

Abstract
Use-dependent modifications of synapses have been well described in the developing visual cortex, but the ability for experience to modify synapses in the adult visual cortex is poorly understood. We found that 10 d of late-onset visual deprivation modifies both presynaptic and postsynaptic elements at the layer 4→2/3 connection in the visual cortex of adult mice, and these changes differ from those observed in juveniles. Although visual deprivation in juvenile mice modifies the subunit composition and increases the current duration of synaptic NMDA receptors (NMDARs), no such effect is observed at synapses between layer 4 and layer 2/3 pyramidal neurons in adult mice. Surprisingly, visual deprivation in adult mice enhances the temporal summation of NMDAR-mediated currents induced by bursts of high-frequency stimulation. The enhanced temporal summation of NMDAR-mediated currents in deprived cortex could not be explained by a reduction in the rate of synaptic depression, because our data indicate that late-onset visual deprivation actually increases the rate of synaptic depression. Biochemical and electrophysiological evidence instead suggest that the enhanced temporal summation in adult mice could be accounted for by a change in the molecular composition of NMDARs at perisynaptic/extrasynaptic sites. Our data demonstrate that the experience-dependent modifications observed in the adult visual cortex are different from those observed during development. These differences may help to explain the unique consequences of sensory deprivation on plasticity in the developing versus mature cortex.
Keywords: NMDA receptor, NR2A, NR2B, metaplasticity, visual cortex, sensory deprivation
Introduction
Sensory experience modifies cortical circuitry by inducing use-dependent changes in synapses (Katz and Shatz, 1996), and these modifications generally are thought to be more dramatic in developing animals than in adults. Monocular deprivation has been a well studied model for critical period plasticity since the pioneering studies of Wiesel and Hubel (1963). Their finding that cortical neurons lose responsiveness to the deprived eye only if the deprivation begins early in life has led to the assumption that the adult cortex lacks the capacity for experience-dependent modifications.
There is a growing consensus, however, that the adult cortex maintains greater plasticity than originally thought (Kaas, 1991; Buonomano and Merzenich, 1998; Gilbert, 1998; Tagawa et al., 2005). For example, monocular deprivation shifts the ocular dominance of neurons in the primary visual cortex of adult mice, although the manner of the shift differs from that observed in juveniles (Sawtell et al., 2003; Frenkel and Bear, 2004; Lickey et al., 2004; Pham et al., 2004). In juvenile mice monocular deprivation causes a rapid reduction of the deprived eye response recorded in the contralateral cortex, followed by a potentiation of the responses driven by the nondeprived eye in the ipsilateral cortex. In contrast, monocular deprivation in adult mice fails to cause a loss of the deprived eye response, although a delayed potentiation of the responses driven by the nondeprived eye is still observed. The basis for the different consequences of sensory deprivation in young and mature animals is poorly understood, and we suggest that key differences may lie in the mechanisms that control the properties of synaptic plasticity.
The ocular dominance plasticity observed in both juvenile and adult mice requires activation of NMDA-type glutamate receptors (NMDARs). NMDARs are required for many forms of synaptic plasticity (Malenka and Bear, 2004), and changes in the attributes of the receptor are likely to influence the properties of synaptic plasticity. The NMDAR complex consists of the obligatory NR1 subunit in combination with NR2A–D and NR3A–B subunits that confer distinct receptor properties (Monyer et al., 1992; McBain and Mayer, 1994; Perez-Otano and Ehlers, 2004). NR1, NR2A, and NR2B subunits predominate in the postnatal visual cortex, and during development the ratio of NR2A- to NR2B-containing NMDARs increases (Quinlan et al., 1999a,b; Roberts and Ramoa, 1999). Because NR2A-containing NMDARs possess shorter current durations than NR2B-containing receptors, NMDAR-mediated current durations shorten over development (Carmignoto and Vicini, 1992; Hestrin, 1992; Monyer et al., 1992; Priestley et al., 1995; Flint et al., 1997; Vicini et al., 1998). The developmental increase in NR2A in the visual cortex is experience-dependent, because dark rearing delays the increase in NR2A in the visual cortex (Nase et al., 1999; Quinlan et al., 1999b).
Given the age-dependent differences in the synaptic consequences of sensory deprivation, we examined whether visual deprivation uniquely affects NMDAR composition and function in the visual cortex of juvenile and adult mice. Our results indicate that intracortical synapses of adult mice are highly plastic but undergo use-dependent modifications in a unique manner as compared with juveniles.
Materials and Methods
Animals. C57BL/6 mice (Charles River, Wilmington, MA) of both genders between postnatal days 21–27 (P21–P27) or P74–P84 were used. These ages represent periods during and after the classically defined critical period for ocular dominance plasticity in mice (Gordon et al., 1996). Control mice were raised on a 12 h light/dark cycle, whereas deprived, dark-reared mice were raised in complete darkness from P2. Late-onset visual deprivation was achieved by placing animals into a completely dark room for ∼10 d starting at ∼P68.
Slice preparation. Mice were anesthetized with an overdose of pentobarbital barbiturate and decapitated after the disappearance of corneal reflexes, in compliance with the U. S. Department of Health and Human Services and the University of North Carolina guidelines. Brains were dissected rapidly, and the visual cortices were cut in 400 μm coronal slices as previously described (Kirkwood et al., 1993; Philpot et al., 2001a), with the exception that the dissection buffer contained the following (in mm): 75 sucrose, 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 7 MgCl2, 0.5 CaCl2, and 1.3 ascorbic acid. We focused our study on the layer 4→2/3 pathway, because this is thought to be the initial site for receptive field plasticity (Trachtenberg et al., 2000).
Voltage-clamp recordings. Slices were allowed to recover for 20 min at 35°C in a submersion chamber containing oxygenated artificial CSF (ACSF) and then moved to room temperature for at least 40 min before use as described (Philpot et al., 2001a). For pharmacologically isolating NMDAR-mediated currents, we placed slices in a submersion chamber, maintained them at 30°C, and perfused them at 2 ml/min with oxygenated ACSF modified to contain the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, 4 MgCl2, 4 CaCl2, 0.001 glycine, 0.05 picrotoxin, and 0.02 CNQX or DNQX. CNQX/DNQX was omitted for recording AMPAR-mediated currents. Cells were visualized with a Nikon E600FN microscope (Tokyo, Japan) equipped with infrared differential interference contrast optics. Patch pipettes were pulled from thick-walled borosilicate glass. Open tip resistances were 3–8 MΩ when pipettes were filled with the internal solution containing the following (in mm): 102 cesium gluconate, 5 TEA-chloride, 3.7 NaCl, 20 HEPES, 0.3 sodium guanosine triphosphate, 4 magnesium adenosine triphosphate, 0.2 EGTA, 10 BAPTA, and 5N-(2,6-dimethylphenyl carboylmethyl) triethylammonium bromide (QX-314) chloride (Alomone Labs, Jerusalem, Israel), with pH adjusted to 7.2 and osmolarity adjusted to ∼300 mmol/kg by the addition of sucrose. Voltage-clamp recordings were performed in the whole-cell configuration with a patch-clamp amplifier (Multiclamp 700A, Molecular Devices, Sunnyvale, CA), and data were acquired and analyzed with pClamp 9.2 software (Molecular Devices). Pipette seal resistances were >1 GΩ, and pipette capacitive transients were minimized before breakthrough. Input and series resistances were determined throughout the experiment by measuring the response to small intermittent test pulses. The recorded series resistance averaged 21.8 ± 0.9 MΩ, and no series resistance compensation was applied. Input resistances recorded at +40 mV did not differ between deprived and control groups at P21–P27 (110.4 ± 6.0 MΩ) or at P74–P84 (92.3 ± 5.4 MΩ). EPSCs were evoked from a stimulating electrode (concentric bipolar; 100 μm tip separation) placed in layer 4, and stimulation was given for 200 μs every 15 s.
To describe the deactivation kinetics of NMDAR-mediated currents recorded at +40 mV, we averaged 30–60 evoked NMDAR EPSCs and described the current decays via the following formula: I(t) = Ifexp(–t/τf) + Isexp(–t/τs), where I is the current amplitude, t is time, If and Is are the peak amplitudes of the fast and slow components, respectively, and τf and τs are their respective time constants. A nonlinear regression in pClamp software was used to fit double exponentials to decay curves. The weighted time constant (τw) was used for quantification purposes and was calculated as τw = τf × (If/(If + Is)) + τs × (Is/(If + Is)).
To examine functional changes in the short-term depression of AMPAR-mediated currents recorded at –70 mV, we applied 11 pulses at 40 Hz every 6 s. The time constant of AMPAR EPSC depression was obtained by fitting the following single exponential formula: Inet(t) = Kexp(–t/τd) + PL, where Inet is the normalized net current amplitude, τd is the time constant of the synaptic depression, PL is the normalized steady-state EPSC amplitude, and K + PL = 1.
To measure the kinetics of (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d]cyclohepten-5,10-imine maleate (MK-801) blockade, we first measured isolated NMDAR EPSCs at +40 mV and adjusted the stimulation intensity to evoke an ∼100 pA response. MK-801 (40 mm) was added to the bath, and responses were evoked every 15 s until the NMDAR-mediated response was abolished. The time constant of MK-801 blockade (τblock) was calculated via the following formula: I(t) = I1exp(–t/τblock), where I is the current amplitude, I1 is the amplitude of the first pulse, and t is time.
Biochemical fractions. Each of the biochemical fractions was prepared by using visual or frontal cortices pooled from three to five brains with a procedure modified from Cho and colleagues (1992). Comparisons were made from fractions run in parallel to minimize variability among preparations. Samples were homogenized in HEPES-buffered sucrose (4 mm HEPES, 0.32 m sucrose, pH 7.4) by using a motor-driven Dounce homogenizer. Postnuclear supernatant (PNS) fractions were prepared by centrifuging the homogenates twice at 1000 × g for 10 min to eliminate nuclei. The PNS fractions were centrifuged at 10,000 × g for 20 min to yield crude synaptic pellets, which then were suspended in HEPES-buffered sucrose and centrifuged. The resulting pellets were lysed in a hypo-osmotic buffer (4 mm HEPES, pH 7.4) with the motor-driven Dounce homogenizer and mixed constantly for 30 min. The lysates were centrifuged at 25,000 × g for 20 min, and pellets were suspended in HEPES-buffered sucrose to obtain lysed synaptosomal membrane (LSM) fractions. The LSM fractions were subjected to density centrifugation (150,000 × g for 2 h) by using a gradient consisting of 0.8, 1.0, and 1.2 m sucrose in 4 mm HEPES, pH 7.4. Synaptic plasma membrane fractions were collected at the 1.0–1.2 m interface, diluted with 4 mm HEPES, and pelleted (150,000 × g for 30 min). These pellets were resuspended in 50 mm HEPES, pH 7.4, containing 0.5% Triton X-100, rotated for 15 min, and centrifuged at 32,000 × g for 20 min. The resulting pellets were resuspended in the 0.5% Triton X-100-containing buffer, rotated for 15 min, and centrifuged at 200,000 × g for 20 min to obtain postsynaptic density (PSD) fractions, which were suspended in 50 mm HEPES containing 0.2% SDS. Complete protease inhibitor mixture tablets (Roche, Mannheim, Germany), 10 μg/ml pepstatin, and phosphatase inhibitor mixtures 1 and 2 (Sigma, St. Louis, MO) were added to all buffers. The above procedures were performed on ice or in a cold room, and the fractions were stored at –80°C. Protein concentrations were measured with the use of Coomassie Plus reagent (Pierce, Rockford, IL).
Immunoblot analysis. PNS, LSM, and PSD fractions (10 μg) were resolved by 7.5% SDS-PAGE (Ready Gels, Bio-Rad, Hercules, CA) and transferred to nitrocellulose membranes. Both blotting and imaging with the Odyssey imaging system (LI-COR, Lincoln, NE) were performed by following the manufacturer's protocols. Primary antibodies were anti-NR2A rabbit antibody (1:500; sc-9056, Santa Cruz, Santa Cruz, CA), anti-NR2B goat antibody (1:20,000; sc-1469, Santa Cruz), anti-PSD-95 monoclonal antibody (1:500; MAB1596, Chemicon, Temecula, CA), and anti-β-tubulin monoclonal antibody (1:3000; MAB3408, Chemicon). The secondary antibodies included Alexa Fluor 680-labeled anti-goat IgG antibody (1:5000; Invitrogen, Eugene, OR), Alexa Fluor 680-labeled anti-mouse IgG antibody (1:5000; Invitrogen), and IRDye 800-labeled anti-rabbit IgG antibody (1:3000; Rockland Immunochemicals, Gilbertsville, PA).
Statistics. Data are expressed as the means ± SEM. ANOVA or Student's t tests were used to test for statistical significance, which was placed at p < 0.05.
Results
Visual deprivation lengthens the decay kinetics of synaptic NMDAR-mediated currents in developing, but not adult, mice
We first determined whether visual deprivation affects NMDARs in the visual cortex of juvenile mice in a manner similar to that observed in rats. Previous studies demonstrated that dark rearing or 5 d of visual deprivation in juvenile rats decreases the relative expression of NR2A- to NR2B-containing NMDARs, resulting in longer NMDAR-mediated currents (Carmignoto and Vicini, 1992; Flint et al., 1997; Philpot et al., 2001a). We found that a similar process occurs in mice. Pharmacologically isolated NMDAR-mediated currents evoked by stimulating layer 4 were measured in layer 2/3 pyramidal neurons in primary visual cortical slices. Dark rearing until P21–P27 significantly increased the duration of NMDAR-mediated currents as compared with controls (Fig. 1) (Deprived, τw = 130.7 ± 6.5 ms and n = 21 cells; Control, τw = 82.1 ± 7.6 ms and n = 16 cells; p < 0.00003). These data suggest that visual deprivation in juvenile mice modifies the composition and function of synaptic NMDARs, as has been observed in rats.
Figure 1.

Visual deprivation in adult mice fails to modify NMDAR EPSCs evoked by single pulses. Shown is a scatter plot of the weighted time constants (τw) of NMDAR-mediated EPSCs recorded from layer 2/3 pyramidal cells after layer 4 is stimulated in the visual cortex of visually deprived and control juvenile mice as well as deprived and control adult mice. Small circles represent individual data points, and larger circles represent the means ± SEM. NMDAR-mediated currents are significantly longer in the visual cortex of deprived juvenile mice as compared with controls. Visual deprivation in adult mice does not alter NMDAR-mediated current duration. Normalized traces are representative of pharmacologically isolated NMDAR EPSCs recorded at +40 mV, and an overlay of the traces (top) is included as a basis for comparisons. *p < 0.05.
To determine whether experience-dependent modifications in synaptic NMDARs could be elicited outside a critical period of development, we examined the consequences of 10 d of lateonset visual deprivation in the visual cortex of adult mice. Unlike in juvenile rodents, we failed to observe a change in NMDAR EPSC decay kinetics between deprived and control adult mice (Fig. 1) (Deprived, τw = 90.5 ± 9.3 ms and n = 13 cells; Control, τw = 87.3 ± 9.8 ms and n = 16 cells; p = 0.82). These data indicate that, in adult mice, visual deprivation does not change the function of NMDARs driven by a single synaptic activation of the layer 4→2/3 pathway.
Visual deprivation enhances the temporal summation of NMDAR EPSCs in adult mice
We previously have demonstrated in juvenile rodents that the temporal summation of NMDAR-mediated currents is correlated tightly with the duration of individual EPSCs: the longer the NMDAR currents, the greater the magnitude of temporal summation (Philpot et al., 2001a). Because of the similar duration of NMDAR EPSCs in visual cortical pyramidal cells from deprived and control mice, we expected that the temporal summation of NMDAR-mediated currents would be nearly identical between the groups. To test this possibility, we delivered bursts of 40 Hz stimulation (11 pulses) to layer 4 and measured the response in layer 2/3 pyramidal cells in deprived and control cortices (Deprived, n = 15 cells; Control, n = 22 cells). We adjusted stimulus intensity to obtain ∼100 pA response on the first pulse (Deprived, 113.5 ± 13.1 pA; Control, 103.4 ± 7.6 pA; p = 0.48). Surprisingly, we observed that visual deprivation greatly enhanced the temporal summation of NMDAR-mediated currents in the adult visual cortex (Fig. 2A) (amplitude of the 11th pulse/1st pulse: Deprived, 1.33 ± 0.15; Control, 0.96 ± 0.005; p < 0.02). The deprivation-induced enhancement of temporal summation was also significant when quantified by averaging the charge transfer (integral) of the normalized currents [Deprived, 354.7 ± 32.1 arbitrary units (a.u.); Control, 259.4 ± 14.0 a.u.; p < 0.005].
Figure 2.
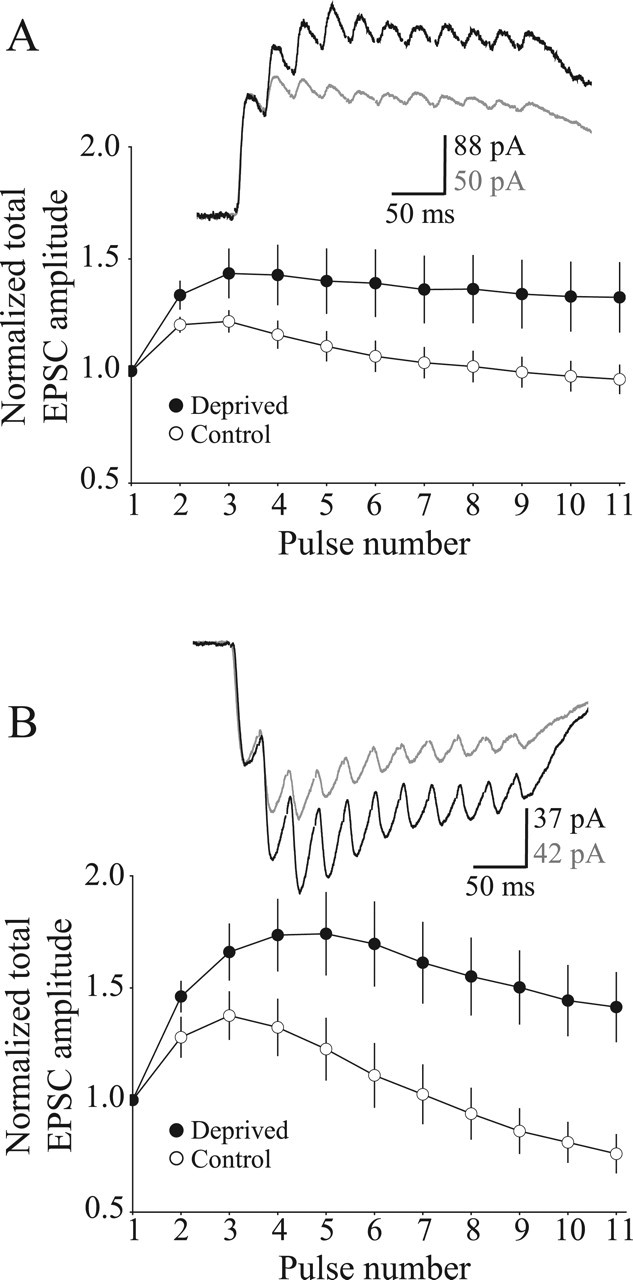
Visual deprivation in adult mice alters the temporal summation of NMDAR EPSCs evoked by burst stimulation in the visual cortex. A, Plot of the normalized and averaged amplitudes of NMDAR EPSCs evoked at 40 Hz in the adult visual cortex at a holding potential of +40 mV. Representative traces of pharmacologically isolated NMDAR EPSCs in response to 40 Hz stimulus trains are shown (dark trace represents response from pyramidal neuron in deprived mice; light trace represents response from pyramidal neuron in control mice). Stimulus artifacts were blanked for clarity. B, Same as in A, but recordings were made at a holding potential of –70 mV in ACSF containing nominal magnesium (0.1 mm MgCl2). Error bars indicate the means ± SEM.
To determine whether the experience-dependent differences in temporal summation arose from recording at a depolarized voltage (+40 mV), we repeated the experiment in nominal magnesium (0.1 mm) while clamping the cells at –70 mV. The deprivation-induced enhancement of temporal summation also was observed when postsynaptic cells were clamped at the hyperpolarized membrane potential (Fig. 2B) (amplitude of the 11th pulse/1st pulse: Deprived, 1.41 ± 0.15 and n = 16 cells; Control, 0.76 ± 0.08 and n = 12 cells; p < 0.003; normalized charge transfer: Deprived, 397.4 ± 36.3 a.u.; Control, 266.5 ± 27.3 a.u.; p < 0.02). This observation suggests that the effects on temporal summation are unlikely to be attributable to an experience-dependent change in an intrinsic membrane current that has voltage-sensitive properties.
Thus although visual deprivation did not alter synaptic NMDAR-mediated currents evoked by a single stimulation, visual deprivation nevertheless enhanced the temporal summation of NMDAR-mediated currents. The enhanced temporal summation could be explained by a change in (1) presynaptic neurotransmitter release or (2) a population of perisynaptic/extrasynaptic NMDARs that is activated with bursts of stimulation.
Visual deprivation increases the release probability of glutamate in adult mice
The temporal summation of NMDAR-mediated currents is determined by both the postsynaptic summation of currents and the properties of presynaptic neurotransmitter release (e.g., the rate of synaptic depression or facilitation) (Zucker and Regehr, 2002). We initially hypothesized that the enhanced temporal summation of NMDAR-mediated currents could be attributable to a reduction in the rate of synaptic depression. Because AMPAR-mediated currents are much faster than NMDAR-mediated currents, there is little, if any, temporal summation of AMPAR EPSCs at frequencies ≤40 Hz. Thus short-term plasticity of AMPAR-mediated currents is a good measure of changes in presynaptic neurotransmitter release. We examined the short-term plasticity of AMPAR-mediated EPSCs recorded at –70 mV in layer 2/3 pyramidal cells by giving 11 pulses of 40 Hz stimulation to layer 4. Contrary to our initial hypothesis, our data indicated that the rate of synaptic depression was increased significantly in the visual cortex of the deprived mice (Fig. 3) (τd: Deprived, 64.9 ± 10.3 ms and n = 18 cells; Control, 129.5 ± 18.2 ms and n = 23 cells; p < 0.007). The normalized steady-state AMPAR EPSC amplitudes were unchanged by visual experience (Deprived, 0.187 ± 0033; Control, 0.130 ± 0.026; p = 0.17). These data suggest that visual deprivation increases the initial probability of neurotransmitter release and limits the relative amount of neurotransmitter available for subsequent release. Thus the enhanced temporal summation of NMDAR-mediated currents in the cortex of deprived mice cannot be explained by a reduction in the rate of short-term synaptic depression.
Figure 3.

Visual deprivation increases the rate of synaptic depression in adult mice. Shown is a plot of the AMPAR EPSC amplitudes in response to a brief 40 Hz stimulation train. Responses were normalized to the first pulse. Traces are representative AMPAR EPSCs recorded at –70 mV in cells from deprived (dark trace) and control (light trace) mice. Stimulus artifacts were blanked for clarity. Error bars indicate the means ± SEM.
Because of the novel and unexpected observation that visual deprivation increases the rate of synaptic depression in the visual cortex of adult mice, we wanted to use an independent assay to verify that deprivation increases the probability of neurotransmitter release. We took advantage of the pharmacological properties of MK-801, an irreversible open-channel NMDAR blocker, to examine neurotransmitter release in control and deprived adult mice. The rate of block of NMDAR-mediated currents by MK-801 is an indicator of the probability of neurotransmitter release: the higher the probability of release, the faster the rate of block by MK-801 (Hessler et al., 1993). Consistent with the increased rate of synaptic depression of AMPAR-mediated currents, we observed that visual deprivation significantly accelerated the rate at which MK-801 blocks pharmacologically isolated NMDAR EPSCs (Fig. 4) (τblock: Deprived, 9.30 ± 0.63 ms and n = 4 cells; Control, 20.00 ± 3.16 ms and n = 7 cells; p < 0.04). These results confirm that visual deprivation increases the probability of release in layer 4→2/3 synapses. Although the enhanced temporal summation of NMDAR EPSCs in adult mice cannot be explained by an increase in the rate of synaptic depression, it is possible that the increased probability of release could facilitate the spillover of glutamate to perisynaptic/extrasynaptic sites (see Discussion).
Figure 4.

Visual deprivation increases the rate of neurotransmitter release in adult mice. Shown is a plot of the normalized amplitude of NMDAR EPSCs in response to repetitive stimulation in the presence of MK-801. Note that NMDAR EPSC blockade by MK-801 occurs faster in pyramidal cells from deprived adult mice than from controls. Error bars indicate the means ± SEM.
Visual deprivation differentially reduces the NR2A/B ratio in biochemical fractions from the visual cortex of juvenile and adult mice
Our data suggest that the change in the rate of synaptic depression could not account for the enhanced temporal summation of NMDAR-mediated currents in pyramidal cells of deprived adult mice. We therefore decided to evaluate possible changes in postsynaptic NMDAR subunit composition. We previously have used the synaptoneurosome preparation to show that there is a correlation between the functional properties of NMDARs observed electrophysiologically and the subunit expression of NMDARs observed biochemically (Quinlan et al., 1999a; Philpot et al., 2001b). The synaptoneurosome preparation, however, cannot distinguish protein expression in the PSD from expression in other compartments near the synapse. Moreover, it was difficult to detect small changes in NMDAR composition with our previously used chemiluminescent immunoblot techniques.
To overcome the limitations of our previous techniques, we produced enriched biochemical fractions that allowed us to differentiate proteins in the PSD from other compartments. We then analyzed these fractions by using a novel immunoblotting technique that used fluorescent secondary antibodies to NR2A and NR2B subunits (see Materials and Methods) (Fig. 5). This method allowed us to achieve a highly quantitative measurement of the NR2A/B ratio because of the elimination of several sources of error. (1) The Odyssey infrared system operates within a very large linear range for quantification; thus errors from working within the small linear range with the use of traditional immunoblots were eliminated. (2) The dual fluorescent labeling of NR2A and NR2B allowed us to compare band intensities within the same gel lane, eliminating errors introduced by variations in sample loading onto SDS-PAGE gels. (3) Membrane stripping was unnecessary, so no error was introduced by incomplete stripping or overstripping. (4) NR2A and NR2B migrate through the SDS-PAGE gel to almost the same distance because of their similar molecular weights, eliminating errors introduced by differential transfer of the proteins from the gel to the nitrocellulose membrane. Although the technique still has the limitation that it is difficult to determine whether differences in the NR2A/B ratio are attributable to changes in NR2A, NR2B, or both, the advantages of the technique allowed us to detect modest differences in the ratio of NR2A/B with high precision and little variability.
Figure 5.
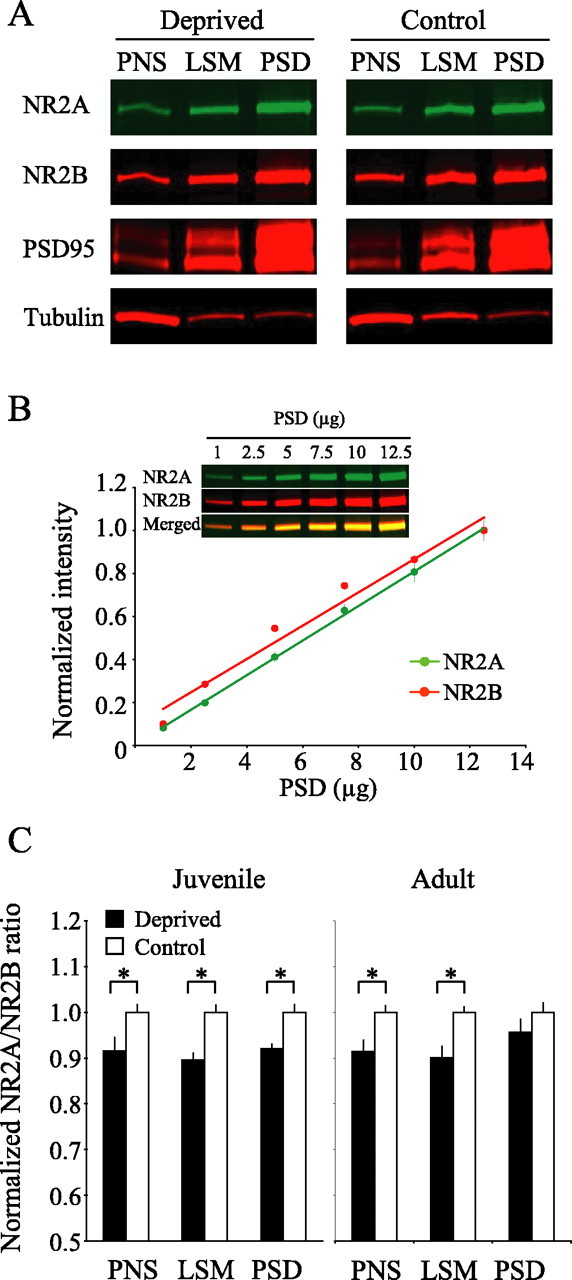
Visual deprivation in juvenile mice alters the NR2A/B ratio in the PSD, but only in PNS and LSM fractions in adult mice. A, Biochemical fractionation progressively enriches NR2A, NR2B, and PSD-95 and eliminates a nonsynaptic protein, β-tubulin, invisual corticals amples from deprived and control adult mice. Samples of 10μg were loaded into each gel lane. B, Quantification of NR2A and NR2B band intensities, which were normalized to the value at 12.5μg. The inset is a representative NR2A/B immunoblot of a PSD fraction. Samples of 1–12μg were loaded into each gel lane; results from three blots were averaged. C, NR2A/B ratios were measured in PNS, LSM, and PSD fractions of visual cortices of deprived and control from both juvenile and adult mice. The values (means±SEM) are normalized to average control values. *p < 0.05.
We first examined NR2A/B expression in the visual cortex from deprived and control juvenile mice (n of each group = 6 pools of five mice each). To evaluate changes in NR2A/B expression, we examined three biochemical fractions: (1) the PNS fraction, containing both cytoplasmic and cell membrane contents, (2) the LSM fractions, which contained both synaptic and extrasynaptic components of the plasma membrane, and (3) the highly enriched PSD. Consistent with previous findings that used synaptoneurosome preparations in rats (Quinlan et al., 1999b), we found that the NR2A/B ratios were significantly lower in the PNS, LSM, and PSD visual cortical fractions of deprived juvenile mice as compared with controls (Fig. 5C) (PNS, p < 0.03; LSM, p < 0.0006; PSD, p < 0.02). To determine the effects of visual deprivation in adults, we compared the NR2A/B ratios in control and deprived adult mice (n of each group = 12 pools of three to five mice each). In contrast to what we observed in the visual cortex of deprived juvenile mice, late-onset visual deprivation in adults failed to modify the composition of NMDARs within the highly enriched PSD (Fig. 5A,C) (p = 0.22). However, late-onset visual deprivation in adults significantly lowered the NR2A/B ratio in the PNS and LSM visual cortical fractions as compared with the controls (Fig. 5C) (PNS, p < 0.006; LSM, p < 0.002). These data indicate that visual deprivation in adults might alter the composition of NMDARs located at perisynaptic/extrasynaptic sites, but not synaptic sites. In both juvenile and adult mice the change in NMDAR subunit composition in control and deprived mice did not appear to be the result of a general stress response, because we failed to observe a change in the NR2A/B ratio in the PNS, LSM, and PSD preparations taken from frontal cortices of deprived mice as compared with controls (p values in all fractions from both juveniles and adults > 0.1; n = 6 pools of tissues for each of the six groups). These data suggest that late-onset visual deprivation alters the composition of perisynaptic/extrasynaptic NMDARs, but not the complement of postsynaptic NMDARs.
Visual deprivation does not alter the temporal summation of NMDAR-mediated currents evoked by minimal stimulation
Because high-frequency stimulation additively can facilitate diffusion of glutamate at synapses and induce activation of perisynaptic/extrasynaptic NMDARs that are not activated by a single pulse (Scimemi et al., 2004), we reasoned that the enhanced temporal summation of NMDAR-mediated currents in deprived mice could be a consequence of glutamate spillover onto a modified population of perisynaptic/extrasynaptic NMDARs. Previous studies demonstrate that glutamate spillover increases with EPSC size (Scimemi et al., 2004); that is, spillover is more likely to occur with an increase in the number of simultaneously activated synapses.
If synaptic spillover contributes to the deprivation-induced enhancement of NMDAR temporal summation, then we reasoned that we would be less likely to observe the effect when activating a lower density of synapses. To test this possibility, we examined the temporal summation of NMDAR EPSCs elicited by minimal stimulation. The assumption in these studies is that minimal stimulation activates one or a small number of afferents. In this experiment the stimulus intensity was adjusted to elicit a response to the first pulse ∼50% of the time. We then delivered 11 pulses at 40 Hz and analyzed only traces in which there was a response to the first pulse (first peak amplitude: Deprived, 14.1 ± 1.0 and n = 14 cells; Control, 15.6 ± 1.6 and n = 16 cells; p = 0.46). With this minimal stimulation protocol the temporal summations of NMDA EPSCs in deprived and control mice were almost identical as measured by the amplitude of the 11th pulse (Fig. 6) (amplitude of 11th pulse/1st pulse: Deprived, 1.15 ± 0.14; Control, 1.12 ± 0.12; p = 0.90) or by the normalized charge transfer (Deprived, 283.1 ± 25.7 a.u.; Control, 288.9 ± 30.3 a.u.; p = 0.89). These data indicate that a critical number of synapses must be activated, reflected by EPSC amplitude, to observe the deprivation-induced enhanced temporal summation. The data are consistent with the idea that the coordinated release of glutamate above a certain threshold of activated synapses can produce glutamate spillover sufficient to reach an extrasynaptic population of NMDARs that is modified by visual deprivation.
Figure 6.

Minimal stimulation fails to reveal deprivation-induced differences in the temporal summation of NMDAR-mediated currents. Shown is a plot of the temporal summation of NMDAR EPSCs in deprived and control mice evoked by minimal stimulation at 40 Hz. Error bars indicate the means ± SEM.
The NR2B-selective antagonist ifenprodil blocks the enhanced temporal summation of NMDAR EPSCs in deprived visual cortex of adult mice
The above data indicated that neither a change in the rate of synaptic depression nor a change in a voltage-sensitive membrane property could account for the enhanced temporal summation of NMDAR-mediated currents in the cortex of deprived mice. However, the data also indicated that (1) visual deprivation in adult mice alters the complement of perisynaptic/extrasynaptic NMDARs without significantly changing the synaptic NMDARs and (2) a critical threshold of synaptic activation was required to observe the deprivation-induced enhancement of temporal summation. These observations are consistent with visual deprivation altering a population of perisynaptic/extrasynaptic NMDARs that is activated by glutamate spillover occurring with coordinated bursts of stimulation. Previous studies suggest that NR2B-containing NMDARs can detect glutamate spillover (Scimemi et al., 2004), likely because of their high affinity for glutamate (Priestley et al., 1995). We used the NR2B-specific antagonist ifenprodil to determine whether the deprivation-induced enhancement of NMDAR temporal summation was mediated via activation of NR2B-containing receptors. Ifenprodil blocked the enhanced temporal summation of NMDAR EPSCs in deprived mice (Fig. 7A) (amplitude of 11th pulse/1st pulse: Deprived, 0.84 ± 0.07 and n = 6 cells; Control, 0.85 ± 0.06 and n = 8 cells; p = 0.98). Although ifenprodil dramatically reduced the temporal summation of NMDAR-mediated currents in the visual cortex of deprived mice, the drug had no noticeable consequence in control mice (Fig. 7B). These observations support our hypothesis that late-onset visual deprivation can increase the relative complement of NR2B-containing NMDARs at perisynaptic/extrasynaptic sites, but not at synaptic sites. An idea consistent with our data is that these perisynaptic/extrasynaptic NMDARs could be activated by glutamate spillover triggered by consecutive pulses, thus helping to explain why we observed a deprivation-induced enhancement of NMDAR-mediated temporal summation as quickly as the second pulse in a train of stimulation (Fig. 2).
Figure 7.
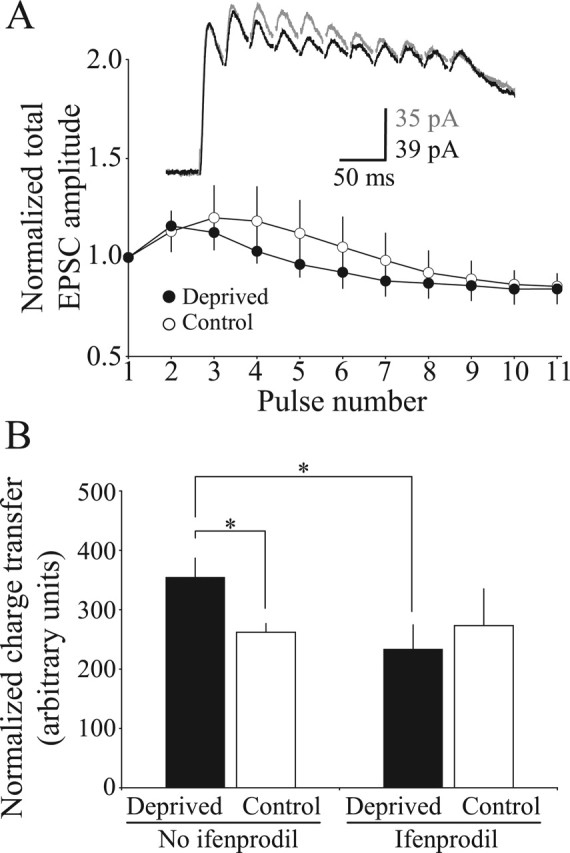
The enhanced temporal summation of NMDAR EPSCs in deprived adult cortex can be blocked by acute administration of ifenprodil, an NR2B-specific NMDAR antagonist. A, Temporal summation of NMDAR EPSCs in deprived and control mice in the presence of ifenprodil. Error bars indicate the means ±SEM. B, The average charge transfer taken from the normalized responses of the 11 pulses evoked at 40 Hz stimulation in the presence or absence of ifenprodil. ANOVA with post hoc analyses; *p < 0.05.
Discussion
We demonstrate that a brief period (10 d) of visual deprivation enhances NMDAR-mediated transmission in the layer 4→2/3 visual cortical synapse by presynaptically increasing the probability of neurotransmitter release and by increasing the relative expression of NR2B-containing NMDARs at perisynaptic/extrasynaptic sites. Both the presynaptic and postsynaptic changes are unique to the mature cortex, because visual deprivation in juvenile rodents does not alter the probability of release but does decrease the expression of NR2A-containing NMDARs at the PSD (Quinlan et al., 1999a,b; Philpot et al., 2001a; present study). These results provide evidence that the history of sensory experience modifies synapses in the mature visual cortex outside of the critical period of receptive field plasticity and that these modifications differ between juvenile and adult animals.
Accumulating evidence indicates that the adult visual cortex is more plastic than previously thought, and our data show that one synaptic basis for adult plasticity is a change in the short-term dynamics of excitatory synaptic responses. The observed deprivation-induced increase in the rate of neurotransmitter release is likely a compensatory mechanism to maintain synaptic drive in the absence of visually evoked activity. This increase in release is reminiscent of what has been observed in culture systems after manipulations that reduce presynaptic activity (Chavis and Westbrook, 2001) or postsynaptic excitability (Murthy et al., 2001). The enhanced neurotransmitter release at vertical intracortical connections after global sensory deprivation appears to be unique to adult sensory cortices, because global reductions in sensory activity in the developing somatosensory or visual cortices fail to modify neurotransmitter release (Finnerty and Connors, 2000; Philpot et al., 2001a). The deprivation-induced increase in release probability in the adult cortex enhances the likelihood that perisynaptic/extrasynaptic NMDARs may be activated by glutamate spillover (Kullmann et al., 1996), and future studies will be needed to investigate whether this spillover could be augmented by a decrease in glutamate reuptake (Shen and Linden, 2005). Nonetheless, our data are consistent with the idea that visual deprivation increases a perisynaptic/extrasynaptic population of ifenprodil-sensitive NMDARs that can be activated by glutamate spillover during bursts of high-frequency stimulation.
In addition to age-dependent differences in the presynaptic consequences of visual deprivation, our data demonstrate that there are also unique postsynaptic consequences to visual deprivation. Deprivation in juvenile animals decreases the NR2A/NR2B ratio at synaptic sites, but late-onset visual deprivation in adults modifies only perisynaptic/extrasynaptic NMDARs. There is a precedent in the literature that NR2A-containing NMDARs are trafficked to synaptic sites, whereas NR2B-containing NMDARs are trafficked preferentially to perisynaptic/extrasynaptic sites. For example, NMDARs appear to be eliminated from the central portion of the synapse in the superior colliculus of mice lacking the NR2A subunit (Townsend et al., 2003). One possibility is that NR2A is trafficked selectively to the synapse, but NR2B-containing NMDARs might be prevented from remaining in the synapse once “slot” proteins for anchoring NR2A-containing NMDARs, such as PSD-95, have been delivered to the synapse (Yoshii et al., 2003). In support of this, manipulations of visual experience in developing mice are known to regulate the expression of synaptic NR2A in the visual cortex bidirectionally (Quinlan et al., 1999b). If, on the other hand, visual experience preferentially modifies NR2B but not NR2A levels in the adult cortex, then changes in NR2B-containing NMDARs might be detected only at perisynaptic/extrasynaptic sites because NR2A-containing NMDARs are entrenched in the central portion of the synapse. Our data provide evidence that this indeed may be the case, because we observe that the NR2B-selective antagonist ifenprodil eliminates the enhanced temporal summation of NMDAR-mediated currents in the visual cortex of deprived mice. In addition, quantitative immunoblot analysis of NR2A and NR2B in synaptoneurosomes suggests that late-onset visual deprivation increases NR2B levels rather than decreasing NR2A (E. Quinlan, unpublished observations).
We suggest that an increase in perisynaptic/extrasynaptic NR2B-containing NMDARs postsynaptically could account for the deprivation-induced increase in the temporal summation of NMDAR currents. This conclusion is supported by three observations. (1) The deprivation-induced enhancement of temporal summation is observed only in conditions that favor glutamate spillover; the effect is not observed with minimal stimulation but is observed with stronger stimulation intensities. (2) Biochemical data indicate that there is an increase in the relative proportion of NR2B-containing NMDARs at perisynaptic/extrasynaptic, but not synaptic, sites. (3) The NR2B-containing NMDAR antagonist ifenprodil blocks the deprivation-induced enhancement of NMDAR EPSC temporal summation.
A possible complication to our interpretation of the data is that presynaptic NR2B-containing NMDARs are known to exist (Aoki et al., 1994) and to enhance neurotransmitter release in the visual cortex (Sjostrom et al., 2003). The deprivation-induced elevation in NR2B proteins within the lysed synaptic membrane fraction (Fig. 5) could be explained by an increase in presynaptic NMDARs, contributing to the observed increase in neurotransmitter release after deprivation. However, an increase in presynaptic NR2B-containing NMDARs is unlikely to account for the deprivation-induced enhancement of NMDAR-mediated temporal summation. If an increase in relative NR2B levels were restricted to presynaptic sites, we should have observed a similar trend in the short-term dynamics of AMPAR- and NMDAR-mediated currents after deprivation (Figs. 2, 3). Specifically, an increase in release by presynaptic NR2B-containing NMDARs with deprivation would be expected to decrease the temporal summation of NMDAR-mediated currents in deprived mice, which was not what we observed. Hence the most parsimonious explanation for our data is that visual deprivation in adult mice increases the relative population of NR2B-containing NMDARs at perisynaptic/extrasynaptic sites, although our data do not rule out a possibility that the increased rate in neurotransmitter release is attributable to an increase in presynaptic NR2B-containing NMDARs.
Although future research is needed to address the physiological importance of the experience-dependent changes in glutamatergic synaptic transmission in the adult visual cortex, some clues may be provided by the very different consequences of monocular deprivation on ocular dominance in juvenile and adult mice (Sawtell et al., 2003; Frenkel and Bear, 2004). Because visual cortex responses in rodents are driven mainly by the contralateral eye, monocular deprivation essentially eliminates visual activity in the contralateral cortex except for a minor input driven by the ipsilateral eye. In juvenile mice monocular deprivation leads to a rapid depression of the contralateral deprived eye inputs, followed by a deprivation-enabled strengthening of the weak ipsilateral inputs from the nondeprived eye. The delayed strengthening of the previously weak inputs might be a consequence of lowering the threshold for synaptic potentiation by increasing the relative expression of NR2B-containing NMDARs in deprived cortex (Quinlan et al., 1999a,b; Philpot et al., 2001a; present study). In contrast to what has been observed in juvenile mice, the synapses in the mature cortex are normally stable and relatively resistant to modifications. One possibility is that the limited plasticity in the adult visual cortex is a consequence of low expression levels of NMDARs, especially the NR2B-containing NMDARs. Although monocular deprivation in the mature cortex fails to depress the inputs driven by the deprived eye, this manipulation can cause a delayed strengthening of the weak ipsilateral eye inputs. Perhaps the increase in the relative expression of NR2B subtypes at perisynaptic/extrasynaptic sites, coupled with an increase in neurotransmitter release that can ensure their activation, provides a synaptic milieu that is permissive for the strengthening of normally weak responses.
The dependence of long-term potentiation and depression on the subunit composition of the NMDAR is debated heavily (Liu et al., 2004; Massey et al., 2004), but a number of recent studies indicate that NR2A- and NR2B-containing NMDARs both can contribute to the induction of long-term depression and potentiation (Hendricson et al., 2002; Berberich et al., 2005; Toyoda et al., 2005; Weitlauf et al., 2005; Zhao et al., 2005). An intriguing possibility is that, under certain conditions, an increase in the relative expression of NR2B-containing NMDARs in the adult visual cortex can reinstate some aspects of synaptic plasticity that normally are lost during development. In support of this hypothesis, 10 d of visual deprivation in adult rats increases NR2B levels and simultaneously reinstates rapid ocular dominance shifts after brief monocular deprivation (E. Quinlan, unpublished observations).
A number of clinical studies suggest that some recovery from amblyopia is possible well past the classically defined critical period in humans, which generally is considered to last up to ∼9 years of age (Birnbaum et al., 1977; Simmers and Gray, 1999; Fronius et al., 2004). These studies indicate that long periods of depriving the dominant eye in amblyopes are needed for recovery of vision in the weak eye. We speculate that patching of the nonamblyopic eye initially mimics visual deprivation, because vision is blurred through the amblyopic eye, and these long periods of patching might be needed to drive an increase in NR2B-containing NMDARs. Our findings could account for the molecular mechanisms behind the clinical observations that visual deprivation in mature humans can facilitate the strengthening of weakened connections from the amblyopic eye.
In summary, our data provide direct evidence that sensory experience differentially modifies synaptic transmission in the cortex of juvenile and mature animals. These differences may provide a synaptic basis to explain why sensory deprivation has unique manifestations across development (supplemental figure, available at www.jneurosci.org as supplemental material).
Footnotes
This work was supported by a grant from the Whitehall Foundation (B.D.P.) and a National Science Foundation graduate student fellowship (R.C.). We thank Lindsey Wilfley, Sushmita Jha, Maile Henson, Roscoe Nicholson, Jacqueline de Marchena, Dr. Manzoor Bhat, and Dr. Anthony Lamantia for helpful discussions and critical readings of this manuscript. We also thank Dr. Michael Ehlers and Dr. Yuanyue Mu for useful discussions and technical advice on the biochemical preparations. We extend special thanks to Dr. Alev Erisir for lending her technical expertise.
Correspondence should be addressed to Benjamin D. Philpot, University of North Carolina, Campus Box 7545, 105 Mason Farm Road, Chapel Hill, NC 27599-7545. E-mail: bphilpot@med.unc.edu.
DOI:10.1523/JNEUROSCI.4362-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511684-09$15.00/0
K.Y. and R.C. contributed equally to this work.
References
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM (1994) Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci 14: 5202–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G (2005) Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci 25: 6907–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum MH, Koslowe K, Sanet R (1977) Success in amblyopia therapy as a function of age: a literature survey. Am J Optom Physiol Opt 54: 269–275. [PubMed] [Google Scholar]
- Buonomano DV, Merzenich MM (1998) Cortical plasticity: from synapses to maps. Annu Rev Neurosci 21: 149–186. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S (1992) Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science 258: 1007–1011. [DOI] [PubMed] [Google Scholar]
- Chavis P, Westbrook G (2001) Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature 411: 317–321. [DOI] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB (1992) The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9: 929–942. [DOI] [PubMed] [Google Scholar]
- Finnerty GT, Connors BW (2000) Sensory deprivation without competition yields modest alterations of short-term synaptic dynamics. Proc Natl Acad Sci USA 97: 12864–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H (1997) NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17: 2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel MY, Bear MF (2004) How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44: 917–923. [DOI] [PubMed] [Google Scholar]
- Fronius M, Cirina L, Cordey A, Ohrloff C (2004) Visual improvement during psychophysical training in an adult amblyopic eye following visual loss in the contralateral eye. Graefes Arch Clin Exp Ophthalmol 243: 278–280. [DOI] [PubMed] [Google Scholar]
- Gilbert CD (1998) Adult cortical dynamics. Physiol Rev 78: 467–485. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Cioffi D, Silva AJ, Stryker MP (1996) Deficient plasticity in the primary visual cortex of α-calcium/calmodulin-dependent protein kinase II mutant mice. Neuron 17: 491–499. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA (2002) Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther 301: 938–944. [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366: 569–572. [DOI] [PubMed] [Google Scholar]
- Hestrin S (1992) Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature 357: 686–689. [DOI] [PubMed] [Google Scholar]
- Kaas JH (1991) Plasticity of sensory and motor maps in adult mammals. Annu Rev Neurosci 14: 137–167. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133–1138. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF (1993) Common forms of synaptic plasticity in the hippocampus and neocortex in vitro Science 260: 1518–1521. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Erdemli G, Asztely F (1996) LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron 17: 461–474. [DOI] [PubMed] [Google Scholar]
- Lickey ME, Pham TA, Gordon B (2004) Swept contrast visual evoked potentials and their plasticity following monocular deprivation in mice. Vision Res 44: 3381–3387. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304: 1021–1024. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5–21. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI (2004) Differential roles of NR2A- and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci 24: 7821–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML (1994) N-methyl-d-aspartic acid receptor structure and function. Physiol Rev 74: 723–760. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH (1992) Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256: 1217–1221. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32: 673–682. [DOI] [PubMed] [Google Scholar]
- Nase G, Weishaupt J, Stern P, Singer W, Monyer H (1999) Genetic and epigenetic regulation of NMDA receptor expression in the rat visual cortex. Eur J Neurosci 11: 4320–4326. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD (2004) Learning from NMDA receptor trafficking: clues to the development and maturation of glutamatergic synapses. Neurosignals 13: 175–189. [DOI] [PubMed] [Google Scholar]
- Pham TA, Graham SJ, Suzuki S, Barco A, Kandel ER, Gordon B, Lickey ME (2004) A semi-persistent adult ocular dominance plasticity in visual cortex is stabilized by activated CREB. Learn Mem 11: 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF (2001a) Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29: 157–169. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Weisberg MP, Ramos MS, Sawtell NB, Tang YP, Tsien JZ, Bear MF (2001b) Effect of transgenic overexpression of NR2B on NMDA receptor function and synaptic plasticity in visual cortex. Neuropharmacology 41: 762–770. [DOI] [PubMed] [Google Scholar]
- Priestley T, Laughton P, Myers J, Le Bourdelles B, Kerby J, Whiting PJ (1995) Pharmacological properties of recombinant human N-methyl-d-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cells. Mol Pharmacol 48: 841–848. [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999a) Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo Nat Neurosci 2: 352–357. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF (1999b) Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci USA 96: 12876–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EB, Ramoa AS (1999) Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol 81: 2587–2591. [DOI] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF (2003) NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 38: 977–985. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA (2004) NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci 24: 4767–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Linden DJ (2005) Long-term potentiation of neuronal glutamate transporters. Neuron 46: 715–722. [DOI] [PubMed] [Google Scholar]
- Simmers AJ, Gray LS (1999) Improvement of visual function in an adult amblyope. Optom Vis Sci 76: 82–87. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654. [DOI] [PubMed] [Google Scholar]
- Tagawa Y, Kanold PO, Majdan M, Shatz CJ (2005) Multiple periods of functional ocular dominance plasticity in mouse visual cortex. Nat Neurosci 8: 380–388. [DOI] [PubMed] [Google Scholar]
- Townsend M, Yoshii A, Mishina M, Constantine-Paton M (2003) Developmental loss of miniature N-methyl-d-aspartate receptor currents in NR2A knockout mice. Proc Natl Acad Sci USA 100: 1340–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Zhao MG, Zhuo M (2005) Roles of NMDA receptor NR2A and NR2B subtypes for long-term depression in the anterior cingulate cortex. Eur J Neurosci 22: 485–494. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Trepel C, Stryker MP (2000) Rapid extragranular plasticity in the absence of thalamocortical plasticity in the developing primary visual cortex. Science 287: 2029–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR (1998) Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol 79: 555–566. [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG (2005) Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci 25: 8386–8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH (1963) Single cell responses in striate cortex of kittens deprived of vision in one eye. J Neurophysiol 26: 1003–1017. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Sheng MH, Constantine-Paton M (2003) Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci USA 100: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M (2005) Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron 47: 859–872. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405. [DOI] [PubMed] [Google Scholar]