

Abstract
The role of blood–brain barrier (BBB) transport in clearance of amyloid β-peptide (Aβ) by Aβ immunotherapy is not fully understood. To address this issue, we studied the effects of peripherally and centrally administered Aβ-specific IgG on BBB influx of circulating Aβ and efflux of brain-derived Aβ in APPsw+/– mice, a model that develops Alzheimer's disease-like amyloid pathology, and wild-type mice. Our data show that anti-Aβ IgG blocks the BBB influx of circulating Aβ in APPsw+/– mice and penetrates into the brain to sequester brain Aβ. In young mice, Aβ–anti-Aβ complexes were cleared from brain to blood by transcytosis across the BBB via the neonatal Fc receptor (FcRn) and the low-density lipoprotein receptor-related protein (LRP), whereas in older mice, there was an age-dependent increase in FcRn-mediated IgG-assisted Aβ BBB efflux and a decrease in LRP-mediated clearance of Aβ-anti-Aβ complexes. Inhibition of the FcRn pathway in older APPsw+/– mice blocked clearance of endogenous Aβ40/42 by centrally administered Aβ immunotherapy. Moreover, deletion of the FcRn gene in wild-type mice inhibited clearance of endogenous mouse Aβ40/42 by systemically administered anti-Aβ. Our data suggest that the FcRn pathway at the BBB plays a crucial role in IgG-assisted Aβ removal from the aging brain.
Keywords: antibody, amyloid β, Alzheimer's disease, blood–brain barrier, amyloid, transport
Introduction
Although the amyloid hypothesis of Alzheimer's disease (AD) remains controversial, many investigators consider accumulation of neurotoxic amyloid β peptide (Aβ) in the brain as a key pathogenic event contributing to neurodegeneration (Hardy and Selkoe, 2002). The levels of Aβ in the brain are controlled by its rates of production from the larger Aβ-precursor protein (APP) and the rates of clearance (Tanzi et al., 2004; Zlokovic, 2005). According to recent studies, the blood–brain barrier (BBB) transport of Aβ critically regulates the levels of brain Aβ and particularly the receptor for advanced glycation end products (RAGE)-mediated influx of circulating Aβ (Deane et al., 2003) and the low-density lipoprotein receptor-related protein 1 (LRP)-mediated efflux of brain-derived Aβ (Shibata et al., 2000; Deane et al., 2004). Under physiological conditions, the LRP-mediated brain efflux of Aβ prevails, whereas in animal models of AD and in AD brains, downregulation of LRP in concert with upregulation of RAGE may create an unfavorable Aβ gradient across the BBB, resulting in Aβ retention in the brain (Tanzi et al., 2004; Zlokovic, 2004).
Vaccination and passive immunization to Aβ lower brain Aβ (Schenk et al., 1999; Sigurdsson et al., 2001; Das et al., 2003) and improve behavior in animal models of AD (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002). Although clinical Aβ immunization trials in AD patients were terminated because of adverse neuroinflammatory effect (Orgogozo et al., 2003), reduced Aβ brain deposition (Nicoll et al., 2003) and slower cognitive decline (Hock et al., 2003) have been reported. How anti-Aβ IgG clears brain Aβ remains debatable. The “sink” theory suggests that the interaction of Aβ with an Aβ-specific IgG in plasma creates a concentration gradient across the BBB that promotes efflux of brain Aβ into blood, as shown by passive and active immunization studies (DeMattos et al., 2001, 2002; Deane et al., 2003; Lemere et al., 2003). Conversely, some circulating anti-Aβ antibodies cross the BBB and activate microglia-mediated Fcγ receptor-dependent (FcRγ) clearance of amyloid (Bard et al., 2000) and/or the FcRγ-independent clearance (Bacskai et al., 2002; Das et al., 2003). Microglia clear amyloid slowly (Frautschy et al., 1992; Paresce et al., 1997; Bacskai et al., 2002), whereas microglia-independent clearance of Aβ seems to be more rapid (DeMattos et al., 2001; Wilcock et al., 2003; Deane et al., 2004) and may require efflux of Aβ across the BBB (Shibata et al., 2000; Banks et al., 2003; Tanzi et al., 2004; Zlokovic, 2004).
Here, we show in APPsw+/– mice (Hsiao et al., 1996), a model that develops AD-like amyloid pathology, that an Aβ-specific IgG prevents RAGE-mediated transport of circulating Aβ across the BBB and enters into the brain to sequester brain Aβ and promote its vigorous clearance to blood. We demonstrated that the major histocompatibility complex class I-related neonatal Fc receptor (FcRn), which is functionally and structurally distinct from the Fcγ receptors (Borvak et al., 1998; Ravetch and Bolland, 2001; Roopenian et al., 2003; Ober et al., 2004; Yoshida et al., 2004), is critical for the IgG clearance from brain and for the elimination of Aβ–anti-Aβ complexes through the BBB. We also show that FcRn-mediated IgG-assisted Aβ efflux across the BBB remains active with aging, in contrast to LRP-mediated clearance of Aβ–anti-Aβ complexes, which decreases with aging. Finally, we demonstrate that inhibition of the FcRn pathway in old APPsw+/– mice (Hsiao et al., 1996) completely blocks rapid clearance of endogenous Aβ40/42 by centrally administered Aβ immunotherapy, whereas deletion of the FcRn gene in mice inhibits clearance of endogenous mouse brain Aβ by systemically administered anti-Aβ.
Materials and Methods
Animals. C57BL/6 and APPsw+/– (transgenic Tg2576) mice were from Taconic Farms (Germantown, NY). β2M–/– mice (neonatal Fc receptor light chain β2-microglobulin null mice), FcRγ–/– mice (Fcγ receptor null mice), RAP–/– (receptor-associated protein null mice), and FcRn–/– mice (neonatal Fc receptor null mice) were from The Jackson Laboratory (Bar Harbor, ME). Mice were anesthetized by ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). All procedures were according to National Institutes of Health guidelines approved by the University Committee on Animal Resources (University of Rochester).
Reagents. Human Aβ1–40 was synthesized at the W. M. Keck Facility (Yale University, New Haven, CT), using solid-phase N-t-butyloxycarbonyl chemistry and purified by HPLC. We used human recombinant RAP (EMD Biosciences, San Diego, CA), monoclonal mouse antibody (ab) against C-terminal domain of human LRP β-chain, which cross reacts with mouse LRP (5A6, 1:350, 5 μg/ml; EMD Biosciences), rat anti-mouse CD31 antibody (1:200; BD PharMingen, Lexington, KY), anti-RAGE IgG F(ab′)2 (20 μg/ml; Dr. Shi Du Yan, Columbia University, New York, NY), rat anti-mouse F4/80, a microglial cell marker (1:100; Serotec, Indianapolis, IN), mouse anti-human GFAP (1: 100; DakoCytomation, Glostrup, Denmark), mouse monoclonal antiphosphotyrosine (1:2500; Sigma, St. Louis, MO), and monoclonal mouse anti-human Aβ (4G8; Signet Laboratories, Dedham, MA) against amino acids 17–24 of Aβ. Monoclonal anti-FcRn (1G3) was obtained as the condition medium of cultured mouse hybridoma cell line CRL-2434 (American Type Culture Collection, Manassas, VA) as described previously (Schlachetzki et al., 2002). The FcRn blocking antibody does not interact with 4G8 in vitro, as indicated by 4G8 ELISA (see below), which showed no change in 4G8 signal in the presence of the FcRn antibody. 4G8 F(ab′)2 was prepared as described previously (Pierce, Rockford, IL) and dialyzed overnight to remove elution buffer and digestion medium. The monoclonal antibody 1560, which recognizes Aβ epitope 1–17, was from Chemicon (Temecula, CA). All chemicals were obtained from Sigma, except protease inhibitor (Roche, Indianapolis, IN) and Triton X-100 (Electron Microscopy Sciences, Gibbstown, NJ). 125I-Na and 14C-inulin were from Amersham Biosciences (Little, Chalfont, UK). 99mTc was obtained from Cardinal Health 414 (Rochester, NY).
Radio-iodinated ligands. Radio-iodination of the Aβ peptide (10 μg) was performed by mild “lactoperoxidase” method (Thorell and Johansson, 1971) using 2 mCi 125I-Na. The mono-iodinated nonoxidized form of Aβ was purified by reverse-phase HPLC separation (Shibata et al., 2000). Typically, the specific activities were in the range of 45–65 μCi/μg peptide. 4G8 (50 μg) and 4G8 F(ab′)2 (10 μg) were radio-iodinated using IODO-BEADS or IODO-GEN (Pierce) and 0.5 and 0.7 mCi 125I-Na, respectively. Typical specific activity was ∼3–5 μCi/μg 4G8 or 4G8 F(ab′)2. 125I-Aβ40–4G8 complex was prepared by incubating 125I-Aβ40 with 4G8 at 5:1 molar ratio for 3 h at 37°C, and excess 125I-Aβ was removed by ultrafiltration. Complex stability was confirmed by Tris-tricine native PAGE and autoradiography. 125I-Aβ–4G8 complex was prepared in a similar manner.
Brain perfusion technique. This method is used to determine influx of radiolabeled ligands across the BBB and has been described in detail previously (LaRue et al., 2004). Briefly, the right common carotid artery was cannulated with a polyethylene tubing (PE10), and the brains were perfused at 1.0 ml/min (Ranin peristaltic pump), with an artificial plasma solution as described previously (LaRue et al., 2004). Radiolabeled test ligands (e.g., 125I–Aβ40, 125I-4G8-Aβ40, and 125I-Aβ40–4G8) and the reference molecules (e.g., 14C-inulin and 99Tc-albumin) were infused simultaneously via a slow-drive syringe pump (Harvard Apparatus, Holliston, MA) at a rate of 0.1 ml/min. Influx of 125I-Aβ40 in APPsw+/– mice was determined at carrier concentrations corresponding to Aβ40 plasma levels in APPsw+/– mice at different ages (Kawarabayashi et al., 2001). The effect of RAGE-specific IgG F(ab′)2 was tested at 20 μg/ml. After a timed perfusion, typically 5–10 min, the brain was rapidly removed, and the ipsilateral hemisphere was homogenized for radioactivity quantification. The perfusion fluid was centrifuged, and supernatant (plasma) was counted. 125I samples were subjected to TCA, SDS-PAGE, native PAGE, and/or HPLC analysis using the procedures we described previously (Shibata et al., 2000; Deane et al., 2003, 2004).
Brain clearance studies. Clearance of 125I-Aβ40, 125I-4G8, or 125I-Aβ–4G8 from brain interstitial fluid (ISF) was determined simultaneously with 14C-inulin (reference marker), using a procedure described previously (Shibata et al., 2000). Briefly, a stainless steel guide cannula was implanted stereotaxically into the right caudate–putamen with the cannula tip coordinates of 0.9 mm anterior and 1.9 mm lateral to bregma and 2.9 mm below the surface of the brain. Animals were recovered after surgery before tracer studies. The experiments were performed before substantial chronic processes occurred, as assessed by histological analysis of tissue, i.e., negative staining for astrocytes (glial fibrillar acidic protein) and activated microglia (anti-phosphotyrosine), but allowing time for the BBB repair for large molecules, as reported previously (Cirrito et al., 2003; Deane et al., 2004). Isotope mixture (0.5 μl) containing 125I-labeled test molecule at 40 nm and 14C-inulin was injected over 5 min via an ultra micropump with a micro4-controller (World Precision Instruments, Sarasota, FL) into brain ISF. The recovery of both radiolabeled inulin and Aβ at zero time was 100%, indicating that 100% of injected material remains present for transport with no loss of tracers via tracking up the cannula. 4G8 was administered by two intraperitoneal injections at 200 μg at 0 and 48 h, and 125I-Aβ40 clearance was measured at 1 and 120 h of 4G8 administration in nontransgenic mice and at 120 h in 18- to 20-month-old APPsw+/– mice. The levels of 4G8 in plasma and brain ISF within 120 h were determined by ELISA (see below). Unlabeled molecular reagents [4G8 (0.5 and 2 μm), anti-FcRn (αFcRn, 60 μg/ml), RAP (0.5 and 5 μm), and fucoidan (1.5 mm)] were infused into brain ISF in control mice 30 min before radiolabeled ligands and then simultaneously with radioligands until the end of experiment. Clearance of 125I-4G8 or 125I-Aβ–4G8 was also studied in FcRn–/–, β2M–/–, RAP–/–, or FcRγ–/– mice. In all studies, brains were sampled within 30 min after tracer injection and prepared for radioactivity analysis. TCA and/or SDS-PAGE/immunoprecipitation analyses were determined to confirm the molecular forms of test tracers in brain and plasma (Deane et al., 2003, 2004).
Brain capillary uptake. Brain capillaries from wild-type and APPsw+/– mice were isolated as described previously (Wu et al., 2003) and incubated with 125I-4G8, 125I-Aβ40–4G8, or 125I-4G8–Aβ40 at 1 nm and 14C-inulin in mock CSF at 37°C for 1 min. The following potential inhibitors were used: 4G8 (0.5 and 2 μm), RAP (0.5 and 5 μm), Fc fragment (1 and 10 μm), and LRP-specific IgG (20 μg/ml). The capillary pellet was separated by centrifugation at 4°C, washed in ice-cold mock CSF, and prepared for radioactivity analysis along with samples of the incubating medium.
Radioactivity measurements. 125I samples and 99Tc-albumin radioactivity analysis were determined by gamma counter analysis (Wallac Vizard Gamma Counter; PerkinElmer, Meriden, CT). 14C samples were solubilized in 0.5 ml of tissue solubilizer (PerkinElmer) overnight, followed by addition of 5 ml of scintillation cocktail (Packard Ultima Gold; PerkinElmer) and analysis on a liquid scintillation counter (Packard Tri-Carb 2100TR Liquid Scintillation Counter; PerkinElmer).
Calculations. The BBB influx was determined as cerebrovascular permeability surface area product (PS) × Cpl, where Cpl was the concentration of the test molecule in plasma. The PS product of [125I]-labeled test molecule was calculated using 14C-inulin correction: PS × T = [(125I cpm/g of brain tissue) × TCA-precipitable radioactivity/(125I cpm/ml of arterial plasma inflow) × TCA-precipitable radioactivity] – (14C dpm/g of brain tissue)/(14C dpm/ml of arterial plasma inflow)] (LaRue et al., 2004), where T is the infusion time and 14C-inulin was infused simultaneously with the test molecule. Influx was expressed per gram brain ISF, assuming the ISF space of 0.1 ml/g of brain (LaRue et al., 2004).
For brain clearance studies, calculations of clearance parameters were as reported previously (Shibata et al., 2000). The percentage of radioactivity of the test ligand and inulin was determined as follows: % recovery in brain = 100 × (Nb/Ni), where, Nb is the radioactivity remaining in the brain at the end of the experiment, and Ni is the radioactivity injected into the brain ISF, i.e., the disintegrations per minute for 14C-inulin and the counts per minute for TCA-precipitable 125I-radioactivity. The percentage of 125I-labeled test ligands (i.e., 4G8, Aβ40–4G8, and Aβ40) cleared through the BBB was calculated as [(1 – Nb(TEST)/Ni(TEST)) – (1 – Nb(inulin)/Ni(inulin))] × 100, using a standard time of 30 min. The loss of tracers via the ISF bulk flow was used to calculate the percentage clearance of 125I-labeled test ligands via the flow of brain ISF and was calculated as (1 – Nb(inulin)/Ni(inulin)) × 100 (Shibata et al., 2000).
Brain capillary uptake of 125I-labeled test molecules [4G8, 4G8–Aβ, and Aβ–4G8–F(ab′)2] were corrected for the distribution of 14C-inulin and determined as the tissue to medium ratio as follows: cpm for TCA-precipitable 125I-radioactivity (g capillary protein)/cpm for TCA-precipitable 125I-radioactivity (ml medium), as reported previously (Deane et al., 2004).
4G8 ELISA. 4G8 titers in plasma and brain were determined within 120 h after two intraperitoneal injections of 4G8 (200 μg/each) at 0 and 48 h by ELISA technique similar to that described previously (Das et al., 2001). Briefly, Aβ42 was coated at 5 μg/well overnight at 4°C in 50 mm carbonate buffer, pH 9.6, and 0.05% sodium azide on 96-well high-binding Stripwell immunoassay plates (Corning Life Sciences, Corning, NY) and blocked with 0.25% bovine serum albumin in PBS overnight at 4°C. After three washes with PBS/0.1% Tween 20, 100 μl of diluted plasma or brain homogenates were added and incubated overnight at 4°C. After washes with PBS/0.1% Tween 20, plasma and brain 4G8 was detected using a goat anti-mouse conjugated with HRP (Sigma) and tetramethylbenzidine (TMB) substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD). 4G8 levels were obtained from the standard curve. The levels of 4G8 in plasma and brain ISF were expressed in nanomolar assuming the ISF volume of distribution of 0.1 ml/g brain.
Immunohistological analysis. Freshly frozen acetone-fixed brain tissues from APPsw+/– and control mice cut in the sagittal plane at 14 μm, and paraffin-embedded human brain tissue from AD patients [Braak stage V–VI, CERAD (for Consortium to Establish a Registry for Alzheimer's Disease), frequent] and age-matched controls (Braak negative; CERAD negative) cut in 6 μm sections were used for double immunostaining for FcRn and CD31 or von Willebrand Factor (vWF) (endothelial cell marker). Anti-FcRn antibodies were prepared from the condition medium from 1G3 cells. Biotinylated anti-mouse IgG was used as a secondary antibody and was detected with fluorescein streptavidin (1:1000; Vector Laboratories, Burlingame, CA). M.O.M kit (Vector Laboratories) was used to block endogenous IgG in studies on mouse brain tissue. For CD31 staining, mouse CD31-specific IgG was used as a primary antibody, and Alexa Fluor 594 donkey anti-rat IgG (1:500; Invitrogen, Carlsbad, CA) was used as a secondary antibody. The anti-vWF primary antibody was detected with fluorescein goat anti-rabbit IgG (1:150; Invitrogen). Image analysis was performed using Olympus Optical (Tokyo, Japan) AX70 microscope equipped with the SPOT digital camera. Ten randomly selected fields in each region from 10 sections spanning the entire hemisphere from four mice per group or from Brodman areas 9/10 in humans were analyzed as we reported previously (Deane et al., 2004).
Western blotting. The cerebrovascular mouse system was washed with 100 ml of ice-cold PBS by cardiac perfusion, and cerebral microvessels were isolated as described previously (Zlokovic et al., 1993). Protein homogenates (5–15 μg) were separated under reducing conditions (FcRn) or nonreducing conditions (GFAP and F4/80), electroblotted in Tris-glycine buffer on nitrocellulose membrane, and probed with anti-FcRn (1:1), anti-GFAP (1:100), or F4/80 (1:100). The signal was detected by enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ).
125I-Aβ40–4G8 complex in brain. After 120 h of the intraperitoneal 4G8 (200 μg) administration at 0 and 48 h, 120 nm 125I-Aβ40 was microinfused into brain ISF in the caudate nucleus. After 30 min, the brain was removed, homogenized in cold PBS containing complete proteinase inhibitor cocktail (Roche), and centrifuged at 18,000 × g for 30 min at 4°C. The supernatant was concentrated by lyophilization and analyzed by 4–20% Tris-tricine native gel electrophoresis, followed by autoradiography.
Aβ immunotherapy and Aβ quantification. Twenty 4-month-old APPsw+/– mice were anesthetized as above (see above, Animals) and placed in a stereotactic apparatus. 4G8 was injected into the right hippocampus (ipsilateral) in mock CSF in a volume of 0.5 μl over 5 min through a 33 gauge injector attached to a 10 μl Hamilton syringe. As a negative control for 4G8, a rabbit anti-mouse IgG against divalent metal transporter-1, which does not interact with Aβ but has an intact Fc region to interact with the FcRn receptor at the BBB, was injected into the contralateral hippocampus in the same animal [nonimmune IgG (NI IgG)]. The coordinates with respect to bregma were –2.7 mm posterior, +2.5 mm lateral, and –3.0 mm ventral to the skull. The following test antibodies were injected: 4G8 (2 μg/0.5 μl); αFcRn (anti-FcRn antibody, 10 μg/0.5 μl) followed by 4G8 (2 μg/0.5 μl) 30 min after αFcRn; NI-IgG (2 μg/0.5 μl) followed by 4G8 (2 μg/0.5 μl) 30 min after NI-IgG; and 4G8 F(ab′)2 (2 μg/0.5 μl). Brains were analyzed after 24 h.
Brain sections were incubated with rabbit polyclonal anti-pan-Aβ (1: 100; BioSource International, Camarillo, CA) overnight at 4°C, washed, and incubated in Alexa Fluor 488-conjugated goat anti-rabbit IgG (1: 200; Invitrogen) for 1 h at a room temperature. Photomicrographs of hippocampi were taken using an AX70 Olympus Optical microscope (Spectra Services, Ontario, NY). The percentage difference between ipsilateral and contralateral hippocampus was determined as [(number of pixels ipsilateral–number of pixels contralateral)/number of pixels contralateral] × 100 (Oddo et al., 2004).
Aβ40 and Aβ42 were quantified by a sandwich ELISA kit (Signet Laboratories) according to the instructions of the manufacturer. Aβ levels were expressed as the percentage change between the ipsilateral compared with the contralateral hippocampus using the following formula: [Aβ (ng/section) in the ipsilateral hemisphere–Aβ (ng/section) in the contralateral hemisphere]/[Aβ (ng/section) in the contralateral hemisphere] × 100.
For soluble brain Aβ oligomer analysis, Aβ was extracted from 10 brain sections from the site of 4G8 or vehicle injection in 200 μl of TBS buffer containing complete protease inhibitor cocktail (Roche), and separated from the insoluble pellet by centrifugation at 20,000 × g for 30 min. Antibody 6E10 (Signet Laboratories) was used as the capturing and detection antibody to determine relative levels of TBS-extracted soluble Aβ oligomers, by using a sandwich ELISA similar to those described previously with different capturing/detecting antibodies (LeVine, 2004; Yang et al., 2005). In this assay, soluble Aβ oligomers captured by 6E10 can only be detected by biotinylated 6E10 (detecting antibody), if there is at least one more accessible epitope for the detecting biotinylated 6E10 antibody on Aβ oligomers. For the detection of biotinylated 6E10 antibody, streptavidin–HRP (BioSource International) was incubated for 2 h at room temperature and visualized using TMB substrate (Kirkegaard & Perry Laboratories). This method did not detect monomeric Aβ but detected soluble Aβ40 or Aβ42 oligomers of different sizes prepared in vitro, as described previously (Kayed et al., 2003) (data not shown). 4G8, at the levels used in intracerebral clearance studies, did not interfere with the 6E10 sandwich ELISA (data not shown). The optical density readings reflecting the number of accessible epitopes on Aβ oligomers were normalized per total TBS-soluble brain protein to estimate the relative levels of oligomers.
The thioflavin S-positive amyloid load (Wilcock et al., 2003) was determined using the Image-Pro-Plus program (Media Cybernetics, Silver Spring, MD).
Mouse endogenous brain Aβ. Mouse endogenous Aβ40 and Aβ42 levels were determined by sandwich ELISA (Best et al., 2005). Cerebral cortex was homogenized in 2% SDS containing complete protease inhibitor cocktail (Roche) (Kawarabayashi et al., 2001). For mouse Aβ40-specific sandwich ELISA, the capturing and biotinylated detecting antibodies were mouse monoclonal mouse Aβ raised against amino acid residues 1–20 (AMB0062; BioSource International) and rabbit polyclonal anti-Aβ40 biotin conjugate (44-3489; BioSource International), respectively. For mouse Aβ42-specific sandwich ELISA, the capturing and detecting antibody were AMB0062 and rabbit polyclonal anti-Aβ42 biotin conjugate (44-3449; BioSource International), respectively. Murine synthetic Aβ40 and Aβ42 standards (American Peptide, Sunnyvale, CA) were prepared in the ELISA buffer (PBS, 0.05% Tween 20, 0.25% BSA, 0.05% sodium azide, and complete protease inhibitor cocktail). The capturing antibody was coated at 5 μg/ml overnight at 4°C in 50 mm carbonate buffer, pH 9.6, and 0.05% sodium azide on 96-well high-binding Stripwell immunoassay plates (Corning Life Sciences), blocked with 0.25% bovine serum albumin in PBS overnight at 4°C, and 100 μlof sample or standard was added and incubated overnight at 4°C. Aβ levels were obtained from the standard curve.
Statistical analysis. The results were compared by multifactorial analysis of variance and Student's t test. The differences were considered to be significant at p < 0.05. All values are mean ± SEM.
Results
Figure 1A shows that 4G8, an Aβ-specific monoclonal IgG2b raised to 17–24 residues of Aβ, reduces by fivefold to sixfold influx of circulating 125I-Aβ40 across the BBB in APPsw+/– (Tg2576) mice at 2–3 and 4–6 months of age. At a later stage, 15- to 20-month-old APPsw+/– mice, the influx of circulating Aβ into brain ISF, although significantly higher than in controls, declines spontaneously. Still, 4G8 produced a significant inhibition of Aβ influx in 15- to 20-month-old and 24- to 36-month-old APPsw+/– mice (Fig. 1A).
Figure 1.
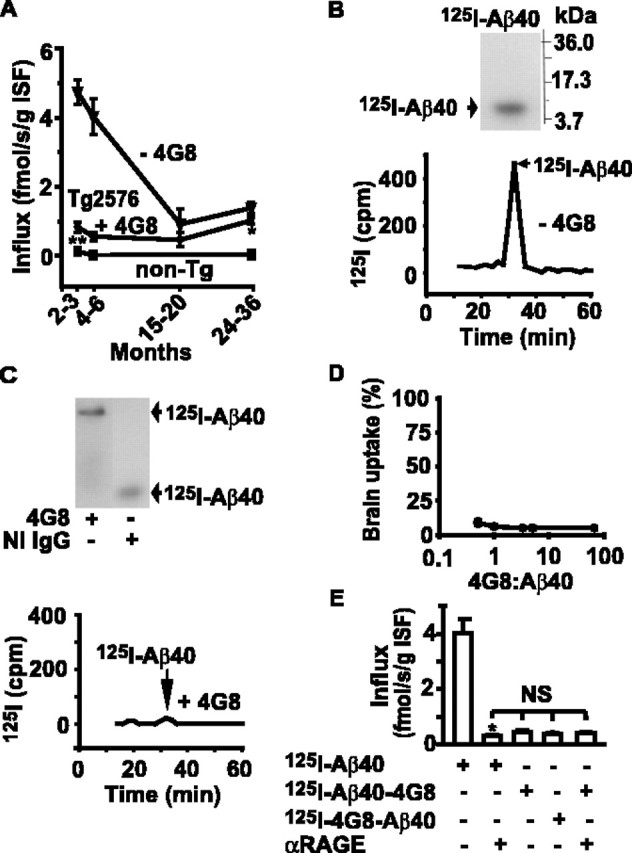
Aβ-specific IgG (4G8) blocks BBB influx of circulating Aβ40. A, Aβ40 BBB influx in APPsw+/– (Tg2576) mice in the presence (circles) and absence (triangles) of a fourfold excess of 4G8 determined with the brain perfusion method (LaRue et al., 2004) with 125I-Aβ40 at carrier concentrations corresponding to Aβ40 plasma levels in Tg2576 mice at different ages (Kawarabayashi et al., 2001), i.e., 4 nm (2–3 and 4–6 months), 3 nm (15–20 months), and 1.5 nm (24–36 months), and in age-matched littermate controls (non-Tg) at physiological Aβ40 plasma levels (50 pm) without 4G8. B, Tris-tricine SDS-PAGE (top) and HPLC (bottom) analysis of 125I-Aβ40 radioactivity in the arterial inflow without 4G8. C, Tris-tricine native PAGE (top) and HPLC (bottom) analysis of 125I-Aβ40 radioactivity in the arterial inflow with a four foldexcess of 4G8 or NI IgG. D, 125I-Aβ40 brain uptake at varying 4G8/Aβ40 plasma ratio in 4- to 6-month-old Tg2576 mice expressed as the percentage of control uptake. E, Transport across the BBB of 125I-Aβ40 and Aβ40–4G8 complexes labeled on either Aβ40 or 4G8 in the absence and presence of RAGE-specific IgG F(ab′)2 in 4- to 6-month-old Tg2576 mice. Mean ± SEM; n = 3–6. *p < 0.001; and **p < 0.05; NS, not significant.
In the cerebral arterial inflow, 125I-Aβ40 alone remains >98.5% in its free monomeric form, as shown by the SDS-PAGE and HPLC analysis (Fig. 1B), as reported previously (Deane et al., 2003). However, in the presence of 4G8, Aβ forms stable complexes in plasma, as demonstrated by the SDS-PAGE and HPLC analysis within 30 min of the cerebral arterial infusion (Fig. 1C), which is longer than typical infusion times of 5–10 min used for influx calculations in Figure 1A. In the present study, the concentrations of 4G8 that block Aβ40 BBB influx (Fig. 1A) were by two orders of magnitude lower than previously reported anti-Aβ IgG plasma levels in actively (Das et al., 2003) or passively (DeMattos et al., 2002) immunized mice. Lowering the 4G8/Aβ40 plasma ratio from 80 to 1 resulted in comparable >95% inhibition of Aβ40 brain uptake (Fig. 1D). Aβ40–4G8 complexes labeled on either Aβ or 4G8 residues did not penetrate across the BBB, and their behavior was not influenced by an RAGE blocking antibody. In contrast, a significant transport of free, circulating Aβ was by >80% inhibited by the blockade of RAGE at the BBB (Fig. 1E), as reported previously (Deane et al., 2003; LaRue et al., 2004). These findings suggest that 4G8 effectively reduces RAGE-mediated increase in circulating Aβ influx across the BBB in APPsw+/– mice by sequestering circulating Aβ in plasma.
Figure 2, A and B, shows time-dependent 4G8 increases in plasma and brain ISF, respectively, within 120 h of two subsequent intraperitoneal injections of 4G8 (200 μg). By using a brain clearance method (Shibata et al., 2000), we showed that efflux of centrally administered 125I-Aβ40 from mouse brain ISF determined over 30 min was moderately but significantly increased by 15% at 1 h of peripheral 4G8 intraperitoneal administration (Fig. 2C). Because the levels of 4G8 in brain ISF were barely detectable after 1 h of peripheral 4G8 injections (Fig. 2B) and intracerebrally administered 125I-Aβ40 does not form complexes with plasma-derived 4G8 after 1 h of peripheral 4G8 administration, as indicated by the autoradiographic analysis of brain aqueous extracts (Fig. 2E), an increase in brain to blood clearance of centrally administered Aβ at this early time point is likely to be mediated by peripheral action of 4G8. Thus, an increased 125I-Aβ40 efflux at 1 h (Fig. 2C) probably reflects the sink effect of 4G8, similar to that reported for other peripheral Aβ sequestering agents by different groups (DeMattos et al., 2002; Deane et al., 2003; Lemere et al., 2003). Namely, after 1 h, plasma levels of 4G8 are already high and ∼5 nm (Fig. 2A), which likely mops up completely the endogenous free plasma Aβ (50–100 pm), alters the Aβ equilibrium at the BBB in favor of Aβ efflux from brain. However, after 120 h of two subsequent 4G8 systemic injections, the BBB efflux of 125I-Aβ40 was substantially increased by 50% (Fig. 2D). Because at 120 h there was a significant increase in 4G8 levels in brain ISF (Fig. 2B), we hypothesized that enhanced clearance of centrally administered 125I-Aβ40 by peripherally administered 4G8 at 120 h (Fig. 2D) is not attributable to peripheral sink action (Fig. 2A). Indeed, the autoradiographic analysis of brain extracts indicated that intracerebrally administered 125I-Aβ40 forms within 30 min complexes with systemically administered 4G8 at 120 h of 4G8 administration (Fig. 2E). This result suggests that peripheral 4G8 may enhance clearance of brain-borne Aβ after its crossing into brain ISF. 125I-Aβ40 clearance via the ISF bulk flow was not altered by 4G8.
Figure 2.

Plasma and brain ISF distribution of systemically administered 4G8 and its effect on 125I-Aβ40 clearance from brain ISF. A, Plasma 4G8 levels determined with ELISA within 120 h in mice that received 4G8 (200μg) intraperitoneally at 0 and 48 h. B, Brain ISF 4G8 levels in mice in A at 1 and 120 h of 4G8 treatment. C, D, Clearance of 125I-Aβ40 (40 nm) via BBB transport and ISF bulk flow within 30 min of 125I-Aβ40/14C-inulin microinjections into brain ISF in mice at 1 h (C) and 120 h (D) of 4G8 treatment as in A. E, Formation of 125I-Aβ40–4G8 complexes in mouse brain ISF determined by Tris-tricine native PAGE of the aqueous brain extracts 30 min after 125I-Aβ40 (120 nm) microinfusion into brain ISF and after either 1 h (left) or 120 h (right) of 4G8 treatment as in A. Mean ± SEM; n = 3–5. *p < 0.001 and **p < 0.05.
To test whether Aβ complexed to an Aβ-specific IgG is cleared from brain, we measured the BBB efflux of centrally administered 4G8 alone and of Aβ40–4G8 and Aβ42–4G8 complexes in wild-type 2- to 3-month-old mice, using our clearance method (Shibata et al., 2000). The BBB clearance of 125I-4G8 was inhibited by the increasing concentrations of centrally administered unlabeled 4G8 (data not shown) and abolished by 2 μm 4G8 (Fig. 3A), suggesting a concentration-dependent IgG transcytosis at the BBB. RAP, which inhibits Aβ BBB clearance via LRP (Shibata et al., 2000; Deane et al., 2004), or fucoidan, an inhibitor of the scavenger receptor on microglia, did not affect 4G8 clearance (Fig. 3A). Immunostaining for activated microglia was negative in the present model (data not shown), as reported previously (Cirrito et al., 2003; Deane et al., 2004).
Figure 3.

FcRn-mediated BBB transcytosis of 125I-4G8, 125I-Aβ40–4G8, and 125I-Aβ42–4G8 from brain to blood. A, The BBB clearance of 40 nm 125I-4G8, 125I-Aβ40–4G8, or 125I-Aβ42–4G8 within 30 min of brain ISF microinjections with and without unlabeled 4G8 (2 μm), RAP (5 μm), fucoidan (1.5 mm), αFcRn (anti-FcRn, 60 μg/ml), and Aβ40 (1 μm) and in FcRn–/–, β2M–/–, RAP–/–, and FcRγ–/– mice. B, C, Serum levels of 125I-4G8 (B) and of 125I-Aβ40–4G8 (C) from experiments in A expressed as the percentage of injected dose (%ID). Western blot analysis of FcRn in brain microvessels in control and β2M–/– mice (inset, C). D, FcRn/CD31 double immunostaining in brains of nontransgenic mice (left) Scale bar, 50 μm. Western blot analysis (right) of FcRn, F4/80 (phagocytic cells), and GFAP (astrocytes) in isolated brain microvessels (lane 1) and capillary-depleted brain (lane 2). E, Uptake of 125I-4G8, 125I-4G8–Aβ40, 125I-Aβ40–4G8, or 125I-Aβ40–4G8-F(ab′)2 (1 nm) at the abluminal side of isolated mouse brain microvessels within 1 min at 37°C. 4G8 (5 μm), RAP (5 μm), Fc (10 μm), and αLRP (20 μg/ml) were applied as potential inhibitors. Mean ± SEM; n = 3–5. *p < 0.05, **p < 0.001, inhibitors or gene deletion versus the corresponding controls; •p < 0.01, 125I-Aβ40–4G8 versus 125I-4G8; NS, not significant.
Because IgG is carried across biological membranes by the FcRn (Roopenian et al., 2003; Ober et al., 2004; Yoshida et al., 2004) and FcRn is expressed in vascular endothelium and at the BBB (Borvak et al., 1998; Schlachetzki et al., 2002), we explored whether deletions of the FcRn gene and the FcRn light chain β2M gene affect 125I-4G8 BBB efflux. Figure 3A shows significantly reduced 4G8 clearance in FcRn–/– mice and β2M–/– mice by 68 and 63%, respectively, whereas deletions of the RAP gene (functional LRP knock-out) (Deane et al., 2004) or the FcRγ gene were without effect. Excess of 4G8 in brain ISF in FcRn–/– mice resulted in complete inhibition of 125I-4G8 BBB efflux (data not shown), consistent with the data demonstrating that unlabeled 4G8 inhibits 125I-4G8 clearance at the BBB in wild-type mice (Fig. 3A). After central administration of 125I-4G8, the radioactivity appearing in plasma was readily detectable and 100% TCA precipitable, suggesting transcytosis of intact 125I-4G8 from brain into blood (Fig. 3B), as confirmed by the autoradiography (data not shown). 125I-4G8 serum levels were significantly reduced (>90%) by central administration of unlabeled excess 4G8 and in FcRn–/– mice (70%) (Fig. 3B), indicating that the FcRn pathway is required for clearance of an anti-Aβ IgG across the BBB.
Next, we showed that 125I-Aβ40–4G8 was cleared across the BBB of 2- to 3-month-old wild-type mice at a rate 2.5-fold faster than 125I-4G8 (Fig. 3A). Unlike 4G8, clearance of Aβ40–4G8 complexes was inhibited by central administration of either unlabeled 4G8 or RAP and was abolished by the combination of both, suggesting that in young mice 125I-Aβ40–4G8 complexes can be cleared by an IgG-dependent mechanism and/or via a RAP-sensitive LRP mechanism (Shibata et al., 2000; Deane et al., 2004). That both the IgG-assisted and LRP-dependent transport are important for clearance of Aβ40–4G8 complexes in young mice has been confirmed by demonstrating ∼60% reductions in 125I-Aβ40–4G8 BBB efflux in β2M–/– mice and RAP–/– mice (Fig. 3A). After central administration of 125I-Aβ–4G8 complexes, the radioactivity appearing in plasma was 100% TCA precipitable (Fig. 3C), suggesting no degradation of 125I-Aβ40 from the complex, as well as transcytosis of intact complexes, which has been confirmed by the autoradiography (data not shown). Serum levels of centrally infused 125I-Aβ40–4G8 complexes were substantially reduced by centrally administered unlabeled 4G8 and/or RAP, as well as by deletion of the β2M gene and RAP gene (Fig. 3C). We also demonstrated that Aβ42–4G8 complexes are cleared by the FcRn pathway (Fig. 3A), as well as Aβ42 bound to anti-Aβ1560 (data not shown).
By using isolated cerebral microvessels positive for FcRn and negative for microglia and astrocytic markers (Fig. 3D), we corroborated our in vivo findings by showing that 125I-4G8 rapid brain capillary uptake at 37°C, which reflects its clearance by brain capillaries, in 2- to 3-month-old mice was dose dependently (data not shown) and completely blocked by excess unlabeled 4G8 (Fig. 3E) but was not affected by RAP. Under the present experimental conditions, uptake of 125I-radiolabeled 4G8 and its complexes with Aβ occurred at the abluminal side of brain capillaries because short, 1 min exposure times precluded a significant diffusion of the studied tracers into the capillary lumen, which is required for the uptake at the luminal side (Deane et al., 2004). This has been confirmed by a negligible uptake of 14C-inulin, which does not interact with the cells and is taken up by the isolated capillaries only via passive diffusion into the lumen. Regarding 125I-labeled 4G8–Aβ40 complexes (with the label on either 4G8 or Aβ40 residues), both the unlabeled Fc fragment and RAP or an LRP-specific IgG blocked clearance of complexes on brain capillaries from 2- to 3-month-old young mice (Fig. 3E). As expected, Aβ40–4G8 F(ab′)2 complexes lacking the Fc domain were cleared by an Fc-independent RAP-sensitive mechanism, confirming the involvement of LRP, as shown for Aβ-mediated clearance (Deane et al., 2004). That Aβ molecule in anti-Aβ–Aβ complexes is critical for interaction with LRP was shown by inhibition of brain capillary clearance of 125I-labeled 4G8–Aβ40 complexes (the label was on 4G8) by excess of unlabeled Aβ40 (data not shown).
Figure 4A shows that 4G8 systemic administration over 120 h increases by approximately threefold efflux of centrally administered 125I-Aβ40 in 18- to 20-month-old APPsw+/– mice (Fig. 4A). To test whether old APPsw+/– mice clear Aβ from brain through an IgG-assisted efflux, we compared clearance of Aβ40 alone and Aβ40–4G8 complexes. Consistent with a significant downregulation of LRP at the BBB with aging, and particularly in transgenic APPsw+/– mice (Deane et al., 2004), the BBB efflux of Aβ40 alone was significantly reduced in old APPsw+/– mice (Fig. 4B) compared with control young mice (Fig. 2C,D) or young APPsw+/– mice (data not shown). However, central infusion of 125I-Aβ40–4G8 complexes in 20- to 23-month-old APPsw+/– mice resulted in an approximately fivefold greater clearance than of 125I-Aβ40 alone (Fig. 4B), suggesting that IgG-assisted efflux of Aβ remains active in old APPsw+/– mice and is even somewhat higher than in young control wild-type mice (Fig. 3A).
Figure 4.

FcRn-dependent 125I-Aβ40–4G8 BBB clearance in old APPsw+/– mice and FcRn BBB expression in AD. A, 125I-Aβ40 (40 nm) BBB and ISF clearance in 18- to 20-month-old APPsw+/– (Tg2576) mice 30 min after brain ISF microinjections and 120 h after vehicle or 4G8 intraperitoneal treatment, as in Figure 2 A. B, The BBB clearance of 125I-Aβ40 or 125I-Aβ40–4G8 complexes (40 nm) in 20- to 23-month-old Tg2576 mice within 30 min of brain ISF microinjections. C, 125I-Aβ40–4G8 (1 nm) uptake by isolated cerebral microvessels of Tg2576 mice at 3–5 and 20–23 months of age determined within 1 min at 37°C without and with Fc fragment (10 μm) and RAP (5 μm). D, FcRn colocalization with vWF (endothelial intracellular marker) in the brain of 10-month-old Tg2576 mouse studied by confocal microscopy. E, Western blot analysis of FcRn in brain microvessels of control and Tg2576 mice at 5–7 and 20–23 months of age (top) and its relative abundance by scanning densitometry corrected for β-actin (bottom). F, FcRn and vWF double immunostaining in brains of AD and age-matched individuals (left) and FcRn-positive vascular profiles (right). Scale bar, 50μm. Mean ± SEM; n = 3–6.
In a next set of studies, we showed that both RAP and Fc fragment effectively block clearance of 4G8–Aβ complexes on isolated cerebral microvessels derived from younger, 3- to 5-month-old APPsw+/– mice (Fig. 4C). In contrast, RAP did not block clearance of 4G8–Aβ complexes on brain capillaries derived from 20- to 23-month-old APPsw+/– mice (Fig. 4C). The loss of inhibitory effect of RAP on capillary clearance in aged APPsw+/– mice is consistent with a significant loss of LRP expression in these animals at an older age (Deane et al., 2004). Conversely, the Fc fragment inhibited significantly by 67% clearance of complexes on aged APPsw+/– mouse brain capillaries (Fig. 4C), confirming that an IgG-dependent clearance of IgG–Aβ complexes remains functional with aging in APPsw+/– mice. These results are consistent with a significant FcRn expression in brain endothelium in older APPsw+/– mice and an age-dependent increase in the FcRn levels in brain capillaries of APPsw+/– mice (Fig. 4D,E). We also observed an age-dependent increase in the FcRn levels in control mice, although the levels of FcRn in APPsw+/– mice were substantially higher than in their corresponding age-matched littermate controls, at both 5–7 and 20–23 months of age, respectively (Fig. 4E). Studies on human tissue indicated an increase in FcRn-positive brain vascular profiles in AD compared with age-matched controls (Fig. 4F).
Next, we determined whether inhibition of the FcRn pathway affects intracerebral 4G8 immunotherapy in 24-month-old APPsw+/– mice. We limited studies to 24 h to minimize possible influence of the antibody on microglia-dependent clearance, because microglia are typically activated in older APPsw+/– mice after 24 h of an anti-Aβ antibody intracerebral injection (Wilcock et al., 2003), as confirmed in this study (data not shown). Our data corroborate findings by the previous studies (Wilcock et al., 2003; Oddo et al., 2004) by showing that centrally administered 2 μg of 4G8 significantly reduces Aβ immunostaining and the levels of human endogenous Aβ40 and Aβ42 in the hippocampus of 24-month-old APPsw+/– mice by 45, 47, and 25%, respectively (Fig. 5A,D–F). These results correlated well with the observed >35% reductions in the levels of TBS-insoluble/SDS-soluble human endogenous Aβ40 and Aβ42 by 4G8. It is noteworthy that the levels of TBS-soluble human Aβ oligomers were also reduced by 25% by 4G8, as demonstrated by an ELISA developed previously for determining the relative levels of Aβ oligomers (Levine, 2004; Yang et al., 2005) (Fig. 5G). However, 4G8 did not affect thioflavin S-positive amyloid load (Fig. 5H), consistent with our finding that 4G8 did not affect the levels of SDS-resistant/formic acid-soluble Aβ40 and Aβ42, using methods described previously (Kawarabayashi et al., 2001). This result corroborates findings by a previous study demonstrating that, within 24 h of intracerebral delivery, the Aβ-specific antibody does not reduce thioflavin S staining (Wilcock et al., 2003). Although 4G8 binds amyloid plaques on tissue sections ex vivo, its binding to plaques after intracerebral in vivo administration was minimal under the present experimental conditions (data not shown).
Figure 5.
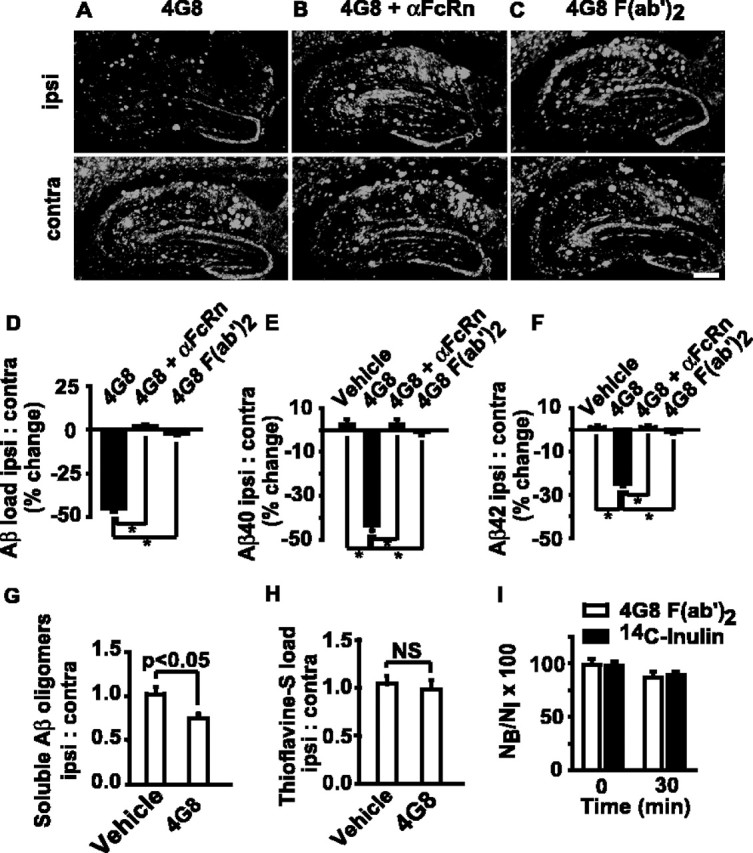
FcRn-dependent clearance of endogenous Aβ in APPsw+/– mice by centrally administered 4G8. A, Intrahippocampal 4G8 (2 μg/0.5 μl) reduces Aβ immunostaining 24 h after injection in 24-month-old Tg2576 mouse (top; ipsi) compared with nonimmune IgG-treated contralateral hippocampus (bottom; contra). B, 4G8 (2 μg/0.5μl) was injected into the ipsilateral hippocampus in 24-month-old Tg2576 mouse 30 min after αFcRn (anti-FcRn antibody; 10 μg/0.5 μl) (top) or vehicle (bottom; contralateral hippocampus). C, 4G8 F(ab′)2 (2 μg/0.5 μl) injected into the hippocampus in 24-month-old Tg2576 mouse (top) versus vehicle-treated contralateral hippocampus (bottom). Scale bar, 350 μm. D–F, Aβ load (D), Aβ40(E), and Aβ42 (F) levels: the percentage change (%) in the ipsilateral versus contralateral hippocampus in Tg2576 mice. *p < 0.05. G, H, TBS-soluble Aβ oligomers (G) and thioflavin S-positive amyloid load (H) in 4G8-treated (ipsilateral) versus vehicle-treated (contralateral) hippocampus in 24-month-old Tg2576 mice at 24 h. Mean ± SEM; n = 5 mice per group and 6–8 sections close to the injection site per mouse. I, 125I-4G8 F(ab′)2 (40 nm) and 14C-inulin brain retention 30 min after brain ISF microinjection of the tracer mixture. Mean ± SEM; n = 3. NS, Not significant.
In contrast to vehicle-treated APPsw+/– mice, pretreatment of APPsw+/– mice with intracerebral infusions of FcRn-specific blocking antibodies 30 min before administration of 4G8 completely abolished Aβ lowering effects of centrally administered 4G8 immunotherapy (Fig. 5B,D–G). A nonimmune IgG did not have any effect on Aβ clearance (Fig. 5A–C, bottom). Similarly, within 24 h, we failed to demonstrate significant reductions in Aβ load and Aβ40 and Aβ42 levels by 4G8 F(ab′)2 in 24-month-old APPsw+/– mice (Fig. 5C–F). Our clearance study with radiolabeled 125I-4G8 F(ab′)2 failed to show clearance of Fab fragments across the BBB (Fig. 5I), in contrast to a rapid removal of the full length 4G8 (Fig. 3A).
To determine whether the FcRn pathway is critical for removal of endogenous brain Aβ by systemically administered 4G8, we compared the effects of peripheral 4G8 immunotherapy (administered as in Fig. 2) on levels of endogenous mouse brain Aβ40 and Aβ42 in FcRn+/+ mice and FcRn–/– mice. To minimize a possible confounding effect of LRP-mediated clearance, we performed these studies in 9-month-old mice, at an age when LRP expression at the BBB is already substantially reduced in mice (Shibata et al., 2000; Deane et al., 2004), as confirmed in this study (data not shown). By using an ELISA similar to that reported recently (Best et al., 2005), we showed that treatment of FcRn+/+ wild-type mice with systemically administered 4G8 results in significant 35 and 40% reductions in brain endogenous mouse Aβ40 and Aβ42 levels, respectively (Fig. 6A,B). In contrast, there was a negligible 5–7% reduction in endogenous Aβ40 and Aβ42 brain levels in FcRn–/– mice (Fig. 6A,B). A reduction in endogenous mouse brain Aβ levels by systemic 4G8 in FcRn+/+ mice compared with FcRn–/– mice was associated with a corresponding significant increase in plasma Aβ, which was >97% G-protein precipitable (G-protein binds the Fc fragment of 4G8) (data not shown), indicating that the majority of Aβ in plasma after systemic therapy with the antibody is bound to 4G8, similarly as shown by previously passive and active immunization studies (DeMattos et al., 2001, 2002; Lemere et al., 2003).
Figure 6.

FcRn-dependent clearance of mouse endogenous brain Aβ by 4G8 systemic administration: studies in FcRn+/+ and FcRn–/– mice. Mice were treated for 5 d with 4G8 (open bar), 200μg intraperitoneally at 0 and 48 h, or vehicle (filled bar), and the levels of endogenous mouse Aβ40 (A) and Aβ42 (B) determined in the cerebral cortex in FcRn+/+ mice and FcRn–/– mice. Mean ± SEM; n = 5. NS, Not significant.
Discussion
Our study shows that an anti-Aβ antibody regulates brain Aβ by exerting important peripheral and central actions on Aβ transport across the BBB. The mechanism of peripheral Aβ sequestration observed in the present study with 4G8 in APPsw+/– mice may be similar to that of other Aβ binding agents, i.e., apolipoproteins E2 and 3 (Martel et al., 1997), ganglioside M and gelsolin (Matsuoka et al., 2003), the soluble forms of RAGE (Deane et al., 2003) or LRP (Deane et al., 2004), and/or other antibodies to Aβ that reduce its influx across the BBB (Pan et al., 2002; Banks et al., 2005). This peripheral binding of Aβ may create a sink effect, providing that the activity of the LRP efflux pathway (Deane et al., 2004) is not lost or is sufficient to maintain elimination of brain Aβ and Aβ–IgG complexes. The central action of certain anti-Aβ IgGs may, however, critically depend on sequestering brain Aβ, followed by the FcRn-mediated rapid BBB clearance. Importantly, the FcRn IgG-dependent clearance for Aβ remains active at the BBB during normal aging and in APPsw+/– mice, and increased FcRn expression at the BBB in AD may likely be of a therapeutic value for Aβ immunotherapy.
Circulating IgG may enter the brain and have widespread extracellular distribution within the CNS and CSF from sites deficient in a BBB such as subarachoind space pial surface in the Wirchow-Robin spaces and subpial cortical gray matter and circumventricular organs (Balin et al., 1986; Kozlowski et al., 1990; Broadwell and Sofroniew, 1993). It has been reported that an antibody directed at 1–17 region of Aβ can use the extracellular pathways for its passive entry into the CNS (Banks et al., 2002). In addition, some IgG species are also transported slowly across the BBB via adsorptive-mediated transcytosis (Zlokovic et al., 1990; Pardridge, 1991). Our results indicate that systemically administered 4G8 exerts a significant clearance effect on endogenous brain Aβ that is abolished in FcRn–/– mice at an age when LRP expression is substantially downregulated (Deane et al., 2004). Although 4G8 recognizes APP in addition to Aβ, it is unlikely that the Aβ-lowering effect of systemically administered 4G8 is attributable to inhibition of APP/Aβ production because this effect is lost in FcRn–/– compared with FcRn+/+ mice. Inhibition of the FcRn pathway influences also the outcome of centrally administered Aβ immunotherapy in old APPsw+/– mice. Although intracerebral immunotherapy has limitations regarding direct insight into what happens with peripheral passive or active approaches, studies using the intracerebral approach have been useful in delineating clearance pathways of Aβ from brain (Tanzi et al., 2004; Zlokovic, 2005) and have been used extensively to demonstrate that antibody in the brain exerts a clearance action on brain Aβ (Bacskai et al., 2001, 2002; Wilcock et al., 2003; Oddo et al., 2004). For example, it has been shown that intracerebral delivery of 2 μg of 4G8 or anti-Aβ1560 (Oddo et al., 2004) and 2 μg of 6E10 (Wilcock et al., 2003), substantially reduces Aβ load in 12-month-old triple-transgenic AD mice and in 20-month-old APPsw+/– mice, respectively. Our data suggest that the FcRn pathway is required for clearance of TBS-insoluble/SDS-soluble endogenous Aβ40 and Aβ42, which possibly represents Aβ in diffuse plaques that is not yet converted into a hard-core thioflavin S-positive amyloid. Although we have not demonstrated FcRn-mediated clearance of thioflavin-S-positive amyloid, it is possible that, over longer periods of time (>24 h), centrally administered 4G8 will reduce thioflavin S-positive load from brain (Wilcock et al., 2003; Oddo et al., 2004), which, at least in part, could involve the FcRn pathway.
Topical administration of Fab fragments alone can reduce Aβ immunostaining in APPsw+/– mice within 3 d (Bacskai et al., 2002). Our data show that this effect could be mediated via LRP efflux system, which can clear Aβ40–4G8 F(ab′)2 complexes. In contrast, Fab fragments alone are eliminated slowly from brain ISF at a rate comparable with that of a reference marker, inulin. However, we also demonstrated that 4G8 F(ab′)2 cannot exert a vigorous effect on Aβ pathology within 24 h in 24-month-old APPsw+/– mice, which could be explained by substantial down-regulation of LRP levels at the BBB in old APPsw+/– mice (Deane et al., 2004).
FcRn plays a central role in delivering IgGs within and across the cells (Ober et al., 2004). Although FcRn was discovered as a developmental IgG receptor, recent studies suggest its expression in different cell types in adult rodents and humans, including polarized epithelia and endothelia, which are able to translocate the FcRn-bound cargo bidirectionally, from either the lumen or tissue spaces into the opposite pole of the cell (Roopenian et al., 2003; Yoshida et al., 2004). FcRn is directly involved in IgG exocytosis (Ober et al., 2004) and binds IgG best at an intracellular pH 6.0–6.5 (Roopenian et al., 2003). It is expressed at low levels in the cell membrane because of its rapid recycling after incomplete fusion with the plasma membrane, known as “kiss and run” (Ober et al., 2004). It is likely that the FcRn-mediated transcytosis of IgG across the BBB may require an additional binding step to another IgG receptor at the abluminal cell membrane, which will facilitate IgG endocytosis before its FcRn-mediated migration to the exocytotic site at the luminal membrane, analogous to the IgG transport across other biological membranes (Roopenian et al., 2003). This hypothesis is supported by our work in progress showing a single 55 kDa band (data not shown) after biotinylation of the abluminal mouse brain capillary membranes and 4G8 affinity purification. This band corresponded to a recently described IgG cell surface receptor in the placental endothelium (Gafencu et al., 2003). The exact cellular mechanisms involved in possible interaction between a putative 55 kDa IgG receptor and FcRn during IgG and Aβ–anti-Aβ complex internalization at the abluminal side of the BBB and exocytosis across the luminal side remain, however, to be investigated.
Based on the present work, we propose two possibilities for enhanced efficacy of immunization therapy directed at an improved clearance of Aβ across the BBB: (1) to increase a peripheral sink of an anti-Aβ antibody and facilitate brain efflux of Aβ and Aβ–IgG complexes by increasing the activity of the BBB LRP efflux pathway at an older age; and (2) to optimize the FcRn pathway for enhanced IgG-mediated Aβ clearance across the BBB in the aging brain.
Footnotes
This research was supported by United States Public Health Service Grants R37AG023084 and NS34467 (B.V.Z.) and AG20222 (D.M.H.). We thank Dr. Derry Roopenian from The Jackson Laboratory for providing FcRn–/– mice, Bristol Myers Squibb for providing us with some aged Tg2576 mice, and Kelly Simons for assistance with animal breeding and maintenance.
Correspondence should be addressed to Berislav V. Zlokovic, Arthur Kornberg Medical Research Building, 601 Elmwood Avenue, Box 670, Rochester, NY 14642. E-mail: berislav_zlokovic@urmc.rochester.edu.
DOI:10.1523/JNEUROSCI.3697-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511495-09$15.00/0
R.D. and A.S. contributed equally to this work.
References
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT (2001) Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7: 369–372. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non-Fc-mediated mechanisms are involved in the clearance of amyloid-β in vivo by immunotherapy. J Neurosci 22: 7873–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin BJ, Broadwell RD, Salcman M, El-Kalliny M (1986) Avenues for entry of peripherally administered protein to the central nervous system in mouse, rat and squirrel monkey. J Comp Neurol 251: 260–280. [DOI] [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE (2002) Passage of amyloid β protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides 23: 2223–2226. [DOI] [PubMed] [Google Scholar]
- Banks WA, Robinson SM, Verma S, Morley JE (2003) Efflux of human and mouse amyloid β proteins 1–40 and 1–42 from brain: impairment in a mouse model of Alzheimer's disease. Neuroscience 121: 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Pagliari P, Nakaoke R, Morley JE (2005) Effects of behaviorally active antibody on the brain uptake and clearance of amyloid beta proteins. Peptides 26: 287–294. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke R, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer's disease. Nat Med 6: 916–919. [DOI] [PubMed] [Google Scholar]
- Best JD, Jay MT, Out F, Ma J, Nadin A, Ellis S, Lewis HD, Pattison C, Reilly M, Harrison T, Shearman MS, Williamson TL, Atack JR (2005) Quantitative measurement of changes in amyloid-β(40) in the rat brain and cerebrospinal fluid following treatment with the γ-secretase inhibitor LY-411575 [N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide]. J Pharmacol Exp Ther 313: 902–908. [DOI] [PubMed] [Google Scholar]
- Borvak J, Richardson J, Medesan C, Antoke F, Radu C, Simionescu M, Ghetie V, Ward S (1998) Functional expression of the MHC class 1-related receptor, FcRn, in endothelial cells of mice. Int Immunology 10: 1289–1298. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Sofroniew MV (1993) Serum proteins bypass the blood-brain fluid barriers to extracellular entry to the central nervous system. Exp Neurol 120: 245–263. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci 23: 8844–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE (2001) Reduced effectiveness of Aβ 1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging 22: 721–727. [DOI] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE (2003) Amyloid-β immunization effectively reduces amyloid deposition in FcRγ–/– knock-out mice. J Neurosci 23: 8532–8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. (2003) RAGE mediates amyloid-β transport across the blood-brain barrier and accumulation in brain. Nat Med 9: 907–913. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004) LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43: 333–344. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart J, Paul SM, Holtzman DM (2001) Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98: 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM (2002) Brain to plasma amyloid-Aβ efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 295: 2264–2267. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat Neurosci 5: 452–457. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Cole GM, Baird A (1992) Phagocytosis and deposition of vascular β-amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol 140: 1389–1399. [PMC free article] [PubMed] [Google Scholar]
- Gafencu A, Heltianu C, Burlacu A, Hunziker W, Simionescu M (2003) Investigation of IgG receptors expression on the surface of human placental endothelial cells. Placenta 24: 664–676. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemka U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, deq Uervain JF, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38: 547–554. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HTJ, Nixon RA, Mercken M, Bergeron C, Fraser PE, St. George-Hyslop P, Westaway D (2000) Aβ peptide immunization reduces behavioral impairment and plaques in a model of Alzheimer's disease. Nature 408: 979–982. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid Aβ protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, Mclntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486–489. [DOI] [PubMed] [Google Scholar]
- Kozlowski GP, Nilaver G, Zlokovic BV (1990) Immunoneurology: a serum protein afferent limb to the CNS. Circulating regulatory factors and neuroendocrine function (Porter JC, ed), pp 345–370. New York: Plenum. [DOI] [PubMed]
- LaRue B, Hogg E, Sagare A, Jovanovic S, Maness L, Maurer C, Deane R, Zlokovic BV (2004) Method for measurement of the blood-brain barrier permeability in the perfused mouse brain: application to amyloid-beta peptide in wild type and Alzheimer's Tg 2576 mice. J Neurosci Methods 138: 233–242. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff KE (2003) Evidence for peripheral clearance of cerebral Aβ protein following chronic, active Aβ immunization in PSAPP mice. Neurobiol Dis 14: 10–18. [DOI] [PubMed] [Google Scholar]
- LeVine III H (2004) Alzheimer's β-peptide oligomer formation at physiologic concentrations. Anal Biochem 335: 81–90. [DOI] [PubMed] [Google Scholar]
- Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, Mc-Comb JG, Frangione B, Ghiso J, Zlokovic BV (1997) Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem 69: 1995–2004. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Saito M, LaFrancois J, Saito M, Taylor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to Aβ-amyloid. J Neurosci 23: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCario G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) Aβ peptide immunization prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982–985. [DOI] [PubMed] [Google Scholar]
- Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med 9: 448–452. [DOI] [PubMed] [Google Scholar]
- Ober RJ, Martinez C, Lai X, Zhou J, Ward SE (2004) Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci USA 101: 11076–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43: 321–332. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubios B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C (2003) Subacute meningeoencephalitis in a subset of patients with AD after Aβ 42 immunization. Neurology 61: 46–54. [DOI] [PubMed] [Google Scholar]
- Pan W, Solomon B, Maness LM, Kastin AJ (2002) Antibodies to β-amyloid decrease the blood-to-brain transfer of β-amyloid peptide. Exp Biol Med 227: 609–615. [DOI] [PubMed] [Google Scholar]
- Pardridge WM (1991) Antibody delivery through the blood-brain barrier. In Peptide drug delivery to the brain (Pardridge WM, ed), pp 219–238. New York: Raven.
- Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer's disease amyloid β-protein by microglia cells. J Biol Chem 272: 29390–29397. [DOI] [PubMed] [Google Scholar]
- Ravetch JV, Bolland S (2001) IgG Fc receptors. Annu Rev Immunol 19: 275–290. [DOI] [PubMed] [Google Scholar]
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, Eden PA, Anderson CL (2003) The MHC1-like IgG receptor controls perinatal IgG transport, IgG homeostasis and fate of IgG-Fc-coupled drugs. J Immunol 170: 3528–3533. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano M, Shopp G, Vasquez G, Vandevert C, et al. (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173–177. [DOI] [PubMed] [Google Scholar]
- Schlachetzki F, Zhu C, Pardridge WM (2002) Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J Neurochem 81: 203–206. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer's amyloid-Aβ 1–40 peptide from brain by low-density lipoprotein receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106: 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T (2001) Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol 159: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL (2004) Clearance of Alzheimer's Aβ peptide; the many roads to perdition. Neuron 43: 605–608. [DOI] [PubMed] [Google Scholar]
- Thorell JI, Johansson BG (1971) Enzymatic iodination of polypeptides with 125I to high specific activity. Biochim Biophys Acta 251: 363–366. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D (2003) Intracranially administered anti-Aβ antibody reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci 23: 3745–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Hofman FM, Zlokovic BV (2003) A simple method for isolation and characterization of mouse brain microvascular endothelial cells. J Neurosci Methods 130: 53–63. [DOI] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen P, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280: 5892–5901. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, Lencer WI, Blumberg RS (2004) Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20: 769–783. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2004) Clearing amyloid through the blood-brain barrier. J Neurochem 89: 807–811. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28: 202–208. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Skundric DS, Segal MB, Lipovac MN, Mackic JB, Davson H (1990) A saturable mechanism for transport of immunoglobulin G across the blood-brain barrier of the guinea pig. Exp Neurol 107: 263–270. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Mackic JB, Wang L, McComb JG, McDonough A (1993) Differential expression of Na,K-ATPase alpha and beta subunit isoforms at the blood-brain barrier and the choroid plexus. J Biol Chem 268: 8019–8025. [PubMed] [Google Scholar]