

Abstract
Biochemical and genetic studies place the amyloid precursor protein (APP) at the center stage of Alzheimer's disease (AD) pathogenesis. Although mutations in the APP gene lead to dominant inheritance of familial AD, the normal function of APP remains elusive. Here, we report that the APP family of proteins plays an essential role in the development of neuromuscular synapses. Mice deficient in APP and its homolog APP-like protein 2 (APLP2) exhibit aberrant apposition of presynaptic marker proteins with postsynaptic acetylcholine receptors and excessive nerve terminal sprouting. The number of synaptic vesicles at presynaptic terminals is dramatically reduced. These structural abnormalities are accompanied by defective neurotransmitter release and a high incidence of synaptic failure. Our results identify APP/APLP2 as key regulators of structure and function of developing neuromuscular synapses.
Keywords: Alzheimer's disease, amyloid precursor protein, neuromuscular junction, synaptic vesicles, synaptic transmission, knock-out mice
Introduction
Alzheimer's disease (AD) is the most common form of dementia occurring in the aged population. The pathology of AD is characterized by the deposition of β-amyloid plaques, of which the principal components are 40 and 42 amino acid β-amyloid peptides derived from the amyloid precursor protein (APP). Although β-amyloid plaques are the hallmark of AD, loss of synapses is closely associated with the duration and severity of cognitive impairment in AD patients (Terry et al., 1991; Masliah and Terry, 1993). These observations lead to the notion that synaptic dysfunction is a critical element of the pathogenesis of AD (Selkoe, 2002). Because of the pivotal role of APP in AD pathogenesis, it is essential to understand its physiological function, particularly its potential activity in synaptic regulation.
APP belongs to a family of conserved type I transmembrane proteins including Apl-1 in Caenorhabditis elegans (Daigle and Li, 1993), Appl in Drosophila (Rosen et al., 1989), and APP (Tanzi et al., 1987), APP-like protein 1 (APLP1) (Wasco et al., 1992), and APLP2 (Wasco et al., 1993; Slunt et al., 1994) in mammals. Within the nervous system, APP can be detected on the membranes of synaptic preparations (Kirazov et al., 2001) and has been shown to localize to the postsynaptic densities, axons, and dendrites (Schubert et al., 1991; Shigematsu et al., 1992). APP undergoes rapid anterograde transport (Koo et al., 1990; Sisodia et al., 1993; Yamazaki et al., 1995) and is targeted to synaptic sites of both central and peripheral nervous systems, including neuromuscular junctions (NMJs) (Schubert et al., 1991; Shigematsu et al., 1992; Akaaboune et al., 2000).
To understand the in vivo function of APP and its family members, individual APP, APLP1, and APLP2 null mutant mice have been generated (Zheng et al., 1995; von Koch et al., 1997; Heber et al., 2000). These single null animals are viable and fertile, although aged mice deficient in APP show impairment in behavior and long-term potentiation (Zheng et al., 1995; Dawson et al., 1999; Phinney, 1999; Seabrook et al., 1999). The subtle phenotypes observed in each of the single null animals could be attributed in part to genetic redundancies, because mice doubly deficient in APP and APLP2 or APLP1 and APLP2 exhibit early postnatal lethality (von Koch et al., 1997; Heber et al., 2000). Previous studies, however, failed to identify any overt anatomic abnormalities in these double null animals (von Koch et al., 1997; Heber et al., 2000).
The expression profile and studies of the Drosophila APP homolog Appl support a potential activity of APP in synaptic modulation (Torroja et al., 1999). We thus sought to investigate the role of the APP family of proteins in mammalian synapses using the NMJ as a model system. Structural and functional analyses of the developing NMJ reveal a critical role of the APP family of proteins in the formation or differentiation of neuromuscular synapses. Mice doubly deficient in APP and APLP2 show an impaired synaptic structure and defective synaptic transmission.
Materials and Methods
Antibodies and reagents. Antibody against SV2 was obtained from Developmental Studies Hybridoma Bank (Iowa City, IA); antisynaptophysin (Syn), anti-neurofilament (NF), and anti-S100 antibodies were purchased from Synaptic Systems, Chemicon (Temecula, CA), and Swant, respectively. α-Bungarotoxin (αBTX) was purchased from Molecular Probes (Eugene, OR). The anti-APP C-terminal antibody was described previously (Xia et al., 2002).
Immunohistochemistry. Sternomastoid and diaphragm muscles were dissected from embryos or newborn pups and fixed in 4% paraformaldehyde. The diaphragm muscle was stained whole mount, and the sternomastoid muscle was sectioned at 20 μm thickness. Tissues were blocked with 5% goat serum, incubated with primary antibodies at 4°C overnight, washed in PBS, incubated with 1:1000 Alexa Fluor-488- or Alexa Fluor-594-conjugated secondary antibody (Molecular Probes) for 1 h at room temperature, washed in PBS, and mounted in glycerol/PBS. Confocal images were obtained with an Axios-kop 2 microscope (Zeiss, Oberkochen, Germany). The quantitation of percentage of coverage was done using the ImageJ program from the National Institutes of Health.
Electron microscopy. Mice were deeply anesthetized with avertin (0.2 ml/10 g body weight, i.p., of a 1.25% solution), perfused with PBS, and fixed in 2.5% glutaraldehyde and 2% paraformaldehyde. Sternomastoid muscles containing nerve terminals were dissected, treated, and serial sectioned. Sections were photographed at 5000-10,000× magnification with a Phillips (Eindhoven, The Netherlands) CM 120 electron microscope, and the developed negatives were scanned at 600 dpi on an MICROTEK NuScan 1200. Twenty randomly selected profiles of various sizes from each mouse and two mice per genotype were selected for quantification.
Electrophysiology. Postnatal day 0 (P0) sternomastoid muscles were studied in a chamber perfused with oxygenated saline containing (in mm) 145 NaCl, 2 KCl, 1 MgSO4, 2 CaCl2, 10 NaHEPES, and 10 glucose, pH 7.1, at 19-22°C. Nerves were stimulated by a suction electrode via an isolation unit (WPI Instruments, Waltham, MA) triggered by pClamp software at low frequency (<0.5/s). Signals from μ-conotoxin GIII-blocked muscle fibers impaled with KCl-filled micropipettes (15-45 MΩ) were amplified (Axoclamp-2A; Axon Instruments, Union City, CA) and digitized at 30 μs (for evoked release) or 100-500 μs (for spontaneous release). Fibers with membrane potential below -50 mV, stable end plate potential (EPP) morphology with rise time ≤1 ms, and low stimulation thresholds for evoking EPPs (typically ≤1 mA) were analyzed.
Results
Impaired apposition of presynaptic proteins with postsynaptic acetylcholine receptors in APP/APLP2 null NMJ
Two sets of breeding were performed: in the first set, the APP heterozygous (APP+/-) mice were intercrossed to generate wild-type (WT) and APP null (APP-/-) animals; in the second set, mice heterozygous for APP and homozygous null for APLP2 (APP+/- APLP2-/-) were interbred to create mice that were wild type for APP and homozygous knock-out for APLP2 (APLP2-/-) or APP and APLP2 double null (APP-/-APLP2-/-, herein referred to as dKO or APP/APLP2 null). At birth, neither the single nor double knock-outs were distinguishable from the WT controls. This stage was chosen for most of the analyses.
The mammalian NMJ is a highly specialized synaptic structure in which the presynaptic terminals of motor neuron axons are closely apposed to postsynaptic acetylcholine receptors (AChRs) of the muscle fiber (Sanes and Lichtman, 1999). To examine NMJ structures, sternomastoid muscle sections of P0 mice were stained with an antibody against Syn, a synaptic vesicle protein that labels presynaptic nerve terminals. For postsynaptic end plate, Texas Red-conjugated α-BTX was used to label AChRs. In the WT NMJ, AChR clusters were formed at the end plate band of the muscle fiber and were closely apposed by Syn-containing synaptic vesicles (Fig. 1Aa-c). A similar staining pattern was observed in both the APP-/- (Fig. 1Ad-f) and APLP2-/- (Fig. 1Ag-i) NMJ. In contrast, the APP/APLP2 dKO NMJ exhibited a significantly sparse pattern of Syn immunoreactivity (Fig. 1Aj) and greatly reduced coverage with postsynaptic AChR clusters (Fig. 1Al). AChR-positive end plates appeared to be normal (Fig. 1Ak).
Figure 1.

Characterization and quantification of P0 sternomastoid muscles. A, Double labeling with the anti-Syn antibody and α-BTX of WT, APP-/-, APLP2-/-, and APP/APLP2 double knock-out (dKO) animals. Scale bar, 5 μm. B, SV2 and α-BTX double staining of littermate APLP2-/- and dKO NMJs. The arrowheads in d and f mark the SV2 staining beyond the end plate. C, Quantification of the percentage of AChR-positive end plates covered by SV2 immunoreactivity (average ± SD of 20 end plates per genotype). *p < 0.01; t test. D, Western blot analysis of P0 spinal cord proteins using an anti-APP C-terminal antibody (APP) or anti-SV2 antibody (SV2). Anti-tubulin (Tubulin) Western blot was used as the loading control. The images were captured by a confocal microscope and displayed either as individual staining or merged images.
To determine whether the sparse presynaptic marker staining in the absence of APP/APLP2 is specific for Syn, muscle sections were stained with antibody to another synaptic vesicle protein, SV2. Similar to Syn staining, the SV2 immunoreactivity was comparable among wild type, APP-/- (data not shown), and APLP2-/- (Fig. 1Ba), whereas the APP/APLP2 dKO NMJ showed sparse SV2 staining at the end plate and abnormal apposition with postsynaptic AChRs (Fig. 1Bd-f). Staining of >50 APP/APLP2 null end plates documented that this phenotype can be invariably identified. Quantitative analysis established that the percentages of coverage of α-BTX-positive end plate by SV2 immunoreactivity in the APLP2 null and APP/APLP2 dKO NMJs were ∼60 and 25%, respectively (Fig. 1C). This reduction at the end plate is not attributable to reduced protein expression because Western blot analysis showed comparable levels of SV2 in the control (APLP2-/-) and dKO samples (Fig. 1D). Similar presynaptic abnormalities were observed with other synaptic marker proteins, including synapsin and synaptotagmin (data not shown). Double labeling using antibodies against SV2 and Syn found that they were colocalized in all four genotypes (data not shown). These observations suggest that the APP family of proteins plays a general role in regulating synaptic vesicles at motor neuron terminals.
In addition to the sparse staining at the end plate, the immunoreactivity of the synaptic vesicle proteins in the APP/APLP2 null NMJ often extended beyond the end plate (Fig. 1B, arrowheads). To substantiate this finding, we performed triple labeling of Syn, NF, and α-BTX of P0 sternomastoid muscle sections (Fig. 2). In WT (Fig. 2a-d), APP-/- (Fig. 2e-h), and APLP2-/- (Fig. 2i-l) preparations, the presynaptic distributions were highly compatible with postsynaptic AChR clusters (Fig. 2, compare a with c, e with g, and i with k), and the NF-positive nerve branches mostly terminated at the end plates (Fig. 2d,h,l). In APP/APLP2 dKO samples (Fig. 2m-p), two abnormalities were readily detectable: (1) the presynaptic distribution was dramatically different from the other genotypes (Fig. 2, compare m with a, e, i), and there was impaired patterning of the presynaptic protein with postsynaptic end plates (Fig. 2, compare m, o); and (2) significant NF-positive nerve terminals, positive for Syn, escaped from the NMJs and extended beyond end plates (Fig. 2m-p, arrowheads). Thus, aberrant presynaptic structure and nerve terminal sprouting are two phenotypes present in APP/APLP2 null NMJs.
Figure 2.

Triple labeling of P0 WT, APP-/-, APLP2-/-, and APP/APLP2 dKO sternomastoid muscles with anti-Syn antibody, anti-NF antibody, and α-BTX. The arrowheads in m, n, and p denote a representative Syn-positive nerve sprouted beyond the end plate. The images were captured by a confocal microscope and displayed either as individual staining or merged images. Scale bar, 20 μm.
Because APLP2-/- animals were indistinguishable from the WT mice as judged by immunostaining methods (Figs. 1, 2), littermate APLP2 null mice were used as controls in future studies.
Reduced synaptic vesicle density in APP/APLP2 null NMJ
Having identified a presynaptic defect in the APP/APLP2 dKO NMJ, we performed serial electron microscopy (EM) analysis of the P0 sternomastoid muscles. Consistent with the sparse staining patterns of presynaptic marker proteins, we observed a dramatic reduction in synaptic vesicle number in presynaptic terminals of APP/APLP2 null mice (Fig. 3A,B). Quantitatively, the vesicle density (number of synaptic vesicles per square micrometer of profile area) was reduced by 2.1-fold (APLP2 null, 80.15 ± 2.02; APP/APLP2 null, 37.83 ± 2.23) (Fig. 3D). Although active zones were formed in the APP/APLP2 null NMJ (Fig. 3C), a reduction in the number of active zones per profile length was observed (APLP2-/-, 0.95 ± 0.05; dKO, 0.497 ± 0.035) (Fig. 3E). The number of docked vesicles (defined as the vesicles within 150 nm from the plasma membrane in an active zone) was similar between the APLP2 null and the APP/APLP2 null NMJ (Fig. 3F). This finding establishes an important role of APP/APLP2 in the control of synaptic vesicles at the presynaptic terminals.
Figure 3.

EM analysis of presynaptic terminal structures of P0 sternomastoid muscles. A, B, Representative EM images showing reduced synaptic vesicles in APP/APLP2 dKO terminals compared with littermate APLP2-/- control terminals of similar sizes. C, Representative electron-dense active zones, which could be identified in both genotypes. D, Quantification of synaptic vesicle densities (number of vesicles per square micrometer of profile area). *p < 0.001; Student's t test. E, Quantification of the number of active zones per micrometer of profile length. *p < 0.001; Student's t test. F, Number of docked vesicles per active zone. All calculations were done on a per section basis. Each column represents mean ± SE of 40 profiles from two animals. Scale bars: A, B, 1 μm; C, 200 μm.
Excessive nerve terminal sprouting in the absence of APP/APLP2
The mammalian NMJ undergoes dynamic changes during development. At embryonic day 14.5 (E14.5), the nerve branches of motor axon bundles initiate contact with muscle fibers. However, specialized interactions between the two cell types have not been established at this developmental time, and the AChR-rich end plates exist in a nerve-independent manner (Lin et al., 2001). In contrast, at E16.5 and onward, the nerve terminals play an active role in stimulating and clustering AChRs at the sites of nerve-muscle contact, leading to close apposition of postsynaptic AChR clusters to presynaptic terminals (Sanes and Lichtman, 1999).
The above analysis of sternomastoid muscle sections revealed a terminal sprouting defect in the APP/APLP2 null NMJ (Figs. 1, 2). To characterize this phenotype on a whole-mount level and to determine the onset of the terminal sprouting, we examined the intramuscular nerves of the diaphragm at various developmental stages by immunostaining with an anti-NF antibody (Fig. 4). At all stages examined, the main nerve structures were comparable between the APLP2-/- control and APP/APLP2 dKO mutant. However, excessive branch sprouting can be seen in APP/APLP2 null samples at P0 (Fig. 4, compare a, b), E18.5 (data not shown), and E16.5 (Fig. 4, compare c, d), stages when neuromuscular synapses have been established. Interestingly, the branch pattern of the nerve was similar in APLP2 null and dKO at E14.5 (Fig. 4e,f), when nerve-muscle contacts were initiated but not specialized (Lin et al., 2001). Similar to results obtained from sternomastoid muscles, comparison of APLP2-/-, APP-/-, and WT animals of the diaphragm by NF staining at all developmental stages revealed indistinguishable patterns (data not shown). We reasoned that, if sprouting were the primary phenotype, it should be independent of synapse specialization (i.e., such a defect is expected to present at all developmental stages). The fact that terminal sprouting only becomes evident after the onset of synapse specialization supports the idea that abnormal synapse formation or maturation might be the underlying trigger for the terminal sprouting phenotype. However, it is also plausible that the two phenotypes arise from independent mechanisms with similar timing.
Figure 4.
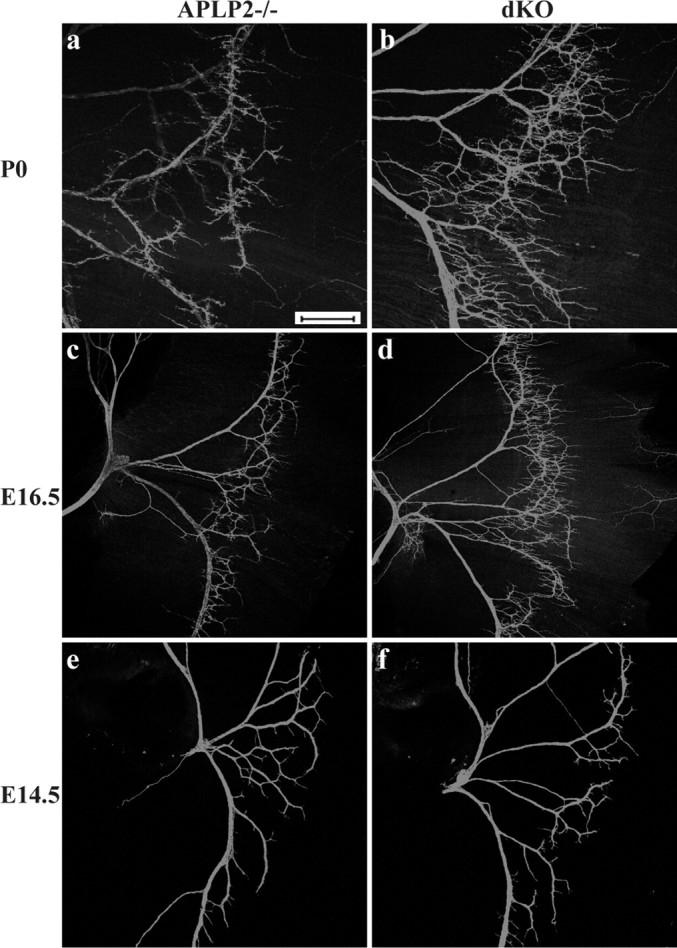
Whole-mount NF staining of P0 (a, b), E16.5 (c, d), and E14.5 (e, f) diaphragm of littermate APLP2-/- control and APP/APLP2 dKO is shown. Nerve terminal sprouting is apparent in E16.5 and P0 but not in E14.5 dKO samples. Scale bar, 400 μm.
Excessive nerve terminal sprouting led to widening of the end plate band in the APP/APLP2 dKO NMJ, as revealed by costaining of NF with α-BTX (Fig. 5A,B, quantified), and diffused presynaptic distribution, shown by Syn immunoreactivity (Fig. 5C). However, synapse numbers quantified by the total number of AChR-positive end plates within 5.0 × 105 μm2 muscle area was not significantly different (218 ± 17 and 245 ± 21 for APLP2 null and APP/APLP2 dKO, respectively; average ± SD of three animals per genotype; p = 0.16; t test). A diffused synaptic staining pattern was in agreement with results obtained from sternomastoid muscles (Fig. 2), indicating that a significant amount of synaptic vesicles extended beyond the synapses to sprouted nerve branches. The abnormal patterning of synapses was further confirmed by acetylcholinesterase histochemistry (Fig. 5D), which serves as a reliable marker for synaptic sites (Brandon et al., 2003).
Figure 5.

Examination of synaptic distribution of the diaphragm muscle. A, Double labeling with the anti-NF antibody (a, d) and α-BTX (b, e), revealing excessive terminal sprouting and widened AChR-positive end plate band in APP/APLP2 dKO animals. B, Quantification of AChR cluster distribution from the medial edge of the diaphragm (mean ± SD of 3 animals per genotype). *p < 0.05; t test. C, Syn and α-BTX double staining, which showed broadened presynaptic distribution. D, Acetylcholine esterase histochemistry, documenting diffused synaptic patterning. The right columns in A and C are the merged images of the first two columns. Scale bars: A, 50 μm; C, D, 100 μm.
The neuromuscular synapses form as a result of complex interactions between nerve terminals, Schwann cells, and muscle fibers. Labeling of Schwann cells with anti-S100 antibody revealed a similar pattern as NF in both the control and the dKO mutant, suggesting that Schwann cells were distributed in close proximity with nerve terminals (data not shown). Muscle histology was comparable in the APP/APLP2 null and the control (data not shown). These data combined support a primary effect of APP/APLP2 at the nerve terminals.
Functional analysis of APP/APLP2 null NMJ
To determine whether the structural abnormalities described above lead to functional impairment in synaptic transmission, intracellular recordings of P0 sternomastoid muscle fibers were performed. Consistent with the immunostaining results, no appreciable differences between WT and APLP2-/- samples could be identified (data not shown). However, compared with littermate APLP2 null animals, dramatically reduced miniature EPP (MEPP) frequency was observed in APP/APLP2 null mice (Fig. 6Aa). Similar differences in MEPP frequency were seen in preparations blocked with μ-conotoxin GIII (data not shown). Resting membrane potentials (data not shown) and MEPP amplitudes (Fig. 6Ab) of sampled fibers were similar. The threshold current applied to the nerve to evoke a muscle response and the latency from the shock artifact to the initiation of the EPP were similar among all groups (data not shown). Significantly, the proportion of fibers in which an EPP could not be evoked by supramaximal nerve stimulation was much greater in APP/APLP2 null mice than in control littermates (Fig. 6B), and these fibers also lacked MEPPs. These findings suggest a defect of presynaptic terminal function at NMJs of APP/APLP2 null mice, with a substantial proportion of nonfunctioning synapses.
Figure 6.

Electrophysiological recordings from P0 sternomastoid muscle fibers of APP/APLP2 double null (dKO) mice and control littermates (APLP2-/-). A, MEPP recordings. Aa, Reduced MEPP frequency (mean ± SD) in dKO (n = 8) compared with control (n = 7); *p < 0.001. Over one-half of the fibers in this analysis did not have detectable MEPPs. Ab, Similar MEPP amplitude in APLP2-/- control and APP/APLP2 null (dKO) mutant (mean ± SD); p = 0.23. B, EPP recordings. Ba, Percentage of fibers in which an evoked response (EPP or action potential) could not be induced by supramaximal nerve stimulation. Control (APLP2-/-) fibers, 1 of 41; double null (dKO) fibers, 8 of 31. *p < 0.005. Bb, Representative EPP evoked in a sternomastoid fiber by stimulation of the muscle nerve, with μ-conotoxin present in the bath. Top trace, EPP from an APLP2-/- control; bottom trace, EPP from a littermate mutant (dKO), at 10 times the threshold required to evoke responses in adjacent muscle fibers.
Discussion
We report here an essential role of the APP family of proteins in developing neuromuscular synapses. We show that APP/APLP null NMJs exhibit abnormal apposition of presynaptic proteins with postsynaptic AChR clusters and a dramatically reduced number of synaptic vesicles at presynaptic terminals. Electrophysiological recordings establish a functional deficit of the mutant in synaptic transmission. In addition to aberrant synapses, excessive nerve terminal sprouting is another feature present in the APP/APLP2 dKO NMJ. These phenotypes are, to a large degree, overlapped with mice deficient in choline acetyltransferase (ChAT), the biosynthetic enzyme for acetylcholine. Analysis of the ChAT knock-out animals documents that lack of synaptic transmission attributable to the absence of neurotransmitter results in nerve terminal sprouting and aberrant synaptic patterning (Misgeld et al., 2002; Brandon et al., 2003). It is hypothesized that the release of acetylcholine from the nerve terminals acts on muscle to arrest nerve growth (Brandon et al., 2003). Thus, it is reasonable to speculate that defective synapse formation in the absence of APP/APLP2 leads to reduced synaptic transmission, followed by nerve terminal sprouting. However, current studies do not prove a cause-effect relationship. It is also possible that impaired synaptic transmission is the primary phenotype for the synapse formation/maturation and nerve terminal sprouting defects seen in APP/APLP2 null NMJs.
Intracellular recording of P0 sternomastoid muscle revealed a dramatic reduction of MEPP frequency and a higher percentage of fibers lacking EPP in the APP/APLP2 null NMJ. These data implicate a presynaptic defect and are consistent with our ultrastructural analysis documenting a decreased number of synaptic vesicles and active zones. Similar MEPP amplitudes in APP/APLP2 null and control littermates suggest that a significant postsynaptic deficit in APP/APLP2 null NMJs is unlikely, although subtle differences cannot be ruled out. Intriguingly, among fibers that showed evoked responses, the EPP amplitude did not differ significantly between the dKO mutant (23 ± 10 mV; n = 17 fibers) and the control (27 ± 8 mV; n = 19 fibers) (p = 0.25), indicating normal quantal content in this subpopulation of fibers. By combining with our EM data, we provide two possible explanations for this phenomenon: (1) a reduction in the number of active zones in the APP/APLP2 null NMJ may lead to a compensatory increase in the release probability, resulting in apparent normal quantal content (Urbano et al., 2003); and (2) the normal number of docked vesicles in APP/APLP2 null active zones may be sufficient to produce normal EPP amplitude after initial stimulation. Additional analysis, such as paired-pulse and repetitive stimulation, is required to allow a definitive answer to this question.
The NMJ phenotypes in APP/APLP2 mutants are somewhat distinct from Appl null Drosophila, because the latter exhibits a subtle reduction of synaptic bouton numbers without structural alterations (Torroja et al., 1999). Whether the mutant fly is functionally impaired is not clear. The relative mild phenotype could be attributed by compensatory mechanisms through a yet unidentified family member. Indeed, neither APP nor APLP2 single knock-out mice show overt NMJ defect. In this regard, it is also interesting to note that both Appl and APP null mutants are viable, whereas mice deficient in APP and APLP2 are early postnatal lethal.
APP has been reported to function as a receptor for kinesin-mediated axonal transport (Kamal et al., 2000, 2001; Gunawardena and Goldstein, 2001). Loss of Appl in Drosophila leads to vesicle “clogging” along the axons. Thus, defective vesicle trafficking can be considered as a candidate mechanism for the reduced synaptic vesicles at the nerve terminals of the APP/APLP2 null NMJ. The early postnatal lethal phenotype of the APP/APLP2 null mice prevents direct measurement of the axonal transport rate using the sciatic ligation method as reported previously (Kamal et al., 2001). Nevertheless, examination of individual motoneuron axons within a main phrenic nerve bundle by NF staining and high-power confocal microscopic imaging revealed uniform and indistinguishable patterns of axonal structures in the control and APP/APLP2 dKO samples across the entire region surveyed (total, 400 μm in length; data not shown), indicating that there was no axonal swelling caused by impaired trafficking. Our findings that total levels of Syn immunoreactivity at the end plate bands were comparable between the control and APP/APLP2 null (Fig. 5C), and that synaptic vesicle proteins were not only transported to the nerve terminals but were also expressed in sprouted nerves beyond end plates in the mutant (Fig. 2), argue against a major function of APP/APLP2 in synaptic vesicle trafficking during NMJ development. Other defects, including vesicle biogenesis, vesicle release, or endocytosis, are possible contributing factors for the presynaptic phenotype in the APP/APLP2 null NMJ.
APP as well as its family members APLP1 and APLP2 undergoes extracellular and presenilin-dependent intramembrane cleavages to generate secreted APP (sAPP), Aβ peptides, and APP intracellular domain (AICD). sAPP has been suggested to exhibit neurotrophic, synaptogenic, and growth-promoting properties (Jin et al., 1994; Ohsawa et al., 1999; Rossjohn et al., 1999), and AICD has been reported to function in transcriptional regulation (Cao and Sudhof, 2001). A potential role of presenilin-dependent APP processing and signaling cannot be tested directly because of the early lethal phenotype of PS1/PS2 double null embryos (Donoviel et al., 1999; Herreman et al., 1999) and a large number of potential PS processing substrates reported (for review, see De Strooper, 2003). Preliminary assessment of the PS1 null NMJ in late embryogenesis did not identify any overt abnormalities (data not shown). Finally, it remains a possibility that the large secreted form of APP is the major functional moiety responsible for the effects seen in this study, because its level coincides with synaptogenesis (Moya et al., 1994).
In summary, we present here an indispensable role of the APP family of proteins in the formation, maturation, and function of neuromuscular synapses. Although the exact mechanism is under intensive study, this is the first report documenting an in vivo physiological function of the APP family of proteins in the mammalian system. In addition to the questions raised for NMJ development, the presynaptic effects have direct implications for a similar role of APP/APLP2 in CNS synapse as well as synaptic dysfunction contributing to AD dementia.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS40039, AG20670, and AG21141 (H.Z.), by NIH Grant NS041846 (W.-B.G.), by the Alzheimer's Association (W.-B.G.), and by a Veterans Health Administration Merit Review Award (D.R.M.). H.Z. is a New Scholar of the Ellison Medical Foundation and a Zenith fellow of the Alzheimer's Association. We are grateful to D. Beasley, S. Memon, and M. An for expert technical support, R. Atkinson for confocal image assistance, and L. Yang for advice.
Correspondence should be addressed to Dr. Hui Zheng, Huffington Center on Aging, Baylor College of Medicine, One Baylor Plaza, M320, Houston, TX 77030. E-mail: huiz@bcm.tmc.edu.
P. Wang's present address: Department of Developmental Biology, Stanford University School of Medicine, Stanford, CA 94305.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251219-07$15.00/0
P.W. and G.Y. contributed equally to this work.
References
- Akaaboune M, Allinquant B, Farza H, Roy K, Magoul R, Fiszman M, Festoff BW, Hantai D (2000) Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol Cell Neurosci 15: 355-367. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23: 539-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Sudhof TC (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293:115-120. [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C (1993) apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 90: 12045-12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ (1999) Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein [In Process Citation]. Neuroscience 90: 1-13. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron 38: 9-12. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A (1999) Mice lacking both presenilin genes exhibit early embryonic patterning defects [In Process Citation]. Genes Dev 13: 2801-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila Neuron 32: 389-401. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U (2000) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20: 7951-7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency [In Process Citation]. Proc Natl Acad Sci USA 96: 11872-11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Ninomiya H, Roch JM, Schubert D, Masliah E, Otero DA, Saitoh T (1994) Peptides containing the RERMS sequence of amyloid beta/A4 protein precursor bind cell surface and promote neurite extension. J Neurosci 14: 5461-5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28: 449-459. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414: 643-648. [DOI] [PubMed] [Google Scholar]
- Kirazov E, Kirazov L, Bigl V, Schliebs R (2001) Ontogenetic changes in protein level of amyloid precursor protein (APP) in growth cones and synaptosomes from rat brain and prenatal expression pattern of APP mRNA isoforms in developing rat embryo. Int J Dev Neurosci 19: 287-296. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 87: 1561-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF (2001) Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410: 1057-1064. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry R (1993) The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol 3: 77-85. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635-648. [DOI] [PubMed] [Google Scholar]
- Moya KL, Benowitz LI, Schneider GE, Allinquant B (1994) The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol 161: 597-603. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Morimoto T, Ishiguro M, Kohsaka S (1999) Aminoterminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci 11: 1907-1913. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M (1999) Aged APP-null mice exhibit a learning impairment which is not mediated by a loss of hippocampal neuron or synaptic bouton number. Neuroscience 90: 1207-1216. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Martin-Morris L, Luo LQ, White K (1989) A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 86: 2478-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW (1999) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6: 327-331. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389-442. [DOI] [PubMed] [Google Scholar]
- Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters CL, Beyreuther K (1991) Localization of Alzheimer beta A4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res 563: 184-194. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG (1999) Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein [In Process Citation]. Neuropharmacology 38: 349-359. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791. [DOI] [PubMed] [Google Scholar]
- Shigematsu K, McGeer PL, McGeer EG (1992) Localization of amyloid precursor protein in selective postsynaptic densities of rat cortical neurons. Brain Res 592: 353-357. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL (1993) Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci 13: 3136-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS (1994) Expression of a ubiquitous, cross-reactive homologue of the mouse betaamyloid precursor protein (APP). J Biol Chem 269: 2637-2644. [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL (1987) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235: 880-884. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572-580. [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V (1999) The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci 19: 7793-7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW (2003) Altered properties of quantal neurotransmitter release at end-plates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci USA 100: 3491-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging 18: 661-669. [DOI] [PubMed] [Google Scholar]
- Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F (1992) Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA 89: 10758-10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE (1993) Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet 5: 95-100. [DOI] [PubMed] [Google Scholar]
- Xia X, Wang P, Sun X, Soriano S, Shum WK, Yamaguchi H, Trumbauer ME, Takashima A, Koo EH, Zheng H (2002) The aspartate-257 of presenilin 1 is indispensable for mouse development and production of {beta}amyloid peptides through {beta}-catenin-independent mechanisms. Proc Natl Acad Sci USA 99: 8760-8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Selkoe DJ, Koo EH (1995) Trafficking of cell surface betaamyloid precursor protein: retrograde and transcytotic transport in cultured neurons. J Cell Biol 129: 431-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, et al. (1995) Betaamyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81: 525-531. [DOI] [PubMed] [Google Scholar]