

Abstract
Presenilin (PS) plays an essential role in intramembranous γ-secretase processing of amyloid precursor protein (APP) and several membrane-bound proteins. Here we report that selective accumulation of a membrane-tethered deleted in colorectal cancer (DCC) derivative (DCC-α) correlates with extensive neurite outgrowth in transfected neuroblastoma cells and axodendritic connectivity associated with increased spine density in cortical neurons derived from PS1-/- embryos, as well as wild-type neurons treated with γ-secretase inhibitors. cAMP-dependent signaling was also increased in both the neuroblastoma and cortical neuron systems. As a physiological consequence of increases in axodendritic connectivity and in the magnitude of cAMP-dependent signaling, short- and long-term glutamatergic synaptic transmission was enhanced in PS-deficient neurons. Together, these results demonstrate for the first time that PS-mediated γ-secretase activity attenuates receptor-mediated intracellular signaling pathways that are critical in regulating glutamatergic synaptic transmission and memory processes.
Keywords: γ-secretase, Alzheimer's disease, deleted in colorectal cancer, glutamatergic synaptic transmission, cAMP/PKA pathway, neurite outgrowth
Introduction
Mutations in PSEN1 and PSEN2 genes, which encode presenilin 1 (PS1) and presenilin 2 (PS2), respectively, cause autosomal dominant early-onset familial Alzheimer's disease (FAD) (Tanzi and Bertram, 2001). Several studies have identified multiple cellular functions for PS1 and PS2 (Fortini, 2002; Sisodia and St. George-Hyslop, 2002; Koo and Kopan, 2004; Thinakaran and Parent, 2004). Particularly, PS is essential for intramembranous γ-secretase cleavage of amyloid precursor protein (APP), a processing event that generates the β-amyloid (Aβ) peptides. FAD-linked PS1 variants enhance the production of highly fibrillogenic Aβ42 peptides (Sisodia and St. George-Hyslop, 2002). It is not clear whether mutant PS1 polypeptides exert pathological effects solely by elevating Aβ42. As proposed previously (Selkoe, 2002; Scheff and Price, 2003; Turner et al., 2003), miscommunication at the synapse during AD pathogenesis might contribute to alter crucial memory processes. Interestingly, synapse loss appears to precede neuronal degeneration. Furthermore, memory and cognitive decline observed in AD patients correlate better with the synaptic pathology than either Aβ plaque load or tangle density. PS1 is expressed ubiquitously in the CNS and peripheral tissues. PS1-related derivatives have been found in small synaptic vesicles, synaptic plasma membranes, synaptic adhesion sites, and growth cones (Thinakaran and Parent, 2004). We and others previously showed an altered magnitude of long-term potentiation in transgenic mice expressing FAD-linked mutant PS1 (Thinakaran and Parent, 2004). Collectively, these observations indicate that PS1 may play a role in regulating aspects of neuronal differentiation, development, or synaptic function.
To extend these observations, we explored the possibility that PS-dependent cleavage of type I transmembrane protein(s), such as deleted in colorectal cancer (DCC), might influence signaling pathways that are critical for the synaptic function underlying memory processes. DCC is a substrate for metalloprotease-dependent proteolytic processing that results in shedding of its ectodomain (Galko and Tessier-Lavigne, 2000). The resulting membrane-tethered DCC derivative (DCC-α), consisting of the transmembrane segment and intracellular domain, is subject to PS-mediated intramembranous γ-secretase processing (Taniguchi et al., 2003). Here we show that DCC is cleaved by PS-dependent γ-secretase activity in cortical neurons. Moreover, DCC-α accumulates in neuroblastoma cells exposed to γ-secretase inhibitor, leading to an increase in neurite outgrowth and forskolin (FSK)-induced phosphorylation of protein kinase A (PKA) substrates. These latter biochemical outcomes are also observed in neurons of embryos harboring targeted deletions in PSEN1 alleles [knock-out (KO)] and wild-type (Wt) neurons treated with γ-secretase inhibitors. To examine the physiological consequence of PS-mediated alteration of cAMP/PKA signaling, we studied the influence of PS on glutamatergic synaptic transmission using cortical neurons cultured from PSEN1 KO and Wt embryos. We report that basal and FSK-induced increases in miniature EPSC (mEPSC) event frequency are enhanced in PSEN1-deficient neurons. This effect was accompanied by an enhancement of synaptic contacts associated with an increase in spiny structures. Together, our results are consistent with the idea that receptor-mediated signal transduction is regulated by PS-mediated intramembranous proteolysis. Specifically, PS-dependent proteolysis of DCC-α modulates the cAMP/PKA cascade, an essential component of synaptic transmission and memory processes.
Materials and Methods
Reagents. The γ-secretase inhibitor {1S-benzyl-4R-[1-(1S-carbamoyl-2-phenylethylcarbamoyl)-1S-3-methylbutylcarbamoyl]-2R-hydroxy-5-phenylpentyl} carbamic acid tert-butyl ester (L685,458) was a generous gift from Merck Research Laboratory (West Point, PA), and Compound E was generously provided by Dr. Todd E. Golde (Mayo Clinic, Jacksonville, FL). 3-[(E)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP) and 6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (NBQX) were purchased from RBI (Natick, MA). KT5720 and lidocaine N-ethyl bromide (QX-314) were obtained from Biomol (Plymouth Meeting, PA) and Alomone Labs (Jerusalem, Israel), respectively. All other compounds were obtained from Sigma (St. Louis, MO). Monoclonal Bassoon and postsynaptic density-95 (PSD-95) antibodies were generously provided by Dr. Craig Garner (University of Stanford, Stanford, CA) and Dr. David Bredt (University of California San Francisco, San Francisco, CA), respectively. Monoclonal DCC (clone G97-449) was purchased from BD Biosciences PharMingen (San Diego, CA). Monoclonal and polyclonal secondary antibodies (Alexa-488, Alexa-555, and Alexa-660) were purchased from Molecular Probes (Eu-gene, OR). pSecTagA/DCC plasmid was generously provided by Dr. Yimin Zou (University of Chicago).
Primary culture. PS1 KO and Wt embryos were generated by inter-crossing C57BL/6J PS1+/- mice as described previously (Wong et al., 1997). Primary neuronal cultures were generated from embryonic day 15 (E15) KO embryos and Wt littermates using standard procedures. Briefly, cortical sections were dissociated by passage through a 10 ml pipette, and the cell suspension was plated on poly-l-lysine-coated 35 or 60 mm dishes in Earle's medium supplemented with 0.15% HCO3, 0.5% glucose, 5% horse serum, 1% glutamine, and nutrients (N3) containing the following: BSA (0.001%), transferrin (20 μg/ml), putrescine (200 μm), sodium selenite (60 nm), triiodothyronine (20 ng/ml), insulin (10 μg/ml), progesterone (40 nm), corticosterone (40 ng/ml), and pyruvate (1 mm). For fluorescence imaging studies, cells are grown in 35 mm glass coverslip dishes (MatTek) coated with 0.1% polyethylenimine diluted in borate buffer (15 mm) adjusted at pH 9.0.
Immunoblot analysis. Primary neurons or N2a cells were lysed in a buffer containing NaCl (150 mm), Tris-HCl (50 mm, pH 7.4), EDTA (5 mm), NP-40 (0.5%), sodium deoxycholate (0.5%), SDS (0.25%), PMSF (2 mm), and protease inhibitor mixture (1:200; Sigma). Lysates from embryonic mouse brain were prepared similarly, except that 1% SDS detergent was used instead of NP-40 and sodium deoxycholate (Thinakaran et al., 1996). Endogenous APP full-length (APP-FL) and APP C-terminal fragments (APP-CTFs) were detected by immunoblotting with CT15, an antisera that recognizes the C-terminal 15 amino acids of APP (Sisodia et al., 1993). Endogenous DCC full-length (DCC-FL) and DCC endoproteolytic fragments (DCC-α, DCC-γ) were detected using G97-449 DCC antibody. PS1 N-terminal fragment (PS1-NTF) was detected using PS1NT antiserum (Thinakaran et al., 1996). DCC and PSD-95 Western blots were quantified by chemiluminescent imaging using a Typhoon multipurpose imager (Molecular Dynamics, Sunnyvale, CA).
Immunofluorescence staining. After FSK treatment (50 μm; 30 min at 37°C), neurons were fixed with 4% paraformaldehyde/4% sucrose for 30 min at 4°C and permeabilized with 0.2% Triton X-100 for 10 min. Dishes were incubated overnight at 4°C with polyclonal PSD-95 (1:100) (Brenman et al., 1996), PS1NT (1:50) (Thinakaran et al., 1996), and phospho-(Ser/Thr) PKA substrate (1:100; Cell Signaling Technology, Beverly, MA) antibodies or monoclonal Bassoon (1:10,000) (Altrock et al., 2003) and DCC (1:1000; BD Biosciences PharMingen) antibodies. Subsequently, cells were incubated with Alexa-488- and Alexa-555- or Alexa-660-conjugated secondary antibodies (1:200; Molecular Probes) for 1 h at room temperature. Confocal images were acquired with a 60× objective using an Olympus (Tokyo, Japan) system (FluoView) or a 100× objective using a Zeiss (Thornwood, NY) LSM Pascal 5 system. The images were processed using MetaMorph software (Universal Imaging, West Chester, PA). Identical photomultiplicator values and parameters of the laser scanning confocal microscope were used to acquire images. To quantify the level of fluorescence, images were first set to a prethreshold level to eliminate nonspecific fluorescence, and then the gray intensity level was determined and divided by the number of pixels. Colocalization was evaluated by the degree of fluorescence-overlapping area. Data are presented as mean ± SEM. Statistical significance was determined by ANOVA with independent post hoc Fisher's multiple comparison test.
Expression plasmids, cell culture, and transfection. Plasmid encoding rat full-length DCC plasmid pSecTagA/DCC has been described previously (Taniguchi et al., 2003). cDNA encoding N-terminally truncated DCC, termed DCC-ΔE, was constructed by fusing the signal sequence of rat DCC to the EcoRI site located 5′ to the sequence encoding the transmembrane region of DCC. This deletes residues 28-1020, eliminating most of the DCC extracellular domain. The signal sequence of DCC was amplified with the primers 5′-GTATGTTCCCATAGTAACGCC-3′, which is complementary to pSecTagA vector, and 5′-GCGAATTCGAAGATGCGCGCTGAGCAAG-3′. To generate the DCC-intracellular domain (IC), the fragment encoding the N-terminal half of the DCC-IC was amplified with primers 5′-GCGGTACCATGTGTACCCGGCGCTCTTCAG-3′ and 5′-CACAGACAGTGTTGGGAATG-3′, digested with KpnI and EcoRV, and ligated to the rest of the intracellular domain in pSecTagA vector. Mouse neuroblastoma N2a cells were maintained in 50% DMEM and 50% Opti-MEM supplemented with 5% fetal bovine serum. Cells were transiently transfected using LipofectAMINE Plus (Invitrogen, Gaithersburg, MD) according to the manufacturer's protocol. Twenty-four hours after transfection, the cells were treated with Compound E (10 nm) and maintained for another 24 h before being lysed for Western blotting or fixed for fluorescence imaging.
Electrophysiology. Whole-cell recordings were obtained from pyramidal-shaped neurons in mixed cortical neuron-astrocyte cultures maintained in vitro for 10-15 d. During recording sessions of mEPSCs, the cells were perfused continuously at room temperature with an external solution of HEPES buffer (10 mm) containing NaCl (140 mm), KCl (5 mm), CaCl2 (3 mm), dextrose (10 mm), strychnine (5 μm), picrotoxin (50 μm), and tetrodotoxin (1 μm), adjusted to pH 7.4. Patch electrodes (3-5 MΩ) were pulled from 1.5-mm-outer diameter glass capillaries and filled with cesium gluconate (100 mm), HEPES (40 mm), MgCl2 (5 mm), EGTA (1 mm), Na2-ATP (5 mm), Na3-GTP (0.5 mm) and QX-314 (5 mm). Whole-cell currents were recorded from individual pyramidal cells under voltage clamp at room temperature using an Axopatch 200A amplifier (Axon Instruments, Union City, CA). Cells were held at -70 mV. The currents were filtered at 2 kHz and digitized at 5 kHz. After recording of a stable baseline for a period of 15-20 min, FSK (50 μm) was bath applied for a period of 5 min to induce presynaptic release. FSK was dissolved in external solution and applied at a rate of ∼0.5 ml/min using a gravity-fed multibarrel perfusion system (DAD-VM from ALAScientific Instruments, Westbury, NY) in which the perfusion solution exited via a single barrel positioned close to the patched cell. For NMDA-independent mEPSC recordings, CPP (10 μm) was added to the perfusion solution supplemented with 1.5 mm MgCl2. To block AMPA receptor-mediated mEPSCs, NBQX (10 μm) was added to the perfusion solution. To inhibit PS function, Wt neurons were pretreated with either L685,458 (1 μm) (Li et al., 2000) or Compound E (10 nm) (Seiffert et al., 2000) for 4-24 h. Because both inhibitors gave similar results, data were pooled and reported as PS inhibitor-treated neurons.
mEPSC analysis. Changes in mEPSC frequency and amplitude were determined using the MiniAnalysis software (Synaptosoft, Decatur, GA). For the basal condition, averages of 250-1000 events for each epoch were manually identified and subsequently analyzed. Amplitude and area thresholds were used to acquire events, and each event was identified visually to protect against software errors. Frequency histograms and cumulative probability curves were generated using a 5 min epoch, binned at 2.5 and 1 pA, respectively. Two-way ANOVA was performed to evaluate the effect of group and time or group and electrophysiological parameters (frequency or amplitude). Statistical significance was determined with independent post hoc Scheffé's multiple comparison test. All experiments were performed in cultures derived from multiple embryos (each n represents an embryo and not multiple dishes of neurons from the same embryo).
Results
PS-dependent proteolysis of DCC
First, we examined DCC protein levels in extracts prepared from brains of E15 Wt and KO embryos (Fig. 1a1). Western blot analysis showed similar levels of ∼250 kDa full-length DCC in Wt and KO embryos, but significantly increased levels of ∼59 and ∼45 kDa DCC derivatives (referred to as DCC-α and DCC-*, respectively) in brains of KO embryos compared with Wt embryos (5.8 ± 1.6- and 4.1 ± 0.6-fold increase in DCC-α and DCC-*, respectively). We also observed accumulation of ∼59 kDa DCC derivative (DCC-α) in cortical neurons cultured from KO embryos in KO neurons and in Wt neurons treated with highly selective γ-secretase inhibitor, termed Compound E (Seiffert et al., 2000) (Fig. 1a2). To verify that Compound E can efficiently inhibit PS-mediated intramembranous cleavage of APP in cultured neurons, we examined the levels of APP-CTFs, the penultimate precursors of Aβ peptides that accumulate in the absence of functional γ-secretase (Li et al., 2000; Seiffert et al., 2000; Shearman et al., 2000). As expected, Western blot analysis revealed that APP-CTFs are not readily detectable in Wt neurons but accumulate to high levels in KO neurons or Wt neurons treated for 24 h with 10 nm Compound E (Fig. 1a2). Similar results were observed in neurons treated with L685,458 (data not shown), another selective inhibitor of PS-mediated γ-secretase activity (Li et al., 2000; Shearman et al., 2000).
Figure 1.
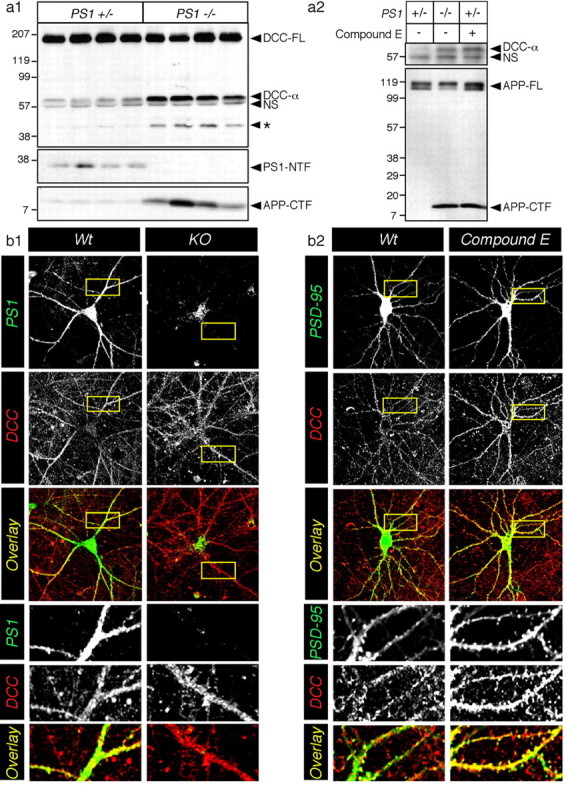
PS-dependent proteolysis of DCC in neurons. a1, Endogenous DCC-FL and proteolytic fragments (DCC-α and DCC-*) were detected by immunoblotting detergent lysates from Wt (PS1+/-) and KO (PS1-/-) mouse embryonic brains harvested at E15. A nonspecific band (NS) was also detected by this antibody. Bottom panels represent immunoblot analysis of PS1-NTF and APP-CTF. a2, In primary cultured neurons, although endogenous DCC expression was low, treatment with the γ-secretase inhibitor Compound E caused accumulation of DCC-α fragment to levels found in KO neurons. The bottom panel shows that endogenous APP-FL and APP-CTF were detected in untreated neurons (lane 1), KO neurons (lane 2), or neurons treated with Compound E (lane 3). Molecular weight standards are indicated on the left. b, Neuronal cultures were also immunostained with monoclonal antibody directed against DCC and polyclonal antibodies directed against PS1 (b1) or PSD-95 (b2). Representative confocal images are shown as a summation of contiguous three-stack images. The bottom panels show enlarged overlay images of the dendritic area. The level of overlap between two antibodies is shown as yellow.
Accumulation of DCC membrane clusters in primary neurons lacking PS function
To assess the functional relevance of PS-dependent intramembranous cleavage of DCC and the accumulation of DCC-α in brain, we examined the subcellular distribution of DCC-related polypeptides in primary neurons (Fig. 1b). We observed that DCC is distributed widely throughout the neuron but localized prominently in dendritic areas (Fig. 1b, enlarged overview) and growth cones (data not shown). In double-labeling studies, we observed considerable colocalization of PS1 and DCC, especially in dendritic areas (Fig. 1b1). We also performed double-immunofluorescence labeling with DCC and the postsynaptic marker PSD-95 and confirmed a close proximity of DCC with PSD-95 synaptic clusters in Wt neurons (Fig. 1b2). Interestingly, the distribution of DCC appears to be affected significantly by the absence of PS expression or loss of γ-secretase activity. We noticed that the DCC staining appears in larger clusters in KO and γ-secretase inhibitor-treated Wt neurons, and this pattern of staining was particularly noticeable on the surface and peripheral membranes in the somatic area (Fig. 1b) [see three-dimensional (3-D) reconstitution images in supplemental Fig. 1S, available at www.jneurosci.org as supplemental material]. Increased spine density as well as costaining of DCC and PSD-95 are particularly striking in reconstituted stacks of confocal images taken from neurons treated with γ-secretase inhibitor (Fig. 1b2) (see also 3-D reconstitution images in supplemental Fig. 1S, available at www.jneurosci.org as supplemental material). These results suggest that in KO and γ-secretase inhibitor-treated neurons, DCC and/or membrane-bound DCC C-terminal derivatives accumulate near or at the cell surface, especially in the somatic area and proximal to dendritic spiny structures.
Abrogation of PS function increases neurite outgrowth in DCC-transfected N2a cells
To study the biochemical significance of the accumulation of DCC-α in cells lacking PS function, we transfected mouse N2a neuroblastoma cells with cDNA encoding DCC-FL, DCC-ΔE that lacks the extracellular domain, or DCC-IC, corresponding to the DCC intracellular domain (Fig. 2a). Consistent with our previous studies (Taniguchi et al., 2003), treatment with Compound E produced a selective accumulation of the membrane-tethered DCC-α in cells transiently expressing DCC-FL, or DCC-ΔE, whereas PS γ-secretase inhibitor did not affect the levels of DCC-IC (Fig. 2b). N2a cells transfected with DCC-FL exhibit extensive neurite outgrowth (Fig. 2c), as reported previously (Shekarabi and Kennedy, 2002), but this phenotype was not observed in N2a cells transiently expressing DCC-ΔE or the DCC-IC. Interestingly, treatment with the γ-secretase inhibitor (Compound E) significantly enhanced levels of unprocessed DCC-ΔE and neurite arborization in cells expressing DCC-ΔE (Fig. 2b,c). Compound E had no effect on cellular morphology or accumulation of the DCC intracellular derivative in cells expressing DCC-IC (Fig. 2b,c). Quantification of DCC immunostaining revealed a 22 ± 9 and 41 ± 7% enhancement in the area occupied by DCC in Compound E-treated cells expressing DCC-FL and DCC-ΔE, respectively, compared with untreated cells (Fig. 2d). We note that by virtue of the fact that the ectodomain and potential ligand binding sites are deleted in DCC-ΔE, the signaling events that lead to the induction of neurite outgrowth in γ-secretase inhibitor-treated cells are most likely mediated by the accumulation of the membrane-tethered DCC derivative.
Figure 2.

Abrogation of PS function increases neurite outgrowth in DCC-transfected N2a cells. a, Schematic representation of DCC and deletion mutants. b, Lysates from N2a cells transfected with DCC-FL, DCC-ΔE, and DCC-IC were analyzed by Western blot before or after treatment with Compound E. DCC-FL and proteolytic fragments, which correspond to the transmembrane domain (TM) and IC, were detected using the monoclonal G97-449 DCC antibody. c, Representative confocal images of untreated cells (top panels) and Compound E-treated cells (bottom panels) expressing DCC-FL, DCC-ΔE, or DCC-IC are shown. d, Quantitative analysis of the morphological changes is represented as relative change in immunostained area compared with untreated cells transfected with DCC-FL. Statistical analysis was performed using ANOVA followed by a post hoc Fisher's analysis. *p < 0.05 or **p < 0.001 compared with untreated cells. The total number of cells used for quantification is shown in parentheses (prepared from at least 3 independent experiments).
Accumulation of membrane-tethered DCC derivatives is associated with an increase in FSK-induced PKA activation
Previous studies have shown that netrin-mediated activation of DCC is intimately related to cAMP-dependent downstream signaling pathways (Song and Poo, 1999; Corset et al., 2000; Ming et al., 2001; Forcet et al., 2002; Shewan et al., 2002; Nishiyama et al., 2003). To examine the extent to which PS-dependent γ-secretase processing of DCC might influence cAMP signaling, we asked whether transfected N2a cells treated with γ-secretase inhibitors exhibited differences in the extent of PKA activation (Fig. 3). As a measure of PKA activation, we examined PKA substrate phosphorylation using an antibody selective for phospho-serine and phospho-threonine epitopes of PKA substrates (Gronborg et al., 2002). As expected, the immunofluorescence intensity of phospho-PKA substrates was increased by approximately twofold in all groups when cells were treated with FSK (Fig. 3b) (mean intensity: control nontransfected, 186 ± 17%; DCC-FL, 206 ± 15%; DCC-ΔE, 170 ± 23%; DCC-IC, 224 ± 24%). After treatment with γ-secretase inhibitor, the overall phosphorylation of PKA substrates was increased further in control nontransfected cells by 82 ± 24%; however, we observed a more profound elevation in PKA substrate phosphorylation in inhibitor-treated cells expressing DCC-FL and DCC-ΔE (175 ± 31 and 173 ± 37%, respectively). Moreover, the intensity of phospho-PKA immunofluorescence was not affected by the inhibitor treatment in DCC-IC-transfected cells (Fig. 3b). N2a cells treated with 1,9-FSK (an inactive analog of FSK) failed to exhibit any increase in phospho-(Ser/Thr) PKA substrate immunostaining, confirming that the FSK-induced increase in phospho-PKA substrates was mediated directly through the activation of adenylate cyclase and the cAMP pathway (Fig. 3b). Together, our results strongly suggest that loss of PS function enhances activation of the cAMP/PKA pathway in N2a cells and that expression of DCC exacerbates this effect. Our results also suggest that accumulation of the transmembrane-bound DCC fragment in γ-secretase inhibitor-treated cells leads to increased phosphorylation of cAMP-dependent substrates.
Figure 3.
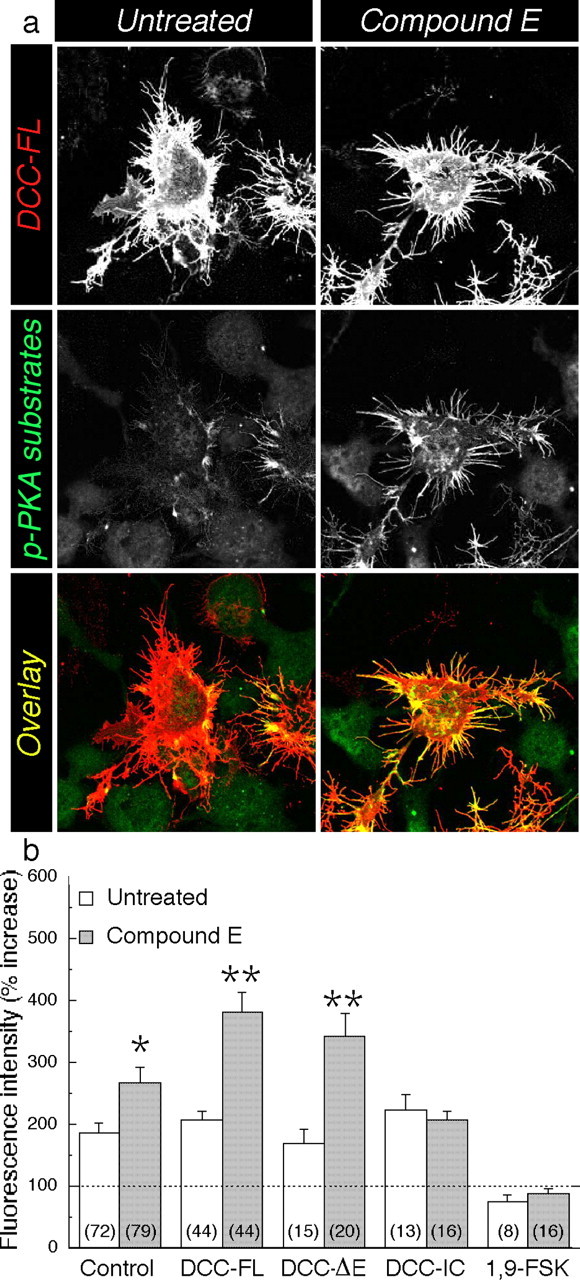
Accumulation of membrane-tethered DCC derivatives is associated with an increase in FSK-induced cAMP-dependent signaling. N2a cells were transfected with DCC-FL, DCC-ΔE, and DCC-IC and immunostained with antibody directed against DCC (red) and phosphorylated Ser/Thr PKA substrates (green). a, Representative confocal images of DCC (top panels) and phospho-(Ser/Thr) PKA substrate (middle panels) staining in DCC-FL-transfected cells (untreated or Compound E treated) after stimulation with FSK. Color overlay (bottom panels) shows strong colocalization (yellow), especially in neurite processes. b, Percentage increase in staining intensity was determined by comparison of fluorescence intensity after application of FSK or 1,9-FSK (an inactive analog of FSK) to a respective unstimulated group (data not shown). Percentage increase is shown in the absence (untreated) or presence (Compound E) of γ-secretase inhibitor. The level of phospho-PKA substrates in untransfected cells is identified as the control group. To normalize the differences in the cell area between transfected conditions, the fluorescence intensity level of phospho-PKA substrates was adjusted to the total area of the cells per field under study. Statistical analysis was performed using ANOVA followed by a post hocFisher's analysis. *p < 0.05 or **p < 0.001 compared with untreated cells in each case. The total number of fields used for quantification is shown in parentheses (prepared from at least 3 independent experiments).
Increase in cAMP/PKA signaling in PS-deficient neurons
Next we sought to identify whether Wt and KO neurons and Wt neurons treated with γ-secretase inhibitors also exhibited differences in the extent of PKA activation. Quantification of immunofluorescence images revealed that the intensity of phospho-(Ser/Thr) PKA substrate staining was comparable among all three groups under basal conditions (Fig. 4a) (intensity/pixel: Wt, 9.2 ± 1.7; KO, 7.2 ± 1.7, p = 0.689; Compound E, 7.0 ± 1.4, p = 0.649). Analysis of secondary antibody staining (no primary antibody) was performed as a control using identical photomultiplicator, laser scanning, and threshold parameters (nonspecific intensity/pixel: 0.2 ± 0.1; n = 16). As expected, the intensity of staining was enhanced after treatment with FSK in all neurons (Fig. 4a). In the absence of PS expression/function, we observed a larger increase in FSK-induced phosphorylation of Ser/Thr PKA substrates, particularly in neuronal soma and large dendrites (Fig. 4a,b1) (Wt, 238 ± 29%; KO, 614 ± 117%, p = 0.002; Compound E, 492 ± 87%, p = 0.031). To establish the specificity of phospho-(Ser/Thr) PKA substrate staining, we pretreated another set of cultures with KT5720 (5 μm), a PKA inhibitor, which blocked the FSK-induced increase in staining of KO and Wt neurons treated with γ-secretase inhibitor (Fig. 4b2). Collectively, these results indicate that in the absence of PS expression/function, PKA-dependent phosphorylation of substrates at Ser/Thr residues is enhanced.
Figure 4.
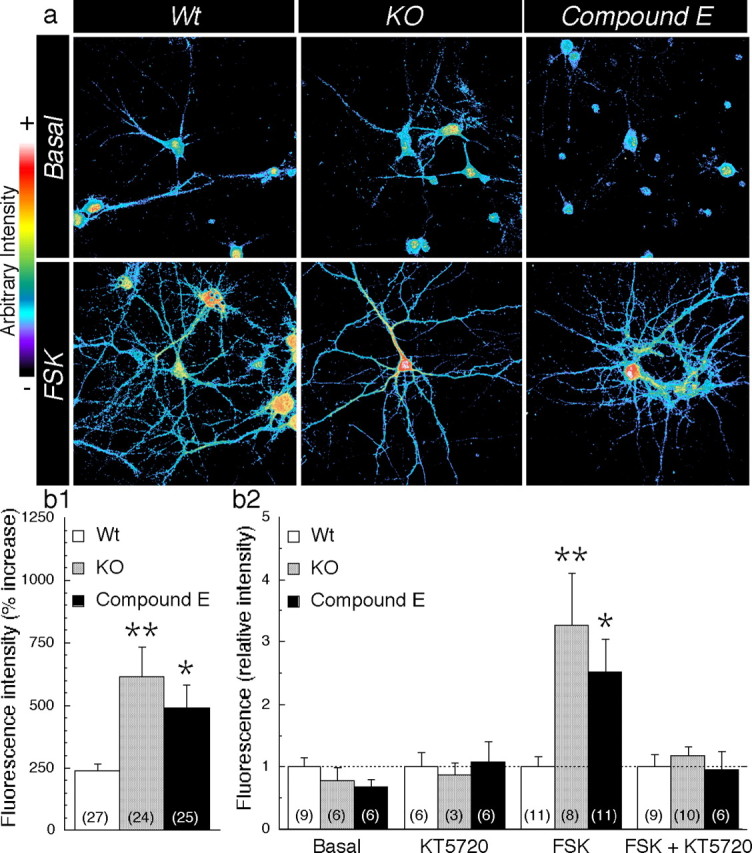
Increase in cAMP/PKA signaling in PS-deficient neurons. a, Representative confocal images of phospho-(Ser/Thr) PKA substrate staining are shown in pseudocolor in the basal condition (top panels) or after FSK application (bottom panels). b1, Quantification of staining intensity is shown as a percentage of increase in fluorescence intensity after FSK treatment. The total number of neurons used for quantification is shown in parentheses (Wt, n = 11 embryos; KO, n = 8 embryos; Compound E, n = 9 embryos). b2, In a separate set of experiments, the relative increase in fluorescence intensity after FSK application is shown in the presence and absence of the PKA inhibitor KT5720 (5 μm). Relative increase was determined by comparison with Wt neurons. Statistical analysis was performed using ANOVA followed by a post hoc Fisher's analysis. *p < 0.05 and **p < 0.005 compared with Wt in each case.
Increase in glutamatergic synaptic transmission in neurons lacking PS expression/function
To evaluate the consequence of the PS-mediated increase in cAMP/PKA signaling in neurons, we examined the glutamatergic synaptic function by recording miniature synaptic events in cortical pyramidal neurons derived from KO embryos and their Wt littermates. mEPSC recordings were performed in HEPES buffer without Mg2+, allowing NMDA receptors to be activated. As shown in Figure 5, a1 and b1, the frequency of mEPSC events is higher in KO neurons and Wt neurons treated with γ-secretase inhibitors; analysis of the cumulative probability curves revealed that the overall mEPSC amplitudes are essentially identical (supplemental Fig. 2S, available at www.jneurosci.org as supplemental material), suggesting that the probability of release is increased in PS-deficient neurons. In addition, we examined mEPSC in neurons after acute application of FSK (50 μm; 5 min). Fifteen minutes after the application of FSK, all groups exhibit an increase in mEPSC frequency, but the increase was more pronounced in KO neurons and in γ-secretase inhibitor-treated neurons compared with Wt neurons (Fig. 5a2,b2-4). We tested the long-lasting enhancement of synaptic release in our system for a period of 45 min after FSK application (Fig. 5c). In Wt neurons, the percentage of total number of mEPSC events reached a plateau between 5 and 15 min after FSK application and declined gradually; however, in KO neurons, the peak of increase in mEPSC events occurred at ∼30 min after FSK application and remained at relatively high levels. The percentage increase in mEPSC frequency was much larger in KO compared with Wt neurons even 45 min after FSK application (p = 0.008). Together, our results suggest that PS regulates a component(s) of the presynaptic release machinery that is sensitive to cAMP/PKA signaling cascade(s), which is consistent with the increase in FSK-induced phospho-PKA substrates seen in these cells (Fig. 4).
Figure 5.

Increase in mEPSC frequency in PS-deficient neurons. a, mEPSC representative traces recorded from Wt, KO, and Wt neurons treated with PS γ-secretase inhibitors are shown before (a1) and after (a2) application of FSK. b, Mean event count histograms of mEPSC are shown in the basal condition (b1) and before and after application of FSK in Wt (b2), KO (b3), and PS inhibitor-treated (b4) neurons (n = 9, n = 7, and n = 7 embryos for Wt, KO, and PS inhibitor, respectively). Statistical analysis was performed using a two-way ANOVA followed by Scheffé's post hoc analysis. *p < 0.05 compared with Wt; #p < 0.05 compared with basal. c, FSK-induced increase in mEPSC frequency, expressed as the percentage of change over basal value, shown in Wt (○) and KO (•) neurons. Statistical analysis was performed using ANOVA followed by a post hoc Fisher's analysis. *p < 0.05 compared with Wt. d, Relative frequency of mEPSC events was analyzed from cortical pyramidal neurons using Mg2+-free HEPES buffer (open bars) or HEPES buffer supplemented with Mg2+ and CPP (filled bars). The number of embryos used for the studies is shown in parentheses.
To evaluate the contribution of NMDA receptors to the observed differences in mEPSC events, we performed a set of experiments in the presence of Mg2+ and CPP (10 μm) in the bath to block NMDA components. Consistent with data shown in Figure 5b, recordings performed in the absence of Mg2+ showed an increase in total mEPSC events in KO and γ-secretase inhibitor-treated Wt neurons, compared with untreated Wt neurons (Fig. 5d). Interestingly, blockade of NMDA receptors did not significantly alter the mEPSC frequency in any of the three groups (no Mg2+ and Mg2+ plus CPP conditions: Wt, 1.00 ± 0.21 vs 0.69 ± 0.24, p = 0.697; KO, 2.31 ± 0.54 vs 1.91 ± 0.76, p = 0.656; PS inhibitor, 2.82 ± 0.90 vs 2.71 ± 0.45, p = 0.913). Addition of NBQX (10 μm) to the perfusion solution eliminated all synaptic events (data not shown). These results support the idea that PS effects mainly AMPA receptor-mediated synaptic transmission and acute blockade of NMDA receptors does not contribute to that process.
Increase in synapse formation in PS-deficient neurons
Based on the observation that the frequency of mEPSCs is increased in KO neurons and Wt neurons treated with γ-secretase inhibitors, we set out to assess whether the absence of PS or inhibition of γ-secretase might alter the communication between the presynaptic and postsynaptic sites (Fig. 6). To evaluate the level of synapse formation, we performed immunofluorescence labeling using antibodies against well characterized presynaptic and postsynaptic markers, Bassoon and PSD-95, respectively (Brenman et al., 1996; Altrock et al., 2003). Using previously described methods (Friedman et al., 2000; Okabe et al., 2001; Prange and Murphy, 2001; Washbourne et al., 2002), we observed that quantification of the immunofluorescence staining by confocal microscopy revealed a twofold increase in axodendritic clusters visualized as an increase in an overlapping area of the two markers in KO neurons and inhibitor-treated Wt neurons compared with untreated Wt neurons (Fig. 6a5,b3). This increase parallels a selective increase of the area (Fig. 6b1) and the number (supplemental Fig. 3Sb1, available at www.jneurosci.org as supplemental material) of dendritic clusters positive for PSD-95, reminiscent of an increase in spiny structures (Fig. 1b2) (for spine visualization, see also supplemental Figs. 1S and 3S, available at www.jneurosci.org as supplemental material). Using glutamate receptor 2 antibody as an independent marker of postsynaptic structures, we were also able to visualize an increase in spine density in KO neurons compared with Wt neurons (supplemental Fig. 3Sd, available at www.jneurosci.org as supplemental material). In addition to the area and number of PSD-95 clusters, we also observed that the intensity of PSD-95 cluster staining was significantly increased in KO and γ-secretase inhibitor-treated Wt neurons (Wt, 1478 ± 18; KO, 1589 ± 28, p = 0.004; Compound E, 1640 ± 30, p < 0.001) (supplemental Fig. 3Sc2, available at www.jneurosci.org as supplemental material), whereas the overall intensity of the PSD-95 staining was not affected (supplemental Fig. 3Sb2, available at www.jneurosci.org as supplemental material). Using Western blot analysis, we also confirmed that the steady-state levels of PSD-95 were not affected by the lack of PS1 expression or γ-secretase activity (supplemental Fig. 3Sa, available at www.jneurosci.org as supplemental material). An increase in the number or intensity of the clusters without changes in the total PSD-95 is consistent with the idea that PSD-95 is redistributed at the postsynaptic membranes in KO or γ-secretase inhibitor-treated neurons. On the other hand, analysis of the fluorescence intensity of the Bassoon clusters revealed that the staining was relatively unchanged (Wt, 1874 ± 16; KO, 1873 ± 23, p = 0.984; Compound E, 1920 ± 18, p = 0.093) (supplemental Fig. 3Sc1, available at www.jneurosci.org as supplemental material); however, the overall distribution of Bassoon-stained clusters was notably more scattered along dendrites of KO neurons (Fig. 6a2), but the relative area was slightly decreased in KO compared with Wt neurons (Fig. 6a4,b2). Together, these results suggest that in the absence of PS expression/function, postsynaptic structures are mainly altered, leading to an increase in synapse formation and consequently an increase in synaptic transmission.
Figure 6.
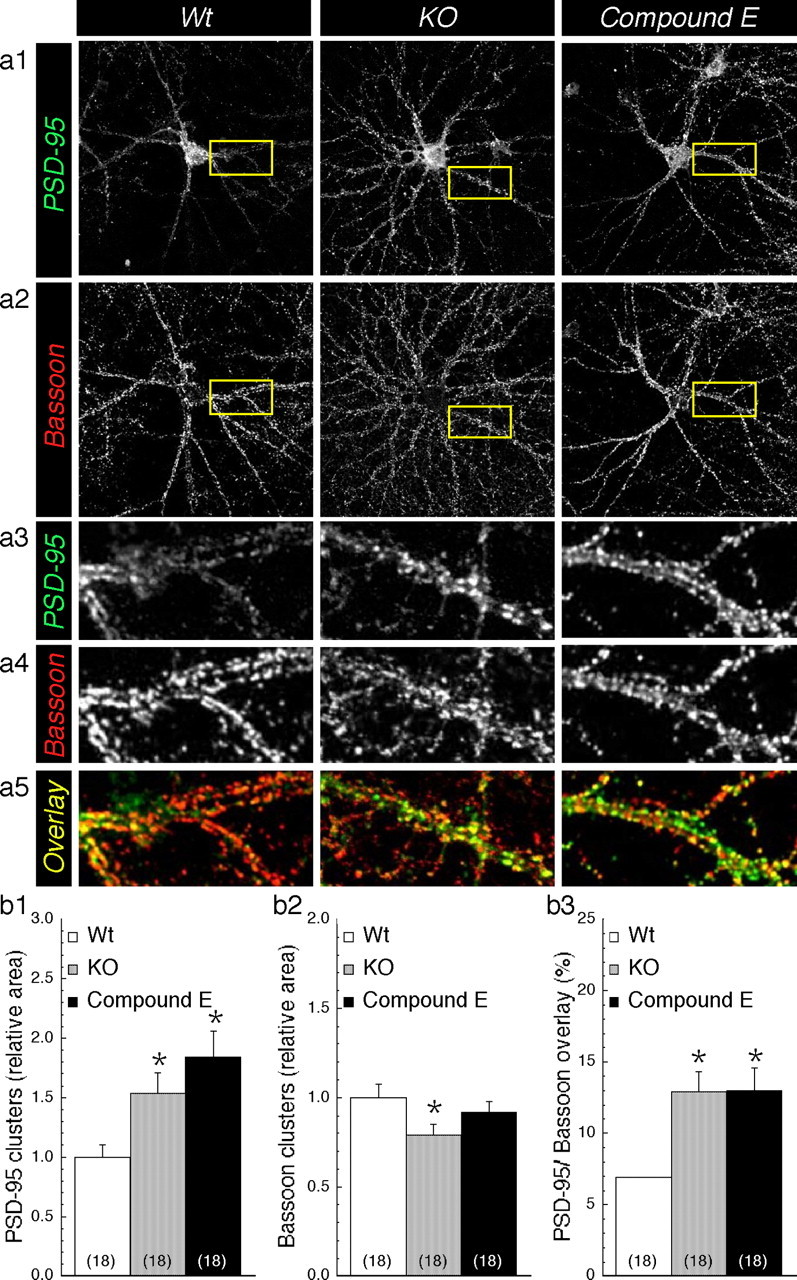
Increase in synaptic clusters in PS-deficient neurons. Representative confocal images of PSD-95 and Bassoon staining are shown in a1 and b2, respectively, for Wt, KO, and Wt neurons treated with Compound E. Enlarged areas are shown in a3 and a4. Synaptic clusters of PSD-95 (green) and Bassoon (red) are visualized in a5 as yellow overlay. Quantification of cluster staining of PSD-95 (b1) and Bassoon (b2) was performed for each field of view by applying a common threshold high enough to subtract the weak staining and isolate the intensely stained clusters. The values are represented as relative change in total cluster area compared with Wt. b3, Change in axodendritic contact is evaluated as percentage of PSD-95/Bassoon overlay staining. Statistical analysis was performed using ANOVA followed by a post hoc Fisher's analysis. *p < 0.05 compared with Wt. The total number of neurons used for quantification is shown in parentheses (cultures prepared from 3 different embryos per group).
Discussion
Several lines of evidence suggest that PS executes multiple cellular functions, in addition to its essential role in the intramembranous processing of APP-CTF and the production of Aβ peptides (Fortini, 2002; Koo and Kopan, 2004; Thinakaran and Parent, 2004). We report that DCC, a receptor known to be intimately associated with cAMP signaling (Song and Poo, 1999; Corset et al., 2000; Ming et al., 2001; Forcet et al., 2002; Shewan et al., 2002; Nishiyama et al., 2003), undergoes PS-dependent proteolysis. Indeed, we observed that loss of PS1 expression caused accumulation of membrane-bound DCC intracellular domain in embryonic brain and cultured primary neurons. We also found that accumulation of this fragment correlates with increased neurite outgrowth and spine density in transfected neuroblastoma cells and cortical neurons, respectively, and increased FSK-induced accumulation of phospho-PKA substrates in both systems. In cortical neurons lacking PS1 expression and γ-secretase activity, we observed an increase in mEPSC frequency, which is enhanced under conditions during which the cAMP/PKA pathway is activated. Changes in synaptic function parallel an enhancement of presynaptic and postsynaptic contact in the axodendritic area, which was associated with an increase in spine density. On the basis of this, we propose that γ-secretase cleavage of DCC may serve to downregulate coupling of DCC to an intracellular signaling cascade. We also propose that one mechanism that links PS function with regulation of the cAMP/PKA signaling pathway and modulation of cellular substrates of memory formation and storage involves, at least to some degree, signaling mediated by DCC.
DCC proteolysis regulates cAMP/PKA signaling
Consistent with our previous study (Taniguchi et al., 2003), we demonstrate that DCC is a substrate for intramembranous proteolysis by a PS-dependent γ-secretase, and a membrane-bound C-terminal fragment of DCC lacking the extracellular domain accumulates in brains of KO embryos and cultured neurons lacking γ-secretase activity. Similar to many receptor tyrosine kinases, which are rendered constitutively active by the deletion of their extracellular domain (Schlessinger, 2000), expression of a membrane-bound UNC-40/DCC molecule lacking an extracellular domain activates cytoplasmic signaling of the UNC-40 pathway in a constitutive, ligand-independent manner (Gitai et al., 2003). Therefore, we propose a model wherein in neurons lacking PS expression/function, enhanced accumulation of membrane-bound DCC intracellular domain facilitates protein interactions that exacerbate phosphorylation of cAMP/PKA-dependent substrates (supplemental Fig. 4S, available at www.jneurosci.org as supplemental material). It was hypothesized recently that PS/γ-secretase complexes might play a role as membrane proteasome either (1) by facilitating the disposal of membrane receptor(s) after extracellular domain shedding or (2) by terminating the signaling cascade induced after receptor activation (Kopan and Ilagan, 2004). Our study provides the first evidence for the second hypothesis because we demonstrates that PS-dependent cleavage of a receptor(s) mitigates downstream second-messenger cascades such as the cAMP/PKA pathway.
cAMP/PKA signaling plays a critical role in regulating short- and long-term synaptic physiology (Milner et al., 1998; Malenka and Nicoll, 1999; Malinow and Malenka, 2002; Bredt and Nicoll, 2003). Therefore, the increase in cAMP/PKA activation associated with the accumulation of DCC-α is likely to impact on synaptic function. Indeed, after stimulation with FSK, we observed a close relationship between increased phosphorylation of PKA substrates and enhanced synaptic transmission. The greater increase in cAMP-dependent phosphorylation of PKA substrates observed in neurons lacking PS function also correlates well with the magnitude of increase in synaptic transmission. On the other hand, an increase in basal synaptic transmission in neurons lacking PS expression/function does not correlate with PKA phosphorylation status. How can we explain the increase in basal synaptic transmission in neurons lacking PS expression/function? It can be speculated that stimulus-induced activation of the PKA pathway might affect synaptic morphology, therefore indirectly affecting basal synaptic transmission (El-Husseini et al., 2000; Murase et al., 2002; De Paola et al., 2003; Kasai et al., 2003; Matsuzaki et al., 2004). In fact, our results illustrate that neurons are morphologically affected by the lack of PS expression/function. KO and γ-secretase inhibitor-treated neurons exhibit an increase in synaptic contact and spine density. Alternatively, other signaling pathways causing neurite outgrowth and spine proliferation might also be involved (El-Husseini et al., 2000; Nakayama et al., 2000; Meyer and Feldman, 2002; Murase et al., 2002; Penzes et al., 2003).
PS attenuates glutamatergic synaptic transmission
The increased frequency of synaptic events observed in KO neurons and Wt neurons treated with γ-secretase inhibitors in our study likely reflects differences in presynaptic release, because the overall amplitude of mEPSC was unaffected either before or after FSK application; however, it has been suggested that the activity-dependent increase in mEPSC frequency is also associated with the conversion of postsynaptically silent synapses, which lack AMPA receptors, to synapses that contain functional AMPA receptors at the cell surface (Malinow and Malenka, 2002; Bredt and Nicoll, 2003). Hence, although our results suggest that PS influences the presynaptic machinery, an alternate hypothesis that might explain the observed increase in mEPSC frequency is that neurons lacking PS function may have fewer silent synapses. In this latter model, loss of PS function results in the “switching on” of silent synapses, thus increasing AMPA receptor localization at the cell surface. Because rapid turnover of silent synapses might be dependent on activation of NMDA receptors, we examined whether the higher frequency of mEPSCs associated with loss of PS expression/function is abolished by NMDA receptor inactivation. Increased synaptic transmission in neurons lacking PS expression/function was observed even under conditions during which NMDA receptors are inactivated, ruling out rapid NMDA-dependent synaptic recruitment of AMPA receptors. In the future it will be interesting to determine whether glutamatergic synaptic events are affected directly by a selective change in AMPA receptor expression or distribution in neurons lacking PS expression/function. It is also unclear how our present findings relate to the altered magnitude of long-term potentiation reported in PS1 heterozygous mice or transgenic mice expressing FAD-linked mutant PS1. In this regard, it remains also to be established whether differences in PS1-dependent proteolysis of developmentally regulated molecules such as DCC might underlie synaptic plasticity in mice expressing FAD-linked PS1 mutations.
We also performed extensive analysis of colocalization between presynaptic and postsynaptic markers in our system because alterations in axodendritic contacts can induce overall changes in synaptic transmission. Indeed, in neurons lacking PS expression/function, we observed a greater level of synaptic contact, as seen specifically as an increase in the spine density, size, and intensity of PSD-95 clusters. Our results are consistent with previous findings in which an increase in spine density and in the size or intensity of PSD-95 clusters paralleled AMPA receptor-mediated increases in mEPSC frequency (El-Husseini et al., 2000; Murase et al., 2002) or current (Kasai et al., 2003; Matsuzaki et al., 2004). Together, as proposed in our model (supplement Fig. 4S, available at www.jneurosci.org as supplemental material), our results indicate that lack of PS function modifies the postsynaptic structures, leading to an increase in synaptic contact associated with enhancement of spine density and ultimately an increase in synaptic transmission.
Regulation of synaptic function by DCC
How does the accumulation of membrane-bound DCC intracellular domain and the parallel increase in cAMP-dependent accumulation of phospho-PKA substrates regulate synaptic function? DCC has been studied mainly for its role in axonal development (Manitt and Kennedy, 2002). Interestingly, it has been demonstrated recently in Drosophila that ligand-activated DCC receptors function as guidance molecules not only for axons but also for dendrites (Furrer et al., 2003). In other systems, growing evidence supports the idea that axodendritic development is intimately related to spine formation (Ziv and Smith, 1996). It has also been reported that functional presynaptic boutons appear after axodendritic contact, and this process is preceded by the assembly of postsynaptic compartment(s) such as clustering of PSD-95 and glutamate receptors (Friedman et al., 2000; Goda and Davis, 2003). Consistent with these findings, we observed that lack of PS function increases axodendritic contacts, which were accompanied by an increase in PSD-95 clusters and spine formation. The magnitude of the DCC-induced increase in synaptic function in our system remains to be established.
In summary, our studies highlight the importance of PS-dependent proteolytic cleavage of putative receptor(s) in regulating signal transduction and, consequently, synaptic function. Our studies are consistent with the functional interaction of PS-mediated γ-secretase activity with the cAMP/PKA pathway via transmembrane substrates. We have presented data on one molecule that fulfills these criteria. As our studies on DCC emphasize, accumulation of membrane-bound DCC intracellular domain affects the cAMP signaling cascade and neurite outgrowth. Consequently, in PS-deficient neurons, accumulation of membrane-bound DCC intracellular domain correlates well with increases in spine density and cAMP/PKA-dependent synaptic transmission and long-term synaptic plasticity. Our results illustrate a novel function of PS as a regulator of the level of membrane-tethered intracellular domain(s) wherein cleavage by PS-mediated γ-secretase terminates intracellular signaling. In addition, the close interaction of PS-mediated regulation of glutamatergic synaptic transmission with cAMP/PKA signaling strongly suggests that PS is a putative regulator of cellular substrates of the memory process.
Footnotes
This work was supported by National Institutes of Health Grants AG021494 (S.S.S.) and AG021495 (G.T.), Alzheimer's Association Grant IIRG-02-3952 (A.T.P.), and the Brain Research Foundation/Women Council (A.T.P.). We thank Michael Ehlers, Aaron Fox, David Linden, and Daniel McGehee for helpful discussions, Vytas Bindokas for advice on confocal imaging, and Su Chu and David Terrano for technical assistance in maintaining the mouse colony and genotyping.
Correspondence should be addressed to Angèle T. Parent, Department of Neurobiology, Pharmacology, and Physiology, University of Chicago, 924 East 57th Street, Chicago, IL 60637. E-mail: aparent@uchicago.edu.
Y. Taniguchi's present address: Department of Radiation Genetics, Kyoto University, Graduate School of Medicine, Kyoto 606-8501, Japan.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251540-10$15.00/0
References
- Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C, Fassler R, Richter K, Boeckers TM, Potschka H, Brandt C, Loscher W, Grimberg D, Dresbach T, Hempelmann A, Hassan H, Balschun D, Frey JU, Brandstatter JH, Garner CC, et al. (2003) Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein Bassoon. Neuron 37: 787-800. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS (1996) Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci 16: 7407-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corset V, Nguyen-Ba-Charvet KT, Forcet C, Moyse E, Chedotal A, Mehlen P (2000) Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 407: 747-750. [DOI] [PubMed] [Google Scholar]
- De Paola V, Arber S, Caroni P (2003) AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat Neurosci 6: 491-500. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Forcet C, Stein E, Pays L, Corset V, Llambi F, Tessier-Lavigne M, Mehlen P (2002) Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 417: 443-447. [DOI] [PubMed] [Google Scholar]
- Fortini ME (2002) Gamma-secretase-mediated proteolysis in cell-surface-receptor signaling. Nat Rev Mol Cell Biol 3: 673-684. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE (2000) Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27: 57-69. [DOI] [PubMed] [Google Scholar]
- Furrer MP, Kim S, Wolf B, Chiba A (2003) Robo and Frazzled/DCC mediate dendritic guidance at the CNS midline. Nat Neurosci 6: 223-230. [DOI] [PubMed] [Google Scholar]
- Galko MJ, Tessier-Lavigne M (2000) Function of an axonal chemoattractant modulated by metalloprotease activity. Science 289: 1365-1367. [DOI] [PubMed] [Google Scholar]
- Gitai Z, Yu TW, Lundquist EA, Tessier-Lavigne M, Bargmann CI (2003) The netrin receptor UNC-40/DCC stimulates axon attraction and outgrowth through enabled and, in parallel, Rac and UNC-115/AbLIM. Neuron 37: 53-65. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW (2003) Mechanisms of synapse assembly and disassembly. Neuron 40: 243-264. [DOI] [PubMed] [Google Scholar]
- Gronborg M, Kristiansen TZ, Stensballe A, Andersen JS, Ohara O, Mann M, Jensen ON, Pandey A (2002) A mass spectrometry-based proteomic approach for identification of serine/threonine-phosphorylated proteins by enrichment with phospho-specific antibodies: identification of a novel protein, Frigg, as a protein kinase A substrate. Mol Cell Proteomics 1: 517-527. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H (2003) Structure-stability-function relationships of dendritic spines. Trends Neurosci 26: 360-368. [DOI] [PubMed] [Google Scholar]
- Koo EH, Kopan R (2004) Potential role of presenilin-regulated signaling pathways in sporadic neurodegeneration. Nat Med 10: S26-33. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX (2004) Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5: 499-504. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689-694. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999) Long-term potentiation—a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Manitt C, Kennedy TE (2002) Where the rubber meets the road: netrin expression and function in developing and adult nervous systems. Prog Brain Res 137: 425-442. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429: 761-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Feldman EL (2002) Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem 83: 490-503. [DOI] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER (1998) Cognitive neuroscience and the study of memory. Neuron 20: 445-468. [DOI] [PubMed] [Google Scholar]
- Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M (2001) Electrical activity modulates growth cone guidance by diffusible factors. Neuron 29: 441-452. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM (2002) Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35: 91-105. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L (2000) Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci 20: 5329-5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 424: 990-995. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H (2001) Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci 21: 6105-6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL (2003) Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 37: 263-274. [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH (2001) Modular transport of postsynaptic density-95 clusters and association with stable spine precursors during early development of cortical neurons. J Neurosci 21: 9325-9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA (2003) Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging 24: 1029-1046. [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211-225. [DOI] [PubMed] [Google Scholar]
- Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith Jr JE, Wang Q, Roach AH, Thompson LA, Spitz SM, Higaki JN, Prakash SR, Combs AP, Copeland RA, Arneric SP, Hartig PR, Robertson DW, Cordell B, Stern AM, Olson RE, et al. (2000) Presenilin-1 and -2 are molecular targets for gamma-secretase inhibitors. J Biol Chem 275: 34086-34091. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791. [DOI] [PubMed] [Google Scholar]
- Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL (2000) L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry 39: 8698-8704. [DOI] [PubMed] [Google Scholar]
- Shekarabi M, Kennedy TE (2002) The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol Cell Neurosci 19: 1-17. [DOI] [PubMed] [Google Scholar]
- Shewan D, Dwivedy A, Anderson R, Holt CE (2002) Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat Neurosci 5: 955-962. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St. George-Hyslop PH (2002) γ-Secretase, Notch, Aβ and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci 3: 281-290. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL (1993) Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci 13: 3136-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Poo MM (1999) Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol 9: 355-363. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Kim SH, Sisodia SS (2003) Presenilin-dependent “gamma-secretase” processing of deleted in colorectal cancer (DCC). J Biol Chem 278: 30425-30428. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2001) New frontiers in Alzheimer's disease genetics. Neuron 32: 181-184. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Parent AT (2004) Identification of the role of presenilins beyond Alzheimer's disease. Pharmacol Res 50: 411-418. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17: 181-190. [DOI] [PubMed] [Google Scholar]
- Turner PR, O'Connor K, Tate WP, Abraham WC (2003) Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70: 1-32. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK (2002) Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5: 751-759. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS (1997) Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387: 288-292. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ (1996) Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17: 91-102. [DOI] [PubMed] [Google Scholar]