

Abstract
Myelin-forming oligodendrocytes facilitate saltatory nerve conduction and support neuronal functions in the mammalian CNS. Although the processes of oligodendrogliogenesis and differentiation from neural progenitor cells have come to light in recent years, the molecular mechanisms underlying oligodendrocyte myelinogenesis are poorly defined. Herein, we demonstrate the pivotal role of the basic helix-loop-helix transcription factor, Olig1, in oligodendrocyte myelinogenesis in brain development. Mice lacking a functional Olig1 gene develop severe neurological deficits and die in the third postnatal week. In the brains of these mice, expression of myelin-specific genes is abolished, whereas the formation of oligodendrocyte progenitors is not affected. Furthermore, multilamellar wrapping of myelin membranes around axons does not occur, despite recognition and contact of axons by oligodendrocytes, and Olig1-null mice develop widespread progressive axonal degeneration and gliosis. In contrast, myelin sheaths are formed in the spinal cord, although the extent of myelination is severely reduced. At the molecular level, we find that Olig1 regulates transcription of the major myelin-specific genes, Mbp, Plp1, and Mag, and suppresses expression of a major astrocyte-specific gene, Gfap. Together, our data indicate that Olig1 is a central regulator of oligodendrocyte myelinogenesis in brain and that axonal recognition and myelination by oligodendrocytes are separable processes.
Keywords: myelin assembly, gene knock-out, mouse models, oligodendrocytes, Olig genes, axonal integrity
Introduction
A characteristic feature of the vertebrate nervous system is ensheathment of axons with myelin (Zalc and Colman, 2000). CNS myelin synthesized by oligodendrocytes (OLs) is a multilayered lipoprotein membrane sheath that functions to insulate axons, facilitate saltatory conduction, and contribute to neuronal function (Raine, 1984). Myelinogenesis is a hallmark of oligodendrocyte differentiation, which begins perinatally in mice after immature oligodendrocytes extend processes to ensheath nearby axons (Raine, 1984). Major components of central myelin include myelin basic protein (MBP), proteolipid protein 1 (PLP1), 2′,3′-cyclic nucleotide 3′-phosphohydrolase (CNP), Claudin 11, myelin-associated glycoprotein (MAG), and glycolipids such as galactocerebrosides (GalC) and gangliosides (Braun, 1984; Lees and Brostoff, 1984; Campagnoni, 1995; Coetzee et al., 1998; Gow et al., 1999). Biosynthesis of these components is tightly regulated and critical for myelin assembly and long-term stability (Stoffel and Bosio, 1997; Werner et al., 1998). Defective or inappropriate maintenance of oligodendrocyte myelination impairs normal neuronal function and axonal integrity and may ultimately contribute to primary demyelinating diseases such as multiple sclerosis and the leukodystrophies (Trapp et al., 1998; Berger et al., 2001).
Transcriptional regulation plays an important role in the differentiation of oligodendrocyte lineage cells from precursors (OPCs) to myelinating oligodendrocytes (Wegner, 2001; Miller, 2002; Ross et al., 2003). Recently, a pair of basic helix-loop-helix (bHLH) transcription factors, Olig1 and Olig2, were identified and shown to be essential for generating oligodendrocyte lineage cells during embryogenesis (Lu et al., 2000; Zhou et al., 2000). Olig1 and Olig2 are tightly linked on mouse chromosome 16. They are expressed not only in OPCs but also in myelinating oligodendrocytes. Importantly, oligodendrocytes are absent in all brain regions of Olig1;Olig2-null double mutants, which demonstrates the critical functions of these genes during development (Lu et al., 2002; Zhou and Anderson, 2002). Additional studies in single null mutants suggest that Olig2 is required for initiation of oligodendrogliogenesis in spinal cord, and Olig1 contributes more to oligodendrocyte differentiation in brain (Lu et al., 2001, 2002; Takebayashi et al., 2002b; Zhou and Anderson, 2002); however, postnatal functions of Olig1 and Olig2 in CNS myelinogenesis have not been elucidated.
The processes of OPC genesis and differentiation from multipotent neural precursors are relatively well characterized (Kagawa et al., 2001; Wegner, 2001; Rowitch, 2004); however, molecular mechanisms governing oligodendrocyte myelinogenesis are poorly understood. This presents a particular challenge for myelin repair in demyelinating diseases, in which OPCs are present within lesions but fail to form myelinating oligodendrocytes (Chang et al., 2002). Herein, we demonstrate that Olig1 plays a pivotal role in oligodendrocyte myelination. Olig1-null mice exhibit severe neurological deficits, including tremors and seizures, and die prematurely around postnatal day 14 (P14). In brain, expression of major myelin genes does not occur, and myelin assembly is arrested despite signs that oligodendrocytes extend processes and contact axons in preparation for myelinogenesis. Furthermore, Olig1-null mice develop prominent axonal swellings, degeneration, and gliosis in the CNS. Together, our studies demonstrate the pivotal role of Olig1 in regulating the onset of oligodendrocyte myelinogenesis and maintaining axonal integrity in developing brain.
Materials and Methods
Generation of Olig1-null mice. Olig1-null;PGKneo mutants previously generated by homologous recombination in embryonic stem cells carry an FRT sequence-flanked PGKneo cassette (Lu et al., 2002). These animals exhibit a mild phenotype, suggesting that Olig1 does not play a major role in CNS development; however, the strong PGK promoter/enhancer is known to influence functions of neighboring genes and regulatory elements and is thought to be responsible for viable or embryonic lethal phenotypes in a number of mutant mice (Ramirez-Solis et al., 1993; Sibilia and Wagner, 1995; Threadgill et al., 1995; Bjornsson et al., 2003). In this regard, the presence of the PGKneo cassette in Olig1-null; PGKneo mice may influence the phenotype of these mutants, and evidence for such a notion comes from the constitutive expression of the Olig1 gene in Olig2;PGKneo mice (Lu et al., 2002). Thus, by analogy, the presence of PGKneo in the Olig1 locus of Olig1-null;PGKneo mutants may induce Olig2 expression and compensate for the Olig1 deficiency. To explore this possibility, we used FLPeR mice expressing Flip recombinase (FLPe) (a gift from Drs. Qiufu Ma and Sue Dymecki, Harvard Medical School, Cambridge, MA) to remove the FRT flanked PGKneo cassette in Olig1-null mice. The resulting mutants were maintained on a 129 and C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) hybrid background and analyzed at various postnatal stages. PCR primers specific for wild-type alleles are as follows: Olig1-1, 5′-tgc tgg gta gct cgc tgc agg agc tgc gcc-3′, and Olig1-2, 5′-agc cag ccc tca ctt gga gaa ctg ggc ctg-3′; for mutant alleles, they are as follows: KiOlig1-1, 5′-cgt tag tga agg gcg ccc cgg gtc gcc cca-3′, and KiOlig1-2, 5′-cgc tag agc ctg ttt tgc acg ttc acc ggc-3′. These animal studies were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center at Dallas.
RNA in situ hybridization. Olig1 control and mutant embryos and brains from postnatal stages were harvested from anesthetized mice, fixed in 4% paraformaldehyde at 4°C overnight, infused with 20% sucrose in PBS overnight, embedded in OCT, and cryosectioned at 16 μm. Digoxigenin-labeled riboprobes were used to perform RNA in situ hybridization, as described previously (Lu et al., 2002), and the probes used were as follows: Olig1, Olig2, platelet-derived growth factor α receptor (PdgfαR), Plp1/Dm-20, and Mbp. The NogoA and oligodendrocytemyelin glycoprotein (Omgp) probes were gifts from Dr. Zhigang He (Harvard Medical School). For in situ hybridization in conjunction with immunostaining, perinatal Olig1-heterozygous and -null littermates were injected with 50 mg/kg 5′-bromo-2′-deoxyuridine (BrdU) (Sigma, St. Louis, MO) 4 h before animals were killed. Sections were hybridized with the PdgfαR riboprobe as described above and then washed with PBS, fixed in 4% paraformaldehyde for 15 min, treated with 2 m HCl in PBS for 30 min at 37°C, rinsed with PBS, and blocked with 0.1% NP-40 and 5% normal goat serum. Immunohistochemistry with anti-BrdU antibodies (1:100 dilution; Sigma) was performed using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), and the HRP was detected with diaminobenzidine (Sigma).
Quantitative real-time-PCR. Quantitative real-time (QRT)-PCR was performed using the ABI Prism 7000 Sequence Detector System (Applied Biosystems, Foster City, CA). RNA was extracted from forebrain and spinal cord of Olig1-heterozygous (+/-) and -null (-/-) mice using Trizol (Invitrogen, Carlsbad, CA). cDNAs were generated using a first-strand cDNA synthesis kit (Amersham Biosciences, Piscataway, NJ). QRT-PCR was performed with an initial denaturation for 10 min at 95°C, followed by 40 cycles of 15 s of denaturation at 95°C, 60 s of annealing at 60°C, and 60 s of elongation at 60°C. The fluorescent dye SYBR green (Applied Biosystems), was included in each assay to measure DNA concentration during each annealing phase. Sizes of PCR products were subsequently confirmed by electrophoresis on 2% agarose gels. Triplicate samples were analyzed for each cDNA with test and internal control primers for the housekeeping gene, glyceraldehyde-3-phosphatase dehydrogenase (Gapdh) (TaqMan kit; Applied Biosystems). Dilution curves were generated by 10-fold serial dilutions of each target cDNA to estimate copy number of the target cDNAs. Fluorescence intensity was plotted against cycle number and standardized using Gapdh to account for sample variability. Primers used for expression analyses are as follows: Mbp, forward 5′-atccaagtacctggccacag-3′, reverse 5′-cctgtcaccgctaaagaagc-3′; Plp1, forward 5′-ctggctgagggcttctacac-3′, reverse 5′-gactgacaggtggtccaggt-3′; Mag, forward 5′-ggaccccatccttaccatct-3′, reverse 5′-cgggttggattttaccacac-3′; UDP-galactose:ceramide galactosyltransferase (Cgt), forward 5′-gaacatggctttgtcctggt-3′, reverse 5′-catggcttaggaaggctctg-3′; Cnp, forward 5′-catcctcaggagcaaaggag-3′, reverse 5′-gtacgcctcggagaagtctg-3′.
Morphometric analyses. PdgfαR+, BrdU+, and CC1+ cells were counted in brain sagittal cryosections using an Olympus (Tokyo, Japan) BX50 microscope with 20× or 40× objective lens. Cell density was estimated in cortex and white matter by determining the average number of PdgfαR+, BrdU+, and CC1+ cells within five nonoverlapping fields per section. The average density of PdgfαR+ and CC1+ cells was determined within the cross section of spinal cords from at least five nonoverlapping fields. For each region of CNS, we counted cells in five sections from three Olig1-heterozygous and -null littermates from two separate litters. Statistical comparisons used Student's t test, and when necessary we used Welch's correction for unequal variances.
Cortical cell cultures. Cortical cell culture was performed as described previously (Lu et al., 2000). Briefly, cortical progenitor cells were obtained from Olig1 mutants (+/- or -/-) or Olig2 mutants (+/- or -/-) at P2 or embryonic day 18.5 (E18.5), respectively, plated on poly-d-lysine-coated plates for 8 d in vitro (DIV) in serum-free medium containing N2 supplement (Invitrogen), 20 ng/ml FGF2, and 10 ng/ml PDGF-AA (Raff et al., 1983; Kondo and Raff, 2000). The medium was then switched to a medium containing 400 ng/ml triiodothyronine (T3) and 20 ng/ml ciliary neurotrophic factor (CNTF) for 14 DIV to promote oligodendrocyte differentiation (Gard and Pfeiffer, 1989; Barres and Raff, 1994). Differentiated oligodendrocytes and Olig1+- and Olig2+-expressing cells were identified by immunostaining with antibodies against CNP, MBP, Cre, and Olig2.
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling. Immunostaining methods with tissue sections from mouse brains have been described previously (Lu et al., 2000). Double immunostaining was performed by simultaneous incubation with antibodies of interest. The following antibodies were used: Olig2 rabbit polyclonal (1:1000; a gift from Chuck Stiles, Harvard Medical School), CNP (1:100; Roche, Indianapolis, IN), rabbit Cre (1:500; Covance, Richmond, CA), and NG2 and MBP (1:500; Chemicon, Temecula, CA). Microscopy was performed using an Olympus BX50 fluorescence microscope. Goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to cyanine 2 (Cy2) and Cy3 (Jackson ImmunoResearch, West Grove, PA) were used for double-labeling experiments. To perform the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assays, paraffin-embedded brain sections from P14 wild-type and Olig1-null mice were deparaffinized, hydrated, and treated with proteinase K. Fragmented DNAs were labeled with biotinylated-dUTP in the presence of terminal deoxynucleotidyl transferase (Invitrogen) and detected by Vectastain kit (Vector Laboratories). Gallyas silver staining was performed according to the method described by Lappe-Siefke et al. (2003).
Electron microscopy. Mice were anesthetized with ketamine and xylazine (9:1) and perfused briefly with PBS followed by 2% glutaraldehyde in 0.1 m cacodylate, pH 7.2. Optic nerves, corpus callosum, and spinal cord were removed and fixed in fresh fixative overnight at 4°C. Tissues were rinsed in PBS, postfixed in 1% OsO4 in PBS for 1 h, dehydrated in a graded ethanol series, infiltrated with propylene oxide, and embedded in Epon. Semithin sections were stained with toluidine blue, and thin sections were stained with lead citrate. To determine myelin g-ratios in cross-sections of optic nerve and spinal cord, we divided outer axon circumference by the circumference of the associated myelin sheath and expressed the ratio as a function of axon diameter (Michailov et al., 2004). Small g-ratios reflect abnormally thick myelin sheaths, and large ratios indicate hypomyelination. Measurements were made on electron micrographs from three pairs of wild-type and Olig1-null mice at P14 (at least 100 myelinated axons per animal and genotype).
Transient transfection and luciferase assay. COS-7 cells were seeded and grown in DMEM with 10% fetal bovine serum 1 d before transfection, and the cells reached 50-70% confluence on the day of transfection. Cells were transfected with 0.2 μg of plasmid using FuGENE6 according to the manufacturer's protocol (Roche) and assayed 46-48 h after transfection. In addition, the pRSV-β-galactosidase (β-Gal) plasmid (0.05 μg) was included to control for variable transfection efficiencies between different experiments. Promoter/enhancer regions for the Mbp and Gfap genes were cloned into pGL3, a vector containing the coding region for luciferase, to generate pGL3Basic-Mbp and pGL3Basic-Gfap reporters. The expression vector, pCS2nlsMT (nuclear localization signal and myc tagged), was used to express Olig1 (pCS2nlsMT-Olig1). The luciferase activity in transfected cells was measured using the Luciferase Assay system (Promega, Madison, WI) in a luminometer (Anthos Labtech, Salzburg, Austria). β-Gal activities were measured using o-nitrophenyl-β-d-galactopyranoside as a substrate.
Results
Olig1-null mice were generated in a previous study by targeted disruption of the Olig1 gene using the Cre recombinase open reading frame and a PGKneo selectable marker gene (Lu et al., 2002). The PGKneo cassette flanked by FRT sequences was removed (Fig. 1A) by breeding with FLPeR-expressing mice (Farley et al., 2000). In subsequent heterozygous crosses, wild-type, Olig1-heterozygous, and Olig1-null mice were generated at Mendelian ratios.
Figure 1.

Targeted mutation of the Olig1 gene and phenotypes of Olig1-null mice. A, Schematic for FLPase-mediated removal of the PGKneo cassette through breeding with FLPeR (Flipper) mice. PGKneo, Neomycin gene driven by the PGK promoter. B, The lifespan of Olig1-null (-/-) mice (46 mice tested) ranges from P13 to P17, which is an active period of oligodendrocyte myelination. All Olig1 heterozygotes (+/-) (95 mice tested) have a normal lifespan, as do their wild-type littermates. C, Olig1-null mice exhibit abnormal clasping of all limbs and are motionless when held upside down, compared with age-matched littermates, which are active and extend their limbs. DTA, Diphtheria toxin.
Olig1-null mice exhibit severe neurological abnormalities
Despite their normal development in utero, Olig1-null mice die prematurely around P14 and have not survived beyond P17 from a total of 46 Olig1-null births (Fig. 1B). On the other hand, Olig1 heterozygotes exhibit normal lifespan without detectable neurological defects. Olig1-null mice develop severe neurological abnormalities, such as abnormal limb clasping, generalized tremors, ataxia, and tonic seizures (Fig. 1C), which are characteristic features of myelin-deficient animals in which oligodendrocyte differentiation occurs normally but the synthesis of myelin sheaths is disrupted to varying degrees (Nave, 1994; Lunn et al., 1995; Gow et al., 1998).
Myelination deficits in the developing brain of Olig1-null mutants
Generalized body tremors and recurrent seizures observed in Olig1-null mice are reminiscent of phenotypes observed in myelin mutants such as shiverer mice, in which most of the Mbp gene is deleted, or jimpy mice, which harbor a frame-shift mutation in the Plp1 gene. The onset of neurological symptoms in these naturally occurring mutants is also around P14 (Sturrock, 1980; Popko et al., 1987; Nave, 1994), which suggests that pathogenesis in Olig1-null mice may involve defective myelin sheath assembly.
To characterize oligodendrocyte development in the brains of Olig1 mutants, we examined expression of major myelin genes by in situ hybridization and immunocytochemistry. In contrast to the intense expression of Mbp and Plp1/Dm-20 in wild-type (data not shown) and Olig1 heterozygotes, expression of these genes in Olig1-null mice is undetectable in corpus callosum, neocortex, and striatum (Fig. 2A). Furthermore, anti-CNP antibodies do not label white matter tracts from these animals (Fig. 2A). Together, these data demonstrate that myelinogenesis in the null mutants is arrested at an early stage. The NogoA and Omgp outer membrane components of mature oligodendrocytes that serve to inhibit axonal regeneration (Huber et al., 2002; Wang et al., 2002) also are not detected in white matter tracts from Olig1-null mice; however, expression of these genes in neurons is comparable with controls (Fig. 2B, red arrow), which indicates that Olig1 is genetically upstream of the NogoA and Omgp genes in oligodendrocytes. Finally, histochemical staining to detect myelin lipid deposition around axons using a Gallyas silver stain (Lappe-Siefke et al., 2003) intensely labels white matter tracts in forebrain and cerebellum from Olig1-heterozygous mice but fails to label sections from Olig1-null mice, indicating a profound myelin sheath deficit in these mutants (Fig. 2C).
Figure 2.
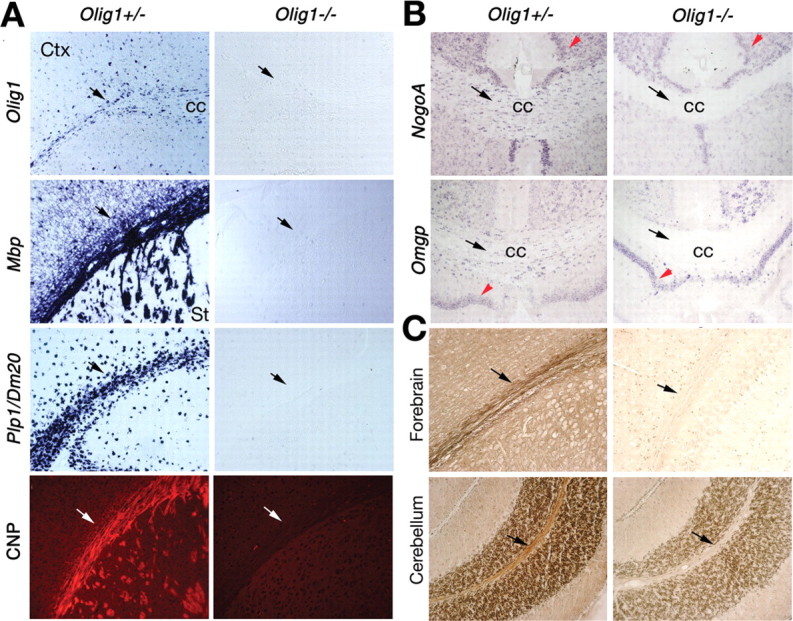
Myelin deficits in Olig1-null mice. A, Expression of myelin genes analyzed by in situ hybridization using probes to Olig1, Mbp, and Plp1/Dm-20 and immunocytochemistry using anti-CNP antibodies on frozen sections of P14 brains from Olig1-heterozygous (+/-) or -null (-/-) mice. Expression of Mbp, Plp1/Dm-20, and CNP is evident in white matter tracts (arrows) of +/- but not -/- mice. B, In situ hybridization signals for mature oligodendrocyte markers, NogoA and Omgp, are absent in -/- mice (black arrows); however, expression in neurons is unaffected (red arrowheads). C, Myelin lipid deposition around axons is detected using Gallyas silver staining in white matter tracts of +/- mice (left column; arrows) but not -/- mice (right column). Coronal and parasagittal sections of forebrain and cerebellum are shown. Ctx, Cortex; cc, corpus callosum; St, striatum.
To quantify expression of the major myelin genes in Olig1-null mice, we performed QRT-PCR for Mbp, Plp1, Mag, Cgt, and Cnp (Table 1). These data demonstrate dramatic reductions in gene expression in excess of 200-fold for Mbp, Plp, and Mag in Olig1-null mice compared with heterozygous mice. Expression of the Cgt and Cnp genes is reduced by only 27- and 10-fold, respectively, possibly because these genes are expressed at low levels by OPCs (Yu et al., 1994; Stoffel and Bosio, 1997) in null mutants and are less dependent on Olig1 transcriptional regulation.
Table 1.
Quantitative analysis of myelin gene expression in the CNS of Olig1-heterozygous and -null mice
|
|
Mbpa |
Maga |
Plp1a |
Cgta |
Cnpa |
|---|---|---|---|---|---|
| Forebrain | |||||
| Olig1+/− | 0.3418 ± 0.1007 | 0.4258 ± 0.1153 | 0.7743 ± 0.1047 | 0.8471 ± 0.1448 | 0.4463 ± 0.0482 |
| Olig1−/− | 0.0005 ± 0.0003 | 0.0015 ± 0.0004 | 0.0029 ± 0.0007 | 0.0309 ± 0.0050 | 0.0456 ± 0.0006 |
| Fold change | 637 | 284 | 267 | 27 | 10 |
| Spinal cord | |||||
| Olig1+/− | 3.1043 ± 0.1475 | 2.4897 ± 0.7200 | 2.5285 ± 0.4615 | 2.6697 ± 0.5169 | 2.6840 ± 0.1880 |
| Olig1−/− | 0.1997 ± 0.0541 | 0.2733 ± 0.1505 | 0.1222 ± 0.0009 | 0.1718 ± 0.0031 | 0.2175 ± 0.0110 |
| Fold change |
15 |
9 |
20 |
15 |
12 |
Gene expression relative to internal control Gapdh. Data are expressed as means ± SD.
Myelin assembly defects in Olig1-null mice
In light of our data demonstrating that myelin gene expression and membrane deposition around axons are negligible in Olig1-null mice, we examined myelin sheath assembly in the brains of these mutants using electron microscopy. The optic nerve, an extensively characterized CNS white matter tract, is composed primarily of myelinating oligodendrocytes, astrocytes, and retinal ganglion cell axons. Optic nerves isolated from Olig1-heterozygous mice at P14 appear white because of the presence of myelin (Fig. 3A). In contrast, optic nerves from Olig1-null littermates are transparent and indicate that myelin in these nerves is essentially absent.
Figure 3.

Myelin sheath assembly in optic nerves from Olig1-null mice. A, Optic nerves from Olig1-heterozygous (+/-) mice at P14 are white because of myelinated axons, whereas optic nerves from Olig1-null (-/-) littermates are translucent. B, Approximately 35% of axons in +/+ and +/- mice are myelinated at P14 (number of axons scored, 800). In contrast, optic nerve axons in Olig1-null (-/-) mice are unmyelinated. C, Electron micrographs of optic nerves in cross sections from +/- mice at P14. Multilamellar myelin sheaths are apparent around many axons. The right panel is shown at higher power (arrow). D, Electron micrographs of optic nerves in cross section from -/- mice at P14. All axons in optic nerves are unmyelinated (red arrow). A large number of intermediate filament-containing astrocytic processes are indicative of gliosis (blue arrows). The right panel is shown at high magnification. E, F, In situ hybridization of PdgfαR and immunohistochemical labeling of CC-1 in optic nerves at P14. PdgfαR+ OPCs (E, arrows) are detected in +/- and -/- mice, but very few CC1+ oligodendrocytes (F, arrows) are observed in -/- mice. G, Electron micrographs of corpus callosum in cross section from +/- and -/- mice at P14. The absence of multilamellar myelin (red arrows) in -/- mice is apparent in the right panel. Scale bars: C, D, left panels, 2 μm; right panels, 500 nm; G, 500 nm.
Myelinated axons are readily observed in optic nerves from wild-type and Olig1 heterozygotes at P14, and ∼35% of axons are ensheathed with multilayer compact myelin (Fig. 3B,C); however, myelin sheaths are not detected around axons in optic nerves and corpus callosum from Olig1-null mutants (Fig. 3B,D,G), indicating that myelin wrapping has not begun in these animals. Nonetheless, OPCs expressing PdgfαR are observed, albeit at a lower density than in Olig1 heterozygotes (Fig. 3E). Strikingly, few mature oligodendrocytes in Olig1-null mice compared with controls are labeled by CC1 monoclonal antibodies (Fig. 3F) (Bhat et al., 1996). These results indicate that OPCs are able to migrate into white matter tracts and differentiate but fail to express myelin-specific genes.
In electron micrographs, the presence of intermediate filament-negative processes around axons indicates that oligodendrocytes extend processes that recognize axons to begin the ensheathment process (Fig. 4A-C) but are arrested at this early stage of myelinogenesis. Axonal swellings, degenerating axons, and accumulations of myelin figures, mitochondria, and fragmented organelles are observed in optic nerves from Olig1-null mice (Fig. 4D-F, arrows). In addition, increases in the numbers of intermediate filament-containing processes in optic nerves from Olig1-null mice are indicative of gliosis (Fig. 3D, arrows), although such pathology is observed in most myelin-mutant animals and is unlikely to be correlated with the absence of myelin-specific gene expression.
Figure 4.
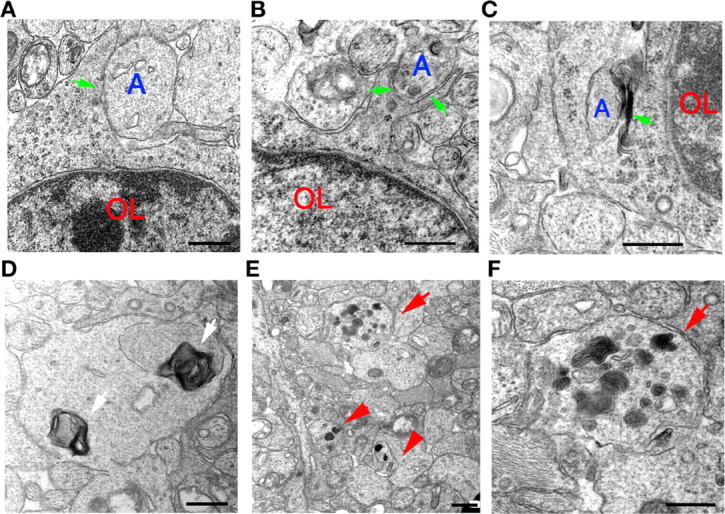
Axon recognition by oligodendrocytes in optic nerves and axonal degeneration in Olig1-null mutants. A-C, Intermediate filament-negative oligodendrocyte processes (arrows) contact (A) and begin to encircle (B, C) axons (A). Formation of a multilayer membrane flap (C, arrow) near an axon is also observed. These ultrastructural analyses indicate that oligodendrocytes recognize axons and begin the process of ensheathment in Olig1-null (-/-) mice. D, Myelin figures (arrows) within optic nerve axons from -/- mice. E, Axonal swellings and degenerating axons (arrows) are observed in optic nerves of -/- mice. F, High-power view of a degenerating axon (arrow) shown in E showing fragmented cellular organelles and mitochondria. Scale bars, 500 nm.
Arrested myelinogenesis in the developing brain of Olig1-null mutants
The possibility that PdgfαR+ OPCs may be reduced in number in optic nerve from Olig1-null mice (Fig. 3E) prompted us to examine other brain regions for quantitative changes. PdgfαR+ OPCs are observed throughout cortical brain regions in P14 Olig1-null mutants but appear to be reduced in white matter tracts (Fig. 5A, arrowheads). Thus, we quantified the number of these cells using in situ hybridization in cerebral cortex and white matter at P1, P7, and P14 (Table 2).
Figure 5.

Oligodendrocyte development in postnatal brain. A, In situ hybridization of PdgfαR shows similar expression levels in cortical regions of Olig1- heterozygotous (+/-) and -null (-/-) mice, but expression in white matter tracts is reduced in -/- mice (arrowheads). Middle and right panels are higher-power views of the left column (20×) and show white matter and cortex, respectively. B, Coronal sections of forebrain from +/- and -/- mice at P14 immunostained with anti-NG2 antibody. The number and morphology of NG2+ cells in the brains of -/- mice are comparable with +/- mice. Panels in the right column are higher-power views of NG2+ cells (arrows). C, In situ hybridization of Olig2 is similar in cortex of +/- and -/- mice, but expression in white matter is reduced in -/- mice (arrowheads). Middle and right panels are higher-power views (20×) showing the external capsule and corpus callosum (cc), respectively. D, Parasagittal brain sections of +/- and -/- mice at P11 immunolabeled with anti-O4 antibody. Intense labeling of mature oligodendrocytes is observed in corpus callosum from +/- mice (top right panel and inset). The numbers of O4+ cells in -/- and +/- cortex are comparable. Many O4+ cells from -/- mice are located at the corpus callosum-cortex boundary (blue arrows); however, these cells are reduced in white matter tracts (bottom panels). Furthermore, O4+ cells in -/- mice exhibit a multipolar ramified morphology (inset in bottom right panel) and fail to ensheath axons. E, Sagittal sections of +/- and -/- cortex at P11 immunolabeled with anti-Cre antibody. The numbers of Cre+ cells (green) from -/- (bottom right panel) and +/- (top right panels) mice are comparable but are reduced in -/- corpus callosum (bottom left panel) compared with +/- (top left panel, arrows). F, Cortical sections from -/- mice at P11 immunolabeled with anti-Cre (red) and anti-NG2 (green, bottom panel) antibodies show colocalization of both antigens (arrows).
Table 2.
Numbers of PdgfαR+ progenitors and CC1+ mature oligodendrocytes in Olig1-heterozygous and -null mice
|
|
PdgfαR+ OPCs |
CC1+ oligodendrocytes |
||||
|---|---|---|---|---|---|---|
|
|
P1 |
P7 |
P14 |
P14 |
||
| Cortexa | ||||||
| Olig1+/− | 38.6 ± 7.2 | 89.6 ± 10.8 | 65.0 ± 5.5 | 24.1 ± 4.5 | ||
| Olig1−/− | 36.7 ± 6.4 | 80.8 ± 8.4 | 83.5 ± 10.7 | 0 | ||
| Corpus callosumb | ||||||
| Olig1+/− | nc | 43.4 ± 3.7 | 38.5 ± 4.1 | 35.6 ± 4.0 | ||
| Olig1−/− | nc | 24.7 ± 4.5 | 20.4 ± 2.6 | 0.3 ± 0.4 | ||
| Spinal cordb | ||||||
| Olig1+/− | 34.6 ± 5.3 | 40.2 ± 4.7 | 28.8 ± 4.2 | 44.5 ± 4.3 | ||
|
Olig1−/− |
32.2 ± 4.5 |
33.0 ± 6.6 |
23.7 ± 4.6 |
10.4 ± 3.2 |
||
Average numbers of PdgfαR+ and CC1+ cells in the indicated CNS regions from three pairs of Olig1-heterozygous and -null mice as described in Materials and Methods. nc, Not counting from P1. Data are expressed as means ± SD.
Average number of cells in 0.1 mm2.
Average number of cells in 0.04 mm2.
In neocortex, the numbers of PdgfαR+ cells are normal in Olig1-null mice at neonatal stages at P1 and P7 (Table 2), but by P14 they increase to 128% of controls. The moderately high density of PdgfαR+ OPCs in Olig1-null mice may be because of the inability of these cells to form mature myelinating oligodendrocytes (Table 2). In contrast, PdgfαR+ cells in the corpus callosum at P7 and P14 are reduced to 57 and 52% of controls (p < 0.01), respectively, which is consistent with our qualitative PdgfαR data in optic tracts. We were unable to perform morphometry in corpus callosum at P1 because this white matter tract is indistinct. Finally, we observed dramatic (∼100-fold) reductions in the numbers of CC1+ cells from cortex and corpus callosum in P14 Olig1-null mice and heterozygotes (one-way ANOVA; p < 0.001), and together with the PdgfαR data, these results demonstrate that the major effect of the absence of Olig1 in mice is at the level of myelinogenesis.
Several other markers have been used to identify OPCs in brain, including the NG2 chondroitin sulfate proteoglycan, Olig1, Olig2, and the O4 ganglioside (Sommer and Schachner, 1981; Levine and Stallcup, 1987; Ellison and de Vellis, 1994; Lu et al., 2000; Zhou et al., 2000). NG2+ OPCs are abundant in brains of Olig1-null mice at P14, and the number of these cells in neocortex is comparable with that in Olig1 heterozygotes (Fig. 5B). Olig2-expressing cells are also widely distributed in the cortex of null mutants (Fig. 5C); however, the number of these cells appears reduced in white matter tracts. In Olig1-null mice, Cre expression is under regulatory control of the Olig1 promoter/enhancer (Fig. 1A) and serves as a marker of oligodendrocyte lineage cells (Fig. 5E); indeed, Cre is coexpressed with NG2+ in OPCs in brain (Fig. 5F). The number of Cre-expressing cells in neocortex from Olig1-null mice is comparable with Olig1 heterozygotes but appears reduced in corpus callosum (Fig. 5E) in a manner similar to the other oligodendrocyte lineage markers examined. Immature oligodendrocytes, identified by O4 labeling, exhibit complex branching morphologies and are present in similar numbers in the cortical regions of Olig1-heterozygous and -null mice but seems reduced in white matter tracts from null mutants (Fig. 5D). Thus, our data consistently indicate lower numbers of OPCs in white matter tracts of Olig1-null mice compared with controls despite similar numbers of these cells in neocortex.
Another noteworthy aspect of O4 staining is apparent in Olig1-null mice. O4 antibodies intensely label myelin sheaths in developing brain at P14 and are apparent as parallel stained fibers in corpus callosum from Olig1 heterozygotes (Fig. 5D). O4 does not label such structures in the null mutants, which demonstrates the absence of myelin-like membranes around axons in these mutants. Interestingly, many cells with an elaborate branching morphology are observed along the cortexcorpus callosum interface as if awaiting some type of signal to initiate myelinogenesis (Fig. 5D, arrows).
Normal oligodendrocyte differentiation from cortical progenitors in vitro
Because OPCs are generated in Olig1-null mice but appear unable to synthesize myelin-specific proteins in vivo, we examined the differentiation of these cells in vitro. Cortices from neonatal brains of Olig1-heterozygous and -null mice were dissociated and cultured in medium containing FGF2 and PDGF-AA to stimulate OPC proliferation (Raff et al., 1983; Kondo and Raff, 2000). After switching to culture medium containing thyroid hormone (T3) and CNTF to promote OPC differentiation (Barres et al., 1993, 1994), immature and mature oligodendrocytes are readily detected in cultures from Olig1-heterozygous pups as CNP+ and MBP+ cells (Fig. 6A,B). These cells also express Cre (Fig. 6A,B, green), which reflects Olig1 expression from the mutant allele. Surprisingly, Cre+ cells from Olig1-null mice exhibit a multipolar web-like morphology and synthesize membrane sheets in mitogen-free medium, which are features typical of differentiated oligodendrocytes (Fig. 6A,B). Moreover, these cells express CNP, in contrast to Olig1-null cortical oligodendrocytes in vivo (Fig. 2A). Very few cells from the Olig1-null cultures express MBP; however, when expressed, this myelin protein is localized to membrane sheets (Fig. 6B,C). In contrast, cortical progenitor cells isolated from Olig2-null embryos (Lu et al., 2002) at E18.5 fail to differentiate into CNP+ oligodendrocytes (Fig. 6D). Together, our results demonstrate that the absence of Olig1 does not prevent dissociated cortical OPCs from differentiating in vitro but rather prevents most cells from expressing myelin-specific genes.
Figure 6.

Oligodendrocyte differentiation in cortical progenitor cell in vitro cultures. A, Cortical cells from P2 Olig1-heterozygous (+/-) and -null (-/-) pups cultured invitro to assess their differentiation potential. Immunolabeling of cultures shows web-like multipolar CNP+ (red) and Cre+ (green) oligodendrocytes (arrowheads) from +/- and -/- mice. B, Cortical OPCs from +/- pups differentiate and express MBP (red, left column, arrowheads); however, few MBP+ oligodendrocytes (arrowheads) are generated from -/- mice (right column). C, Bar graph (mean ± SD) showing comparable numbers of CNP+ oligodendrocytes generated from +/- and -/- cultures and a 20-fold reduction in MBP+ oligodendrocytes from -/- cultures. D, Cortical OPCs from E18.5 Olig2 +/- and -/- embryos cultured to assess their differentiation potential. Cells in -/- cultures fail to express Olig2 (green) or CNP; however, cells from +/- cultures generate Olig2+, CNP+ oligodendrocytes (red). E, Induction of Mbp- but not Gfap-promoter constructs in transiently transfected cells expressing Olig1. Transfection efficiency is normalized using β-galactosidase activity from a cotransfected reporter construct.
Induction of myelin gene promoter by Olig1 in vitro
To determine whether Olig1 directly regulates expression of glial-specific genes, we examined the effects of this transcription factor on the activities of the Mbp and Gfap promoter/enhancer regions. A 1.9 kb fragment of the upstream region of murine Mbp has been shown to direct expression of a reporter specifically in oligodendrocytes (Gow et al., 1992). The Gfap promoter/enhancer has been identified previously as a 1.9 kb upstream region that directs reporter gene expression specifically in astrocytes (Brenner et al., 1994; Quintana et al., 1998; Sun et al., 2001). To generate reporter constructs, the promoter regions of these glial-specific genes were fused upstream of luciferase, and expression levels were measured in the presence or absence of Olig1 in transfected COS-7 cells. As shown in Figure 6E, Mbp promoter activity is increased approximately fourfold by Olig1 expression in these assays. In contrast, luciferase activity driven by the Gfap promoter is reduced twofold in the presence of Olig1, which indicates that this transcription factor induces expression of oligodendrocyte-specific genes and represses astrocyte-specific genes. Thus, Olig1 may function as a transcriptional regulator for myelin-specific gene expression.
Oligodendrocytes differentiate and express myelin-specific genes in spinal cords of Olig1-null mice
In view of the profound absence of myelin and myelin-specific gene expression in the brains of Olig1-null mice, we examined spinal cords from these mutants to determine whether other regions of CNS are severely affected. Strong Mbp and Plp1 expression is observed in gray and white matter regions of developing spinal cords from Olig1 heterozygotes at P14, and three other markers of oligodendrocyte lineage cells, Olig2, Sox10, and PdgfαR, are also broadly expressed (Fig. 7A, Table 2). Cells expressing Mbp and Plp1 in spinal cords from Olig1-null mice are present, although the number of these cells is markedly reduced. Olig2- and Sox10-expressing cells are also reduced in number in Olig1-null mutants, particularly in presumptive white matter tracts (Fig. 7A, white arrows). Importantly, most oligodendrocyte lineage cells are localized to white matter tracts (Fig. 7A, white arrows), which indicates that OPCs arising in the ventral ventricular zone respond normally to migration and differentiation cues and take up appropriate positions for myelinating axons in the postnatal period. The absence of myelin gene expression in gray matter oligodendrocytes indicates that these cells are nonmyelinating (Fig. 7A, red arrows).
Figure 7.
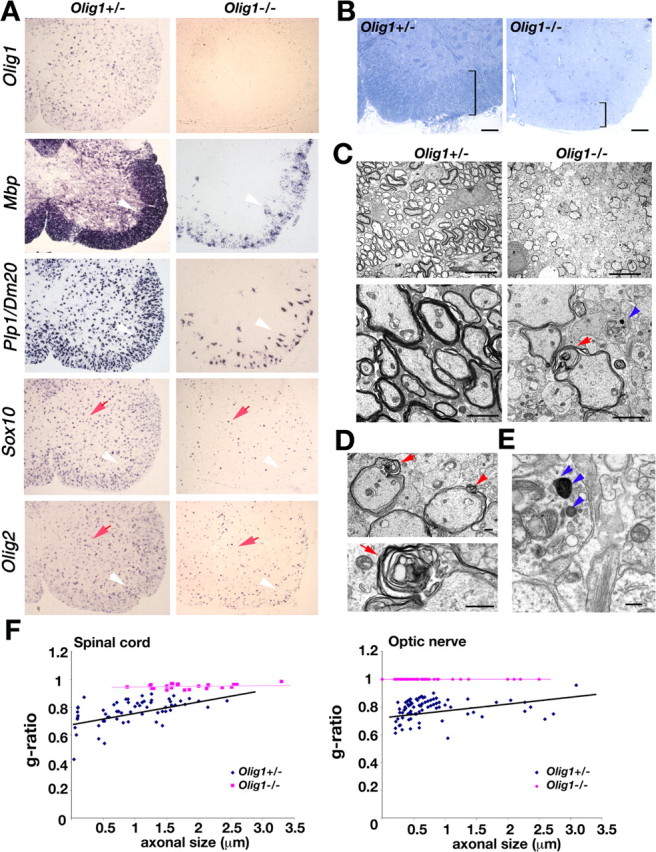
Oligodendrocyte myelination in spinal cord from Olig1-null mice. A, In situ hybridization of Olig1, Mbp, Plp1, Sox10, and Olig2 analyzed in frozen sections from Olig1-heterozygous (+/-) and -null (-/-) mice at P14. Mbp, Plp1, Sox10, and Olig2 are strongly expressed in spinal cord from +/- mice but only weakly expressed in -/- mutants (arrowheads). Olig2- and Sox10-expressing cells are found in gray matter from -/- mice (arrows). B, Spinal cords from +/- and -/- mice stained with toluidine blue dye to visualize white and gray matter regions. White matter thickness is significantly reduced in -/- (right panel) compared with +/- (left panel) sections (square brackets). C, Electron micrographs show fewer myelinated axons in -/- mice compared with +/- mice. Bottom panels show high-power views. Myelin sheaths from -/- mice are thinner compared with littermate controls (arrows). Degenerating axons are also detected in these mice (arrowheads). D, Abnormal splitting of myelins heaths (top panel, arrows) and Wallerian myelin degeneration (bottom panel) from -/- mice. E, Vesicle and organelle inclusions in degenerating axons from -/- mice (arrowheads). F, Scatter plots showing g-ratios as a function of axon diameter for spinal cord (left panel) and optic nerve (right panel) in +/- (blue symbols) and -/- (purple symbols) mice. Scale bars: B, 50 μm; C, top panels, 10 μm; C, bottom panels, 2 μm; D, E, 500 nm.
The number of PdgfαR+ OPCs is quantified in spinal cord at P1, P7, and P14 (Table 2). We observed no significant differences in OPC numbers between heterozygous and null mutants (p > 0.05), which is consistent with a minor role for Olig1 in OPC lineage cells in this region of the CNS. Nonetheless, the number of CC1+ cells in spinal cord is reduced in Olig1-null mice to <23% of controls. Notably, reductions in major myelin gene expression (Mbp, Mag, and Plp1) are not as dramatic in spinal cord as in brain when determined by QRT-PCR (Table 1).
To examine myelinogenesis in Olig1-null spinal cords in detail, we stained transverse sections with toluidine blue dye, which clearly delineates gray and white matter regions in controls (Fig. 7B, square brackets). Gray matter is well defined in sections from Olig1-null mice by strong labeling of neuron cell bodies, but the white matter tracts in these animals are significantly thinner at P14. Ultrastructural analysis reveals few myelin sheaths around axons, which are compacted but significantly thinner than for comparably sized axons in Olig1-heterozygous mice (Fig. 7C). Redundant myelin sheaths, myelin splitting, and disruption are prominent features in Olig1-null mice (Fig. 7D, red arrows), as is myelin vacuolation and Wallerian degeneration (Fig. 7E, arrows). Nevertheless, compact myelin sheaths are synthesized, which demonstrates that myelinogenesis in spinal cord is less dependent on Olig1 than in brain. To quantify the degree of myelin sheath formation in spinal cord, we determined the thickness of axons and their myelin sheaths for Olig1-heterozygous and -null mice by calculating g-ratios. In Olig1 heterozygous mice, the average g-ratio at P14 is 0.77 ± 0.06 (Fig. 7F) and is in close agreement with published data (Hildebrand and Hahn, 1978; Lunn et al., 1997). In contrast, the average g-ratio for Olig1-null mice (0.95 ± 0.02) is significantly higher than that of control mice (p < 0.01) and demonstrates that myelin sheaths are significantly thinner in Olig1-null mice compared with controls. To examine regional differences in CNS, we determined g-ratios from optic nerves of Olig1 heterozygous and null mice. In this white matter tract, the average myelin g-ratio is 0.79 ± 0.04 at P14 for heterozygotes. In contrast, there is essentially no myelin around optic nerve axons in Olig1-null mice, and the average g-ratio = 1. Thus, these data clearly demonstrate regional differences in myelin sheath formation in the CNS of Olig1-null mice.
Normal neural development in brains from Olig1-null mice
In view of severe neurological abnormalities, myelinogenesis defects, and premature death of Olig1-null mutants, we examined overall brain development in these mutant mice. The numbers of cells and cortical layers in neocortex from Olig1-null mice labeled by the neuronal marker, neuronal-specific nuclear protein (NeuN), are comparable with those of Olig1-heterozygous mice (Fig. 8A). Moreover, we do not detect significant neural cell death in cortex from Olig1 heterozygotes or Olig1-null mice by TUNEL (Fig. 8B). Thus, these data indicate that neurogenesis is relatively normal and that cell loss is not a significant factor in the phenotype of Olig1-null mutants.
Figure 8.
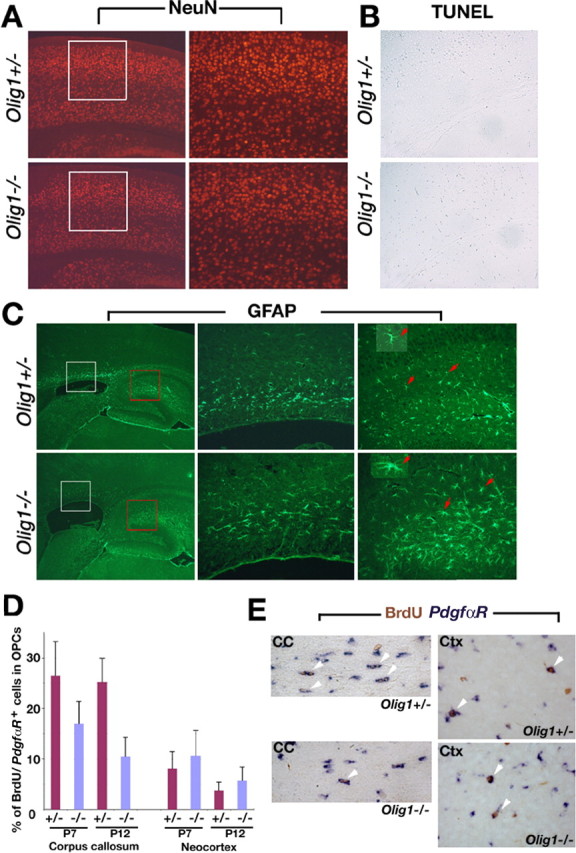
Neuronal development in brain from Olig1-null mice. A, NeuN expression in coronal sections of cortex from Olig1-heterozygous (+/-) and -null (-/-) mice at P14 to examine neuronal development. White-boxed areas in left panels are shown at higher power (20×) in the right panels. Neurogenesis and lamination are comparable in +/- and -/- mice. B, TUNEL of coronal sections from cortex indicates that there are few apoptotic cells in +/- and -/- mice. C, Astrocyte development examined in parasagittal brain sections using GFAP immunocytochemistry. White-boxed (corpus callosum) and red-boxed (hippocampus) areas (left panels) are shown at higher power (20×) in middle and right panels, respectively. GFAP+-reactive astrocytes (arrows) from -/- mice exhibit large cell bodies and bulky processes (bottom inset); however, astrocytes (arrows) from control mice have thin processes and relatively small cell bodies (top inset). D, The numbers of cycling cells in brain measured by double in situ hybridization and immunohistochemistry 4 h after BrdU administration to animals at P7 and P12. The proportion of PdgfαR+ OPCs in the cell cycle at the time of the BrdU injections in cortex is comparable between +/- and -/- mice but reduced in white matter from the null mutants. E, Examples of BrdU+/PdgfαR+ cells (arrowheads) are shown in the white matter and the cortex of Olig1 +/+ and -/- mice. CC, Corpus callosum; Ctx, cortex.
Reactive astrogliosis is a common feature of pathophysiology in most myelin mutants (Lappe-Siefke et al., 2003), and we observed increased numbers of intermediate filament-containing processes in optic nerves from Olig1-null mice by electron microscopy. Accordingly, we examined GFAP expression in hippocampus and corpus callosum by immunocytochemistry to determine whether gliosis is widespread in these mutants. GFAP+ astrocytes appear to develop normally in Olig1-null mutants, and there is no evidence of abnormal cell death; however, we observed a moderate increase in GFAP+ astrocytes in the hippocampus of these animals (Fig. 8C), suggesting that gliosis occurs in the brains of Olig1-null mice in a manner similar to optic nerve. In addition, astrocytes appear to have larger cell bodies and processes than those in Olig1 heterozygotes.
Using several markers, we have demonstrated that a paucity of OPCs in white matter tracts from Olig1-null mice does not appear to be associated with cell death, because we did not detect abnormally high numbers of TUNEL+ cells in white matter tracts (Fig. 8B). These observations suggest that OPC proliferation may be perturbed in the null mutants; to explore this possibility, we used a short BrdU pulse in animals at P7 and P12 to label proliferating PdgfαR+ OPCs. Analysis of double-labeled PdgfαR+ and BrdU+ cells in the brain of Olig1-null mice indicates that the proportion of OPCs in S-phase during the BrdU pulse in the cortex of Olig1-null mice at P7 and P12 is comparable with heterozygous animals but is decreased in corpus callosum 1.6- and 2.4-fold, respectively (p < 0.01), compared with controls (Fig. 8D,E). These data are consistent with the reduced numbers of PdgfαR+ cells that we observed in the corpus callosum of Olig1-null mice and suggest that the cell cycle of white matter tract OPCs may be prolonged in the absence of Olig1.
Discussion
Myelin sheath formation by oligodendrocytes in the CNS is critical for normal function of neuronal circuits during development, and in mice, most of these cells become terminally differentiated in the first few postnatal weeks. OPCs arising in several subventricular proliferative zones divide and migrate throughout the brain and spinal cord but eventually come to rest, differentiate, and extend multiple cellular processes. When one of these processes recognizes and contacts an axon, the oligodendrocyte induces myelin-specific protein and lipid synthesis and ensheaths the axon with myelin. Hitherto, the molecular mechanism governing this recognition and gene induction program was unknown (Wegner, 2001; Miller, 2002; Rowitch, 2004); however, the data presented herein demonstrate that Olig1 is obligatory for the regulatory switch that triggers myelination at the cellular level for oligodendrocytes arising in brain. Curiously, this critical developmental switch exhibits some degree of diversity because spinal cord-derived oligodendrocytes are much less dependent on Olig1 to myelinate axons.
Axonal recognition and myelin assembly by oligodendrocytes in Olig1 mutants
Despite the successful generation, migration, and proliferation of OPCs in the brains of Olig1-null mice, the process of myelinogenesis in these mutants is arrested at a stage just before myelin-specific gene expression, as summarized in Figure 9. Previous studies have demonstrated the importance of Olig2 transcriptional activity for the generation of OPCs in vivo and their subsequent differentiation into immature oligodendrocytes in developing neural tube (Lu et al., 2002). In Olig1-null mutants, brain-derived OPCs differentiate and extend processes, adopt an elaborate branched morphology, recognize and contact axons, and begin the process of axonal ensheathment but fail to progress beyond this point. The absence of myelin membrane structural proteins is consistent with a failure of compact sheath formation and demonstrates that axonal recognition and contact can be uncoupled from subsequent myelin sheath assembly, at least in brain.
Figure 9.

Major roles of Olig1 and Olig2 during oligodendrocyte maturation in brain. Oligodendrocyte maturation involves the progression of cells through a series of defined developmental stages including OPCs, immature OLs, differentiated premyelinating OLs, and mature myelinating OLs. Olig2 is essential for initiation of oligodendrogliogenesis from neural progenitor cells and the formation of immature OLs. Olig1 is critical for the appearance of myelinating OLs in developing brain, and there are likely to be spatiotemporally specific and nonredundant roles of bHLH proteins Olig1 and Olig2 in oligodendrocyte development. At present, the requirement of Olig2 for oligodendrocyte myelination in brain cannot be assessed formally. NPC, Neural progenitor cell.
Olig1-null mice exhibit severe neurological deficits, including generalized tremors and recurrent tonic seizures. This phenotype is markedly more severe than for other myelin mutants harboring disruptions in the Mbp, Plp1, Cnp, Cgt, Cst, and Claudin 11 genes in which myelin sheaths are elaborated to some extent or are synthesized without gross abnormalities (Popko et al., 1987; Coetzee et al., 1996; Rosenbluth et al., 1996; Fruttiger et al., 1999; Stecca et al., 2000; Honke et al., 2002; Lappe-Siefke et al., 2003). In contrast to these single gene mutants and several compound derivatives (Billings-Gagliardi et al., 1987, 2001; Wolf et al., 1999), ablation of Olig1 leads to the absence of all major myelin-specific gene expression in brain and complete failure of myelin sheath assembly. Thus, the current study is consistent with the notion that disease severity is inversely correlated with the degree of myelination.
Oligodendrocyte differentiation in brains of Olig1-null mice
A number of “differentiation” stages in the oligodendrocyte lineage have been defined in the literature according to the expression of specific marker proteins and lipids (Schachner, 1982; Pfeiffer et al., 1993). The presence of these markers has been extremely valuable for determining the extent to which oligodendrocyte lineage cells differentiate under various in vitro and in vivo conditions; however, the absence of any one or combination of these markers is less informative about the extent of differentiation. For example, oligodendrocytes from Cgt-null mice fail to express galactocerebrosides and their sulfated derivatives and are not labeled by anti-O1 or anti-RmAb antibodies but, nonetheless, are able to differentiate and synthesize compact myelin (Coetzee et al., 1996). In the same vein, Plp1-null mice do not express Dm-20 during embryogenesis or Plp1/Dm-20 postnatally but still synthesize normal amounts of compact myelin (Rosenbluth et al., 1996; Griffiths et al., 1998; Stecca et al., 2000). Indeed, the same can be said for almost every myelin-specific gene that has been ablated to date. In this light, we do not regard the lack of expression of Cnp, Plp1, and Mbp in brain-derived oligodendrocytes from Olig1-null as indicative of the failure of OPCs to differentiate. This notion is confirmed by our ultrastructural data, which demonstrate that differentiated oligodendrocytes in optic nerve begin myelinogenesis by recognizing and contacting axons before Olig1 function becomes critical. Thus, Olig1 can be considered a stage-specific marker for oligodendrocytes in the brain, the primary function of which is to serve as a key regulator for induction of the myelin-specific gene expression and the program of myelinogenesis.
Spatiotemporal specificity of oligodendrocyte development in the CNS
Despite many similarities in oligodendrocyte morphology and function throughout the CNS, our data indicate that oligodendrogenesis during brain development is more complex than for spinal cord in several respects. First, a single gene, Olig2, is essential for oligodendrogliogenesis in spinal cord; however, the presence of OPCs in several brain regions of Olig2-null embryos during development, and their complete absence in Olig1-null; Olig2-null compound mutants, demonstrates a unique role for Olig1 in brain (Lu et al., 2002; Zhou and Anderson, 2002). Second, several studies indicate that OPCs normally arise from proliferative zones at multiple sites in brain, with a second wave of oligodendrogenesis in cortex (Thomas et al., 2000; Ivanova et al., 2003; Liu et al., 2003); however, OPCs in spinal cord arise from a single region of the ventral neural tube (Fu et al., 2002; Lu et al., 2002; Zhou and Anderson, 2002). Finally, our data demonstrate that myelinogenesis in the brains of Olig1-null mice is arrested at an early stage; however, compact myelin sheaths are synthesized in the spinal cords of these mutants. Thus, our data demonstrate the region-specific regulation of myelin gene expression and provide direct evidence for at least two populations of OPCs in the CNS that regulate myelinogenesis through distinct mechanisms.
The limited myelinogenesis that we observe in spinal cords of Olig1-null mice demonstrates the partial functional compensation for the absence of this transcription factor by Olig2 because OPCs do not arise in Olig1-null;Olig2-null mice; however, we cannot formally exclude an additional contribution from Olig3, for which little functional data are available (Takebayashi et al., 2002a). Nonetheless, Olig1 and Olig2 clearly serve nonredundant functions in OPC proliferation, differentiation, and myelination in a specific spatial and temporal sequence (Fig. 9). Currently, the role of Olig2 in oligodendrocyte myelinogenesis cannot be assessed in detail because of the neonatal lethality of Olig2-null mutants, which stems from the absence of motor neurons.
Oligodendrocyte differentiation in vitro and in vivo
The elaboration of large membrane sheets and the expression of CNP and MBP in oligodendrocytes cultured in vitro from the cortex of Olig1-null mice are unexpected findings in the absence of comparable behavior in vivo. Nonetheless, precedence for such altered differentiation potential of progenitor populations in culture has been established in other cell types (Hasty et al., 1993; Nabeshima et al., 1993). For example, the bHLH transcription factor Myogenin is essential for differentiation of myoblasts to form myotubes in vivo; however, muscle cell differentiation still occurs in myoblasts cultured from Myogenin-null mice in vitro. In this vein, our in vitro data indicate that extrinsic components in the culture medium, or the absence of inhibitory signals that are normally present in brain, enable oligodendrocytes to bypass the in vitro requirement for Olig1 transcriptional activity, which demonstrates that myelin gene expression is regulated more tightly in vivo than in a culture dish.
The myelin-deficient phenotype in Olig1-null mice is likely attributable to the absence of Olig1 function rather than expression of Cre from the Olig1 mutant allele in oligodendrocytes, because Olig1 heterozygotes also express this recombinase and are indistinguishable from wild-type littermates. Furthermore, we do not observe significant levels of cell death in the brains of Olig1-null mice compared with controls. Finally, Cre-expressing neural progenitors from the cortex of Olig1 heterozygotes and Olig1-null mice differentiate, exhibit a mature morphology, and express differentiated oligodendrocyte markers (CNP and MBP).
Axonal degeneration in Olig1-null mice
Olig1-null mice exhibit not only severe hypomyelination in the CNS but also axonal degeneration. The failure of myelinogenesis in brain, coupled with axonal loss in these mutants, likely contributes to the neurological phenotype, which is characterized by generalized tremors, tonic seizures, and premature death at ∼2 weeks of age. In spinal cords from null mutants, the preponderance of naked axons, the prevalence of degenerating myelin sheaths, and widespread Wallerian degeneration indicate that Olig1 plays a critical role in maintaining myelin sheaths and protecting axons in the CNS after the onset of myelination. Animal models of hypomyelination and axonal degeneration are of clinical importance, particularly for human demyelinating diseases, because remyelination is limited not only by OPC differentiation at demyelinated lesion sites but also by axonal loss (Wolswijk, 1998; Chang et al., 2002). Thus, Olig1 transgenic mice will provide a powerful model system in which to explore the mechanisms of oligodendrocyte myelinogenesis and remyelination after myelin damage.
Footnotes
These studies were funded by grants from the Wadsworth Foundation (Q.R.L.), the National Multiple Sclerosis Society (JF2115 to Q.R.L. and RG2891 to A.G.), and the National Institutes of Health-National Institute of Neurological Disorders and Stroke (5RO1 NS43783 to A.G.). Q.R.L. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society and a Southwestern Medical Foundation Scholar for biomedical research. We thank Dr. Michael K. Racke, Dr. Jane Johnson, and Edward Hurlock for critical comments on this manuscript and Dr. Mike Henke and Irena Malkovska for stimulating discussions. We are grateful to Drs. Chuck Stiles, David Rowitch, and Brian Popko for support and advice. We thank Wen-hui Cai, Kendy Xian, Jens Fielitz, and Meritxell Garcia-Alzamora for technical assistance.
Correspondence should be addressed to Q. Richard Lu, Center for Developmental Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390. E-mail: qrichard.lu@utsouthwestern.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251354-12$15.00/0
M.X., T.Y., and Z.M. contributed equally to this work.
References
- Barres BA, Raff MC (1994) Control of oligodendrocyte number in the developing rat optic nerve. Neuron 12: 935-942. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC (1993) Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118: 283-295. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC (1994) A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120: 1097-1108. [DOI] [PubMed] [Google Scholar]
- Berger J, Moser HW, Forss-Petter S (2001) Leukodystrophies: recent developments in genetics, molecular biology, pathogenesis and treatment. Curr Opin Neurol 14: 305-312. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM (1996) Expression of the APC tumor suppressor protein in oligodendroglia. Glia 17: 169-174. [DOI] [PubMed] [Google Scholar]
- Billings-Gagliardi S, Kerner AL, Kirschner DA, Wolf MK (1987) Shiverer jimpy double mutant mice. III. Comparison of shimld, jpmsd and shi/jp phenotypes demonstrates dissimilar interactions of allelic mutations. Brain Res 388: 199-214. [PubMed] [Google Scholar]
- Billings-Gagliardi S, Nunnari JJ, Wolf MK (2001) Rumpshaker behaves like juvenile-lethal Plp mutations when combined with shiverer in double mutant mice. Dev Neurosci 23: 7-16. [DOI] [PubMed] [Google Scholar]
- Bjornsson JM, Larsson N, Brun AC, Magnusson M, Andersson E, Lundstrom P, Larsson J, Repetowska E, Ehinger M, Humphries RK, Karlsson S (2003) Reduced proliferative capacity of hematopoietic stem cells deficient in Hoxb3 and Hoxb4. Mol Cell Biol 23: 3872-3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun PE (1984) Molecular organization of myelin. In: Myelin, Ed 2 (Morell P, ed), pp 97-116. New York: Plenum.
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A (1994) GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci 14: 1030-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnoni AT (1995) Molecular biology of myelination. In: Neuroglia (Kettenmann H, Ransom BR, eds), pp 555-570. New York: Oxford UP.
- Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346: 165-173. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B (1996) Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 86: 209-219. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Suzuki K, Popko B (1998) New perspectives on the function of myelin galactolipids. Trends Neurosci 21: 126-130. [DOI] [PubMed] [Google Scholar]
- Ellison JA, de Vellis J (1994) Platelet-derived growth factor receptor is expressed by cells in the early oligodendrocyte lineage. J Neurosci Res 37: 116-128. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM (2000) Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28: 106-110. [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD (1999) Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development 126: 457-467. [DOI] [PubMed] [Google Scholar]
- Fu H, Qi Y, Tan M, Cai J, Takebayashi H, Nakafuku M, Richardson W, Qiu M (2002) Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development 129: 681-693. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE (1989) Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development 106: 119-132. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA (1992) Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol 119: 605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA (1998) Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol 140: 925-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA (1999) CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 99: 649-659. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave K-A (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280: 1610-1613. [DOI] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364: 501-506. [DOI] [PubMed] [Google Scholar]
- Hildebrand C, Hahn R (1978) Relation between myelin sheath thickness and axon size in spinal cord white matter of some vertebrate species. J Neurol Sci 38: 421-434. [DOI] [PubMed] [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N (2002) Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci USA 99: 4227-4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME (2002) Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci 22: 3553-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova A, Nakahira E, Kagawa T, Oba A, Wada T, Takebayashi H, Spassky N, Levine J, Zalc B, Ikenaka K (2003) Evidence for a second wave of oligodendrogenesis in the postnatal cerebral cortex of the mouse. J Neurosci Res 73: 581-592. [DOI] [PubMed] [Google Scholar]
- Kagawa T, Wada T, Ikenaka K (2001) Regulation of oligodendrocyte development. Microsc Res Tech 52: 740-745. [DOI] [PubMed] [Google Scholar]
- Kondo T, Raff M (2000) Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science 289: 1754-1757. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366-374. [DOI] [PubMed] [Google Scholar]
- Lees MB, Brostoff SW (1984) Proteins in myelin. In: Myelin, Ed 2 (Morell P, ed), pp 197-224. New York: Plenum.
- Levine JM, Stallcup WB (1987) Plasticity of developing cerebellar cells in vitro studied with antibodies against the NG2 antigen. J Neurosci 7: 2721-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Cai J, Hu X, Tan M, Qi Y, German M, Rubenstein J, Sander M, Qiu M (2003) Region-specific and stage-dependent regulation of Olig gene expression and oligodendrogenesis by Nkx6.1 homeodomain transcription factor. Development 130: 6209-6220. [DOI] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, Rowitch DH (2000) Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25: 317-329. [DOI] [PubMed] [Google Scholar]
- Lu QR, Cai L, Rowitch D, Cepko CL, Stiles CD (2001) Ectopic expression of Olig1 promotes oligodendrocyte formation and reduces neuronal survival in developing mouse cortex. Nat Neurosci 4: 973-974. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH (2002) Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75-86. [DOI] [PubMed] [Google Scholar]
- Lunn KF, Fanarraga ML, Duncan ID (1995) Myelin mutants: new models and new observations. Microsc Res Tech 32: 183-203. [DOI] [PubMed] [Google Scholar]
- Lunn KF, Clayton MK, Duncan ID (1997) The temporal progression of the myelination defect in the taiep rat. J Neurocytol 26: 267-281. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA (2004) Axonal neuregulin-1 regulates myelin sheath thickness. Science 304: 700-703. [DOI] [PubMed] [Google Scholar]
- Miller RH (2002) Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol 67: 451-467. [DOI] [PubMed] [Google Scholar]
- Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S, Nonaka I (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364: 532-535. [DOI] [PubMed] [Google Scholar]
- Nave KA (1994) Neurological mouse mutants and the genes of myelin. J Neurosci Res 38: 607-612. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R (1993) The oligodendrocyte and its many cellular processes. Trends Cell Biol 3: 191-197. [DOI] [PubMed] [Google Scholar]
- Popko B, Puckett C, Lai E, Shine HD, Readhead C, Takahashi N, Hunt III SW, Sidman RL, Hood L (1987) Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell 48: 713-721. [DOI] [PubMed] [Google Scholar]
- Quintana JG, Lopez-Colberg I, Cunningham LA (1998) Use of GFAP-lacZ transgenic mice to determine astrocyte fate in grafts of embryonic ventral midbrain. Brain Res Dev Brain Res 105: 147-151. [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M (1983) A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303: 390-396. [DOI] [PubMed] [Google Scholar]
- Raine CS (1984) Morphology of myelin and myelination. In: Myelin, Ed 2 (Morell P, ed), pp 1-50. New York: Plenum.
- Ramirez-Solis R, Zheng H, Whiting J, Krumlauf R, Bradley A (1993) Hoxb-4 (Hox-2.6) mutant mice show homeotic transformation of a cervical vertebra and defects in the closure of the sternal rudiments. Cell 73: 279-294. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Stoffel W, Schiff R (1996) Myelin structure in proteolipid protein (PLP)-null mouse spinal cord. J Comp Neurol 371: 336-344. [DOI] [PubMed] [Google Scholar]
- Ross SE, Greenberg ME, Stiles CD (2003) Basic helix-loop-helix factors in cortical development. Neuron 39: 13-25. [DOI] [PubMed] [Google Scholar]
- Rowitch DH (2004) Glial specification in the vertebrate neural tube. Nat Rev Neurosci 5: 409-419. [DOI] [PubMed] [Google Scholar]
- Schachner M (1982) Cell type-specific surface antigens in the mammalian nervous system. J Neurochem 39: 1-8. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Wagner EF (1995) Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269: 234-238. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M (1981) Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83: 311-327. [DOI] [PubMed] [Google Scholar]
- Stecca B, Southwood CM, Gragerov A, Kelley KA, Friedrich VLJ, Gow A (2000) The evolution of lipophilin genes from invertebrates to tetrapods: DM-20 cannot replace PLP in CNS myelin. J Neurosci 20: 4002-4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel W, Bosio A (1997) Myelin glycolipids and their functions. Curr Opin Neurobiol 7: 654-661. [DOI] [PubMed] [Google Scholar]
- Sturrock RR (1980) Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol 6: 415-420. [DOI] [PubMed] [Google Scholar]
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME (2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104: 365-376. [DOI] [PubMed] [Google Scholar]
- Takebayashi H, Ohtsuki T, Uchida T, Kawamoto S, Okubo K, Ikenaka K, Takeichi M, Chisaka O, Nabeshima Y (2002a) Non-overlapping expression of Olig3 and Olig2 in the embryonic neural tube. Mech Dev 113: 169-174. [DOI] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K (2002b) The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol 12: 1157-1163. [DOI] [PubMed] [Google Scholar]
- Thomas JL, Spassky N, Perez Villegas EM, Olivier C, Cobos I, Goujet-Zalc C, Martinez S, Zalc B (2000) Spatiotemporal development of oligodendrocytes in the embryonic brain. J Neurosci Res 59: 471-476. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269: 230-234. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338: 278-285. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417: 941-944. [DOI] [PubMed] [Google Scholar]
- Wegner M (2001) Expression of transcription factors during oligodendroglial development. Microsc Res Tech 52: 746-752. [DOI] [PubMed] [Google Scholar]
- Werner H, Jung M, Klugmann M, Sereda M, Griffiths IR, Nave KA (1998) Mouse models of myelin diseases. Brain Pathol 8: 771-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MK, Nunnari JN, Billings-Gagliardi S (1999) Quaking*shiverer double-mutant mice survive for at least 100 days with no CNS myelin. Dev Neurosci 21: 483-490. [DOI] [PubMed] [Google Scholar]
- Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18: 601-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WP, Collarini EJ, Pringle NP, Richardson WD (1994) Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron 12: 1353-1362. [DOI] [PubMed] [Google Scholar]
- Zalc B, Colman DR (2000) Origins of vertebrate success. Science 288: 271-272. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ (2002) The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109: 61-73. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang S, Anderson DJ (2000) Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 25: 331-343. [DOI] [PubMed] [Google Scholar]