

Abstract
The dual nature of the NMDA receptor as a mediator of excitotoxic cell death and activity-dependent cell survival likely results from divergent patterns of kinase activation, transcription factor activation, and gene expression. To begin to address this divergence, we examined cellular and molecular signaling events that couple excitotoxic and nontoxic levels of NMDA receptor stimulation to activation of the cAMP response element-binding protein (CREB)/cAMP response element (CRE) pathway in cultured cortical neurons. Pulses (10 min) of NMDA receptor-mediated synaptic activity (nontoxic) triggered sustained (up to 3 h) CREB phosphorylation (pCREB) at serine 133. In contrast, brief stimulation with an excitotoxic concentration of NMDA (50 μm) triggered transient pCREB. The duration of pCREB was dependent on calcineurin activity. Excitotoxic levels of NMDA stimulated calcineurin activity, whereas synaptic activity did not. Calcineurin inhibition reduced NMDA toxicity and converted the transient increase in pCREB into a sustained increase. In accordance with these observations, sustained pCREB (up to 3 h) did not require persistent kinase pathway activity. The sequence of stimulation with excitotoxic levels of NMDA and neuroprotective synaptic activity determined which stimulus exerted control over pCREB duration. Constitutively active and dominant-negative CREB constructs were used to implicate CREB in synaptic activity-dependent neuroprotection against NMDA-induced excitotoxicity. Together these data provide a framework to begin to understand how the neuroprotective and excitotoxic effects of NMDA receptor activity function in an antagonistic manner at the level of the CREB/CRE transcriptional pathway.
Keywords: calcium, CREB, apoptosis, MAPK, CaMK, neuron, neuroprotection
Introduction
Excitatory neurotransmission is a critical regulator of both the development and maintenance of normal nervous system physiology. For example, neuronal activity facilitates cell survival, suppresses apoptotic cell death (Catsicas et al., 1992; Sherrard and Bower, 1998; West et al., 2002), functions as a guidance cue during neuronal migration, and refines synaptic circuitry (Katz and Shatz, 1996; Behar et al., 1999; Cohen-Cory, 2002; Cancedda et al., 2003). As the dominant fast excitatory neurotransmitter, glutamate is a key regulator of these processes (Barry and O'Donovan, 1987; Barger and Mattson, 1995). Its capacity to drive synaptic communication is derived in part by the NMDA subtype of ionotropic glutamate receptor. The NMDA receptor functions as a cellular “gate” that allows Ca2+ to enter the cell only under the correct spatial and temporal pattern of synaptic activity (for review, see Nakanishi, 1992; Malenka and Nicoll, 1993; Cull-Candy et al., 2001). In turn, elevated intracellular Ca2+ triggers the expression of new genes, thus converting brief NMDA receptor stimulation into long-term changes in CNS physiology.
NMDA receptor stimulation can protect against cell death resulting from both the withdrawal of trophic factors and excitotoxic insults (Balazs et al., 1988; Chuang et al., 1992; Rocha et al., 1999; Raval et al., 2003). NMDA receptor-mediated neuroprotection is a transcriptionally dependent process (Marini and Paul, 1992) that appears to be dependent, in part, on the expression of both the anti-apoptotic gene BCL-2 (Mabuchi et al., 2001; Alavez et al., 2003) and the neurotrophin BDNF (Aliaga et al., 1998; Rocha et al., 1999). A number of studies have shown that the cAMP response element-binding protein (CREB)/cAMP response element (CRE) transcriptional pathway regulates the expression of both BCL-2 and BDNF (Wilson et al., 1996; Shieh et al., 1998; Tao et al., 1998; Riccio et al., 1999). With respect to the role of the CREB/CRE transcriptional pathway in neuroprotection, Hara et al. (2003) found that CRE-mediated gene expression is necessary for the induction of ischemic tolerance. Likewise, Mabuchi et al. (2001) showed that the neuroprotective effects of an ischemic preconditioning stimulus involve both NMDA receptor stimulation and CREB-dependent transcription. These observations suggest that a signaling cassette formed by the NMDA receptor and the CREB/CRE transcriptional pathway contributes to activity-dependent neuroprotection.
These neuroprotective effects appear to be mediated specifically by synaptic NMDA receptors; activation of extrasynaptic NMDA receptors has been shown to trigger a transient form of CREB phosphorylation and apoptotic cell death (Hardingham et al., 2002). Although these studies suggest a role for both synaptic activity and CREB-dependent transcription in neuroprotection, issues regarding the signaling events that regulate the induction and duration of NMDA receptor-evoked CREB phosphorylation, as well as how stimulus intensity triggers protection or cell death, have yet to be addressed. The data presented here provide new mechanistic insights into the connections between the NMDA receptor/CREB signaling cassette and the induction of excitotoxic tolerance.
Materials and Methods
Tissue culture and transfection. Embryonic day 19-20 Sprague Dawley rat pups were decapitated, their brains were removed, and the cortices were isolated and placed in dissociation media [DM; containing the following (in mm): 90 Na2SO4, 30 K2SO4, 16 MgCl2, 0.25 CaCl2, 32 HEPES, and 0.01% phenol red (Sigma, St. Louis, MO), pH 7.7]. Tissue then was washed three times in DM, finely minced with a razor blade, and digested with a mild protease solution [100 U/ml papain latex (Worthington, Freehold, NJ) and 4.5 mg of cysteine (Sigma) in DM] at 37°C for 30 min. After removal of the digestion solution, the tissue was washed three times in DM and transferred to tissue culture medium [minimal essential medium (MEM); Invitrogen, San Diego, CA] containing 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. Then the tissue was triturated into a single-cell suspension via a 5 ml serological pipette and plated at a density of 2.2 × 105 cells/cm2 on poly-l-lysine-coated (>540 kDa; Sigma) glass coverslips or tissue culture dishes. At 2 h after plating, the medium was replaced with MEM containing 2% B-27 (Invitrogen), 1% fetal bovine serum, and 100 U/ml penicillin/streptomycin. Cultures were maintained at 37°C and 5% CO2 in a Napco (Winchester, VA) 6100 incubator. Experiments were performed after 10-12 d in vitro (DIV). All animal procedures were in accordance with The Ohio State University animal welfare guidelines.
Fura-2 calcium digital imaging. Time-lapse ratiometric fluorescent digital microscopy was performed as described previously (Obrietan and van den Pol, 1998). Ratiometric single-cell imaging data were collected with MetaFluor software (Universal Imaging, West Chester, PA). Data acquisition was performed every 5 s. Cells were maintained in a HEPES-based buffer containing the following (in mm): 137 NaCl, 25 glucose, 10 HEPES, 5 KCl, 1 MgCl2, and 3 CaCl2, pH 7.4. To facilitate NMDA receptor activity during synaptic activity assays and NMDA administration, we added glycine (2 μm) and removed Mg2+ from the perfusion medium. Tetrodotoxin (TTX; 1 μm) was added to the perfusion solution to block spontaneous synaptic activity during NMDA administration.
Western blotting. Cultured cells were transferred from tissue culture medium to a HEPES perfusion solution containing 1 μm TTX 30 min before stimulation. Cells were stimulated for 10 min and lysed in 200 μl of hot (95°C) 3× SDS sample buffer. Lysates were stored at -80°C until use. Before being loaded, the lysates were heated to 95°C for 10 min, vortexed (10 s), and centrifuged for 7 min at 15,000 × g. Extracts (20 μl/lane) were electrophoresed into a 10% SDS polyacrylamide gel and transblotted onto polyvinylidene fluoride (Immobilon P; Millipore, Bed-ford, MA). Next the membranes were washed with 5% (w/v) powdered milk dissolved in PBS with 0.1% Triton X-100 (PBST), followed by incubation (4°C overnight) with one of the following antibodies: affinity-purified rabbit polyclonal anti-CREB phosphorylated at Ser133 (1:1000 final dilution; Cell Signaling Technology, Beverly, MA), mouse monoclonal anti-extracellular signal-regulated kinase-1 (ERK-1) and anti-ERK-2 phosphorylated at Thr202 and Tyr204 (1:5000; Sigma), or mouse monoclonal anti-Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) phosphorylated at Thr196 (acquired from Dr. Thomas R. Soderling, Vollum Institute, Portland, OR). Next the membranes were washed and incubated [4 h at room temperature (RT)] with horseradish peroxidase (HRP)-conjugated secondary antibodies directed against the IgG domains of the primary antibodies (1:2000; PerkinElmer Life Sciences, Norwalk, CT). The signal was visualized by using Renaissance chemiluminescent HRP substrate (PerkinElmer Life Sciences). As a control for equal loading of protein across the gel, the membranes were stripped and probed for total ERK expression with a rabbit polyclonal antibody against ERK-1 and ERK-2 (1:1000 final dilution; Santa Cruz Biotechnology, Santa Cruz, CA). ERK expression was revealed with a goat anti-rabbit IgG antibody (1:2000 final dilution; PerkinElmer Life Sciences) conjugated to HRP, followed by visualization as described above. Blots were washed a minimum of six times (5 min per wash) in PBST with 5% milk after each antibody treatment. Quantitation of band intensity was performed with Scion Image analysis software (Frederick, MD). Band intensity was normalized to total ERK-1 for the corresponding lane. Each experiment was repeated a minimum of three times.
Immunostaining. After stimulation, the cells were fixed with 4% (w/v) formaldehyde for 15 min, washed five times in PBST, and then blocked (2 h at RT) with 1% normal goat serum and 10% bovine serum albumin in PBST containing 0.02% azide. Cultures were immunolabeled (overnight at 4°C) with an affinity-purified rabbit polyclonal CREB phosphorylation (pCREB) antibody (1:500; Cell Signaling Technology), a monoclonal antibody raised against the neuronal nuclear marker neuronal-specific nuclear protein (NeuN) (1:500; Chemicon, Temecula, CA), or a rabbit polyclonal antibody raised against green fluorescent protein (GFP; 1:5000 final dilution; acquired from Dr. Luc Berthiaume, University of Alberta, Edmonton, Canada). Next the cells were washed and incubated (4 h at RT) with an Alexa 488- or 594-conjugated secondary antibody (1:250 final dilution; Molecular Probes, Eugene, OR). To examine cell health, we also incubated cultures (10 min) with the DNA stain Hoechst 33342 (Hoechst, 1 μg/ml; Molecular Probes) before mounting them with gel mount (Biomedia, Foster City, CA). Photomicrographs were captured by a 16-bit digital camera (Micromax YHS 1300; Princeton Instruments, Trenton, NJ) connected to an inverted epifluorescence microscope [Leica (Nussloch, Germany) DM IRB at 200× magnification]. All tabulated data are expressed as the mean ± SEM. Significance was determined by using the two-tailed Student's t test.
Transfection and reporter gene assays. Primary cultured cortical neurons were transfected after 8 DIV (1.5 μg of DNA per well) by using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. The following constructs were used: pcDNA3 VP16-CREB, pcDNA3 A-CREB, pcDNA3.1 (empty vector), pGL3-CRE-luciferase, and pEGFP-N3. Cells were assayed 48 h after transfection. Luciferase activity was measured by using the Bright-Glo Luciferase Assay System (Promega, Madison, WI).
Full-length NF-ATc4 was amplified by PCR from a mouse brain cDNA library and cloned in frame into the pcDNA3.1/CT-GFP expression vector (Invitrogen). Cells were assayed 48 h after transfection. Cells were immunostained for GFP and for the neuronal marker MAP2 (1:2000; Sigma). Fluorescent images were captured with a Zeiss (Oberkochen, Germany) 510 Meta confocal microscope (2-μm-thick optical section). The nuclear/somatic GFP signal was determined for each neuron and expressed as the mean ratio for each condition.
Cell toxicity assay. Cell toxicity was determined by measuring lactate dehydrogenase (LDH) release as described by Koh and Choi (1987). The percentage of LDH release was calculated as LDH in the culture medium divided by total LDH (cellular plus medium LDH). Toxicity also was measured by labeling with the DNA stain Hoechst as described above. For the quantification of apoptotic cell death, the regions were selected randomly as described above, and the viability of all neurons (NeuN-positive) within the region was determined. Neurons with fragmented or condensed nuclei were scored as dead or dying (apoptotic).
Materials. Pyruvate, nicotinamide adenine dinucleotide, NMDA, glutamate, nimodipine, bicuculline, 4-aminopyridine (4-AP), TTX, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) disodium salt, and 2-amino-5-phosphonopentanoic acid (APV) were acquired from Sigma. Bisindolylmaleimide, U0126, KN62, KN93, SP600125, and staurosporine were acquired from Calbiochem (La Jolla, CA). H89, FK506, LY294002, PD98059, Rp-cAMPs, and SB203580 were acquired from LC Laboratories (Woburn, MA). Hoechst 33342 and fura-2 were acquired from Molecular Probes.
Results
The NMDA receptor couples synaptic activity to CREB phosphorylation
To begin to examine the receptors that mediate synaptically evoked CREB phosphorylation, we prepared cortical neuronal cultures from embryonic day 20 rats and maintained them in vitro for 10-12 d. Several groups have shown that 10 DIV cortical neurons form functional synaptic connections and secrete both excitatory and inhibitory amino acid neurotransmitters (Segal and Barker, 1984; Wilcox et al., 1994; Obrietan and van den Pol, 1995; Craig et al., 1996). Fura-2 timelapse digital microscopy was used to monitor neuronal Ca2+ changes mediated by synaptic transmitter release. In the presence of the GABAA receptor antagonist bicuculline (50 μm), the withdrawal of the sodium channel blocker TTX (1 μm) from the perfusion solution triggered a series of rapid Ca2+ transients (Fig. 1A). The majority of neurons within an imaging region (> 70%) exhibited synchronized Ca2+ transients. The readministration of TTX at the end of the recording returned Ca2+ to basal levels, indicating that the rise in Ca2+ was mediated by the action potential-dependent release of an excitatory neurotransmitter. In parallel with this effect on Ca2+, spontaneous synaptic activity triggered an increase in the Ser133 phosphorylated form of CREB (Fig. 1A).
Figure 1.

The NMDA receptor couples synaptic activity to the Ser133 phosphorylated form of CREB. A, Top, Initially, cortical neurons cultured for 10 d were placed in a HEPES-based buffer containing TTX (1 μm), and internal Ca2+ levels were monitored by using fura-2 time-lapse digital microscopy. In the presence of bicuculline (50 μm), the withdrawal of the TTX from the buffer elicited rapid and reproducible Ca2+ transients. A, Bottom, Relative to cortical cultures maintained in TTX, 10 min of synaptic activity elicited by the removal of TTX trigged a marked increase in pCREB expression. As a protein loading control, this blot was stripped and probed for total ERK expression. Duplicate determinations are shown for each condition. B, Top, Driving the synaptic network via activation of the NMDA receptor. To examine the necessity of L-type Ca2+ channels and ionotropic glutamate receptors in synaptic activity-dependent CREB phosphorylation, we placed cortical cultures in HEPES buffer containing bicuculline (50 μm), 4-AP (200 μm), and nimodipine (5 μm). In the majority of neurons, the basal Ca2+ levels were maintained by the withdrawal of TTX (1 μm) and the addition of APV (100 μm) to the perfusion medium. Washout of APV and the non-NMDA ionotropic receptor antagonist CNQX (10 μm) triggered a robust and sustained increase in Ca2+ levels. Ca2+ levels also were increased by the withdrawal of APV, indicating that the synaptic release of glutamate stimulated Ca2+ influx via an NMDA receptor-mediated mechanism. B, Bottom, Western blot analysis for pCREB revealed that the NMDA receptor coupled synaptic activity to pCREB expression. C, Synaptic activity attenuates NMDA-induced cell death. Hoechst labeling was used to monitor the effects that synaptic activity has on NMDA-evoked neurotoxicity. A bout (15 min) of synaptic activity was elicited by the application of bicuculline and 4-AP (bic+4AP) in the presence of nimodipine and CNQX at time 0 (t = 0). Then 24 h later (t = 24), the cultures were exposed to NMDA (50 μm, 15 min); the number of dead and dying cells was scored 8 h later (t = 24 + 8). Arrowheads denote the location of nuclei from healthy cells; arrows indicate apoptotic cells with condensed or fragmented nuclei. D, In the absence of pretreatment with synaptic activity, relatively high numbers (∼40%) of apoptotic cells were observed. Pretreatment with synaptic activity significantly (*p < 0.05) reduced the apoptotic effects of NMDA. Control data are from cells that were stimulated neither with synaptic activity nor with NMDA. Numbers above each bar indicate the number of cells assayed. E, The neuroprotective effects of synaptic activity also were monitored via LDH release. Cell stimulation was performed as described in C. *Significant difference (p < 0.05) from the NMDA treatment condition. NMDA administration was performed in media containing TTX (1 μm).
Next we examined whether signaling via the NMDA receptor was sufficient to drive the synaptic network and trigger CREB phosphorylation. In this experiment, synaptic activity was elicited by the withdrawal of TTX from the perfusion media. To augment network-dependent neuronal activity, we added bicuculline (50 μm) and the potassium channel blocker 4-AP (200 μm) to the perfusion media. Nimodipine (5 μm) was added to the perfusion media to eliminate the potential contribution of L-type voltage-activated Ca2+ channels (VACC) to synaptically evoked Ca2+ transients and activity-dependent CREB phosphorylation. Under these conditions, network-driven neuronal activity elicited robust Ca2+ transients and CREB phosphorylation (Fig. 1 B). When the NMDA receptor antagonist APV was added to the perfusion media, synaptic activity was mostly blocked (we did note a small percentage of cells with increased Ca2+ levels even in the presence of NMDA receptor blockade) (data not shown), and CREB phosphorylation was attenuated markedly. The administration of the non-NMDA ionotropic receptor antagonist CNQX did not inhibit the synaptically evoked Ca2+ rise, nor was it able to block CREB phosphorylation (Fig. 1 B). Collectively, these data show that a synaptic network can be driven specifically by the NMDA receptor and that NMDA receptor signaling is sufficient to drive CREB phosphorylation.
Synaptic/NMDA receptor stimulation, CREB, and excitotoxic cell death
NMDA receptor stimulation has been shown to be both neuroprotective and excitotoxic. Although the mechanism or mechanisms by which the NMDA receptor exerts this dualist character are not clear, receptor location, the magnitude of the Ca2+ response, and CREB activation may be critical factors. Initially, we tested whether NMDA receptor stimulation mediated by synaptic glutamate release attenuated the apoptotic effects of exogenous NMDA (50 μm) application. For these experiments, NMDA receptor-dependent synaptic activity was elicited by the application of bicuculline and 4-AP in the presence of nimodipine and CNQX for 15 min. Then 24 h later, NMDA (50 μm) was administered for 15 min. Hoechst labeling and LDH release were examined 8 h after NMDA administration. In the absence of a pretreatment with synaptic activity, NMDA elicited a marked increase in the number of dead and dying cells (Fig. 1C-E). In contrast, synaptic activity 24 h before NMDA application significantly decreased both the number of apoptotic cells and LDH release. These results indicate that excitatory neurotransmission mediated by NMDA receptor activation protects against NMDA-induced excitotoxic cell death.
Upregulation of the neurotrophin BDNF and the anti-apoptotic protein BCL-2 may be a mechanism by which synaptic activity confers neuroprotection against excitotoxic insults. Given that both BDNF and BCL-2 are CREB-regulated genes (Shieh et al., 1998; Tao et al., 1998), these results suggest that neuroprotective (synaptic activity) and toxic (50 μm NMDA) stimuli differentially regulate signaling via the CREB/CRE transcriptional pathway. To begin to address this issue, we examined the effects that brief stimulations (15 min) with synaptic activity and NMDA have on the activation of CRE-mediated gene expression. For these experiments, the neurons were transfected with a CRE-luciferase reporter construct after 8 DIV and stimulated (48 h later) with either synaptic activity driven specifically by the NMDA receptor (bicuculline, 4-AP, nimodipine, and CNQX) or the exogenous application of NMDA (1, 5, or 50 μm). Luciferase activity measurements revealed that both synaptic activity and low concentrations of NMDA (1 and 5 μm) stimulated significant increases in CRE-mediated gene expression, whereas the application of 50 μm NMDA did not stimulate luciferase expression (Fig. 2A). In an attempt to determine why these stimuli have differential effects on CRE-mediated gene expression, we monitored the activation state of CREB. A number of studies have shown that the duration of CREB phosphorylation correlates with transcriptional activation (Bito et al., 1996; Liu and Graybiel, 1996; Impey et al., 1998). The exogenous application of 50 μm NMDA triggered a robust but transient increase in pCREB; levels declined by 15 min after stimulation (25 min time point) and returned to basal levels by 45 min after stimulation (Fig. 2D, 45 min time point). A significant increase in NMDA-induced cell death, as assessed by Hoechst labeling (data not shown) and LDH release (Fig. 3C, 2 h time point), was not observed during the period of CREB dephosphorylation, indicating that the rapid drop in pCREB levels was not the result of acute cell death. In contrast to the transient pCREB response elicited by 50 μm NMDA, the administration of bicuculline elicited a robust increase in CREB phosphorylation that persisted for ≥90 min after stimulus onset (Fig. 2B). Typically, synaptically evoked CREB phosphorylation persisted from 90 to >180 min. Long-lasting CREB phosphorylation was mediated by NMDA receptor activation; in the presence of nimodipine and CNQX, a brief period of synaptic activity triggered long-lasting CREB phosphorylation (Fig. 2C, 180 min time point). The transient nature of CREB phosphorylation mediated by exogenous NMDA administration raised the possibility that a process of dephosphorylation also had been activated by the stimulus. Several studies have shown that the phosphatase calcineurin regulates the duration of CREB phosphorylation (Bito et al., 1996; Liu and Graybiel, 1996; Wu et al., 2001). To address the potential role of calcineurin, we incubated cultures with the calcineurin inhibitor FK506 (1 μm). Under this condition, the administration of NMDA (50 μm, 10 min) led to a sustained increase in CREB phosphorylation that persisted for 180 min (Fig. 2E, top), paralleling the long-lasting increase in CREB phosphorylation mediated by synaptic activity. In addition, FK506 significantly attenuated the toxic effects of exogenous NMDA administration (Fig. 3E, bottom). These data suggest that strong, potentially neurotoxic levels of NMDA receptor stimulation specifically may activate calcineurin. To assess the relative responsiveness of calcineurin to synaptic activity and NMDA (50 μm), we monitored the subcellular localization of the transcription factor NF-ATc4. Nuclear translocation of NF-ATc4 is elicited by calcineurin-mediated dephosphorylation (Crabtree, 1999; Graef et al., 1999). Thus by monitoring the nuclear/cytoplasmic NF-ATc4 ratio, one may infer the activation state of calcineurin. For these studies, the cortical neurons were transfected with an NF-ATc4-GFP fusion protein and stimulated (10 min) with NMDA (50 μm) or synaptic activity (bicuculline, 4-AP, CNQX, nimodipine); then they were fixed and immunolabeled for GFP and the neuronal marker protein MAP2. NMDA elicited marked NF-ATc4-GFP nuclear translocation, whereas synaptic activity did not increase the nuclear expression of the transcription factor, relative to control conditions (Fig. 2F). Pretreatment with FK506 blocked NMDA-induced NF-ATc4 translocation, indicating that calcineurin activity is essential to couple NMDA to NF-ATc4 nuclear accumulation. Interestingly, FK506 reduced NMDA-induced NF-ATc4 nuclear localization to a level that was lower than that observed in unstimulated (control) neurons, indicating that there is tonic calcineurin activity under control conditions. Together these data reveal that NMDA (50 μm) stimulates calcineurin activity but that synaptic activity is not effective. These differential response profiles provide mechanistic insight into how the duration of CREB phosphorylation is affected differentially by these two stimuli.
Figure 2.

NMDA, the CREB/CRE pathway, and cell toxicity. A, Neurons transfected with a CRE-luciferase reporter gene construct were stimulated (15 min) with NMDA (1, 5, 50 μm), forskolin (5 μm), or NMDA receptor-dependent synaptic activity (as described in Fig. 1C) and assayed 8 h later. *Significant difference (p < 0.05) relative to control buffer treatment. Data were averaged from triplicate determinations. B, Synaptic activity triggered a long-lasting form of CREB phosphorylation. In this example, the phosphorylated form of CREB was stimulated after a brief (10 min) administration of bicuculline (bic; 20 μm). Cultures were collected at the poststimulus onset time points indicated. C, Long-lasting CREB phosphorylation was mediated by NMDA receptor activation. In this experiment, synaptic activity was stimulated (10 min) in the presence of CNQX (10 μm) and nimodipine (nim; 5 μm). D, The exogenous application of NMDA (50 μm) triggered a transient form of CREB phosphorylation. Initially, cortical neurons were stimulated for 10 min. Then NMDA was washed from the medium, and the cultures were collected at the poststimulus onset time points indicated. E, Top, Preincubation (120 min) with the calcineurin antagonist FK506 (1 μm) converted the NMDA-evoked (50 μm) transient increase in CREB phosphorylation into a sustained elevation. Basal levels of CREB phosphorylation were not altered by FK506 pretreatment (Con t = 0 and t = 180 vs FK506 t = 0 and t = 180). Western blots are representative of at least three independent experiments. E, Bottom, FK506 pretreatment (1 μm, 120 min) attenuated the toxic effects of NMDA (50 μm, 15 min stimulation). LDH release was examined 8 h after NMDA administration. For the NMDA administration experiments, all solutions contained TTX. *Significant difference (p < 0.05) relative to both control and NMDA administration. F, Neurons were transfected with an NF-ATc4-GFP fusion protein, and the effects of NMDA (50 μm, 10 min stimulation) and synaptic activity (bicuculline plus 4-AP in the presence of nimodipine and CNQX, 10 min stimulation) on NF-ATc4-GFP subcellular localization were examined. F, Top, Representative confocal images of NF-ATc4-GFP expression under the four conditions. Cells were incubated (120 min) with FK506 (1 μm) before NMDA stimulation. Note the marked nuclear translocation after NMDA administration. F, Bottom, Mean nuclear/cytoplasmic NF-ATc4-GFP ratio under the four treatment conditions. Error bars denote the SEM. Numbers above each bar indicate the number of neurons analyzed. *p < 0.005, relative to all other conditions. Con, Control.
Figure 3.

The duration of CREB phosphorylation depends on NMDA concentration. A, In contrast to the transient increase in pCREB elicited by 50 μm NMDA (Fig. 2), brief stimulation (10 min) with low concentrations of NMDA (1 and 5 μm) triggered a sustained (90-180 min) increase in CREB phosphorylation. As a protein-loading control, blots were stripped and probed for total ERK expression. B, Quantitation of the duration of NMDA-evoked (1-50 μm) CREB phosphorylation. pCREB values were normalized to ERK 1 expression, and control conditions were set equal to 100%. Note that low concentrations of NMDA (1 and 5 μm) elicited longer-lasting pCREB than the high (50 μm) concentration of NMDA. *Significant differences (p < 0.05) between low (1 and 5 μm) and high (50 μm) NMDA concentrations from the 25-180 min time points. Data are from triplicate determinations. C, The toxic effects of NMDA were correlated inversely with the duration of CREB phosphorylation. LDH assays performed 2 and 8 h after NMDA stimulation (15 min) revealed that low levels of NMDA (1 and 5 μm) are relatively nontoxic, whereas 50 μm NMDA led to a pronounced increase in LDH release at the 8 h time point. As a relative comparison for the toxic effects of NMDA, cortical neurons also were treated with glutamate (1 mm, 15 min). Data are from triplicate determinations. *Significant difference (p < 0.05) relative to control conditions. D, Pretreatment with a low concentration of NMDA (2.5 μm) significantly attenuated the excitotoxic effects of NMDA (50 μm). In this assay, the cortical neurons were stimulated at time 0 (t = 0) with 2.5 μm NMDA for 15 min and then stimulated 24 h later (t = 24) with 50 μm NMDA; LDH release was assayed 8 h later. *p < 0.05, relative to 50 μm NMDA administration. Con, Control; bic, bicuculline.
The transient CREB phosphorylation mediated by exogenous NMDA application was in striking contrast to the sustained level of CREB phosphorylation mediated by synaptic NMDA receptor activation. These results raise the possibility that distinct pools of NMDA receptors (synaptic and nonsynaptic) and/or the intensity of the stimulus may govern pCREB duration. To gain insight into whether pCREB duration is related to NMDA concentration, we monitored the pCREB time course after exogenous stimulation with relatively low concentrations of NMDA. In contrast to the transient increase in pCREB mediated by 50 μm NMDA, brief (10 min) treatment with either 1 or 5 μm NMDA (in the presence of TTX) led to a persistent (∼90-180 min) increase in CREB phosphorylation (Fig. 3A,B).
To address the issue of whether the duration of CREB phosphorylation corresponds with the toxicity of the treatment, we analyzed LDH release from neurons treated with low and high concentrations of NMDA. Consistent with the idea that the prolonged CREB phosphorylation is associated with nontoxic NMDA stimulation, we found that low concentrations of NMDA (1 and 5 μm) did not trigger significant cell death, whereas 50 μm NMDA stimulated a significant increase in cell death (Fig. 3D). In an additional parallel to the results observed by using the synaptic activity paradigm, pretreatment with a low concentration of NMDA (2.5 μm) significantly attenuated excitotoxic effects of 50 μm NMDA (Fig. 3D). Collectively, these data indicate that NMDA concentration determines the duration of CREB phosphorylation and determines whether NMDA is protective or toxic.
Kinase coupling to CREB
A panel of pharmacological inhibitors was used to identify the signaling events that couple synaptic/NMDA receptor activity to CREB phosphorylation. Initially, cortical neurons were treated with the broad-spectrum CaMK inhibitor KN62 (10 μm), the mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor U0126 (10 μm), the PKA inhibitor H89 (5 μm), the inactive cAMP analog Rp-cAMPs (300 μm), or the PKC inhibitor bisindolylmaleimide (1 μm). Western blot analysis and immunofluorescent labeling for pCREB revealed that KN62 and U0126 significantly attenuated the capacity of synaptic activity to stimulate CREB phosphorylation (Fig. 4A,B). None of the other inhibitors were effective. As a control experiment, fura-2 Ca2+ imaging was used to examine whether KN62 and U0126 inhibit neurotransmission and thereby nonspecifically affect the ability of synaptic input to regulate CREB phosphorylation. Representative recordings (Fig. 4C) revealed that synaptic/NMDA receptor-mediated Ca2+ transients persisted in the presence of the CaMK and mitogen-activated protein kinase (MAPK) inhibitors. Western blot analysis was used to show that ERK phosphorylation at Thr202 and Tyr204 (a marker of ERK activation) was blocked by U0126 and that CaMKIV phosphorylation at Thr196 (a marker of CaMKIV activation) was inhibited by KN62 (Fig. 4C).
Figure 4.
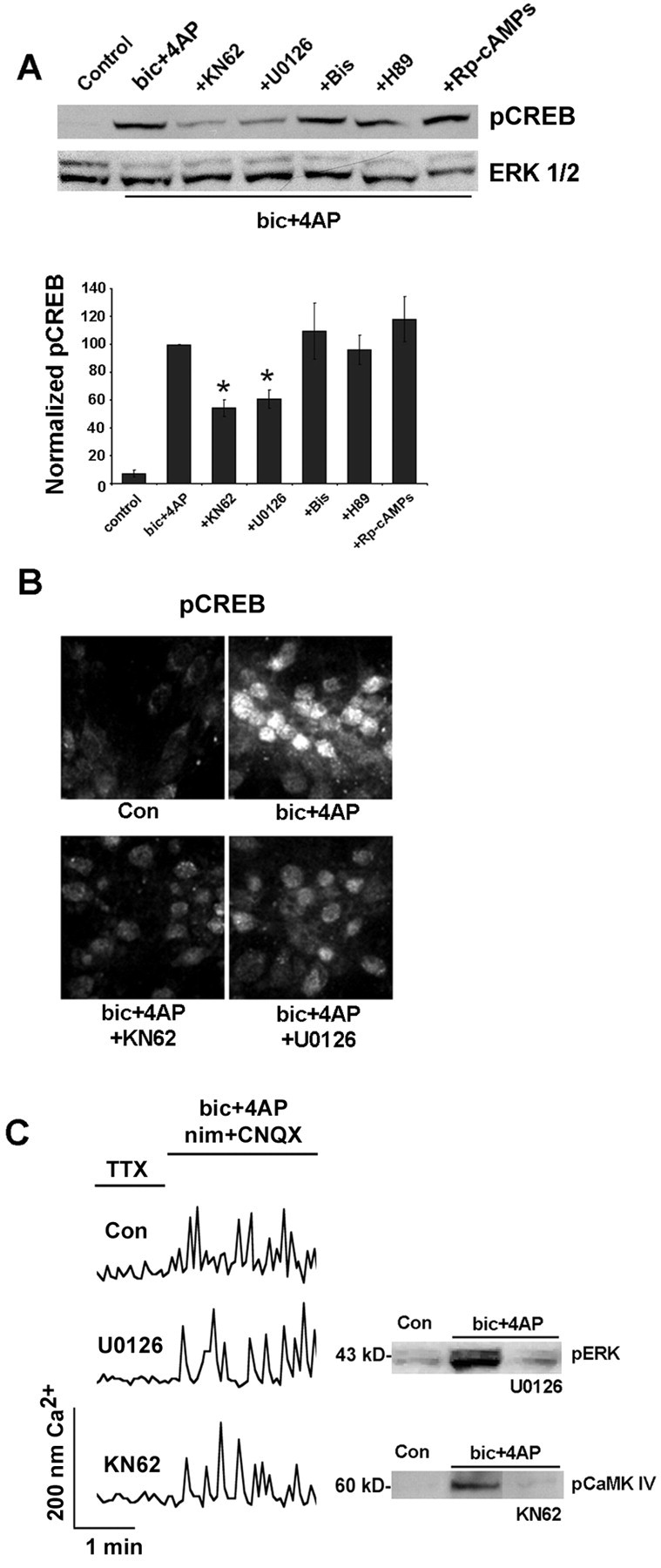
The MAPK and CaMK pathways couple synaptically evoked NMDA receptor activation to CREB phosphorylation. A, Initially, cortical cultures were pretreated (for 30 min) with U0126 (10 μm), KN62 (10 μm), H89 (5 μm), Rp-cAMPs (300 μm), or bisindolylmaleimide (Bis; 1 μm). Then NMDA receptor-mediated synaptic activity was elicited (10 min) by bicuculline (bic) plus 4-AP in the presence of nimodipine and CNQX. Both U0126 and KN62 attenuated CREB phosphorylation. Mean densitometric band analysis is shown in the bars below. Data are representative of triplicate determinations. *Significant difference (p < 0.05) relative to the bicuculline plus 4-AP condition. Intensity values were normalized to the bicuculline plus 4-AP treatment condition, which was set equal to 100%. Data are representative of triplicate determinations. B, Immunofluorescent staining for pCREB was used to compliment the Western blot analysis. Relative to control cultures, the administration of bicuculline increased pCREB expression; pretreatment with KN62 or U0126 attenuated the synaptic activity-dependent increase in CREB phosphorylation. C, To determine whether inhibition of MAPK and CaMK signaling disrupts Ca2+ response characteristics, we loaded cortical cultures with fura-2, pretreated them (10 min) with 10 μm KN6 or 10 μm U0126, and examined the capacity of NMDA receptor-mediated synaptic activity to stimulate Ca2+ transients. Representative examples show that robust Ca2+ transients were generated in the presence of these inhibitors. As an additional control, Western blot analysis was used to examine the potency of these inhibitors. U0126 (10 μm) attenuated synaptically evoked ERK phosphorylation; KN62 (10 μm) attenuated synaptically evoked CaMKIV phosphorylation. Con, Control.
In contrast to the role the MAPK and CaMK pathways play in synaptically evoked CREB activation, neither pathway appeared to contribute to CREB phosphorylation mediated by high levels of NMDA (50 μm) (Fig. 5A,C). Control experiments (Fig. 5B) validated the capacity of KN62 and U0126 to attenuate NMDA-evoked (50 μm) CaMKIV phosphorylation and MAPK activation, respectively. We also tested the capacity of a number of other pathways to couple excitotoxic levels of NMDA to CREB phosphorylation. Inhibitors of cAMP/PKA-dependent signaling (5 μm H89; 300 μm Rp-cAMPs) as well as the PKC inhibitor bisindolylmaleimide (1 μm) had no effect on pCREB levels (Fig. 5A). Because 50 μm NMDA is neurotoxic, we assessed whether two stress-responsive pathways, the p38 MAPK pathway and c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK), regulated CREB phosphorylation. Both pathways were activated by NMDA (data not shown); however, neither the disruption of p38 MAPK activity with SB203580 (10-50 μm) nor addition of the JNK/SAPK inhibitor SP600125 (30 μm) had an effect on the NMDA-evoked CREB phosphorylation (Fig. 5D,E). In addition, the phosphatidylinositol kinase pathway inhibitor LY294002 (30 μm) and the broad-spectrum protein kinase inhibitor staurosporine (200 nm) had no effect on CREB activation (data not shown). In initial tests of ERK-5, we found that NMDA (50 μm) did not elicit its phospho-activation (data not shown). Finally, kinase coupling to CREB phosphorylation was tested by using a potent mixture of inhibitors (U0126, KN62, bisindolylmaleimide, H89, SB203580, SP600125, LY294002, and staurosporine). Under this condition, we noted a modest ∼40% decrease in CREB phosphorylation (Fig. 5F). Together, these data indicate that high, potentially toxic levels of NMDA couple to CREB phosphorylation via an atypical signaling event.
Figure 5.

Coupling excitotoxic levels of NMDA receptor stimulation to CREB phosphorylation. A, The small molecule inhibitor panel used in Figure 4 A had no effect on CREB phosphorylation mediated by the application of 50 μm NMDA. Intensity values were normalized to the NMDA treatment condition, which was set equal to 100%. Data are representative of triplicate determinations. Bis, Bisindolylmaleimide. B, The capacity of KN62 to disrupt NMDA-evoked CaMKIV activation and of U0126 to disrupt NMDA-evoked MEK-dependent ERK phosphorylation was verified. C, To test for potential redundancy within the MAPK and CaMK pathways, we incubated cells in both 10 μm KN62 and 10 μm U0126 and assayed the effects of NMDA. Treatment with another broad-spectrum CaMK inhibitor, KN93 (10 μm), as well as another MEK inhibitor, PD98059 (50 μm), had no discernible effect on NMDA-evoked CREB phosphorylation. D, E, Inhibition of JNK/SAPK with SP600125 (30 μm) and disruption of p38 MAPK with SB203580 (10-50 μm) did not affect NMDA-evoked CREB phosphorylation. F, Pretreatment with an inhibitor mixture attenuated, but did not block, NMDA-evoked CREB phosphorylation. In this experiment, the neurons were incubated with U0126 (10 μm), KN62 (10 μm), bisindolylmaleimide (1 μm), H89 (5 μm), SB203580 (20 μm), SP600125 (30 μm), LY294002 (30 μm), and staurosporine (200 nm). The number above each lane indicates the pCREB/ERK ratio, which was normalized to the first control condition, which was set equal to 1. Inhibitors were applied 30 min before NMDA stimulation and maintained in the culture media until the cells were lysed. All buffers contained TTX (1 μm). Con, Control.
The MAPK and CaMK pathways do not determine the duration of CREB phosphorylation
Next we examined the potential signaling events that govern the duration of CREB phosphorylation. Given the data showing that U0126 and KN62 attenuated synaptic/NMDA receptor-mediated CREB phosphorylation, we focused attention on the MAPK and CaMK cascades. To examine the duration of kinase activation, we collected cultures after 10-60 min of persistent synaptic/NMDA receptor activation and processed them for the phosphorylated form of ERK and for the phosphorylated form of CaMKIV. As expected, synaptic activity led to CREB phosphorylation throughout the stimulus period (Fig. 6A). In contrast, CaMKIV activation was transient, peaking 10 min after stimulus onset and returning to near-basal levels before the end of the stimulus period. Given its transient activation profile, these results suggest that CaMK signaling may not contribute to the sustained increase in CREB activation. Indeed, treatment of cortical neurons with KN62 after 10 min of synaptic activity did not decrease the duration of CREB phosphorylation (Fig. 6B); in the presence of KN62, elevated pCREB levels were observed 90-180 min after stimulation termination, paralleling the pCREB duration after synaptic stimulation in the absence of inhibitor administration. Persistent synaptic/NMDA receptor activation led to a sustained increase in ERK phosphorylation, paralleling the pCREB activation profile (Fig. 6A). Surprisingly, the administration of U0126 (10 μm) after the onset of synaptically mediated CREB phosphorylation also failed to reduce the duration of CREB activation (Fig. 6B). In control experiments, synaptic activity (10 min) led to a sustained increase in ERK phosphorylation, and the administration of U0126 blocked sustained ERK phosphorylation (Fig. 6C). These data suggest that MAPK and CaMK signaling are required for the onset of synaptically evoked CREB phosphorylation but that neither pathway regulates the duration of CREB activation.
Figure 6.
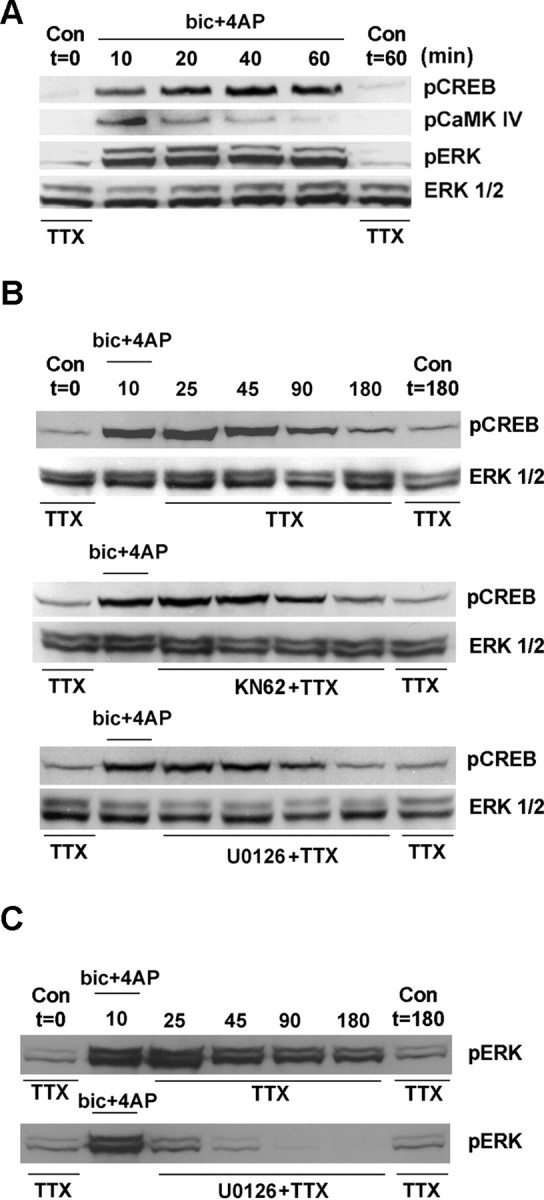
The MAPK pathway and CaMKIV do not regulate the duration of CREB phosphorylation. A, The duration of CREB, CaMKIV, and ERK phosphorylation was monitored during a period of persistent NMDA receptor-mediated synaptic activity. Cultures were lysed 10-60 min after the onset of synaptic stimulation [bicuculline (bic) plus 4-AP containing nimodipine and CNQX]. Control cultures (no stimulation) were collected at the beginning and the end of the stimulus period. Robust increases in both CREB and ERK phosphorylation were observed for the duration of the stimulation. However, CaMKIV phosphorylation was transient, showing maximal activation at the 10 min time point. B, To examine the contribution of CaMK and MAPK signaling to the duration of CREB phosphorylation, we transferred cells to media containing KN62 (10 μm) or U0126 (10 μm) immediately after synaptic stimulation. Samples were collected at the prestimulus and poststimulus time points indicated. Relative to the control synaptic activity condition (no inhibitor), disruption of CaMK signaling and MAPK signaling did not alter the duration of CREB phosphorylation. C, As a control experiment, the duration of ERK phosphorylation was examined. Synaptic activity triggered sustained ERK phosphorylation; the administration of U0126 after stimulation dramatically attenuated ERK activation. Con, Control.
Temporal regulation of CREB “shut-off”
Hardingham et al. (2002) found that exogenous glutamate applied after 30 min of persistent synaptic activity triggered a rapid decrease in pCREB levels. Under this stimulus paradigm, the extrasynaptic CREB shut-off mechanism determines pCREB duration. In light of our data above showing that the duration of synaptically evoked CREB phosphorylation is independent of persistent kinase activity, we wanted to identify specifically when shut-off occurs: during the induction of synaptically evoked pCREB and/or during the period when persistent CREB phosphorylation has become independent of the MAPK and CaMK pathways. To test whether shut-off occurs during induction, we stimulated synaptic activity while we applied exogenous NMDA (50 μm). After 10 min, the medium was changed to a TTX-containing solution, and the pCREB duration was determined. Under these conditions pCREB levels persisted for up to 90 min, consistent with the pCREB time course elicited by synaptic activity alone (Fig. 7A). These data indicate that, when both synaptic and exogenous stimuli are applied simultaneously, synaptic activity dominates and thus determines pCREB duration. In agreement with the work of Hardingham et al. (2002), we found that if synaptically mediated CREB phosphorylation was elicited before exogenous NMDA receptor stimulation, pCREB shut-off occurred (Fig. 7B). These data indicate that the sequence of stimulation determines which pathway exerts control over pCREB duration.
Figure 7.

Temporal regulation of CREB shut-off. A, Cultures were costimulated with NMDA (50 μm) and synaptic activity [bicuculline (bic) plus 4-AP with nimodipine and CNQX] from t = 0-10 min. After stimulation, the medium was changed to a TTX-containing solution, and the pCREB expression was determined at the time points outlined. Mock stimulation is shown for the t = 0 time point. Under this 10 min stimulus condition, pCREB expression persisted for 90 min. B, Cultures initially were stimulated (10 min) with synaptic activity (bicuculline plus 4-AP with nimodipine and CNQX from -40 to -30 min) and then placed in a TTX/HEPES buffer (30 min; from -30 to 0 min). After this treatment, NMDA (50 μm) was administered for 10 min (from 0 to 10 min), and the cells were then returned to a TTX/HEPES buffer. pCREB expression was determined at the time points outlined. pCREB levels for both synaptic stimulation (bicuculline plus 4-AP in the presence of nimodipine and CNQX) and mock stimulation [control (Con)] are shown for the t = 0 time point (before NMDA administration). Under this sequential stimulus paradigm, pCREB expression returned to basal levels by 25-45 min after NMDA administration. Data are representative of triplicate determinations.
Synaptic activity, CRE-mediated transcription, and neuroprotection
Finally, to form a definitive link between CREB and attenuation of NMDA-mediated toxicity, we transfected cortical neurons with a constitutively active CREB construct (VP16-CREB) and a GFP reporter construct and then, 48 h later, stimulated them with 50 μm NMDA. GFP immunofluorescence was used to identify transfected cells, NeuN was used to verify that the cells were neurons, and Hoechst labeling was used to examine cell health. Relative to empty vector-transfected neurons, transfection of VP16-CREB significantly attenuated NMDA-induced cell death (Fig. 7A,B). In the absence of NMDA administration, VP16-CREB did not affect cell survival (data not shown). As a control, we show that VP16-CREB stimulated the expression of a CRE-luciferase reporter gene (Fig. 7C). A-CREB, a dominant-negative inhibitor of CREB (Ahn et al., 1998), also was used to test the role of CREB in synaptic activity-dependent neuroprotection. In this set of experiments the neurons were transfected with A-CREB and GFP; synaptic activity was elicited 24 h later (at t = 0) for 15 min (Fig. 8D). At 24 h after synaptic stimulation, the cells were exposed to NMDA (50 μm, 15 min), and the apoptotic effects were assayed 8 h later (Fig. 8D). In the absence of NMDA administration, A-CREB did not increase the number of apoptotic cells, relative to empty vector (pcDNA3.1) transfection. However, A-CREB did exacerbate the excitotoxic effects of NMDA and significantly reduce the neuroprotective effects of synaptic activity (Fig. 8E). Thus under control conditions (empty vector transfection), synaptic activity reduced the toxic effects of NMDA by 71%, whereas in the presence of A-CREB, the synaptic activity reduced the toxic effects of NMDA by 36%, an approximately two-fold diminution of the neuroprotective effects of synaptic activity. Collectively, these data indicate that the CREB/CRE signaling pathway contributes to the neuroprotective effects of synaptic/NMDA receptor activity.
Figure 8.
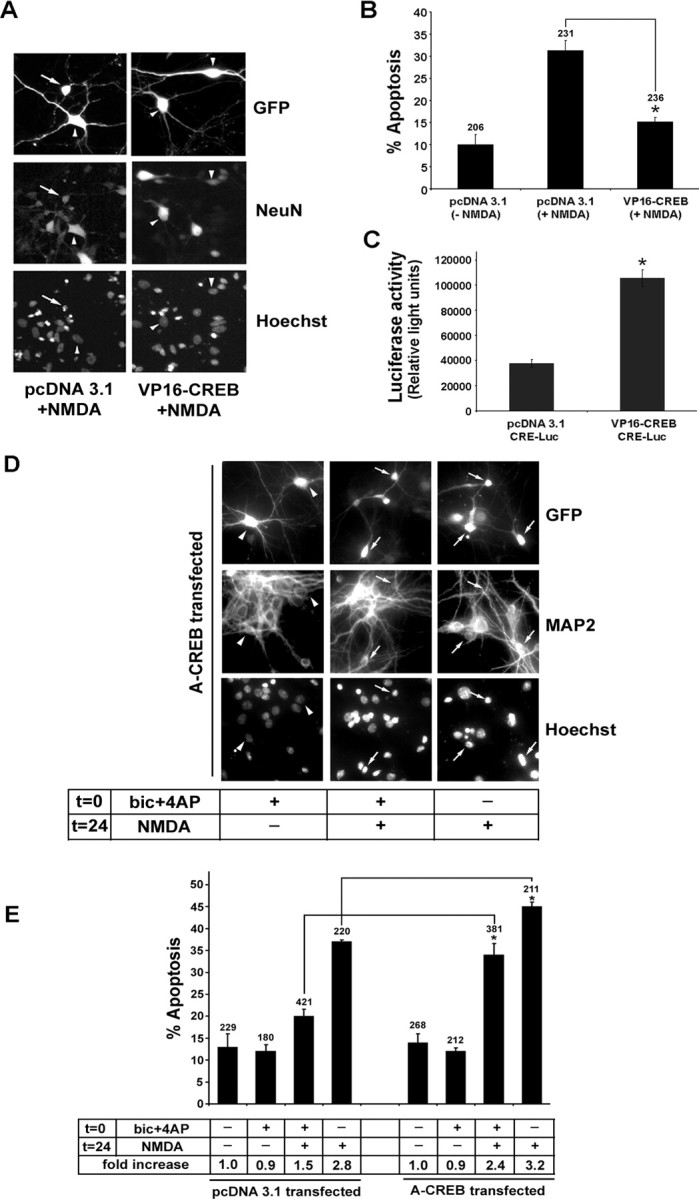
Synaptic activity attenuates NMDA-mediated cell death via a CREB-dependent mechanism. A, Neurons were transfected with VP16-CREB or the control vector pcDNA3.1 and stimulated 48 h later with NMDA (50 μm, 15 min). Cell viability was scored 8 h later. Cotransfection with GFP was used to identify transfected cells, NeuN was used to verify that the cells were neurons, and Hoechst labeling was used to assess toxicity. The arrow identifies a representative transfected cell that is dying; arrowheads denote healthy cells. B, Quantitation of the percentage of transfected neurons undergoing apoptosis. Relative to vector transfection, the transfection with VP16-CREB attenuated (*p < 0.05, significantly different from control vector transfection) the toxic effects of NMDA. Error bars denote SEM. Numbers above the bars indicate the number of neurons assayed. C, A CRE-luciferase reporter construct was cotransfected with VP16-CREB or pcDNA3.1 to validate that the constitutively active CREB construct stimulated CRE-dependent transcription (*p < 0.05, significantly different from control vector transfection). D, A-CREB attenuates the neuroprotective effects of synaptic activity. Neurons were cotransfected with GFP and A-CREB expression vectors. Then 24 h later, neuronal activity was elicited [t = 0, bicuculline (bic) plus 4-AP with nimodipine and CNQX, 15 min]. NMDA (50 μm) was administered 24 h after bicuculline stimulation (t = 24, 15 min), and cell viability was scored 8 h later. GFP immunofluorescence was used to identify transfected cells, MAP2 was used to verify that the cells were neurons, and Hoechst labeling was used to assess toxicity. Arrows identify transfected cells that were dead or dying; arrowheads denote healthy cells. E, Quantitation of transfected neurons undergoing apoptosis. Relative to control expression vector transfection (pcDNA3.1), transfection with A-CREB significantly attenuated (*p < 0.05, relative to control vector transfection) the neuroprotective effects of synaptic activity and augmented the neurotoxic effects of NMDA. Numbers above each bar indicate the number of neurons assayed. In the table shown below the bars, the percentage of apoptotic neurons was normalized to control conditions (no stimulation), which was set equal to 1. Error bars denote SEM. Data are the mean of triplicate determinations.
Discussion
Work over the past several years has revealed that the CREB/CRE transcriptional pathway is a complex regulator of a vast array of activity-dependent physiological processes. Of particular prominence have been data examining the role of CREB as a regulator of neuronal health and viability (Walton et al., 1996, 1999; Bonni et al., 1999; Mabuchi et al., 2001; Lonze and Ginty, 2002; Jaworski et al., 2003). In this study, we focused on the interplay between the neuroprotective and cytotoxic effects of NMDA receptor stimulation and on how these extremes in cellular responsiveness affect the activation state of CREB. The data presented here reveal that (1) the magnitude of NMDA receptor stimulation (toxic vs nontoxic) determines the duration of CREB phosphorylation via the selective recruitment of calcineurin activity, (2) permissive and toxic levels of NMDA receptor activation regulate CREB phosphorylation via distinct cellular signaling pathways, (3) the sequence of stimulation with neurotoxic levels of NMDA and neuroprotective synaptic activity determines which pathway exerts control over pCREB duration, and (4) the neuroprotective effects of NMDA receptor activation are mediated, in part, via activation of CREB-dependent transcription.
We have shown previously that cortical neurons cultured from embryonic day 18 rat pups begin to form functional synaptic connections by 5 DIV and secrete both glutamate and GABA (van den Pol et al., 1995; Obrietan and van den Pol, 1998). To observe robust levels of synaptic activity after 10 DIV, we cultured our neurons under relatively dense conditions. The density of the cultures affects both the rate of synapse formation and the number of functional synapses. For example, van den Pol et al. (1998) found that presynaptic release of glutamate occurred after 3-4 DIV in high-density cultures but was absent in low-density cultures at this time. Furthermore, neurites grow three times faster in high-density cultures than in low-density cultures (van den Pol et al., 1998). Thus the withdrawal of tetrodotoxin from the perfusion media elicited a rapid rise in neuronal Ca2+ levels. This increase was blocked by the application of APV, indicating that the action potential-dependent release of glutamate drove activation of the network. Synaptic activity triggered a marked increase in the Ser133 phosphorylated form of CREB. This result was expected, given the work showing that synaptic transmission in cultured neurons drives CREB phosphorylation (Bito et al., 1996; Hardingham et al., 2002). However, in the work by Bito et al. (1996) and Hardingham et al. (2002), the receptor subtypes that couple synaptic activity to CREB phosphorylation were not addressed. In an attempt to create a simplified synaptic network that relied specifically on the NMDA receptor, we added antagonists of both non-NMDA ionotropic glutamate receptors and L-type VACC to the bath media. Under this condition, we found that the application of bicuculline and 4-AP stimulated robust Ca2+ transients and a marked increase in CREB phosphorylation, indicating that L-type VACC and the AMPA/kainate subtype of glutamate receptors were dispensable. Many of the effects of synaptic/NMDA receptor activation (persistent CREB phosphorylation, neuroprotection) also were elicited by exogenous application of low concentrations of NMDA (1-5 μm), suggesting that the effects of synaptic activity were mediated by NMDA receptor-dependent signaling. However, it should be noted that synaptic activity also stimulates trophic, hormonal, or metabotropic processes that, in conjunction with NMDA receptor stimulation, might affect CREB phosphorylation. Thus additional work will be required to assess the relative contribution of these other pathways to synaptic/NMDA receptor-mediated CREB phosphorylation and neuroprotection.
The cellular signaling events that couple neuronal activity to CREB phosphorylation vary, depending on the developmental state of the cells as well as the source and intensity of the stimulus (Herdegen et al., 1997; Xing et al., 1998; Sala et al., 2000; Hardingham et al., 2002; Obrietan et al., 2002). Two of the best characterized intermediate-signaling pathways are the MAPK pathway and CaMKIV (Enslen et al., 1994; Sun et al., 1994; Bito et al., 1996; Xing et al., 1996; Impey et al., 1998). The MAPK pathway is composed of the kinases RAF, MEK, and ERK. The MAPK cascade stimulates the Ser133 phosphorylated form of CREB via a kinase functioning downstream of ERK. Several studies have identified RSK (ribosomal S6 kinase) and MSK (mitogen- and stress-activated protein kinase) family members as ERK-activated CREB kinases (Xing et al., 1996; De Cesare et al., 1998; Impey et al., 1998; Arthur and Cohen, 2000). The enzymatic activity of CaMKIV is stimulated by an array of activity-dependent signaling events, including NMDA receptor activation and membrane depolarization (Bito et al., 1996; Kasahara et al., 2000; Soderling, 2000). CaMKIV is enriched in neuronal nuclei (Jensen et al., 1991; Miyano et al., 1992), and its activation is mediated in part by its phosphorylation at Thr196 by CaMK (Selbert et al., 1995). Both the MAPK cascade and CaMKIV were activated by neuronal stimulation and found to elicit CREB phosphorylation. Although it is unclear how both pathways contribute to the phosphorylation of CREB at the same site (Ser133), it has been shown that CaMKIV mediates a rapid and transient phosphorylation of CREB, whereas an ERK-activated kinase mediates a subsequent, prolonged phosphorylation of CREB (Impey et al., 1998; Wu et al., 2001).
Although both the MAPK and CaMK pathways stimulate CREB phosphorylation, the duration of CREB phosphorylation is not dependent on the persistent activation of either pathway. These data suggest that other cellular signaling events (or the lack thereof) determine the duration of CREB phosphorylation.
Along these lines, Bito et al. (1996) found that calcineurin inhibition with FK506 significantly enhanced the duration of CREB phosphorylation elicited by high-frequency stimulation. In some systems, calcineurin has been shown to function as a Ca2+-activated negative regulator of CREB-dependent transcription by limiting the duration of CREB phosphorylation (Chang and Berg, 2001). To address the activation state of calcineurin, we monitored the subcellular localization of NF-ATc4. The nuclear translocation of NF-ATc4 is regulated by calcineurin-dependent dephosphorylation (Crabtree, 1999; Graef et al., 1999); thus, by measuring the cytosolic/nuclear ratio of an NF-ATc4-GFP fusion protein, we were able to infer relative levels of calcineurin activity. With this approach, we found that our two stimulus paradigms had strikingly dissimilar effects on calcineurin activity; NMDA (50 μm) elicited a marked increase in calcineurin-dependent NF-ATc4 nuclear accumulation, whereas synaptic activity was relatively ineffective. Furthermore, our data revealed that transient CREB phosphorylation triggered by the exogenous application of a high concentration of NMDA was reversed by FK506 pretreatment, thus supporting the idea that calcineurin is recruited specifically by, and limits the duration of, CREB phosphorylation elicited by excitotoxic levels of NMDA receptor stimulation.
Calcineurin activity may account in part for the CREB shut-off (Hardingham et al., 2002) effect elicited by the bath application of a high concentration of NMDA, whereas the lack of synaptically evoked calcineurin activity may contribute to persistent CREB phosphorylation. Interestingly, the induction of CREB shut-off was dependent on the sequence of stimulation. For example, when NMDA (50 μm) was applied and synaptic activity was elicited simultaneously, CREB phosphorylation persisted. Conversely, CREB shut-off occurred after synaptically evoked pCREB had become kinase-independent. Thus when both synaptically evoked kinase activity and NMDA-evoked (50 μm) phosphatase activity are stimulated simultaneously, kinase activity dominates and CREB phosphorylation becomes long-lasting (>90 min).
NMDA-mediated toxicity was related inversely to the duration of CREB phosphorylation. Thus a high concentration of NMDA (50 μm) led to transient CREB phosphorylation and significant cell death, whereas low concentrations of NMDA or synaptic activity were nontoxic and triggered long-lasting CREB phosphorylation. Interestingly, long-lasting CREB phosphorylation is observed in cells resistant to toxic insults (Hu et al., 1999; Tanaka et al., 2001; Hara et al., 2003). In our analysis, we did not attempt to address whether neuroprotection with low concentrations of NMDA was mediated by activation of synaptic NMDA receptors or by a combination of synaptic and extrasynaptic receptors. Additional analysis using receptor subtype-specific antagonists, analogous to the approach used by Lu et al. (2003) and Hardingham et al. (2002), will be required to address this issue.
In addition to finding that synaptic activity led to persistent CREB phosphorylation, we observed that synaptic activity triggered a robust increase in CRE-dependent transcription. Extending the duration of CREB phosphorylation may be neuroprotective by increasing the expression of CREB-regulated cell survival genes. Along these lines, a number of studies have shown that the CREB-regulated genes BCL-2 and BDNF are neuroprotective (Martinou et al., 1994; Kitagawa et al., 1996; Lawrence et al., 1996; Bonni et al., 1999; Riccio et al., 1999; Mabuchi et al., 2001; Sugiura et al., 2004). In our model, synaptic activity and low concentrations of NMDA, but not high concentrations of NMDA, stimulated BDNF expression (data not shown). Furthermore, we found that overexpression of constitutively active CREB reduced the toxic effects of high concentrations of NMDA and that A-CREB blocked the neuroprotective effects of synaptic activity. These results raise the possibility that a targeted approach to upregulate CREB-dependent transcription may confer resistance against the neurotoxic effects of excessive excitatory neurotransmission.
Footnotes
This work was supported by National Institutes of Health Grants MH62335 and NS47176 to K.O. and NS41003 to K.R.H. G.Q.B. is a National Research Service Award trainee (MH073374).
Correspondence should be addressed to Karl Obrietan, Department of Neuroscience, The Ohio State University, Graves Hall, Room 4118, 333 West 10th Avenue, Columbus, OH 43210. E-mail: obrietan.1@osu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251137-12$15.00/0
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos Mol Cell Biol 18: 967-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavez S, Pedroza D, Moran J (2003) Mechanisms of cell death by deprivation of depolarizing conditions during cerebellar granule neurons maturation. Neurochem Int 43: 581-590. [DOI] [PubMed] [Google Scholar]
- Aliaga E, Rage F, Bustos G, Tapia-Arancibia L (1998) BDNF gene transcripts in mesencephalic neurons and its differential regulation by NMDA. NeuroReport 9: 1959-1962. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Cohen P (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett 482: 44-48. [DOI] [PubMed] [Google Scholar]
- Balazs R, Hack N, Jorgensen OS (1988) N-methyl-d-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience 27: 437-451. [DOI] [PubMed] [Google Scholar]
- Barger SW, Mattson MP (1995) Excitatory amino acids and growth factors: biological and molecular interactions regulating neuronal survival and plasticity. In: CNS neurotransmitters and neuromodulators, Vol 2, Glutamate (Stone TW, ed), pp 273-294. Boca Raton, FL: CRC.
- Barry MA, O'Donovan MJ (1987) The effects of excitatory amino acids and their antagonists on the generation of motor activity in the isolated chick spinal cord. Brain Res Dev Brain Res 36: 271-276. [DOI] [PubMed] [Google Scholar]
- Behar TN, Scott CA, Greene CL, Wen X, Smith SV, Maric D, Liu QY, Colton CA, Barker JL (1999) Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J Neurosci 19: 4449-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca2+ and stimulus duration-dependent switch for hippocampal gene expression. Cell 87: 1203-1214. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358-1362. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Putignano E, Impey S, Maffei L, Ratto GM, Pizzorusso T (2003) Patterned vision causes CRE-mediated gene expression in the visual cortex through PKA and ERK. J Neurosci 23: 7012-7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catsicas M, Pequignot Y, Clarke PG (1992) Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci 12: 4642-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Berg DK (2001) Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron 32: 855-865. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Gao XM, Paul SM (1992) N-methyl-d-aspartate exposure blocks glutamate toxicity in cultured cerebellar granule cells. Mol Pharmacol 42: 210-216. [PubMed] [Google Scholar]
- Cohen-Cory S (2002) The developing synapse: construction and modulation of synaptic structures and circuits. Science 298: 770-776. [DOI] [PubMed] [Google Scholar]
- Crabtree GR (1999) Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell 96: 611-614. [DOI] [PubMed] [Google Scholar]
- Craig AM, Banker G, Chang W, McGrath ME, Serpinskaya AS (1996) Clustering of gephyrin at GABAergic but not glutamatergic synapses in cultured rat hippocampal neurons. J Neurosci 16: 3166-3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327-335. [DOI] [PubMed] [Google Scholar]
- De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P (1998) Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci USA 95: 1202-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR (1994) Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem 269: 15520-15527. [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR (1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401: 703-708. [DOI] [PubMed] [Google Scholar]
- Hara T, Hamada J, Yano S, Morioka M, Kai Y, Ushio Y (2003) CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J Neurochem 86: 805-814. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathway. Nat Neurosci 5: 405-414. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Blume A, Buschmann T, Georgakopoulos E, Winter C, Schmid W, Hsieh TF, Zimmermann M, Gass P (1997) Expression of activating transcription factor-2, serum response factor and cAMP/Ca response element binding protein in the adult rat brain following generalized seizures, nerve fibre lesion and ultraviolet irradiation. Neuroscience 81: 199-212. [DOI] [PubMed] [Google Scholar]
- Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH (1999) Persistent phosphorylation of cyclic AMP responsive element-binding protein and activating transcription factor-2 transcription factors following transient cerebral ischemia in rat brain. Neuroscience 89: 437-452. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR (1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1: 595-601. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Mioduszewska B, Sanchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L (2003) Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro J Neurosci 23: 4519-4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KF, Ohmstede CA, Fisher RS, Sahyoun N (1991) Nuclear and axonal localization of Ca2+/calmodulin-dependent protein kinase type Gr in rat cerebellar cortex. Proc Natl Acad Sci USA 88: 2850-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara J, Fukunaga K, Miyamoto E (2000) Activation of Ca2+/calmodulin-dependent protein kinase IV in cultured rat hippocampal neurons. J Neurosci Res 59: 594-600. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133-1138. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Wong F, Lo P, Elliott M, Verburgt LM, Hogg JC, Daya M (1996) Overexpression of Bcl-2 and mutations in p53 and K-ras in resected human non-small cell lung cancers. Am J Respir Cell Mol Biol 15: 45-54. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 1: 83-90. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM (1996) Overexpression of Bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo J Neurosci 16: 486-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FC, Graybiel AM (1996) Spatiotemporal dynamics of CREB phosphorylation: transient versus sustained phosphorylation in the developing striatum. Neuron 17: 1133-1144. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Lu J, Goula D, Sousa N, Almeida OF (2003) Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-d-aspartate receptors. Neuroscience 121: 123-131. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M (2001) Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo J Neurosci 21: 9204-9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1993) NMDA receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16: 521-527. [DOI] [PubMed] [Google Scholar]
- Marini AM, Paul SM (1992) N-methyl-d-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA 89: 6555-6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C (1994) Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13: 1017-1030. [DOI] [PubMed] [Google Scholar]
- Miyano O, Kameshita I, Fujisawa H (1992) Purification and characterization of a brain-specific multifunctional calmodulin-dependent protein kinase from rat cerebellum. J Biol Chem 267: 1198-1203. [PubMed] [Google Scholar]
- Nakanishi S (1992) Molecular diversity of glutamate receptors and implications for brain function. Science 258: 597-603. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN (1995) GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci 15: 5065-5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN (1998) GABAB receptor-mediated inhibition of GABAA receptor calcium elevations in developing hypothalamic neurons. J Neurophysiol 79: 1360-1370. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Gao XB, van Den Pol AN (2002) Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism—a positive feedback circuit in developing neurons. J Neurophysiol 88: 1005-1015. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA (2003) ϵPKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci 23: 384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358-2361. [DOI] [PubMed] [Google Scholar]
- Rocha M, Martins RA, Linden R (1999) Activation of NMDA receptors protects against glutamate neurotoxicity in the retina: evidence for the involvement of neurotrophins. Brain Res 827: 79-92. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M (2000) Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci 20: 3529-3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Barker JL (1984) Rat hippocampal neurons in culture: voltage-clamp analysis of inhibitory synaptic connections. J Neurophysiol 52: 469-487. [DOI] [PubMed] [Google Scholar]
- Selbert MA, Anderson KA, Huang QH, Goldstein EG, Means AR, Edelman AM (1995) Phosphorylation and activation of Ca2+-calmodulin-dependent protein kinase IV by Ca2+-calmodulin-dependent protein kinase Ia kinase. Phosphorylation of threonine 196 is essential for activation. J Biol Chem 270: 17616-17621. [DOI] [PubMed] [Google Scholar]
- Sherrard RM, Bower AJ (1998) Role of afferents in the development and cell survival of the vertebrate nervous system. Clin Exp Pharmacol Physiol 25: 487-495. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A (1998) Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20: 727-740. [DOI] [PubMed] [Google Scholar]
- Soderling TR (2000) CaM kinases: modulators of synaptic plasticity. Curr Opin Neurobiol 10: 375-380. [DOI] [PubMed] [Google Scholar]
- Sugiura S, Kitagawa K, Omura-Matsuoka E, Sasaki T, Tanaka S, Yagita Y, Matsushita K, Storm DR, Hori M (2004) CRE-mediated gene transcription in the peri-infarct area after focal cerebral ischemia in mice. J Neurosci Res 75: 401-407. [DOI] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8: 2527-2539. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nogawa S, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y (2001) Phosphorylation of cyclic adenosine monophosphate response element binding protein in oligodendrocytes in the corpus callosum after focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 21: 1177-1188. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20: 709-726. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Cao V, Trombley PQ (1995) Embryonic hypothalamic expression of functional glutamate receptors. Neuroscience 67: 419-439. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Belousov AB, Yang Y, Heller HC (1998) Early synaptogenesis in vitro: role of axon target distance. J Comp Neurol 399: 541-560. [DOI] [PubMed] [Google Scholar]
- Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M (1996) The role of the cyclic AMP-responsive element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Brain Res Mol Brain Res 43: 21-29. [DOI] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M (1999) CREB phosphorylation promotes nerve cell survival. J Neurochem 73: 1836-1842. [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3: 921-931. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Buchhalter J, Dichter MA (1994) Properties of inhibitory and excitatory synapses between hippocampal neurons in very low density cultures. Synapse 18: 128-151. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM (1996) Induction of Bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 16: 5546-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 98: 2808-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273: 959-963. [DOI] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 18: 1946-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]