

Abstract
In relation to expectation and delivery of reward, pyramidal neurons of the prefrontal cortex either switch from a single spiking mode to transient phasic bursting, or gradually increase their sustained tonic activity. Here, we examined how switching between firing modes affects information processing at the corticoaccumbens synapse. We report that increasing presynaptic firing frequency in a tonic manner either depresses or facilitates synaptic transmission, depending on initial probability of release. In contrast, repeated bursts of stimulation of cortical afferents trigger a new form of short-term potentiation of synaptic transmission (RB-STP) in the nucleus accumbens (NAc). RB-STP involves the regulation of axonal excitability mediated by 4-AP-sensitive potassium channels in afferent cortical neurons. Thus, in a tonic mode, information flow is tightly controlled by regulatory mechanisms at the level of presynaptic terminals, whereas switching to a bursting mode reliably enhances efficacy of information processing for all cortical afferents to NAc neurons.
Keywords: nucleus accumbens, short-term synaptic plasticity, presynaptic, glutamate, K+ channels, axonal excitability
Introduction
The nucleus accumbens (NAc) has been proposed to serve as an interface between the limbic and the motor systems and is involved in motivation, attention, and reward (Mogenson et al., 1980; Kalivas and Nakamura, 1999). The output pathway of the NAc consists of GABAergic medium spiny neurons (MSNs) that depend on glutamatergic excitatory afferents to generate an action potential (Pennartz et al., 1994). The NAc receives a dense glutamatergic innervation from the prefrontal cortex (PFC) and from various limbic structures, including the hippocampal formation (Groenewegen et al., 1991). Cortical projection onto the NAc principally arises from intrinsic bursting pyramidal neurons of layers V–VI in the prelimbic and infralimbic cortical areas (Yang et al., 1996). These neurons, depending on their membrane potential, display either a tonic action potential discharge or a bursting pattern of activity (Yang et al., 1996). In vivo, subpopulations of PFC neurons respond in different manners during expectation and delivery of reward (Schultz et al., 2000; Cooper, 2002); some neurons switch from a single spiking mode to transient phasic bursting, whereas other neurons gradually increase their sustained tonic activity.
The switch in the pattern of spike discharge may lead to different forms of plastic changes in synaptic transmission in target NAc neurons through several forms of short-term synaptic plasticity (Zucker and Regehr, 2002). Short-term changes in synaptic strength induced by presynaptic burst activity are commonly thought to be mediated by presynaptic mechanisms such as Ca2+ control of release probability or presynaptic vesicle replenishment (Stevens and Wesseling, 1998; Wang and Kaczmarek, 1998; Fortune and Rose, 2001; Zucker and Regehr, 2002). It has recently emerged that action potential firing driving synaptic transmission in the CNS may be controlled by regulation of K+ channels in the axon and synaptic terminal (Debanne et al., 1997; Dodson et al., 2003; Ishikawa et al., 2003; Debanne, 2004; Meeks and Mennerick, 2004), but it is not clear whether this regulation can be involved in short-term plasticity induced by physiologically relevant stimuli.
In this study, we examined whether and how switching from a tonic single-spiking mode to a phasic-bursting mode may influence synaptic transmission in NAc neurons, with physiologically relevant firing patterns of the corticoaccumbens pathway. Our data demonstrate that presynaptic-firing mode controls the cortical excitatory input to the MSNs of the NAc through independent forms of short-term plasticity.
Materials and Methods
Parasagittal brain slices (300–400 μm thick) were prepared from C57BL/6 mice aged 15–25 d. Slices were transferred to a recording chamber in which they were continuously perfused with the oxygenated extracellular medium (95% O2 and 5% CO2) containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 16 glucose, pH 7.4. In some experiments, the extracellular Ca2+ concentration was changed to either 0.5 or 4 mm, as mentioned in Results, without changing the Mg2+ concentration. However, we checked that the observed effects were not attributable to a change in the overall excitability by repeating the experiments with similar Ca2+/Mg2+ ratio changes, but maintaining the extracellular divalent concentration constant. These controls have not been mentioned in the text, for clarity. Whole-cell voltage-clamp recordings of visually identified medium-sized neurons in the NAc were made at room temperature with patch electrodes filled with a solution containing the following (in mm): 120 K-gluconate, 20 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES, and 2 Na2-ATP, pH 7.3. Electrode resistance was 3.5–5.5 MΩ. Recordings were performed with a computer-driven EPC9 amplifier (Heka, Lambrecht, Germany). Neurons were voltage clamped at a holding potential of –80 mV for NAc neurons and –70 mV for PFC pyramidal cells. Recordings of layer V–VI neurons of the prefrontal cortex were performed on frontal slices cut in an oblique plane (10°), as first described by Yang et al. (1996). Stability of series resistance was controlled throughout the duration of the experiments.
EPSCs were evoked by electrical stimulation (100–150 μs duration) through a bipolar tungsten electrode placed at the prefrontal cortex–NAc border (Pennartz et al., 1993). Stimulation intensity was set to evoke stable EPSCs over a control period of 5–10 min. In some experiments, minimal intensity stimulations were used to reveal failures of EPSCs. In prefrontal cortex experiments, antidromic responses were evoked using the same stimulation electrode placed in the NAc in the vicinity of the anterior commissure. Bicuculline (10 μm) was present throughout all experiments. Data were acquired using the Pulse program (Heka) and analyzed using macros written for Igor (Wavemetrics, Lake Oswego, OR). The statistical significance of the results was assessed by using, in the appropriate case, a Wilcoxon test or a Mann–Whitney test (for paired and unpaired observations, respectively) and ANOVA in the case of multiple comparisons.
All drugs were applied in the perfusion solution. d(-)-2-Amino-5-phosphonopentanoic acid (d-AP-5), bicuculline, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium (NBQX), and 4-aminopyridine (4-AP) were purchased from Fischer Bioblock Scientific (Illkirch, France). Margatoxin, cyclothiazide, and 1,2-bis(2-aminophenoxy)ethane-N, N,N′,N′-tetra-acetic acid tetrakis AM (BAPTA-AM) were obtained from Sigma-Aldrich Chimie (Saint Quentin-Fallavier, France).
All experiments were conducted with respect to the European directives and the French law on animal experimentation (authorization number 02345/33).
Results
Tonic stimulation triggers a distinct form of short-term synaptic plasticity
Pyramidal PFC neurons fire in a tonic mode at various frequencies. We thus examined how a gradual increase in their sustained tonic activity affected synaptic transmission in target NAc neurons. Increasing tonic stimulation of cortical fibers in the low frequency range (from 0.1 to 2 Hz) reversibly affected the amplitude of EPSCs recorded in identified MSNs. Depending on the recorded neuron, this protocol induced either depression or facilitation of EPSCs (range, –58 to +144%; n = 44) (Fig. 1). These effects became significant at 0.5 Hz and increased to a plateau value at 1 Hz. For a given neuron, the direction and extent of EPSC amplitude variation was highly reproducible with repetition of the protocol. Similar observations were made when recordings were performed at 33°C (range, –41 to + 76%; n = 10). Interestingly, increasing the frequency of stimulation from 0.1 to 1 Hz led to a significant decrease in the coefficient of variation (CV) for neurons displaying frequency facilitation (control CV, 0.47 ± 0.05; 1 Hz, 0.37 ± 0.03; n = 22 cells; p = 0.022), although, conversely, it led to a significant increase in the CV for neurons showing depression (control CV, 0.35 ± 0.06; 1 Hz, 0.46 ± 0.08; n = 22; p = 0.025). These data suggest that short-term frequency modulation has a presynaptic origin.
Figure 1.
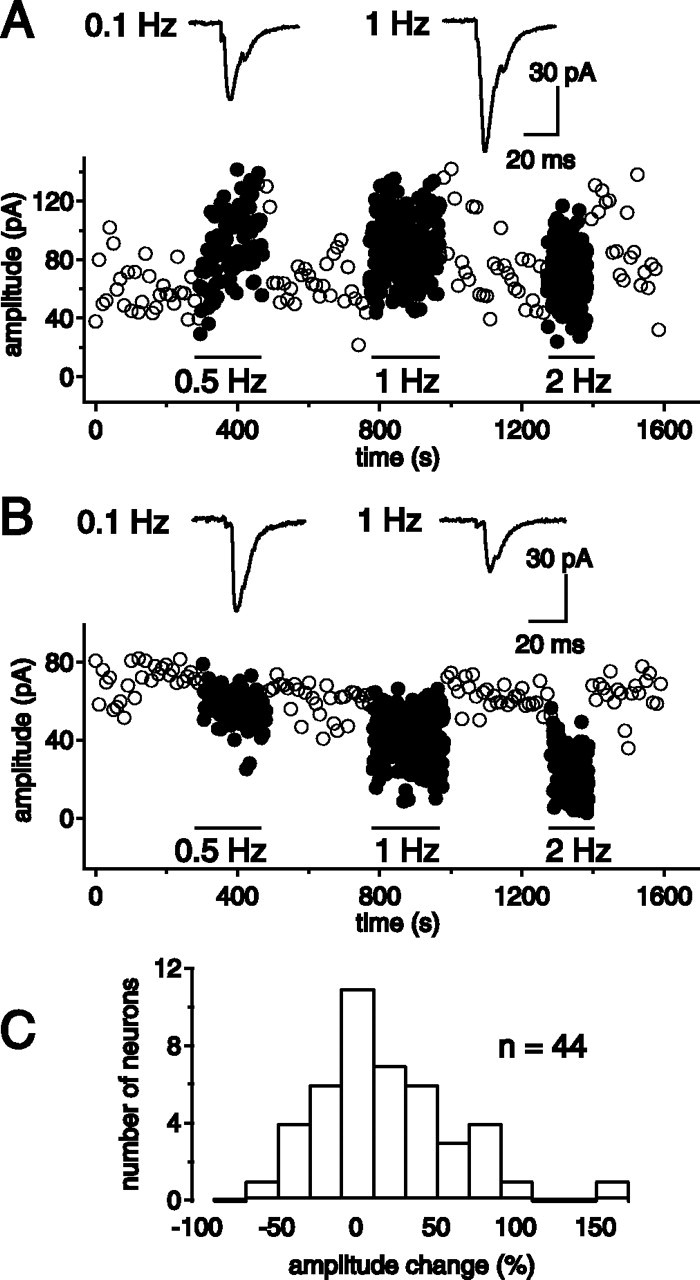
Tonic frequency increase in presynaptic activity triggers short-term plasticity. A, B, Increasing cortical-stimulation frequency from 0.1 to 0.5, 1, and 2 Hz triggered either facilitation or depression of EPSCs in different NAc neurons. A,B, Sample experiments from two recorded neurons illustrating facilitation (A) and depression (B). C, Distribution histogram of the extent and direction of tonic short-term plasticity in 44 NAc neurons (frequency shift from 0.1 to 1 Hz).
We further investigated whether the direction of frequency modulation was correlated with presynaptic release probability. Shifting frequency from 0.2 to 1 Hz led to facilitation in low release probability conditions (0.5 mm Ca2+; mean facilitation, +169 ± 51%; n = 6; p = 0.028), and to depression in high release probability conditions (4 mm Ca2+; mean depression, –29 ± 6%; n = 6; p = 0.028) (Fig. 2A,B). CV values were significantly higher for low (CV = 1.3 ± 0.2; n = 6) than for high release probability conditions (CV = 0.15 ± 0.01; n = 6; p = 0.002), with a linear correlation between the extent of frequency modulation and initial CV (p < 0.001) (Fig. 2C). To confirm that frequency facilitation or depression in normal conditions (2 mm Ca2+) depended on the initial presynaptic release probability state, we directly compared both parameters in single cells. Presynaptic release probability was determined according to the method described by Clements (2003). The variance and the mean of EPSC amplitudes were determined under different extracellular Ca2+ concentrations to calculate the initial presynaptic release probability in 2 mm Ca2+ (Fig. 2D,E). The extent of frequency facilitation or depression was also measured in the same cells. Plotting frequency modulation as a function of initial presynaptic release probability for all cells tested (Fig. 2F) confirmed that the direction of tonic low-frequency modulation (facilitation or depression) depended on initial presynaptic release probability. Considering this, the various levels of short-term plasticity expressed in control conditions (Fig. 1C) seem to reflect a large heterogeneity in release probability at corticoaccumbens synapses. Similar frequency-dependent short-term plasticity was also observed for a higher stimulation rate. Short trains of stimuli at a frequency of 12.5 Hz led to a facilitation that stabilized at +210 ± 58% (n = 8) in low release probability conditions (0.5 mm Ca2+) and to a depression in high release probability conditions (4 mm Ca2+) (mean depression, –72 ± 3%; n = 4) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 2.
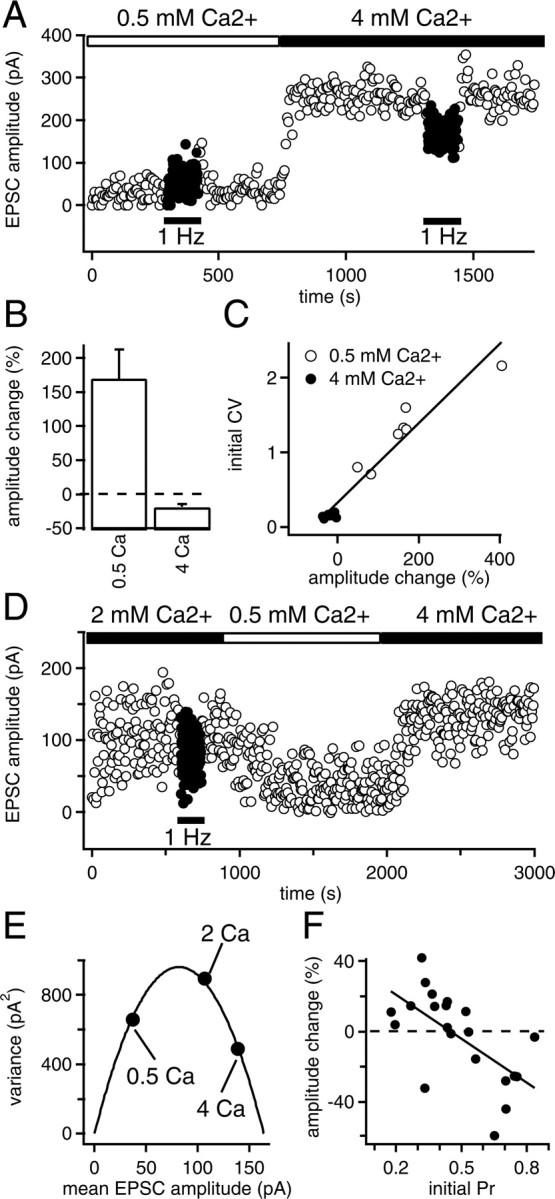
Tonic short-term plasticity direction depends on initial presynaptic release probability. A, In a given neuron, increasing stimulation frequency from 0.2 to 1 Hz led to facilitation of EPSCs in low release probability conditions (extracellular Ca2+, 0.5 mm) and to depression in high release probability conditions (extracellular Ca2+, 4 mm). B, Average facilitation or depression obtained for six neurons in the two release probability conditions. Error bars indicate SE. C, The extent of changes in EPSC amplitude during tonic short-term plasticity strongly correlated with the initial CV value (measured before shifting frequency to 1 Hz). D, Sample experiment showing the protocol used to determine the relationship between frequency modulation of evoked EPSC amplitude and initial release probability. Frequency modulation was first determined in 2 mm extracellular Ca2+ (depression in this example). EPSC amplitude means and variances were subsequently measured in various release probability conditions (here, 0.5 and 4 mm extracellular Ca2+, both leading to a decrease in EPSC amplitude variance). E, From the experiment shown in D, variances were plotted as a function of EPSC mean amplitude for each extracellular Ca2+ condition and fitted with a parabola of the following equation: y =–(QP/μ)x2 + Q(1 + CV2)x, where Q is the weighted averaged quantal response, and CV, μ, and P are the coefficient of variation, the mean amplitude, and the probability of release in 2 mm Ca2+ conditions, respectively (Clements, 2003). In the sample experiment shown here, Q = 22 pA, CV = 1.08, μ = 107 pA, and P = 0.704. F, EPSC amplitude changes induced by increasing frequency stimulation from 0.2 to 1 Hz were plotted as a function of initial probability of release for all cells tested. Data points were fitted with a linear regression (r =–0.63).
Short-term synaptic potentiation induced by repetitive bursts of cortical stimulation
PFC pyramidal neurons can also fire in bursts with an intraburst frequency ranging between 10 and 15 Hz (Yang et al., 1996). To investigate how this physiologically relevant presynaptic activity may alter synaptic efficacy in postsynaptic MSNs, we stimulated cortical afferent fibers with bursts of six stimuli evoked at 12.5 Hz. Test EPSCs evoked 5–20 s after the end of the burst were significantly larger, compared with the first EPSC of the burst (Fig. 3). This increase in EPSC amplitude peaked at 5–10 s after the end of the train and recovered to control values within 30 s (mean EPSC amplitude increase at 5 s, +27 ± 9%; n = 8; p = 0.017) (Fig. 3B). In control conditions, EPSC amplitude within bursts either facilitated or depressed, as observed with tonic low-frequency modulation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, EPSC amplitude displayed a positive rebound 5 s after the end of the burst, whatever the direction of intraburst modulation (Fig. 3A). In presynaptic pyramidal cells, bursts of activity occur in a repetitive manner. Repetition of the bursts every 5 s led to a prominent cumulative increase in synaptic transmission (Fig. 4A) that saturated after 5–10 bursts to reach a level of facilitation of +56 ± 13% (n = 44), and fully recovered to control values after 2–10 min (Fig. 4A). This form of short-term potentiation caused by repetitive bursts (named RB-STP in the rest of the article) was maximal for a 5 s interval between the bursts (Fig. 4C). Similar observations were made at 33°C (+51 ± 14% facilitation; n = 6), except for the recovery, which never exceeded 2 min. Cumulative increase of the amplitude of the first EPSC in the train was accompanied with a concomitant decrease in paired-pulse ratio (PPR) measured on the two first EPSCs of the train (Fig. 4B). These results, in addition to the analysis of the CV of EPSC amplitude (data not shown), support the idea of a presynaptic locus for this form of short-term plasticity (Faber and Korn, 1991; Casassus and Mulle, 2002).
Figure 3.

Burst conditioning of cortical afferent fibers induces positive rebound of excitatory synaptic transmission. A, EPSCs evoked by a conditioning burst of six cortical stimuli (intraburst frequency, 12.5 Hz) and by single test stimuli at variable time intervals after the end of the burst (average of 30 traces). Single bursts of cortical stimulation induced a positive rebound of synaptic transmission independently of the direction of intraburst short-term plasticity (top traces, depression; bottom traces, facilitation). B, Time course for increased EPSC amplitude induced by a burst of stimuli. Mean increase in EPSC amplitude is expressed as a percentage of the amplitude of the first EPSC in the conditioning burst. The rebound was maximal at 5–10 s and recovered to control values 40 s after the end of the burst. Error bars indicate SE.
Figure 4.

Repetitive bursts of cortical stimulation induce short-term synaptic plasticity in NAc neurons (RB-STP). A, Repetitive bursts of cortical stimuli induced a cumulative increase in the amplitude of the first EPSC of the burst (filled circle), compared with control EPSCs evoked at a frequency of 0.1 Hz (open circle). Maximal increase in synaptic transmission was reached after 5–10 repetitive bursts and recovered 2–10 min after the end of the burst stimulation protocol. B, Repetitive burst stimulations led to decreased PPR in parallel with an increase in EPSC amplitude. Sample traces of paired EPSCs at the first (left) and the last train (right) of the repetitive burst protocol are shown (inset). C, The amplitude of RB-STP strongly depended on intervals between the successive bursts. RB-STP was maximal for burst repetition each 5 s (mean change, 145 ± 11%; n = 11) and decreased with increasing the inter-burst interval. No synaptic facilitation was observed with bursts separated by 40 s (mean change, 101 ± 5%; n = 11). Error bars indicate SE.
We examined whether RB-STP was correlated with initial presynaptic release probability by varying extracellular Ca2+ concentration. The extent of RB-STP was not different in high (4 mm Ca2+) or low (0.5 mm Ca2+) release probability conditions (mean facilitation, low Ca2+, +56 ± 14%, n = 6; high Ca2+, +66 ± 35%, n = 7) (Fig. 5A). In addition, preincubating the slices for 1 h with BAPTA-AM (50 μm) to lower intracellular Ca2+ and to prevent Ca2+ accumulation in presynaptic terminals, failed to inhibit RB-STP (mean facilitation, +53 ± 21%; n = 7) (Fig. 5A). Thus, RB-STP does not appear to depend on initial release probability and Ca2+ influx.
Figure 5.

RB-STP involves regulation of K+ channels but not Ca2+ influx. A, Mean change in the amplitude of EPSCs induced by RB-STP was not affected by changing extracellular Ca2+ concentration (0.5, 2, or 4 mm) or by buffering presynaptic Ca2+ (BAPTA-AM; 50 μm). RB-STP was not inhibited by the K+-channel blockers TEA (10 mm), margatoxin (100 nm), and tityustoxin (100 nm). B, Pretreatment of the slices with 4-AP (100 μm) abolished RB-STP (24 consecutive bursts; 5 s interval; averaged every 15 s; filled circles). Sample average EPSCs recorded during control (left), repetitive bursts (center), and recovery (right) periods are shown (inset). C, Dose–response curve for 4-AP inhibition of RB-STP revealed an IC50 value of 23 μm. Error bars indicate SE.
RB-STP involves inhibition of 4-AP-sensitive K+ channels
We investigated the potential role of K+ channels by incubating the slices with the broad spectrum K+-channel blocker 4-AP. Experiments were conducted with 0.5 mm Ca2+ in the extracellular medium to prevent overactivation of synaptic transmission. Bathing the slices with 4-AP (100 μm) totally prevented RB-STP (Fig. 5B) (in 4-AP, mean EPSC amplitude vs control, 85 ± 8%; n = 7; ANOVA; p = 0.91). These experiments point to a critical role of K+ channels. To further examine the nature of K+ channels involved, we applied varying concentrations of 4-AP and found that RB-STP was highly sensitive to 4-AP, with an IC50 value of 23 μm (Fig. 5C). K+ channels containing either Kv1 or Kv3 α subunits families display a relatively high sensitivity to 4-AP (submillimolar) (Coetzee et al., 1999). Among them, Kv1.5 and Kv3.1 are the only subunits reported to be sensitive to 4-AP with an IC50 < 100 μm. To distinguish between these two subunits, we used tetraethylammonium (TEA), which potently inhibits Kv3 subunits (as well as Kv1.1). TEA, at a concentration of 10 mm, was ineffective in inhibiting RB-STP (Fig. 5A). Similarly, margatoxin (100 nm), a selective blocker of Kv1.3 and Kv1.6, and tityustoxin (100 nm), a selective blocker of Kv1.2, did not affect RB-STP. Therefore, Kv1.5-containing potassium channels, which are highly sensitive to 4-AP but insensitive to TEA, appear as likely candidates involved in this form of short-term plasticity.
RB-STP involves an increase in the excitability of presynaptic axons
To investigate whether RB-STP involved regulation of axonal excitability, we directly recorded from PFC pyramidal neurons of layers V–VI projecting to the NAc. We used frontal cortical slices cut in an oblique plane (10°) that preserve PFC neurons and their afference to the NAc (Yang et al., 1996). A bipolar stimulation electrode placed in the NAc evoked antidromic action currents in PFC neurons in the presence of the ionotropic glutamate receptor antagonists NBQX (20 μm) and d-AP-5 (50 μm). PFC neurons with antidromic responses were intrinsic bursting pyramidal neurons with an initial spike doublet and a depolarizing afterpotential in response to a depolarizing pulse (Fig. 6A) (Yang et al., 1996). Stimulation intensity in control conditions (1 pulse, 0.2 Hz) was adjusted to evoke a low success rate of antidromic action currents (Fig. 6B,C). Repetitive bursts of stimuli in the NAc (6 pulses; 12.5 Hz; 5 s interval) reversibly increased the success rate of antidromic action currents measured at the first pulse of the burst from 22 ± 5 to 55 ± 15% (n = 6; ANOVA; p = 0.036) (Fig. 6C). In addition, RB-STP significantly reduced the variability of the latency of antidromic action potentials (CV control, 0.040 ± 0.018; CV during RB-STP, 0.022 ± 0.004; n = 6; p = 0.02), suggesting an enhancement of excitability or a change in the reliability of action potential propagation during RB-STP. Incubation of the slices with 4-AP (100 μm) had a direct effect on axonal excitability (Fig. 6D), increasing the success rate from 29 ± 4 to 81 ± 11% (n = 3; ANOVA; p = 0.027), thus mimicking RB-STP. The effect of 4-AP was not reversible within the time of the experiment. In similar conditions, TEA (10 mm) had no effect (data not shown). In the presence of 4-AP, a series of bursts of stimulation applied in the NAc was ineffective in increasing the success rate of action currents (Fig. 6E). Instead, a decrease of the success rate from 39 ± 11 to 18 ± 9% (n = 5) was observed.
Figure 6.

K+ channels regulate afferent axonal excitability in response to repetitive bursts of antidromic stimuli. A, Electrophysiological identification of intrinsic bursting neurons in the PFC displaying a characteristic depolarizing afterpotential (left; black arrow) and a spike doublet at the beginning of the discharge. The current injected through the patch pipette is indicated under each trace. B, Sample of 10 successive antidromic action currents evoked in intrinsic bursting neurons by bipolar stimulation in the NAc during low-frequency stimulation (0.1 Hz) (left) and repetitive bursts (right). B–E, In all experiments with recordings of antidromic action potentials, electrical stimulation was adjusted to obtain a low success rate in control conditions (in the range between 20 and 40%). This allowed us to monitor a potential increase in success rate after RB-STP protocol or acute application of 4-AP. C, Average success rate of antidromic action currents increased during repetitive burst stimulations (filled circle) compared with single control stimulations (open circle). D, This effect was mimicked by bath application of 4-AP (100μm) (4 min). E, In the continuous presence of 4-AP (100μm), repetitive burst stimulations (black circle) did not induce an increase in antidromic success rate. Error bars indicate SE.
Application of 4-AP did not broaden the recorded action current, did not affect the threshold for spike discharge, and did not change miniature EPSC frequency recorded in NAc neurons (supplemental Fig. 2A–E, available at www.jneurosci.org as supplemental material), as might be expected if this effect was mediated by depolarization of the synaptic terminal. However, 4-AP increased spontaneous EPSC frequency and evoked EPSC amplitude, as expected from the effects on action potential occurrence in the presynaptic axons (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material). In contrast to RB-STP, the acute effects of 4-AP on evoked EPSC rise and decay time appeared to depend on external Ca2+ concentrations (data not shown), suggesting that 4-AP affects synaptic transmission by several distinct mechanisms. Nevertheless, in the presence of 4 mm Ca2+, 4-AP (30 μm) increased evoked EPSC amplitude (+42 ± 14%; n = 5; p = 0.03) and occluded RB-STP, in the absence of a detectable effect on the rise and decay time of EPSCs (control vs 4-AP, 10–90% rise time = 2.2 ± 0.5 ms vs 2.3 ± 0.5 ms, n = 5, p = 0.53; decay time, τ = 5.9 ± 0.6 vs 5.9 ± 0.6 ms, n = 5, p = 0.86) (supplemental Fig. 2 E, available at www.jneurosci.org as supplemental material). Surprisingly, we observed a slight lengthening of evoked EPSC latency (+0.2–0.5 ms; p = 0.003), an effect that may be related to partial sodium-channel inactivation in the axon. Together, these data suggest that RB-STP is mediated by an inhibition of K+ channels and results from an increase in axonal excitability rather than from a change in the presynaptic action potential waveform. To test whether RB-STP decreased spike propagation failures rather than favored spike triggering at the site of stimulation, we examined RB-STP in minimal stimulation conditions. If RB-STP increased spike triggering, it should induce the recruitment of additional fibers and should lead to a concomitant decrease in the failure rate and an increase in synaptic potency (amplitude of EPSCs excluding failures). As shown in Figure 7, RB-STP protocols led to a decrease in failures (–43 ± 8%; n = 9; p < 0.001). However, no significant change in synaptic potency (+6 ± 6%; n = 9; p = 0.36) was observed. Thus, this result is in favor of a decrease in failure of spike propagation as the mechanism of RB-STP rather than an increase of spike triggering at the site of stimulation.
Figure 7.

RB-STP induces a decrease in EPSC failures without changes in synaptic potency. A, Sample experiment in which cortical fiber stimulation intensity was adjusted to obtain ∼50% of EPSCs but all with similar amplitudes. In these minimal stimulation conditions, RB-STP induces a decrease in the number of failures but EPSCs remained all in the same range of amplitude (i.e., synaptic potency remained constant). The frequency of stimulation was 0.2 Hz, and controls were performed to ascertain that decreasing and increasing stimulation intensity led to failures only and increased synaptic potency, respectively. B, Percentage of EPSC failures under minimal stimulation conditions before (control), during (RB-STP), and after (recovery) RB-STP, for each of the nine cells tested. C, Mean EPSC potencies under minimal stimulation conditions before (control), during (RB-STP), and after (recovery) RB-STP, for each of the nine cells tested.
Functional consequences of mode switching versus gradual increase in tonic activity
We compared, in the same cells, the effects of increasing stimulation frequency from a tonic 0.2 Hz stimulation to an average stimulation of 1 Hz in two different conditions: either with a shift to a regular tonic pattern or with a switch to a phasic burst pattern. As described above, a tonic shift of stimulation frequency from 0.2 to 1 Hz induced either facilitation (Fig. 8A) or depression (Fig. 8B), and the direction of the changes likely depends on initial release probability. In contrast, regardless of the initial release probability, switching from a tonic to a phasic burst pattern (with the same average stimulation frequency) induced a cumulative enhancement of synaptic transmission (Fig. 8A,B). Thus, both tonic short-term plasticity and RB-STP can occur independently in the same neuron. As a result, tonic short-term plasticity tends to level synaptic strength (high-probability synapses being decreased and low-probability synapses being increased), whereas RB-STP increases synaptic transmission in all neurons, whatever the initial state of release probability (Fig. 8C).
Figure 8.

Functional consequences of mode switching to phasic stimulation versus gradual increase in tonic frequency. A, B, Cortical stimulation frequency was first increased from a tonic 0.2 Hz stimulation to a tonic 1 Hz stimulation. Then cortical stimulation was switched to a phasic mode while maintaining an average 1 Hz stimulation. This protocol was successively applied in the same neuron in low (Ca2+; 0.5 mm) and high (Ca2+; 4m m) release probability conditions. Shifting tonic stimulation from 0.2 Hz (“0.2 Hz tonic”; open circles) to 1 Hz (“1Hz tonic”; filled squares) led to facilitation or depression depending on release probability conditions. In contrast, for the same average frequency, switching to repetitive burst stimulations (“1 Hz phasic”; filled circles) increased EPSC amplitude in both release probability conditions. C, Scheme summarizing how the pattern of presynaptic discharge drives changes in synaptic strength in the corticoaccumbens pathway. Error bars indicate SE.
Discussion
In the present study, we described two new forms of short-term synaptic plasticity at the corticoaccumbens pathway. These two forms of plasticity, which occur within the same physiological range of firing frequency, function independently. Increasing presynaptic activity in a tonic manner either depresses or facilitates synaptic transmission. Inversely, repetitive bursts of presynaptic activity enhance corticoaccumbens synaptic transmission in all neurons, likely through the regulation of axonal potassium channels. Thus, we provide evidence that short-term synaptic plasticity at this synapse not only depends on the global rate of firing of presynaptic afferents, but is critically conditioned by the pattern of presynaptic activity.
Tonic presynaptic activity controls calcium-dependent short-term plasticity at the corticoaccumbens synapse
We report that tonic stimulation of cortical afferents triggers either frequency facilitation or depression in medium-sized neurons of the NAc. This short-term synaptic modulation occurs within a physiological range of frequencies for cortical pyramidal neurons. As previously reported for 10 Hz stimulation (Lape and Dani, 2004), this form of plasticity is likely presynaptic in origin, and it is sensitive to the concentration of extracellular Ca2+. As in the majority of other synapses studied, frequency facilitation probably results from accumulation of residual Ca2+ induced by previous action potential invasion in the synaptic terminal. Simple models of presynaptic release events predict that short-term synaptic facilitation generally occurs at synapses exhibiting low release probability, whereas high release probability synapses frequently present activity-dependent depression (Fortune and Rose, 2001). Release probability is mostly heterogeneous among individual synapses in the CNS (Hessler et al., 1993; Murthy et al., 1997), as also shown here for corticoaccumbens synapses. Our results indicate that the extent and direction of facilitation depends on the initial release probability of individual corticoaccumbens synapses. The functional consequence of a tonic increase in presynaptic afferent firing rate will be to level input strength from populations of synapses with different release probability states, because active synapses will tend to be depressed and less active synapses will tend to be facilitated.
A novel form of short-term plasticity involving axonal K+ channels
We described a new form of short-term plasticity, RB-STP, which relies on the regulation of axonal K+ channels. RB-STP is triggered by repetitive bursts of presynaptic activity within a physiological range of firing frequencies. RB-STP is presynaptic in origin, as indicated by changes in CV and paired-pulse ratio. RB-STP appears independent of extracellular Ca2+ concentration and overall excitability because of changes in total divalent concentrations, and is not affected by preincubation with the Ca2+ chelator BAPTA-AM. However, RB-STP is highly sensitive to 4-AP, suggesting an involvement of K+ channels. Repetitive burst firing prominently increases the success rate of antidromic action potentials recorded in corticoaccumbens neurons and significantly reduces the variability of the latency of antidromic action potentials, indicating an increase in axonal excitability. This effect is mimicked by 4-AP, which occludes RB-STP. Although this does not imply that RB-STP directly relies on 4-AP-sensitive channel activity, we were not able to detect any sign of indirect 4-AP effects that may explain RB-STP occlusion. For instance, 4-AP does not change the threshold and duration of action potentials recorded in the soma of presynaptic neurons, nor does it affect miniature EPSC frequency. We propose that RB-STP reflects changes in the reliability of axonal propagation. An alternative explanation might be that 4-AP and RB-STP increase spike initiation successes at the site of stimulation. Although recruitment of additional fibers cannot be formally ruled out, our data showing that RB-STP was related to a decrease in EPSC failure rate without significant changes in EPSC potency rather suggest an increase in spike propagation reliability. Lape and Dani (2004) recently reported strong reduction of the glutamatergic frequency facilitation in the NAc by 4-AP, similar to those observed in high extracellular Ca2+ condition. These results are consistent with a presynaptic locus of 4-AP action; however, they do not discriminate between direct changes in release probability and upstream effects on axonal properties. Likewise, we cannot directly rule out that RB-STP affects the action potential waveform in synaptic terminals. However, it would then be expected that the level of extracellular Ca2+ influences RB-STP. In addition, 4-AP affects evoked EPSC amplitude without changing rise and decay times when recorded in high extracellular Ca2+. Overall, our data provide evidence for a form of short-term plasticity induced by regulation of axonal K+ channels, which had been previously hypothesized (Debanne, 2004). We propose that repetitive stimulation of corticoaccumbens axons leads to cumulative K+-channel inactivation. In hippocampal slice cultures, A-type K+ channels control axonal action potential propagation (Debanne, 2004). Inactivation of A-type K+ channels increases spike occurrence at the presynaptic terminal and therefore decreases the paired-pulse ratio (Debanne et al., 1999), as observed with RB-STP. Short, repetitive depolarization pulses result in cumulative inactivation of Kv1.4 A-type K+ channels in the frequency range used within bursts (Roeper et al., 1997). Recovery from inactivation of Kv1.4 channels is known to comprise a fast (millisecond time range) and a slow (several seconds) component resulting from N-type and C-type inactivated states recovery (Baukrowitz and Yellen, 1995; Roeper et al., 1997). The biophysical properties of A-type K+ channels are thus compatible with an involvement in RB-STP. However, recovery from RB-STP can take up to several minutes, a time range that appears different from that of recovery from inactivation of A-type K+ channels. Activation of A-type K+ channels with repetitive bursts, such as used in the present study, might lead to more profound inactivation with a longer time course of recovery. Additional mechanisms, such as phosphorylation (Roeper et al., 1997) or binding of Kv β subunit to the K+ channel (Castellino et al., 1995), might also slow down recovery from inactivation. We do not exclude that other mechanisms might also contribute to RB-STP.
Although the identity of the K+ channels involved is not precisely known, high sensitivity of RB-STP to 4-AP and insensitivity to TEA strongly suggest an involvement of Kv1.5 α-subunit-containing K+ channels (Coetzee et al., 1999). Transcripts for these channels are detected in pyramidal neurons (Pan et al., 2004) and the NAc is labeled with a Kv1.5-specific antibody (Chung et al., 2000). However, homomeric Kv1.5 channels form functional, delayed rectifier K+ channels presenting a slowinactivating rate with poor cumulative properties (Coetzee et al., 1999; Kurata et al., 2001; Song, 2002). In a heterologous system, heteromeric assembly of Kv1.5 and Kv1.4 α subunits forms functional channels expressing A-type-like inactivation and recovery from inactivation properties. Kv β subunit binding to homomeric Kv1.5 also confers rapid A-type inactivation properties on this noninactivating K+ channel (Sewing et al., 1996). Finally, Kv β targets Kv1-containing K+ channels to axons (Gu et al., 2003). Based on our pharmacological data and on previous studies with recombinant K+ channels, we propose that axonal A-type-like K+ channels composed of Kv1.5 assembled with either Kv1.4 or with a Kv β subunit are involved in RB-STP.
We propose that repetitive burst-induced inactivation of A-type K+ channels leads to synaptic potentiation through an increased reliability of spike propagation. Whether naturally triggered action potentials at the axon initial segment may be subject to conduction failures is, however, a matter of debate, and this probably varies between pathways, physiological conditions, and axonal geometry (Debanne et al., 1999; Debanne, 2004). To our knowledge, there is no evidence demonstrating that action potential propagation in corticoaccumbens fibers is highly secure or not. An additional potential consequence of axonal A-type K+-channel inactivation by an RB-STP-type mechanism is a change in the delay of action potential initiation, leading to increased temporal fidelity (Mahon et al., 2003). In support of this notion, we observed that RB-STP significantly reduced the variability of the latency of antidromically recorded action potentials.
Mode-switching versus gradual increase in tonic activity
The results presented here provide new insight into mechanisms by which glutamatergic cortical inputs to the NAc are modulated by the temporal pattern of presynaptic action potential discharge. The PFC plays a key role in working-memory maintenance and reward processes (Miller and Cohen, 2001). In vivo recordings of prefrontal neurons during goal-directed tasks reveal tonic and phasic changes in activity, depending on prediction, presentation, or delivery of reward (Schultz et al., 2000; Cooper, 2002). Consistent with a high functional interplay between PFC and the NAc region in reward-related processes, simultaneous recording of PFC and NAc neurons reveals a strong correlated activity during cocaine-seeking behavior (Chang et al., 2000). Considering that a large majority of PFC neurons contacting the NAc display intrinsic bursting properties (Yang et al., 1996), switching presynaptic cortical activity from a tonic to a phasic burst pattern will play a major role in driving NAc activity by rapid and reversible changes in the efficacy of corticoaccumbens synaptic transmission. The NAc has been proposed as a collection of neuronal “ensembles” synchronized by a specific set of excitatory inputs (Pennartz et al., 1994). Functionally segregated circuits link the basal ganglia and neocortex, and processing of information within and between these circuits is largely parallel in nature. For instance, separate populations of NAc neurons differentially encode information about natural reinforcers (food and water) and cocaine (Carelli et al., 2000). Because of this functional segregation, reliable enhancement of sets of excitatory synaptic inputs might contribute to activation of functionally coherent neuronal populations and help encode specific information. At the corticoaccumbens synapse, switching from tonic activity to repetitive bursts reliably increases the efficacy of synaptic transmission for all afferent fibers, independently of the initial presynaptic state. Thus, RB-STP might be involved in the overall increase in synaptic transmission in sets of functionally correlated neurons, if presynaptic neurons fire in a bursting mode. In contrast, an increase in tonic stimulation of cortical afferents triggers, in NAc neurons, either frequency facilitation or depression, depending on initial presynaptic release probability. A tonic increase in presynaptic afferent firing rate will thus level input strength from populations of synapses with different release probability states. Tonic activity-dependent plasticity might also favor the activation of a neuronal subpopulation presenting initial low release probability of excitatory input. Then, hypothetically, the two short-term synaptic-plasticity processes described here could contribute in different ways to the functional response of the NAc in reward-related processes. Recent data revealed a critical implication of the corticoaccumbens pathway in addiction processes. Reinstatement of cocaine-seeking behavior by cocaine priming was demonstrated to result from an increase in corticoaccumbens AMPA-mediated glutamate transmission (Cornish and Kalivas, 2000; Park et al., 2002; McFarland et al., 2003). A thorough evaluation of dynamic changes in glutamatergic corticoaccumbens transmission is thus essential to understanding addictive processes.
Footnotes
This work was supported by grants from Centre National de la Recherche Scientifique, Ministère de la Recherche of France, and Conseil Régional d'Aquitaine. We thank Olivier Manzoni, Dominique Debanne, and Olivier George for useful comments on this manuscript.
Correspondence should be addressed to Christophe Mulle, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 5091, Institut François Magendie, Université Victor Segalen-Bordeaux II, rue Camille Saint-Saëns, 33077 Bordeaux Cedex, France. E-mail: mulle@u-bordeaux2.fr.
DOI:10.1523/JNEUROSCI.2466-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511504-09$15.00/0
References
- Baukrowitz T, Yellen G (1995) Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron 15: 951–960. [DOI] [PubMed] [Google Scholar]
- Carelli RM, Ijames SG, Crumling AJ (2000) Evidence that separate neural circuits in the nucleus accumbens encode cocaine versus “natural” (water and food) reward. J Neurosci 20: 4255–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casassus G, Mulle C (2002) Functional characterization of kainate receptors in the mouse nucleus accumbens. Neuropharmacology 42: 603–611. [DOI] [PubMed] [Google Scholar]
- Castellino RC, Morales MJ, Strauss HC, Rasmusson RL (1995) Time- and voltage-dependent modulation of a Kv1.4 channel by a beta-subunit (Kv beta 3) cloned from ferret ventricle. Am J Physiol 269: H385–H391. [DOI] [PubMed] [Google Scholar]
- Chang JY, Janak PH, Woodward DJ (2000) Neuronal and behavioral correlations in the medial prefrontal cortex and nucleus accumbens during cocaine self-administration by rats. Neuroscience 99: 433–443. [DOI] [PubMed] [Google Scholar]
- Chung YH, Shin CM, Kim MJ, Cha CI (2000) Immunohistochemical study on the distribution of six members of the Kv1 channel subunits in the rat basal ganglia. Brain Res 875: 164–170. [DOI] [PubMed] [Google Scholar]
- Clements JD (2003) Variance-mean analysis: a simple and reliable approach for investigating synaptic transmission and modulation. J Neurosci Methods 130: 115–125. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B (1999) Molecular diversity of K+ channels. Ann NY Acad Sci 868: 233–285. [DOI] [PubMed] [Google Scholar]
- Cooper DC (2002) The significance of action potential bursting in the brain reward circuit. Neurochem Int 41: 333–340. [DOI] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW (2000) Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci 20: RC89(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D (2004) Information processing in the axon. Nat Rev Neurosci 5: 304–316. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM (1997) Action-potential propagation gated by an axonal I(A)-like K+ conductance in hippocampus. Nature 389: 286–289. [DOI] [PubMed] [Google Scholar]
- Debanne D, Kopysova IL, Bras H, Ferrand N (1999) Gating of action potential propagation by an axonal A-like potassium conductance in the hippocampus: a new type of non-synaptic plasticity. J Physiol (Paris) 93: 285–296. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Billups B, Rusznak Z, Szucs G, Barker MC, Forsythe ID (2003) Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol (Lond) 550: 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber DS, Korn H (1991) Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J 60: 1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune ES, Rose GJ (2001) Short-term synaptic plasticity as a temporal filter. Trends Neurosci 24: 381–385. [DOI] [PubMed] [Google Scholar]
- Groenewegen H, Berendse H, Meredith G, Haber S, Voorn P, Wolters J, Lohman A (1991) Functional anatomy of the ventral limbic system innervated striatum. In: The mesolimbic dopamine system, pp 19–59. New York: Wiley.
- Gu C, Jan YN, Jan LY (2003) A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science 301: 646–649. [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366: 569–572. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, Takahashi T (2003) Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci 23: 10445–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Nakamura M (1999) Neural systems for behavioral activation and reward. Curr Opin Neurobiol 9: 223–227. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Soon GS, Fedida D (2001) Altered state dependence of c-type inactivation in the long and short forms of human Kv1.5. J Gen Physiol 118: 315–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R, Dani JA (2004) Complex response to afferent excitatory bursts by nucleus accumbens medium spiny projection neurons. J Neurophysiol 92: 1276–1284. [DOI] [PubMed] [Google Scholar]
- Mahon S, Casassus G, Mulle C, Charpier S (2003) Spike-dependent intrinsic plasticity increases firing probability in rat striatal neurons in vivo. J Physiol (Lond) 550: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S (2004) Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci 24: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001) An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24: 167–202. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14: 69–97. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF (1997) Heterogeneous release properties of visualized individual hippocampal synapses. Neuron 18: 599–612. [DOI] [PubMed] [Google Scholar]
- Pan Y, Xu X, Tong X, Wang X (2004) Messenger RNA and protein expression analysis of voltage-gated potassium channels in the brain of Abeta(25–35)-treated rats. J Neurosci Res 77: 94–99. [DOI] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC (2002) Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci 22: 2916–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CM, Ameerun RF, Groenewegen HJ, Lopes da Silva FH (1993) Synaptic plasticity in an in vitro slice preparation of the rat nucleus accumbens. Eur J Neurosci 5: 107–117. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Groenewegen HJ, Lopes da Silva FH (1994) The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological, and anatomical data. Prog Neurobiol 42: 719–761. [DOI] [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O (1997) Frequency-dependent inactivation of mammalian A-type K+ channel Kv1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci 17: 3379–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Tremblay L, Hollerman JR (2000) Reward processing in primate orbitofrontal cortex and basal ganglia. Cereb Cortex 10: 272–284. [DOI] [PubMed] [Google Scholar]
- Sewing S, Roeper J, Pongs O (1996) Kv beta 1 subunit binding specific for shaker-related potassium channel alpha subunits. Neuron 16: 455–463. [DOI] [PubMed] [Google Scholar]
- Song WJ (2002) Genes responsible for native depolarization-activated K+ currents in neurons. Neurosci Res 42: 7–14. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF (1998) Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron 21: 415–424. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK (1998) High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394: 384–388. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK, Gorelova N (1996) Electrophysiological and morphological properties of layers V–VI principal pyramidal cells in rat prefrontal cortex in vitro J Neurosci 16: 1904–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405. [DOI] [PubMed] [Google Scholar]