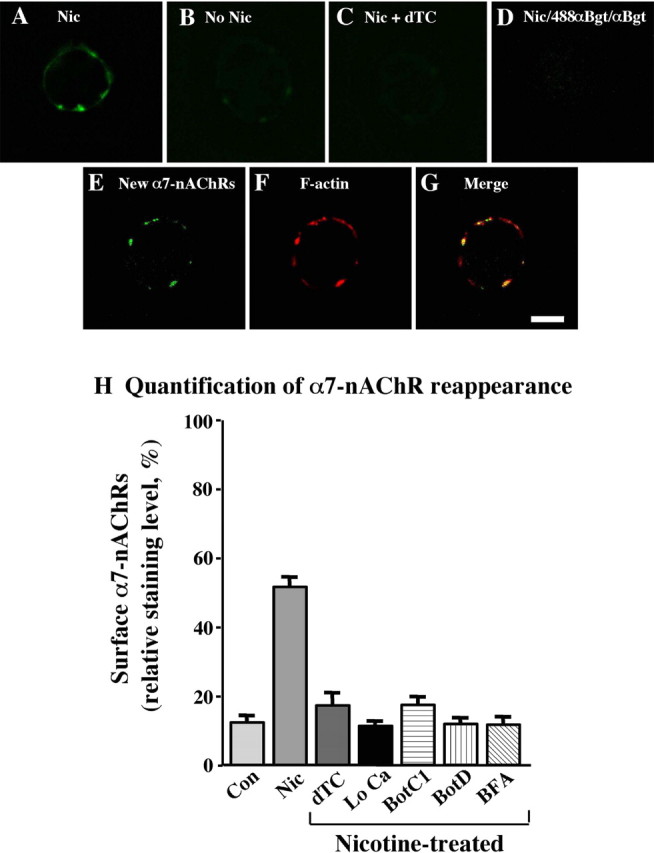
Nicotine-driven SNARE-dependent recruitment of internal α7-nAChRs onto spine surfaces. E14 CG neurons were stimulated with nicotine (10 μm, 1 min), rinsed, incubated with αBgt (4 μm, 3 min) to block surface α7-nAChRs, rinsed again, and then incubated with AlexaαBgt for the next 15 min to visualize receptors newly appearing on the surface. Distinctive clusters appeared on such cells (A) that were absent when nicotine was omitted (B), dTC was included with the nicotine (C), or αBgt was included with the AlexaαBgt (D). The newly appearing α7-nAChR clusters (E) colocalized with spine mats as defined by costaining with phalloidin for F-actin (F) and merging the images (G). Scale bar, 10 μm. H, Relative levels of staining were quantified as described in Materials and Methods. Values represent the mean ± SEM of results from ≥60 cells compiled from three or more experiments per condition and have been normalized to levels seen on untreated cells before blockade of surface receptors with unlabeled αBgt (defined as 100%). Background values, representing the fluorescence observed over noncellular regions of the substratum, have been subtracted. Nicotine (Nic) induces a substantial increase in the number of α7-nAChRs seen on the surface compared with untreated controls (Con), and the effect is blocked by dTC, removal of extracellular calcium (Lo Ca), botulinum toxins C1 (BotC1) and D (Bot D), and BFA.
The extent of α7-nAChR reappearance was quantified by measuring the mean intensity of specific fluorescent signal arising from individual receptor clusters, averaging the results, and comparing the mean to that obtained from untreated control cells. The net levels of receptor that appeared on the surface after nicotinic stimulation (and blockade of preexisting surface receptors with αBgt as outlined above) was over one-third as great as that initially present (Fig. 6H). Blockade of the stimulation with dTC, omission of calcium from the medium, or treatment with either botulinum C1 or botulinum D prevented the appearance. Heat-inactivated botulinum toxins were ineffective (data not shown). The results demonstrate rapid SNARE-dependent trafficking of α7-nAChRs to the cell surface.
Not surprisingly, the appearance of receptors was also blocked by BFA, a compound that prevents vesicle trafficking from the Golgi (Fig. 6H). Interestingly, BFA also blocked nicotineinduced vesicular endocytosis as monitored by FM4-64 uptake (Fig. 4) (43 ± 3% for nicotine; 4 ± 3% for nicotine plus BFA). Similarly, BFA blocked α7-nAChR internalization as monitored by internalization of the fusion protein α7-nAChR-GFP (as in Fig. 5) (data not shown). These latter results suggest that either vesicle trafficking is required for surface delivery of components needed for endocytosis (e.g., competent SNAREs) or that nicotine-induced endocytosis may be coupled with exocytosis.
Nicotine-induced trafficking does not alter α7-nAChR numbers or whole-cell response
Although a major function of activity-driven trafficking for AMPA receptors is regulating the number of receptors on the cell surface, this does not seem to be the primary function of the α7-nAChR trafficking observed here. Quantifying α7-nAChRs on neurons with (125I)αBgt before and after nicotinic stimulation revealed approximately equivalent numbers (Fig. 7A). Blocking preexisting surface α7-nAChRs with unlabeled αBgt showed that the nicotine treatment (immediately before blockade with αBgt) recruited unblocked receptors to the surface, equaling approximately one-third of those initially present. This is in good agreement with the fluorescence measurements above. BFA prevented the appearance, again in agreement with the fluorescence measurements, but did not change the total number of surface α7-nAChRs. That the neurons normally contain substantial internal pools of α7-nAChRs was shown by comparing the number of (125I)αBgt-binding sites found for saponin-permeabilized cells that had previously been incubated with and without unlabeled αBgt to block surface sites. The internal pool represented 45 ± 4% of the total (mean of two experiments).
Figure 7.

Effects of trafficking on the surface pool of α7-nAChRs. A, Quantification of surface α7-nAChRs using 125I-αBgt binding. E14 neurons were incubated for 1 min with culture medium only (Control), with medium containing 10 μm nicotine (Nic) or 10 μm nicotine plus 10 μg/ml BFA (Nic/BFA), and then rinsed and incubated an additional 20 min with 125I-αBgt to label surface α7-nAChRs. BFA, when present, was added 1 h before the nicotine. For measurement of α7-nAChRs newly trafficked to the surface, cells were preblocked (+) with αBgt (4 μm, 3 min) immediately after removing the nicotine and then rinsed further before exposure to 125I-αBgt. For total surface α7-nAChRs, the preblock with αBgt was omitted (-). Values represent the mean ± SEM of results from nine cultures compiled from three experiments per condition. Nicotine treatment recruited α7-nAChRs to the surface in a manner that required vesicular trafficking. The amount was equivalent to nearly one-third of that initially present on the surface but did not increase the total number significantly. Under the seconditions, nicotine-induced receptor recruitment to the surface approximately equaled receptor internalization. B, Whole-cell patch-clamp recording in voltage-clamp mode showing typical responses in CG neurons to rapid application of 20 μm nicotine for 1 s from a large bore pipette. The responses were elicited 20 min after the cells were incubated with nicotine (Nic) or nicotine plus BFA (Nic/BFA) as in A but omitting preblock with αBgt. Blockade of receptor trafficking with BFA had no obvious effect on the response.
In agreement with the (125I)αBgt-binding studies, patch-clamp recording indicated that the nicotine-induced whole-cell response did not depend on nicotine-induced SNARE-dependent trafficking. This was demonstrated by dividing cells into control and BFA-treated groups and then stimulating with 10 μm nicotine for 1 min, followed by a 20 min incubation to allow trafficking to occur. Then, testing the whole-cell response to rapid application of nicotine for 1 s revealed no significant difference between control and BFA-treated neurons in time course or amplitude of the response (Fig. 7B). Mean peak amplitudes, which reflect primarily the α7-nAChR component (Liu and Berg, 1999), were 216 ± 20 and 217 ± 13 pA/pF (mean ± SEM; n = 6-11 cells) for control and BFA-treated cells, respectively.
SNARE-dependent trafficking is needed to sustain receptor coupling with downstream events
Surprisingly, blocking nicotine-induced SNARE-dependent trafficking had a profound effect on the ability of α7-nAChRs to activate the transcription factor CREB when rechallenged with nicotine after an initial exposure. Normally, nicotinic stimulation of E14 CG neurons produces long-term activation of CREB and changes in gene expression when VGCCs are silent (Chang and Berg, 2001). VGCCs can be silenced by including either cadmium or nifedipine with the nicotine. In this case, activated CREB in the nucleus, as seen by immunostaining for phosphorylation on ser 133 (pCREB), remains long after the nicotine is removed. If instead VGCCs are allowed to remain active, the pCREB is rapidly lost after nicotine removal and does not correlate with changes in gene expression (Chang and Berg, 2001). These features can be used to test the role of SNARE-dependent trafficking in α7-nAChR-mediated induction of long-term pCREB.
The protocol was initiated by a 1 min exposure to nicotine, followed by a 20 min recovery to allow trafficking to occur (Fig. 8A). Then, to induce long-term pCREB, nicotine was applied a second time (5 min), along with cadmium to inhibit VGCCs. After a second 20 min recovery, the cells were immunostained for nuclear pCREB (Fig. 8A). The initial nicotine incubation did not itself produce long-term pCREB because VGCCs were not blocked at that time; nor did it have any effect on the ability of nicotine (plus cadmium) in the second incubation to induce long-term pCREB (Fig. 8B,C, compare Med for No-Stim and Nic conditions). Blockade of SNARE-dependent trafficking by botulinum toxin also had no effect on the pCREB assay (Fig. 8B, compare Med and BotC1). If, however, α7-nAChRs had previously been exposed to nicotine, then and only then did botulinum toxin prevent subsequent nicotine from inducing long-term pCREB (Fig. 8C, asterisk). Only the α7-nAChR component was affected (i.e., the portion of the long-term pCREB signal that could be blocked by αBgt). Botulinum toxin had no effect if the α7-nAChRs were blocked with MLA during the first nicotine exposure (Fig. 8D) or if nicotine was replaced by KCl in the first exposure (Fig. 8E). Substituting BFA for the botulinum toxin produced equivalent results (data not shown). Thus, SNARE-dependent trafficking became critical for subsequent long-term pCREB induction by α7-nAChRs if the receptors had previously been exposed to agonist in a way that would induce trafficking. Although the α7-nAChRs remained capable of generating inward current after the first incubation with nicotine (Fig. 7B), they apparently became uncoupled from the signaling pathway that would enable them to activate CREB. SNARE-dependent trafficking was required to reestablish the link.
Figure 8.

Effect of trafficking on nicotine-induced long-term pCREB. A, The protocol started with a 1 min incubation of culture medium only (No-Stim; B), 10 μm nicotine (Nic; C), 10 μm nicotine plus 50 nm MLA added 1 min earlier to block α7-nAChRs (Nic/MLA; D), or KCl (E). After rinsing, the cells were given a first recovery period of 20 min to allow trafficking. Then, to induce pCREB, the cells received a 5 min second incubation in culture medium with cadmium (to block VGCCs) and no additives (control), nicotine (nic), or nicotine with αBgt applied 10 min earlier (nic/αBgt) to block α7-nAChRs, followed by rinsing and a second 20 min recovery period before immunostaining for nuclear pCREB. Cells were pretreated either with medium alone (Med) or with botulinum toxin C1 (BotC1) to block SNARE-dependent trafficking. Values represent the mean ± SEM of results compiled from three experiments. Blockade of SNARE-dependent trafficking by botulinum toxin specifically inhibited the ability of α7-nAChRS to drive CREB activation during the second incubation, if and only if the receptors had been exposed to nicotine in the first incubation (*p < 0.001 comparing BotC1 and Med for Nic-induced pCREB in C). Neither the blockade of trafficking nor the first incubation with nicotine or KCl had any other effect in the pCREB assay.
Discussion
We show here that α7-nAChRs on neurons are subject to rapid SNARE-dependent trafficking. The trafficking can be bidirectional, involving both internalization of activated receptors to remove them from the cell surface and recruitment of new receptors from intracellular pools to repopulate the surface. It is initiated by receptor activation and requires extracellular calcium, calcium release from internal stores, and CamKII. The underlying cellular machinery includes tSNAREs and vSNAREs with the likely involvement of NSF. The trafficking can be substantial, replacing one-third of the total surface pool within minutes, and does so without producing dramatic changes in receptor number or responsiveness. Instead, the trafficking appears essential for maintaining coupling between receptor activation and downstream signaling pathways. This is the first demonstration, to our knowledge, of rapid activity-induced trafficking of ionotropic receptors at nonglutamatergic synapses, and it suggests a novel role for such trafficking.
Previously described forms of trafficking for nicotinic receptors operate on slower time scales. Several hours are required for constitutive receptor synthesis, transport, and insertion into the plasma membrane. Agonist-induced downregulation of nicotinic receptors also occurs over hours and involves a significant reduction in the complement of receptors on the neuron surface (Messing, 1982; Stollberg and Berg, 1987). At the neuromuscular junction, receptor blockade can accelerate the rate of nicotinic receptor removal, but both the mechanism and time course differ significantly from that seen here for α7-nAChRs (Akaaboune et al., 1999).
The rapid trafficking of α7-nAChRs induced here by nicotine was dependent on SNARE proteins both for receptor internalization and receptor recruitment to the cell surface. This was illustrated by showing sensitivity of both processes to botulinum toxin. SNARE-dependent mechanisms are known to deliver components to the plasma membrane and to remove them as well (Pelham, 1999; Jahn et al., 2003). The ability of BFA to block both α7-nAChR internalization and recruitment suggests that either the two processes are coupled or that vesicle trafficking to the surface is required for maintaining endocytic capability in this case. The large intracellular pools of α7-nAChR are certainly sufficient to provide a source of receptors for rapid recruitment to the cell surface, but internalized receptors may recycle as well. Recycling is known to occur for AMPA receptors (Ehlers, 2000).
Essentially all of the CG neurons examined here appeared capable of nicotine-induced SNARE-dependent α7-nAChR internalization and recruitment. Ciliary neurons, which comprise half of the neurons in the ganglion, display large α7-nAChR clusters that represent groups of somatic spines or spine mats heavily endowed with receptors (Shoop et al., 1999, 2002). Choroid neurons, which make up the other half, tend to be smaller and have smaller α7-nAChR clusters; their associated postsynaptic specializations have yet to be described at the ultrastructural level. Choroid and ciliary neurons are difficult to distinguish at the lightmicroscopic level in dissociated cell preparations; no effort was made to resolve them here. It is possible that the two neuronal populations respond somewhat differently to nicotine, perhaps accounting for some of the cellular heterogeneity in the FM4-64 uptake experiments. The fact that receptor internalization and reappearance was prominent at sites demarcated by large α7-nAChR clusters and F-actin staining provided reassurance that receptors on somatic spines participated in the trafficking.
Ultrastructural analysis identified small HRP-filled vesicles appearing within spines on ciliary neurons after stimulation with nicotine and presumably contained the internalized α7-nAChRs. The internalization was unlikely to depend on clathrin-mediated endocytosis because it was not affected by high sucrose concentrations. This differs from one type of AMPA receptor trafficking that relies on diffusion of receptors to juxtasynaptic sites and then internalization via clathrin-mediated endocytosis (Carroll et al., 1999; Luscher et al., 1999). A second difference is that nicotine-induced internalization of α7-nAChRs was not dependent on calcineurin, whereas NMDA-induced internalization of AMPA receptors requires calcineurin (Beattie et al., 2000; Ehlers, 2000). Nicotine-induced receptor internalization did not appear to extend to α3*-nAChRs, perhaps because α3*-nAChRs are tethered in postsynaptic densities and interact with different intracellular components (Jacob et al., 1984; Williams et al., 1998; Conroy et al., 2003).
It was surprising that the trafficking of α7-nAChRs seen here did not produce a notable difference in the number of surface receptors. Activity-induced trafficking of AMPA receptors specifically alters the number of receptors available at postsynaptic sites and provides the basis for several forms of synaptic plasticity at glutamatergic synapses (Bredt and Nicoll, 2003). The methods of quantification used in the present studies for α7-nAChRs revealed no effect of trafficking on the number or distribution of surface receptors: total binding was comparable, and newly recruited receptors appeared to be localized at spine mats as defined by F-actin costaining, as were the original receptors. In fact, lateral movement of receptors may not offer an effective mechanism for regulating synaptic response, at least in this case of ciliary neurons, because nearly the entire surface of the cell is enveloped by a presynaptic calyx that releases transmitter at numerous sites (Dryer, 1994; Shoop et al., 1999). It remains possible, however, that changes in receptor number occurred at specific sites but went undetected by the assays used. Alternatively, more physiological methods of inducing trafficking may redistribute nicotinic receptors on the surface.
The finding that nicotine-induced trafficking was required to sustain α7-nAChR coupling with downstream pathways was unexpected. The coupling requirement was revealed by the all-or-none pCREB assay in which nicotinic induction of long-term pCREB in a subpopulation of the cells (15-20%) has a specific requirement for α7-nAChR activation. Trafficking of α7-nAChRs very likely becomes uncoupled in the other cells as well but is not apparent in the assay either because long-term pCREB formation is limited by some other feature (nonresponders) or because α3*-nAChR activation is sufficient by itself in those cases (responders in the presence of αBgt). The basis for this cellular variation is unknown but may reflect differences in choroid versus ciliary neurons or in the viability of cells after dissociation of the ganglion.
Vesicular trafficking was not a general requirement for pCREB appearance; nor was it needed to permit α7-nAChR internalization as a possible obligatory first step in pCREB activation. These conclusions follow from the observations that neither botulinum toxin nor BFA prevented α7-nAChRs from inducing long-term pCREB when the receptors were stimulated de novo. The requirement for trafficking only manifested when α7-nAChRs had previously been activated. If α7-nAChRs were pharmacologically protected during the initial incubation with nicotine, they remained capable of inducing long-term pCREB when subsequently challenged with nicotine, even with trafficking having been blocked. Depolarization with KCl also failed to impose a trafficking requirement on long-term pCREB induction by α7-nAChRs, and the trafficking requirement applied only to the α7-nAChR component of the pCREB response. The α3*-nAChRs were not trafficked, did not induce trafficking, and did not require trafficking for pCREB induction, even when repeatedly challenged with agonist. Thus, the α7-nAChR trafficking reported here appears to be specific in its target and effects.
Rapid agonist-induced internalization of G-protein-coupled receptors is well documented and was initially thought to represent a form of receptor desensitization. Subsequently, it became clear that, in many cases, the internalized receptors are either rapidly replaced on the surface by preexisting intracellular receptors or are directly recycled to the surface themselves. This process of replacement/recycling provides a resensitization of the signaling pathway after an initial desensitization of the receptor (Ferguson, 2001; Pierce et al., 2002). In some cases, the resensitization results from changes in the receptor itself during the recycling, as found for β2-adrenergic receptors that undergo phosphorylation state changes. A similar process has not been identified yet for ligand-gated ion channels. Activity-dependent recycling of AMPA receptors requires protein kinase A activity and correlates with changes in receptor phosphorylation, but the process appears designed to regulate the number of AMPA receptors at synapses rather than to resensitize signaling pathways or recouple receptors to intracellular components (Ehlers, 2000).
The rapid and extensive α7-nAChR turnover seen here may represent a recovery response triggered by extensive nicotine-induced desensitization. Although the receptors recover the ability to pass current, they may remain modified in such a way as to prevent recoupling with downstream signaling machinery. Internalization followed either by replacement or by recycling could then provide a mechanism for the cell to reestablish coupling between functional receptors and internal machinery. A candidate mechanism for the initial decoupling might be calcium-driven changes in receptor phosphorylation, caused by calcium influx associated with α7-nAChR activation. Neuronal nicotinic receptors, including α7-nAChRs on somatic spines, are associated with postsynaptic scaffolds that mediate downstream signaling (Conroy et al., 2003; Parker et al., 2004; Temburni et al., 2004). How the scaffold components are affected by receptor activity is unknown. Additional, as of yet, unidentified components may also need to traffic with α7-nAChRs to reestablish the link.
Footnotes
This work was supported by National Institutes of Health Grants NS12601 and NS35469. Z.L. is an American Heart Association Postdoctoral Fellow. A.W.T. and Q.N. are Tobacco-Related Disease Research Program Postdoctoral Fellows. We thank Xiao-Yun Wang and Lynn Ogden for expert technical assistance.
Correspondence should be addressed to Dr. Darwin K. Berg, Neurobiology Section, Division of Biology, 0357, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0357. E-mail: dberg@ucsd.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251159-10$15.00/0
References
- Adams CE, Broide RS, Chen Y, Winzer-Serham UH, Henderson TA, Leslie FM, Freedman R (2002) Development of the α7 nicotinic cholinergic receptor in rat hippocampal formation. Dev Brain Res 139: 175-187. [DOI] [PubMed] [Google Scholar]
- Akaaboune M, Culican SM, Turney SG, Lichtman JW (1999) Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo Science 286: 503-507. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Barbosa CTF, Albuquerque EX (1997) Neuronal nicotinic acetylcholine receptor activation modulates γ-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther 283: 1396-1411. [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Albuquerque EX (1998) α-Bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res 810: 257-263. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291-1300. [DOI] [PubMed] [Google Scholar]
- Berg DK, Conroy WG (2002) Nicotinic α7 receptors: synaptic options and down-stream signaling in neurons. J Neurobiol 53: 512-523. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP (1993) Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal α7 nicotinic receptor. Proc Natl Acad Sci USA 90: 6971-6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE (1988) Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci USA 85: 7852-7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379. [DOI] [PubMed] [Google Scholar]
- Broide RS, Leslie FM (1999) The α7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol 20: 1-16. [DOI] [PubMed] [Google Scholar]
- Brusés JL, Chauvet N, Rutishauser U (2001) Membrane lipid rafts are necessary for the maintenance of the α7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci 21: 504-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M (1999) Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA 96: 14112-14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Berg DK (2001) Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron 32: 855-865. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK (2003) PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron 38: 759-771. [DOI] [PubMed] [Google Scholar]
- Dryer S (1994) Functional development of the parasympathetic neurons of the avian ciliary ganglion: a classic model system for the study of neuronal differentiation and development. Prog Neurobiol 43: 281-322. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A (2001) Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci 21: 7993-8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk L, Nordberg A, Seiger A, Kjaeldgaard A, Hellstrom-Lindahl E (2003) Higher expression of alpha7 nicotinic acetylcholine receptors in human fetal compared to adult brain. Dev Brain Res 142: 151-160. [DOI] [PubMed] [Google Scholar]
- Ferguson SSG (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role of in receptor desensitization and signaling. Pharmacol Rev 53: 1-24. [PubMed] [Google Scholar]
- Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV (1998) Synaptic potentials mediated via α-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci 18: 8228-8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquiaux H, Lecat S, Gaire M, Dieterlen A, Mely Y, Takeda K, Bucher B, Galzi J-L (2002) Rapid internalization and recycling of the human neuropeptide Y Y1 receptor. J Biol Chem 277: 6645-6655. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383: 713-716. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB (2001) Signal transduction in the postsynaptic neuron. Activity-dependent regulation of gene expression. In: Synapses (Cowan WM, Sudhof TC, Stevens CF, eds), pp 357-391. Baltimore: Johns Hopkins UP.
- Hatton GI, Yang QZ (2002) Synaptic potentials mediated by α7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci 22: 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Hulo S, Bertrand D, Muller D (1999) Synaptic transmission at nicotinic acetylcholine receptors in rat hippocampal organotypic cultures and slices. J Physiol (Lond) 515: 769-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu Q-s, Chang KT, Berg DK (2002) Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci 21: 616-625. [DOI] [PubMed] [Google Scholar]
- Jacob MH, Berg DK, Lindstrom JM (1984) Shared antigenic determinant between the Electrophorus acetylcholine receptor and a synaptic component on chicken ciliary ganglion neurons. Proc Natl Acad Sci USA 81: 3223-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112: 519-533. [DOI] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL (1999) Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci 22: 555-561. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK (2002) Nicotinic α7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci 22: 7903-7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RB, Aoki C (2002) α7 Nicotinic acetylcholine receptors occur at postsynaptic densities of AMPA receptor-positive and -negative excitatory synapses in rat sensory cortex. J Neurosci 22: 5001-5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q-s, Berg DK (1999) Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating α7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci 19: 10280-10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA (1999) Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24: 649-658. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- McGehee D, Heath M, Gelber S, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269: 1692-1697. [DOI] [PubMed] [Google Scholar]
- Messing A (1982) Cholinergic agonist-induced down-regulation of neuronal alpha-bungarotoxin receptors. Brain Res 232: 479-484. [DOI] [PubMed] [Google Scholar]
- Nishi R, Berg DK (1981) Two components from eye tissue that differentially stimulate the growth and development of ciliary ganglion neurons in cell culture. J Neurosci 1: 505-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Zhao S, Bredt DS, Sanes JR, Feng G (2004) PSD-93 regulates synaptic stability at neuronal cholinergic synapses. J Neurosci 24: 378-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HRB (1999) SNAREs and the secretory pathway—lessons from yeast. Exp Cell Res 247: 1-8. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639-650. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW (1993) Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13: 596-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Martone ME, Yamada N, Ellisman MH, Berg DK (1999) Neuronal acetylcholine receptors with α7 subunits are concentrated on somatic spines for synaptic signaling in embryonic chick ciliary ganglia. J Neurosci 19: 692-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK (2001) Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci 21: 771-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Esquenazi E, Yamada N, Ellisman MH, Berg DK (2002) Ultrastructure of a somatic spine mat for nicotinic signaling in neurons. J Neurosci 22: 748-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Huganir RL (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25: 758-788. [DOI] [PubMed] [Google Scholar]
- Stollberg J, Berg DK (1987) Neuronal acetylcholine receptors: fate of surface and internal pools in cell culture. J Neurosci 7: 1809-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Scheller RS (2001) Mechanism and regulation of neurotransmitter release. In: Synapses (Cowan WM, Sudhof TC, Stevens CF, eds), pp 217-274. Baltimore: Johns Hopkins UP.
- Temburni MK, Rosenberg MM, Pathak N, McConnell R, Jacob MH (2004) Neuronal nicotinic synapse assembly requires the adenomatous polyposis coli tumor suppressor protein. J Neurosci 24: 6776-6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS (2003) Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol 161: 805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Bading H (2003) Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signaling and BDNF gene regulation. Curr Opin Neurobiol 13: 366-371. [DOI] [PubMed] [Google Scholar]
- Williams BM, Tamburni MK, Schwartz Levey M, Bertrand S, Bertrand D, Jacob MH (1998) The long internal loop of the α3 subunit targets nAChRs to subdomains within individual synapses on neurons in vivo Nat Neurosci 1: 557-562. [DOI] [PubMed] [Google Scholar]
- Zhang Z-w, Vijayaraghavan S, Berg DK (1994) Neuronal acetylcholine receptors that bind α-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron 12: 167-177. [DOI] [PubMed] [Google Scholar]