

Abstract
Alterations of long-term synaptic plasticity have been proposed to participate in the development of addiction. To preserve synaptic functions, homeostatic processes must be engaged after exposure to abused drugs. At the mouse cortico-accumbens synapses, a single in vivo injection of Δ9-tetrahydrocannabinol (THC) suppresses endocannabinoid-mediated long-term depression. Using biochemical and electrophysiological approaches, we now report that 1 week of repeated in vivo THC treatment reduces the coupling efficiency of cannabinoid CB1 receptors (CB1Rs) to Gi/o transduction proteins, as well as CB1R-mediated inhibition of excitatory synaptic transmission at the excitatory synapses between the prefrontal cortex and the nucleus accumbens (NAc). Nonetheless, we found that cortico-accumbens synapses unexpectedly express normal long-term depression because of a reversible switch in its underlying mechanisms. The present data show that, in THC-treated mice, long-term depression is expressed because a presynaptic mGluR2/3 (metabotropic glutamate receptor 2/3)-dependent mechanism replaces the impaired endocannabinoid system. Thus, in the NAc, a novel form of presynaptic homeostasis rescues synaptic plasticity from THC-induced deficits.
Keywords: accumbens, cannabinoids, metabotropic glutamate receptor, glutamate, mice, LTD, long-term depression
Introduction
Neural circuits and their elements adapt to changes in their environment (Burrone and Murthy, 2003). Drug addiction is a remarkable example of this concept: a large body of evidence has shown that neurotransmitter receptors, signaling cascades, neuronal morphology, synaptic transmission/plasticity, and gene expression can be substantially altered in response to single or repeated exposure to addictive drugs (Hyman and Malenka, 2001; Nestler, 2002). Drugs of abuse potently alter excitatory glutamatergic signaling in the brain-reward pathway, and it has been proposed that such protracted changes are at the origin of addiction-related behaviors (Williams et al., 2001; Tzschentke and Schmidt, 2003). In neuronal circuits, when synaptic activity is enhanced (or reduced) beyond a normal level of activity, compensatory changes are engaged to return the system to its functional range (Burrone and Murthy, 2003). Until now, there has been no data of similar compensatory changes in synaptic functions after exposure to an abused drug. Cannabis sativa L. derivatives (e.g., marijuana, hashish...) have been used for centuries for both therapeutic and recreational purposes and remain the most widely used illegal drugs. We reported recently that a single exposure to a low dose of Δ9-tetrahydrocannabinol (THC) induces functional tolerance of cannabinoid CB1 receptors (CB1Rs) and blocks endocannabinoid (eCB)-mediated long-term depression (LTD) in the nucleus accumbens (NAc) (Mato et al., 2004), a structure of the mesocorticolimbic dopaminergic system essential for the reinforcing properties of addictive drugs (Koob, 1996). Because Cannabis derivatives are often used over extended periods of time, we decided to evaluate the consequences of repeated in vivo treatment with a low nonaversive dose of THC on eCB–LTD in mice prelimbic-cortex–accumbens synapses. The present data show that, at the prefrontal cortex (PF-Cx)–NAc synapses of THC-treated mice, it is still possible to induce LTD in response to physiologically relevant cortical stimulation despite impaired CB1R functions. Our data indicate a key role for presynaptic metabotropic glutamate receptor 2/3 (mGluR2/3) in the homeostatic rescue of LTD in THC-treated mice.
Materials and Methods
Animal treatment. Mice (male, 4 weeks of age, C57B1/6 strain) were housed, grouped, and acclimatized to laboratory conditions (12 h light/ dark cycles) 1 week before the experiment and had ad libitum food and water access. Animals were injected once per day with THC (3 mg/kg, i.p.) (Mato et al., 2004) or vehicle until the day before the experiment. Mice were killed 15–20 h after the last injection to avoid any interference caused by the in vivo prebound drug. The vehicle for THC was 5% cremophor-el/5% ethanol in NaCl 0.9%.
Slice preparation and electrophysiology. Whole-cell patch-clamp and extracellular field recordings were made from medium spiny neurons (MSNs) in parasagittal slices of mouse NAc. These methods have been described in detail previously (Robbe et al., 2001). In brief, mice were anesthetized with isoflurane and decapitated (all experimental procedures were in accordance with the Society for Neuroscience and European Union guidelines and were approved by the institutional animal care and use committees). The brain was sliced (300 μm) in the parasagittal plane using a vibratome (Integraslice; Campden Instruments, Loughborough, UK) and maintained in physiological saline at 4°C. Slices containing the accumbens and the PFCx were stored for at least 1 h at room temperature before being placed in the recording chamber and superfused (2 ml/min) with artificial CSF (ACSF) that contained (in mm) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, 1.2 NaH2PO4, and 11 glucose, and was equilibrated with 95% O2/5% CO2. All experiments were performed at room temperature. The superfusion medium contained picrotoxin (100 μm) to block GABAA receptors. All drugs were added at the final concentration to the superfusion medium.
For extracellular field recordings, the pipette was filled with ACSF. To evoke synaptic currents, stimuli (100–150 μs duration) were delivered at 0.05 Hz through a glass electrode filled with ACSF and placed at the PFCx–NAc border. Both the field EPSP (fEPSP) slope (calculated with a least square method) and amplitude were measured (graphs depict amplitudes). The glutamatergic nature of the extracellular fEPSP was confirmed at the end of the experiments through the application of the non-NMDA ionotropic glutamate receptor (GluR) antagonist CNQX (20 μm), which completely blocked the synaptic N2 component without altering the nonsynaptic N1 component (Hoffman et al., 2003).
For whole-cell patch-clamp experiments, neurons were visualized using an upright microscope with infrared illumination, and recordings were made with whole-cell electrodes containing the following (in mm): 128 K-gluconate, 20 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 2 Na2+-ATP, 0.3 Na+-GTP, and 10 glucose, buffered with 10 HEPES, pH 7.3 (osmolarity, 290 mOsm). Electrode resistance was 4–6 MΩ. Access-resistance compensation was not ordinarily used, and acceptable access resistance was <30 MΩ. The potential reference of the amplifier was adjusted to zero before breaking into the cell. In current-clamp experiments, the resting membrane potential was measured as soon as the whole-cell configuration was achieved, and a 13 mV estimated junction potential was subtracted from all membrane potential measurements (the junction potential was also measured in each experiment and matched the theoretical value that was calculated using the junction potential tool of the Clampex 9.2 software suite; Molecular Devices, Union City, CA). For current–voltage (I–V) curve analysis, voltages were measured at the end of 400 ms current-step episodes starting from –200 pA. The membrane input resistance was calculated in the linear range of the I–V curve around the resting membrane potential. The amount of inward rectification was estimated by subtracting the input resistance measured in the hyperpolarized range (at the voltage response evoked by a –200 pA current pulse) from the input resistance calculated around the resting membrane potential. The membrane decay time constant was calculated by fitting a simple exponential to a hyperpolarizing current pulse (–25 pA), which evoked voltage responses <5 mV to minimize the activation of voltage-gated ion channels (Clampfit 9.2; Molecular Devices). The hyperpolarization SAG amplitude was measured as the difference between the dip voltage of the SAG and the stable voltage response evoked by a –200 pA hyperpolarizing current pulse. The amplitude and latency of the first spike afterhyperpolarization (AHP) were defined as the voltage and time differences, respectively, from action potential threshold to the dip voltage of the AHP. An Axopatch 1D (Molecular Devices) was used to record the data, which were filtered at 1–2 kHz, digitized at 5 kHz on a DigiData 1200 interface (Molecular Devices) and collected on a personal computer using Clampex 9.2 (Molecular Devices) and analyzed using Clampfit 9.2 (Molecular Devices). Spontaneous miniature EPSCs (sEPSCs) were recorded in the whole-cell voltage-clamp configuration using Axoscope 9.2 (Molecular Devices). sEPSC amplitude and interinterval time were detected and analyzed using Clampfit 9.2 (Molecular Devices). For this analysis, a template of sEPSCs generated from averaging several typical synaptic events was slid along the data trace one point at a time. At each position, this template is optimally scaled and offset to fit the data, and a detection criterion is calculated. The detection criterion is the template-scaling factor divided by the goodness-of-fit at each position. An event is detected when this criterion exceeds a threshold and reaches a sharp maximum. Input–output curves in the whole-cell patch-clamp configuration were built by delivering stimuli (120 μs duration; stimulus intensity from 0 to 0.4 mA) at 0.2 Hz through a glass electrode filled with ACSF and placed at the PFCx–NAc border. For the estimation of the paired-pulse ratio (PPR), two stimuli were delivered with a 50 ms interval at stimulus intensity ∼40–60% of the one eliciting the maximal response (∼0.2 mA). The PPR was calculated from 30 consecutive episodes by dividing the amplitude of the EPSC evoked by the second stimulus by the amplitude of the first EPSC. A –2 mV hyperpolarizing pulse was applied before each evoked EPSC to evaluate the access resistance, and those experiments in which this parameter changed >20% were rejected. Both the EPSC area and amplitude were measured (graphs depict amplitudes). Recordings were made in the rostral–medial dorsal accumbens close to the anterior commissure.
Biochemical assays. For autoradiographic studies, the brains were rapidly removed and immersed in isopentane at –35°C. Twenty coronal sections (20 μm) were obtained in a cryostat, mounted onto gelatincoated slides, and stored at –20°C until use.
CB1R autoradiography. Slides were incubated for 2 h at 37°C in a buffer containing 50 mm Tris-HCl, 5% BSA, pH 7.4, and 3 nm (–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]trans-4-(3-hydroxypropyl)cyclohexanol ([3H]CP55,940) (specific activity, 165 Ci/mmol). Nonspecific binding was determined in adjacent sections by coincubation with 10 μm R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl)methanone monomethane-sulfonate (WIN55,212-2). Unbound radioligand was removed by washing the slides twice for 2 h each at 4°C in a buffer containing 50 mm Tris-HCl and 1% BSA, pH 7.4. After drying on a cold air stream, auto-radiograms were generated by apposing the tissues for 15 d at 4°C to tritium-sensitive films (Hyperfilm- 3H; GE Healthcare, Little Chalfont, UK) together with 3H-polymer standards. The films were scanned, and images were analyzed with NIH ImageJ 1.29 software. Optical densities were then evaluated by comparison with the microscale standards and expressed in femtomoles per milligram tissue equivalent.
CB1R agonist-stimulated [35S]GTPγS autoradiography. Slides were incubated in the assay buffer that contained the following (in mm): 50 Tris-HCl, 0.2 EGTA, 3 MgCl2, 100 NaCl, 1 DTT, 2 GDP, and 0.5% BSA, pH 7.7, for 30 min at 25°C. Basal [35S]GTPγS binding was defined by subsequent incubation of the slides for 120 min at 25°C in the same buffer containing 0.04 nm [35S]GTPγS. CB1R-stimulated [35S]GTPγS binding was evaluated by incubation of consecutive sections in the presence of WIN55,212-2 (0.01–100 μm). Additional sections were coincubated in the presence of 100 μm WIN55,212-2 and 10 μm N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide (SR141716A) for the antagonism condition or with 1 μm SR141716A to assess the presence of residual THC. Nonspecific binding was determined using 10 μm GTPγS. The slides were washed twice for 15 min each in 50 mm Tris-HCl buffer, pH 7.4, at 4°C, dried, and exposed for 48 h at 4°C to β-radiation-sensitive films (Hyperfilm-βmax; GE Healthcare) together with 14C-polymer standards (14C microscale standards). Autoradiograms were scanned, and images were analyzed using NIH ImageJ 1.29 software. Optical densities were corrected to nanocurie per gram tissue equivalent by comparison with the microscale standards, and WIN55,212-2 stimulation values were expressed as percentage of over basal activity.
Confocal immunofluorescence. For these experiments, a particular fixative was used to protect from suppression of immunoreactivity (Neki et al., 1996). Two male C57BL/6 mice were anesthetized with 4% chloral hydrate (1 ml/100 g body weight) and then transcardiacally perfused with 200 ml of 10 mm phosphate-buffered 0.9% saline (PBS), followed by 300 ml of a mixture containing 0.2% formaldehyde and 75% saturated picric acid in 0.1 m PBS, pH 7.3, at room temperature (20–25°C). The brains were then removed and fixed for3dat4°Cina mixture containing 2% formaldehyde and 75% saturated picric acid in 0.1 m PBS, pH 7.3.
Parasagittal vibratome sections (100 μm) containing the nucleus accumbens were cut and collected in 0.1 m PBS at room temperature. The slices were washed and blocked in 0.1 m PBS containing 3% calf serum, 0.5% Triton X-100, and 0.025% azide for ∼60 min and incubated with primary antibody for 2 d at 4°C. A monoclonal mouse antibody against mGluR2 (a generous gift from Dr. Ryuichi Shigemoto, National Institute for Physiological Sciences, Myodaiji, Okazaki, Japan) was used at 1:500, prepared in PBS blocking solution. Polyclonal rabbit antibodies against CB1Rs (Chemicon, Temecula, CA) were used at 1.5 μg/ml. Slices were then washed in 0.1 m PBS and incubated with Alexa dye-conjugated secondary antibodies (Invitrogen, Eugene, OR) for 24 h at 4°C at 1:650. Slices were then washed in 0.1 m PBS and then mounted in hard-set mounting medium (Vector Laboratories, Burlingame, CA), cover-slipped, and imaged on a laser-scanning confocal microscope [Leica Microsystems (Nussloch, Germany) TCS sp2 AOBS]. Photomicrographs were taken and presented using Adobe Photoshop 8 (Adobe Systems, San Jose, CA).
Data analysis and materials. All values are given as mean ± SEM. For biochemical and field recording experiments, the data (slices/sections) obtained from each animal tested were averaged so that n corresponds to the number of mice tested in each set of experiments. In patch-clamp experiments, n corresponds to the number of cells analyzed, with at least four animals included in each condition. Statistical significance between groups was tested using unpaired Student's t tests. Kolmogorov–Smirnov test was used for the statistical comparison of the cumulative distributions. All statistical tests were performed with Kyplotβ13 (Koichi Yoshioka, Department of Biochemistry and Biophysics, Tokyo Medical and Dental University, Tokyo, Japan) using a critical probability of p < 0.05.
Drugs used. THC, picrotoxin, GDP, GTPγS, and Cremophor EL were from Sigma (St. Louis, MO); CNQX, CP55,940, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 2-(S)-α-ethylglutamic acid (eGlu), and 2-amino-2-(2-carboxycyclopropan-1-yl)-3-(dibenzopyran-4-yl) propanoic acid (LY341495) were from Tocris Cookson (Ellisville, MO). [3H]CP55,940 and [35S]GTPγS were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Radiation-sensitive films and microscale standards were purchased from GE Healthcare. SR141716A was a generous gift from Sanofi Recherche (Montpellier, France). (+)-2-aminobicyclo-[3.1.0]hexane-2,6-dicarboxylic acid (LY354740) was a gift from Drs. Darryl D. Schoepp and Jim A. Monn (Eli Lilly, Indianapolis, IN). Other chemicals were from the highest commercial grade available.
Results
LTD is abolished after a single in vivo THC exposure but recovers after repeated treatment
Repetitive stimulation of PFCx afferents to the NAc (10 min at 13 Hz) induces an endocannabinoid/CB1R-mediated LTD of glutamatergic inputs that can be observed when recording MSNs in the whole-cell patch-clamp configuration, as well as when recording extracellular field responses in the NAc (Robbe et al., 2002a). Previous data indicate that this form of long-term plasticity is completely abolished when CB1Rs were desensitized in response to a single in vivo THC exposure (3 mg/kg) (Mato et al., 2004). Cannabis derivatives are often abused for long periods of time, and LTD in the NAc may play an important role in the adaptations induced by long-term exposure to addictive drugs. Thus, we decided to investigate the consequences of a more prolonged exposure to THC on this form of endocannabinoid-mediated synaptic plasticity. In contrast to the absence of LTD after a single in vivo exposure to THC (Fig. 1A, 1d THC), LTD was readily observed in the NAc of mice injected with THC for 7 consecutive days (Fig. 1B,C, 7d THC), showing that repetitive, but not unique, exposure to THC triggers compensatory mechanisms. This latest finding is in apparent contrast with a recent report showing the absence of the LTD induced by a milder stimulation protocol (5 min at 10 Hz) in the NAc of rats chronically exposed to THC (Hoffman et al., 2003). Nevertheless, and in agreement with the findings of Hoffman et al. (2003), the LTD induced in response to this shorter cortical stimulation (5 min at 10 Hz) was also completely abolished in mice treated for 1 week with 3 mg/kg THC (Fig. 1D, 7d THC), showing a correlation between the intensity of cortical afferent activity and the apparent level of recovery.
Figure 1.

Activity-dependent LTD in the NAc is blocked after a single in vivo exposure to THC but recovers after repeated treatment. A, Representative experiments showing blockade of activity-dependent LTD in the accumbens after a single in vivo exposure to THC (1d THC). Stimulation of PFCx afferents to the NAc (10 min at 13Hz) induced LTD of the fEPSP in vehicle-treated mice (sham) that was absent after a single in vivo exposure to THC (1d THC, 3 mg/kg). Averaged traces taken at the times indicated by numbers are shown in the insets. Calibration: 0.2 mV (y-axis), 10 ms (x-axis). B, Typical experiments and sample traces showing recovery of 13 Hz-induced LTD in the accumbens of mice treated in vivo with THC for 7 d (7d THC). Calibration: 0.2 mV (y-axis), 20 ms (x-axis). C, Summary data of all of the experiments performed: the LTD induced in the NAc by 10 min at 13Hz stimulation of PFCx afferents does not differ between mice treated for 7 d with THC (7d THC, n = 8) or vehicle (sham, n = 7, p > 0.05). D, Compensatory adaptation of LTD is apparent in response to long, but not short, cortical stimulation. The data show the summary of all of the experiments performed: LTD induced by a 5 min at 10 Hz tetanus did not recover in slices prepared from mice repeatedly exposed to THC (7d THC, n = 4) compared with vehicle-treated mice (sham, n = 3).
Repeated in vivo THC exposure leads to functional and biochemical reduction of CB1R-mediated functions
A single exposure to THC induces functional tolerance of CB1Rs both in the NAc and the hippocampus, providing a mechanism explaining the abolishment of endocannabinoid-mediated synaptic plasticity in these areas (Mato et al., 2004). Functional tolerance to the synaptic effects of cannabinoids has also been shown to occur in the rat NAc after 1 week of THC exposure (Hoffman et al., 2003). Consistent with these data, we found that the maximal ability of the cannabinoid agonist CP55,940 to inhibit synaptic transmission was clearly reduced in NAc slices from mice chronically exposed to THC exposure (Fig. 2A, 7d THC). This functional tolerance to the inhibitory effects of cannabinoid agonist on synaptic transmission could be explained by the presence of residual THC prebound to CB1Rs. This possibility was excluded based on the fact that bath application of the cannabinoid CB1R antagonist SR141716A did not increase synaptic transmission in the accumbens of THC-treated mice (Fig. 2 B).
Figure 2.

Repeated THC exposure desensitizes CB1R in the NAc. A, Dose–response curves showing the inhibition of the fEPSP amplitude by the cannabinoid agonist CP55,940 in the NAc of mice repeatedly exposed to THC (n = 3–5) or vehicle (n = 3–5). Reduced CP55,940-mediated inhibition of accumbens fEPSP was observed in THC-treated mice (*p < 0.05). B, Perfusion of SR141716A (SR-1; 1μm) did not affect baseline synaptic transmission in the NAc of THC-treated (black circles; n = 3) or vehicle-treated (white circles; n = 3) mice, indicating the absence of residual THC prebound to CB1R. C, CB1R autoradiographic labeling in the accumbens after repeated exposure to THC or vehicle. D, Stimulation of [35S]GTPγS binding by the cannabinoid agonist WIN55,212-2 in the NAc of mice treated with THC (n = 9) or vehicle (n = 10) for 7 d. Impaired ability of the cannabinoid agonist to stimulate [35S]GTPγS binding was observed after repeated exposure to THC (*p<0.05).E, Absence of residual THC in brain sections after 7 d exposure. Similar basal [35S]GTPγS binding levels were measured in the NAc of sham (white bar; n = 10) and 7 d THC-treated (black bar; n = 9) mice, and incubation with the CB1R antagonist SR-1 (1 μm) did not increase basal [35S] GTPγS binding in the NAc of THC-injected mice (gray bar; n = 8).
The uncoupling and downregulation of brain CB1Rs in response to prolonged THC exposure play a key role in the development of cannabinoid tolerance (Maldonado, 2002; Martin et al., 2004), and these mechanisms could be underlying the impaired functionality of CB1Rs in the NAc of mice chronically exposed to THC. To test this possibility, CB1R density was first analyzed in the NAc of THC-treated mice by autoradiography with the cannabinoid agonist [3H]CP55,940. As shown in Figure 2C, the administration of THC for 7 d produced a slight decrease of CB1R binding in the mouse NAc (16%), which was close to statistical significance (p = 0.051). Consistent with these data, 1 week of THC treatment markedly reduced the ability of the cannabinoid agonist WIN55,212-2 to stimulate [35S]GTPγS binding in the mouse NAc (Fig. 2D). The possibility that this finding could be explained by the presence of residual THC in brain sections was excluded because basal [35S]GTPγS binding was not increased in the NAc of THC-treated mice, and incubation with the CB1R antagonist SR141716A (1 μm) did not reduce basal [35S]GTPγS binding (Fig. 2E). Finally, the stimulatory effect of WIN55,212-2 (100 μm) in this assay was fully prevented by coincubation with SR141716A (10 μm) in brain sections from both sham and THC-treated mice. In the NAc, the percentage of stimulation of [35S]GTPγS binding by 100 μm WIN55,212-2 was 100.1 ± 7.3% versus 9.78 ± 3.36% in the presence of 10 μm SR141716A in sham (p < 0.05) and 69.72 ± 10.32% versus 8.7 ± 2.3% in THC-treated mice (p < 0.05), indicating the involvement of CB1Rs in the cannabinoid agonist effect. Together, these data indicate that reduced coupling efficiency of CB1Rs to Gi/o transduction proteins may underlie the observed loss of CB1R-mediated synaptic function in the NAc after prolonged exposure to THC.
Basal synaptic strength and release probability at the PFCx–NAc synapses and the electrophysiological properties of MSNs are not altered by repeated in vivo exposure to THC
Altered slice viability after prolonged THC treatment might also play a role in the observed tolerance to the synaptic effects of cannabinoid agonists in the mouse NAc. To test this possibility, NAc MSNs were recorded in the whole-cell voltage-clamp configuration to evaluate the consequences of the 7 d in vivo treatment THC on basal synaptic transmission and synaptic strength at the PFCx-MSNs synapses. Input-output curves for evoked EPSCs were similar between the two groups of mice, suggesting that in the NAc, the efficacy of synaptic transmission is unchanged after prolonged THC-exposure (Fig. 3A) (for similar experiments using extracellular field recordings, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Subsequently, we compared the paired-pulse ratio (PPR), a form of plasticity highly sensitive to presynaptic modulations, in sham and treated mice. Similar baseline PPR were measured after repeated in vivo exposure to vehicle (3.07 ± 0.34, n = 14) and THC (2.85 ± 0.23, n = 14), suggesting that THC treatment does not alter the basal release probability at the PFCx-NAc synapses (Fig. 3B). Finally, we found no differences in the frequency or the amplitude of miniature sEPSCs recorded from MSNs of both animal groups (Fig. 3C–E). Together, these data strongly suggest that repeated THC treatment does not modify the basic properties of synaptic transmission at the glutamatergic synapses between the PFCx and the NAc.
Figure 3.
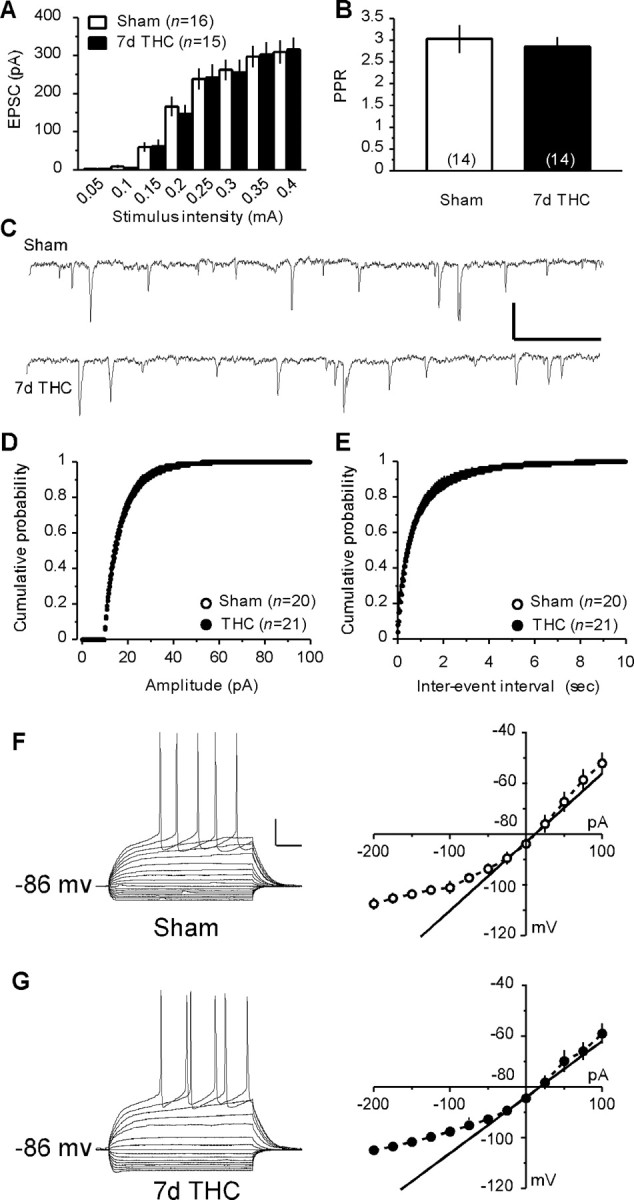
Repeated exposure to THC alters neither basal synaptic strength and release probability at the PFCx–NAc synapses nor the basic electrophysiological properties of MSNs. A, Similar input–output curves were obtained when stimulating PFCx afferences and recording the EPSCs evoked in accumbens MSNs from vehicle-treated (sham, n=16) and THC-treated (7d THC, n=15) mice.B, The PPR of the EPSCs did not differ between the MSNs from sham (n=14) and 7 d THC-treated (n = 14) mice. C, Representative 3 s sweeps showing the sEPSCs recorded from MSNs in the NAc of sham and 7 d THC-treated mice. Calibration: 20 pA (y-axis), 200 ms (x-axis). D, E, The distribution of the sEPSCs amplitude (D) and interevent interval (E) did not differ between MSNs from sham (n = 20) and 7 d THC-treated (n = 21) mice. F, G, Typical responses to hyperpolarizing and depolarizing somatic current pulses of an MSN in the NAc of a sham (F) and a 7 d THC-treated (G) mouse. Calibration: 20 pA (y-axis), 50 ms (x-axis). Similar average I–V curves were calculated from MSNs recorded in the NAc of sham (n = 8) and THC-treated (n = 11) mice.
Although anatomical and functional evidence suggest a general presynaptic localization of CB1Rs in the brain, including the NAc (Manzoni and Bockaert, 2001; Robbe et al., 2001, 2002a), postsynaptic actions of cannabinoids have also been proposed (Hoffman and Lupica, 2001; Bacci et al., 2004; Pickel et al., 2004). Therefore, the possibility that long-term exposure to cannabinoids modified the basic electrophysiological characteristics of MSNs in the mouse NAc core could not be completely excluded. To address this question, we decided to compare the basic electrophysiological features of MSNs in the NAc core from mice chronically treated with THC or vehicle. The neurons recorded in this study were identified as medium spiny based on their hyperpolarized resting membrane potentials (–75 to –85 mV), slowly depolarizing ramps in response to subthreshold depolarizing current pulses and marked inward rectification (Kawaguchi, 1997). As shown in Figure 3, F and G, the average I–V curves obtained in MSNs from both animal groups were similar. Detailed analysis of the passive membrane properties (resting membrane potential/membrane input resistance/inward rectification/membrane decay time constant/SAG amplitude) yielded similar values in both groups of animals (Table 1). Similarly, there was no difference in the discharge properties (firing threshold/first spike amplitude/first spike AHP amplitude/latency of first spike AHP) of MSNs between both groups (Table 1). These data strongly suggest that repeated in vivo exposure to THC does not alter the basic electrophysiological properties of MSNs in the mouse NAc.
Table 1.
Repeated exposure to THC does not alter the basic electrophysiological properties of NAc core MSNs
|
|
Sham (n = 8) |
7 d THC (n = 11) |
p |
|---|---|---|---|
| Resting membrane potential (mV) | -83.6 | -84.5 | 0.78 |
| Membrane input resistance (MΩ) | 269.8 | 221.3 | 0.40 |
| Inward rectification (MΩ) | 150.8 | 119.8 | 0.45 |
| Membrane decay time constant (ms) | 22.2 | 20.3 | 0.68 |
| SAG amplitude (mV) | 1.22 | 1.38 | 0.67 |
| Firing threshold (mV) | -44.7 | -43.9 | 0.69 |
| First spike amplitude (mV) | 73.1 | 75.5 | 0.39 |
| First spike AHP amplitude (mV) | 11.9 | 11.2 | 0.85 |
| Latency of first spike AHP (ms) |
10.7 |
9.4 |
0.14 |
Detailed comparison of passive membrane properties and discharge properties of MSNs revealed no difference between sham and THC-treated mice (p value is given for each parameter analyzed).
CB1Rs do not mediate LTD in THC-treated mice
Thus, in marked contrast to the absence of endocannabinoid-mediated LTD after a single in vivo exposure to THC, long-term depression can still be observed in the NAc after a prolonged exposure to the drug, which induces marked CB1R desensitization. How can a form of plasticity normally mediated by CB1Rs be observed in a structure in which these proteins display a substantial loss of function (Fig. 3)? To address this apparent discrepancy, we decided to test the involvement of CB1Rs in the LTD observed after tetanic stimulation in the NAc of THC-treated mice. As described previously in naive and 1 d sham animals (Robbe et al., 2002a; Mato et al., 2004), bath application of the CB1R antagonist SR141716A (1 μm) prevented LTD induction in 1 week sham-injected mice (Fig. 4A, 1d THC). In contrast, the CB1R antagonist was without effect in NAc slices prepared from chronic THC-treated mice (Fig. 4A, 7d THC), demonstrating that, in these animals, LTD at PFCx–NAc synapses does not rely on CB1Rs (Fig. 4B). We concluded that a consequence of long-term exposure to THC is to engage compensatory mechanisms, allowing LTD expression despite the reduction of CB1R functionality. Finally, we found that the compensatory mechanism was reversible during withdrawal, as perfusion of SR141716A (1 μm) abolished LTD expression in NAc slices prepared 1 week after the last injection of THC (Fig. 4C).
Figure 4.

CB1Rs do not mediate activity-dependent LTD in the NAc of 1 week THC-treated mice. A, The CB1R antagonist SR141716A (SR-1; 1 μm) abolished 10 min at 13 Hz-induced LTD in the NAc of mice treated for 7 d with vehicle but not in the THC-treated group (*p < 0.05). Sample fEPSPs taken at the times indicated by numbers are shown in the insets. Calibration: 0.2 mV (y-axis), 20 ms (x-axis). B, Summary bar graph showing the effects of CB1R blockade on the LTD induced by 10 min at 13 Hz-stimulation of PFCx afferences in the NAc of vehicle-treated mice (*p < 0.05) and after repeated exposure to THC. C, Recovery of CB1R-mediated LTD in the NAc 1 week after repeated THC treatment. SR-1 (1 μm) abolished 10 min at 13 Hz-induced LTD in slices from animals taken 1 week after the last THC injection (*p < 0.05).
mGluR2/3 metabotropic glutamate receptors mediate LTD in THC-treated mice
The PFCx–accumbens synapses express postsynaptic NMDA receptor-dependent LTD (Thomas et al., 2000) as well as two other different forms of presynaptic LTD, mediated by either CB1Rs or metabotropic glutamate mGluR2/3 receptors (Robbe et al., 2002a,b). The LTD induced by pharmacological activation of presynaptic mGluR2/3 can be observed when recording MSNs in the whole-cell patch-clamp configuration as well as when recording extracellular field responses in the NAc (Robbe et al., 2002b). The fact that both CB1R- and mGluR2/3-dependent LTDs are expressed presynaptically at PFCx–NAc synapses and that both receptors are coupled to Gi/o-proteins (Conn and Pin, 1997; Howlett, 2002) prompted us to test for possible interactions between these two forms of LTD in slices prepared from naive mice. As shown in Figure 5A, the 10 min at 13 Hz protocol failed to induce LTD in slices in which mGluR2/3–LTD has already been induced pharmacologically by bath-application of the specific mGluR2/3 agonist LY354740 (200 nm; 10 min) (Robbe et al., 2002b). Conversely, the mGluR2/3 agonist was unable to induce LTD after saturation of eCB–LTD (Fig. 5B). Together, these data show for the first time that eCB–LTD and mGluR2/3–LTD in the NAc can occlude each other, despite being mediated by different receptors.
Figure 5.

Endocannabinoid-mediated LTD and mGluR2/3-mediated LTD occlude each other in naive animals. A, The data show the summary of all of the experiments performed as follows: mGluR2/3-mediated LTD was first induced by perfusion with the mGluR2/3 agonist LY354740 (200 nm, 10 min) (Robbe et al., 2002c), and the 10 min at 13 Hz-tetanus (Robbe et al., 2002b) was given 70 min thereafter. In these conditions, bath application of LY354740 reliably induced LTD of evoked transmission (fEPSP was 73 ± 3% 50–55 min after LY354740; n = 5) but endocannabinoid-mediated (10 min at 13 Hz) LTD was never observed afterward [fEPSP was 105 ± 8.1% 50–55 min after tetanus (n = 5) in experiments in which mGluR2/3-LTD was induced first with LY354740 versus 78.9 ± 5.6% in control (n = 10); p < 0.05], showing the complete occlusion between both forms of plasticity. B, Conversely, mGluR2/3-dependent LTD was also occluded in slices in which endocannabinoid-mediated LTD was induced first. The data show the summary of all of the experiments performed as follows: perfusion of LY354740 (200 nm, 10 min) after saturation of 10 min at 13 Hz-induced LTD (fEPSP was 83 ± 2.3% 50–55 min after tetanus; n = 11) results in acute inhibition but not in LTD (fEPSP was 94.8 ± 3% 50–55 min after LY354740 in experiments in which CB1R–LTD was induced first by tetanus saturation; n = 11 vs 78.9 ± 5.6%; n = 10 in control; p < 0.05), showing the occlusion of mGluR2/3-dependent LTD after saturation of endocannabinoid-mediated LTD.
The mutual occlusion observed between eCB–LTD and mGluR2/3–LTD prompted us to test the hypothesis of a colocalization of these receptors at PFCx–MSN synapses by confocal microscopy. Previous studies have shown mRNAs coding for CB1R (Mailleux and Vanderhaeghen, 1992), mGluR2 (Ohishi et al., 1993a), and mGluR3 (Ohishi et al., 1993b) in projecting frontal pyramidal neurons. Indeed, we observed CB1R and mGluR2 immunoreactivities colocalizing in layers V–VI cortical neurons of the prefrontal cortex (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In addition, numerous CB1R- and mGluR2-immunopositive axon-like fibers in the neuropil crossed the cortical layers perpendicularly to the labeled cell bodies (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Thus, projecting prefrontal cortical fibers were likely to be main contributors to the CB1R (Fig. 6, left, green) and mGluR2 (Fig. 6, middle, red) immunoreactivities observed in the core of the nucleus accumbens. Actually, CB1R and mGluR2 were colocalized in both axons and in small dots reminiscent of synaptic terminals (Fig. 6, right, merged image). Both receptors were also expressed in neuronal cell bodies and dendrites in the nucleus.
Figure 6.

Immunolocalization of CB1R and mGluR2 in the core of the nucleus accumbens. Representative confocal images showing labeling for CB1R (green, left) and mGluR2 (red, middle) in the neuropil of the core of the accumbens. As seen in the merged picture (right), both receptors colocalize in axons (arrow) and punctates reminiscent of synaptic terminals (arrowheads). Scale bar, 10 μm.
Based on these observations, we decided to test the possibility that mGluR2/3 substituted for the desensitized CB1Rs and mediated LTD in THC-treated mice. As reported previously in naive mice (Robbe et al., 2002a), LTD induced by the 10 min at 13 Hz protocol was not sensitive to the highly specific mGluR2/3 antagonist LY341495 in sham mice (200 nm) (Fig. 7A,B). In marked contrast, the mGluR2/3 antagonist completely abolished LTD in slices prepared from animals repeatedly exposed to THC (Fig. 7A,B). Similar results were obtained with eGlu (200 μm), another selective mGluR2/3 antagonist (Robbe et al., 2002b) (Fig. 7B). To evaluate whether other presynaptic Gi/o-protein-coupled receptors play the same role as mGluR2/3, we tested the role of A1 receptors (described previously at these synapses) (Manzoni et al., 1998) on LTD in sham and 1 week THC-treated mice. As shown in supplemental Figure 4 (available at www.jneurosci.org as supplemental material), the specific A1 antagonist DPCPX (200 nm) had no effect of 13 Hz-induced LTD in both sham and THC-treated mice in agreement with a specific role of mGluR2/3 in THC-induced homeostatic plasticity. Together, our data are consistent with the idea that in THC-treated mice mGluR2/3 replace CB1Rs to induce LTD in response to sustained, but not mild (Fig. 1), cortical activity.
Figure 7.

mGluR2/3 mediates activity-dependent LTD in the NAc after repeated in vivo THC administration. A, The mGluR2/3 antagonist LY341495 (200 nm) prevented the expression of 10 min at 13 Hz-induced LTD in NAc slices from THC-treated mice (n = 7) but not in the vehicle-treated group (n = 6) (*p < 0.01). B, Summary bar graph of the effects of the mGluR2/3 antagonists LY341495 (200 nm, black bars) and eGlu (200 μm, gray bars) on 10 min at 13 Hz-induced LTD in the NAc of mice treated with THC or vehicle for 7 d(*p < 0.05).
Discussion
The present study reports several novel findings regarding the adaptations induced by prolonged cannabinoid exposure on the physiology of the NAc. First, repeated in vivo treatment with a nonaversive dose of THC induces functional tolerance and desensitization of CB1Rs in the NAc core without altering the basic electrophysiological properties of MSNs in this area. Second, although a single exposure to THC completely abolishes endocannabinoid-mediated LTD in the NAc (Mato et al., 2004), prolonged treatment engages compensatory mechanisms, allowing the recovery of activity-dependent LTD at PFCx–MSNs synapses despite compromised CB1R function. Third, these compensatory mechanisms involve mGluR2/3. Together, these data reveal a new form of synaptic adaptation in response to prolonged exposure to cannabinoids.
The observation that the ability of cannabinoid agonist to inhibit glutamatergic transmission at PFCx–NAc synapses is reduced after repeated exposure to THC suggests that this treatment induces functional tolerance of CB1Rs. This tolerance could be explained by the presence of residual THC prebound to CB1Rs, the existence of deficits in the slices from THC-treated animals, or by THC-induced downregulation and/or desensitization of CB1Rs. Our data argue against the first hypothesis, because bath application of the CB1R antagonist SR141716A did not increase baseline synaptic responses and did not reduce the basal binding of [35S]GTPγS in THC-treated mice. Furthermore, no difference was detected when comparing basal [35S]GTPγS binding levels in the NAc of THC-treated and sham mice. It is also unlikely that tolerance to cannabinoids is attributable to compromised viability of the slices after repeated in vivo THC because neither baseline synaptic transmission nor the basic electrophysiological properties of NAc MSNs were altered after chronic THC.
Downregulation and desensitization of CB1Rs have been consistently involved in development of cannabinoid tolerance (Maldonado, 2002; Martin et al., 2004). Here, in vivo administration of a low THC dose resulted in decreased CB1R stimulation of [35S]GTPγS binding, indicating a loss of CB1R ability to activate Gi/o-proteins. Because the activation of Gi/o-proteins is the first step in the CB1R signaling cascade, the reduced ability of the cannabinoid agonist to stimulate [35S]GTPγS binding indicates the functional uncoupling of CB1Rs from their whole transduction pathway, which may underlie the functional tolerance to the synaptic effects of CB1R activation in the NAc. The observed uncoupling of CB1R from Gi/o-proteins was accompanied by little reduction of CB1R binding, consistent with the established concept that desensitization and downregulation are separate processes and that desensitization precedes downregulation. Alternatively, a small loss of CB1R resulting from repeated cannabinoid exposure could theoretically contribute to the observed decrease in CB1R-activated Gi/o-proteins, which would indicate the absence of receptor reserve for CB1Rs in the NAc, as suggested previously for other brain regions (Breivogel et al., 1999). Nevertheless, the percentage of putative internalized receptors (16%) does not account by itself for the observed amount of desensitization (30%), suggesting that the loss of CB1R functionality does not reflect only receptor downregulation.
Although detailed studies addressing CB1R expression and coupling in mouse brain after repeated exposure to low THC doses have not been published, our data are in general agreement with previous work. First, the slight reduction in CB1R density is in concordance with a report showing comparable decreases of CB1R binding in the rat NAc after exposure to a dose of THC (5 mg/kg) close to the one used here (Romero et al., 1998). Second, the fact that repeated exposure to THC induced a clear desensitization of CB1Rs in the NAc and only a marginal reduction of CB1R density is in agreement with previous data showing a smaller decrease in CB1R binding than in CB1R coupling to Gi/o-proteins in the rat brain (Breivogel et al., 1999).
Single in vivo exposure to THC blocks endocannabinoid LTD by inducing functional tolerance of CB1Rs (Mato et al., 2004). Consistent with the existence of functional tolerance and desensitization of CB1Rs after repeated drug exposure, a CB1R antagonist did not prevent expression of LTD in THC-treated mice, indicating that CB1Rs are no longer involved in this form of plasticity. A plausible candidate for the mediation of this activity-dependent plasticity in THC-treated mice was mGluR2/3, based on the fact that these receptors are coupled to Gi/o-proteins, presynaptically expressed at PFCx afferents to the NAc, and that their activation induces LTD (Robbe et al., 2002b). Furthermore, the LTDs induced by activation of CB1Rs and mGluR2/3 at PFCx–NAc synapses are mutually occlusive, and immunohistochemical data indicate that both receptor types are coexpressed on the same axons.
The mechanisms by which mGluR2/3 replaces CB1R, allowing the induction of LTD at PFCx–NAc synapses, remain unknown. A putative upregulation of mGluR2/3 after prolonged THC exposure could account for the observed result. Indeed, increases in the mGluR2/3 content have been reported previously in the NAc after chronic exposure to cocaine (Xi et al., 2002). This possibility was tested by Western immunoblotting of mGluR2/3 in NAc homogenates, with negative results (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Biotinylation assays were subsequently performed in an attempt to compare the membrane expression of mGluR2/3 in the NAc of sham and THC-treated mice, but consistent labeling of mGluR2/3 could not be obtained using this method, probably because of a low level of membrane expression. Moreover, increased mGluR2/3 protein expression in the NAc after chronic cocaine exposure is accompanied by decreased mGluR2/3 function, suggesting that the changes in mGluR2/3 expression level may not always reflect the alterations in their functional activity (Xi et al., 2002).
A second attractive hypothesis is that desensitization of CB1Rs allows enhanced signaling through mGluR2/3. Indeed, and in contrast to mGluR2/3, CB1R can sequester Gi/o-proteins and thus prevent signaling from neighboring Gi/o-protein-coupled receptors (Vasquez and Lewis, 1999). Both CB1Rs and mGluR2/3 are presynaptically located on the cortical afferents to the NAc (Robbe et al., 2001, 2002b), they colocalize at the same axon terminals in the NAc (present report), their activation induces presynaptic forms of LTD at PFCx-NAc synapses, and both forms of plasticity occlude each other (present report), suggesting that both receptors use a common pathway for the induction of LTD. Although the level of the signaling pathway at which occlusion takes place is not clear, it is possible that competition for Gi/o-proteins might be involved (Vasquez and Lewis, 1999). A tempting hypothesis is that the uncoupling/downregulation of CB1Rs that follows prolonged THC exposure enhances the amount of Gi/o-proteins available for mGluR2/3 and that the after facilitation of mGluR2/3 signaling rescues LTD.
It is particularly interesting to note that the rescue mechanism described here only functions in response to sustained cortical activities: eCB–LTD induced by the 5 min at 10 Hz protocol is not rescued by mGluR2/3 receptors (the present study and Hoffman et al., 2003). One possibility is that mild cortical stimulations do not release enough glutamate to allow the activation of presynaptic mGluR2/3 and engage the rescue mechanism. In support of this idea, presynaptic mGluR2/3 are usually located in membrane compartments distant from active release sites (Shigemoto et al., 1997; Tamaru et al., 2001), and electrophysiological studies support the concept that glutamate spillover is necessary for synaptic activation of mGluR2/3-mediated negative feedback on glutamate release. In this line of evidence, Scanziani et al. (1997) showed that the occupancy of presynaptic mGluR2 in the rat mossy fiber pathway is frequency dependent, because selective blockade of these receptors enhanced excitatory transmission only in conditions of high-frequency stimulation. Nevertheless, conditions for the occupancy of presynaptic mGluR2/3 on glutamatergic terminals may depend on the synapses involved, because these receptors seem to be tonically active at synapses onto layer V pyramidal neurons in the rat prefrontal cortex (Marek et al., 2000). Although the extrasynaptic localization of presynaptic mGluR2/3 on excitatory afferents from the PFCx has not been thoroughly demonstrated, electrophysiological data support the concept that glutamate spillover is required for the synaptic activation of these receptors in the mouse NAc: bath application of the selective mGluR2/3 antagonist LY341495 (200 nm) did not increase baseline fEPSP responses in the NAc under conditions of low-frequency stimulation (0.05 Hz) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In contrast, and reminiscent of the study by Scanziani et al. (1997), perfusion of LY341495 drives PFCx–NAc synapses to express LTP, instead of LTD, in response to tetanic stimulation (Robbe et al., 2002b). These data indicate that synaptic activation mGluR2/3 at PLCx–NAc synapses is frequency dependent, supporting the idea that only when large amounts of glutamate are released in response to sustained cortical stimulation will these receptors be able to induce LTD.
The idea that an important consequence of exposure to addictive drugs is to modify synaptic plasticity in reward and learning-related brain areas is supported by studies in the accumbens, the ventral tegmental area, and the hippocampus (Tamaru et al., 2001; Thomas et al., 2001; Ungless et al., 2001; Robbe et al., 2002c; Thompson et al., 2002; Hoffman et al., 2003; Fourgeaud et al., 2004; Mato et al., 2004). Our data extend this concept by showing that alterations of synaptic plasticity induced by addictive drugs can be compensated by homeostatic processes that, at least in the case of the psychoactive ingredient of marijuana, rescue synaptic functions up to a certain point. How this new form of homeostatic synaptic plasticity relates to the behavioral aspects of addiction is beyond the goal of this study, but it is remarkable that pharmacological manipulations of both CB1R and mGluR2/3 modulate addictive-drug-evoked behaviors (Fundytus et al., 1997; Helton et al., 1997; Ledent et al., 1999; Vandergriff and Rasmussen, 1999; Cartmell et al., 2000; Cartmell and Schoepp, 2000; Racz et al., 2003).
In addition to its potential involvement in the brain response to prolonged cannabis abuse, the compensatory adaptation described here could be relevant in other situations leading to CB1R desensitization and/or downregulation, such as prolonged therapeutic use of drugs activating the cannabinoid system [for examples of exogenous cannabinoid agonist and inhibitors of endocannabinoid uptake and/or degradation, see Makriyannis et al. (2005)]. Following this line of reasoning, the homeostatic phenomenon that we report here might equally be important in physiological and/or pathological situations involving both mGluR2/3 (e.g., chronic stress) (Schoepp et al., 2003) and a reduction of CB1R functions.
Footnotes
Work in O.J.M.'s laboratory was supported by grants from Institut National de la Santé et de la Recherche Médicale, Fondation pour la Recherche Médicale, and Région Aquitaine. P.G.'s laboratory was supported by Ministerio de Ciencia y Tecnología Grant BFI2002-01474. We thank Drs. Monn and Shoepp at Eli Lilly (Indianapolis, IN) for their generous gift of LY354740, Dr. Ryuichi Shigemoto (National Institute for Physiological Sciences, Myodaiji, Okazaki, Japan) for the generous gift of the monoclonal mouse mGluR2 antibody, Dr. Angel Pazos (University of Cantabria, Spain) for his support with the autoradiography experiments, and G. Casassus and Dr. J. T. Williams for critical reading of a previous version of this manuscript.
Correspondence should be addressed to Olivier J. Manzoni, Institut National de la Santé et de la Recherche Médicale, U378 Equipe Avenir, Institut Magendie des Neurosciences, 146 rue Léo-Saignat, 33077 Bordeaux Cedex, France. E-mail: manzoni@bordeaux.inserm.fr.
D. Robbe's present address: Center for Molecular and Behavioral Neuroscience, Rutgers University, Newark, NJ 07102.
DOI:10.1523/JNEUROSCI.2294-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511619-09$15.00/0
References
- Bacci A, Huguenard JR, Prince DA (2004) Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431: 312–316. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ (1999) Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem 73: 2447–2459. [DOI] [PubMed] [Google Scholar]
- Burrone J, Murthy VN (2003) Synaptic gain control and homeostasis. Curr Opin Neurobiol 13: 560–567. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75: 889–907. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Monn JA, Schoepp DD (2000) The mGlu(2/3) receptor agonist LY379268 selectively blocks amphetamine ambulations and rearing. Eur J Pharmacol 400: 221–224. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hémar A, Worley PF, Manzoni OJ (2004) A single in vivo exposure to cocaine abolishes endocannabinoid mediated long-term depression in the nucleus accumbens. J Neurosci 24: 6939–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fundytus ME, Ritchie J, Coderre TJ (1997) Attenuation of morphine withdrawal symptoms by subtype-selective metabotropic glutamate receptor antagonists. Br J Pharmacol 120: 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ (1997) LY354740: a metabotropic glutamate receptor agonist which ameliorates symptoms of nicotine withdrawal in rats. Neuropharmacology 36: 1511–1516. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR (2001) Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol 85: 72–83. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Caulder T, Lupica CR (2003) Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci 23: 4815–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediat 68–69: 619–631. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC (2001) Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2: 695–703. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y (1997) Neostriatal cell subtypes and their functional roles. Neurosci Res 27: 1–8. [DOI] [PubMed] [Google Scholar]
- Koob GF (1996) Drug addiction: the yin and yang of hedonic homeostasis. Neuron 16: 893–896. [DOI] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283: 401–404. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ (1992) Localization of cannabinoid receptor in the human developing and adult basal ganglia. Higher levels in the striatonigral neurons. Neurosci Lett 148: 173–176. [DOI] [PubMed] [Google Scholar]
- Makriyannis A, Mechoulam R, Piomelli D (2005) Therapeutic opportunities through modulation of the endocannabinoid system. Neuropharmacology 48: 1068–1071. [DOI] [PubMed] [Google Scholar]
- Maldonado R (2002) Study of cannabinoid dependence in animals. Pharmacol Ther 95: 153–164. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Pujalte D, Williams J, Bockaert J (1998) Decreased presynaptic sensitivity to adenosine after cocaine withdrawal. J Neurosci 18: 7996–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Bockaert J (2001) Cannabinoids inhibit GABAergic synaptic transmission in mice nucleus accumbens. Eur J Pharmacol 412: R3–R5. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK (2000) Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J Pharmacol Exp Ther 292: 76–87. [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ, Selley DE (2004) Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci 25: 325–330. [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ (2004) A single in-vivo exposure to delta9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci 7: 585–586. [DOI] [PubMed] [Google Scholar]
- Neki A, Ohishi H, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N (1996) Pre- and postsynaptic localization of a metabotropic glutamate receptor, mGluR2, in the rat brain: an immunohistochemical study with a monoclonal antibody. Neurosci Lett 202: 197–200. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2002) Common molecular and cellular substrates of addiction and memory. Neurobiol Learn Mem 78: 637–647. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993a) Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience 53: 1009–1018. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993b) Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol 335: 252–266. [DOI] [PubMed] [Google Scholar]
- Pickel VM, Chan J, Kash TL, Rodriguez JJ, MacKie K (2004) Compartment-specific localization of cannabinoid 1 (CB1) and mu-opioid receptors in rat nucleus accumbens. Neuroscience 127: 101–112. [DOI] [PubMed] [Google Scholar]
- Racz I, Bilkei-Gorzo A, Toth ZE, Michel K, Palkovits M, Zimmer A (2003) A critical role for the cannabinoid CB1 receptors in alcohol dependence and stress-stimulated ethanol drinking. J Neurosci 23: 2453–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002a) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99: 8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni OJ (2002b) Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci 22: 4346–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Bockaert J, Manzoni OJ (2002c) Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci 16: 2231–2235. [DOI] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Manzanares J, Perez A, Corchero J, Fuentes JA, Fernandez-Ruiz JJ, Ramos JA (1998) Time-course of the cannabinoid receptor down-regulation in the adult rat brain caused by repeated exposure to delta9-tetrahydrocannabinol. Synapse 30: 298–308. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA (1997) Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 385: 630–634. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ (2003) LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6: 189–197. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N (1997) Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17: 7503–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R (2001) Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience 106: 481–503. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Malenka RC, Bonci A (2000) Modulation of long-term depression by dopamine in the mesolimbic system. J Neurosci 20: 5581–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217–1223. [DOI] [PubMed] [Google Scholar]
- Thompson AM, Gosnell BA, Wagner JJ (2002) Enhancement of long-term potentiation in the rat hippocampus following cocaine exposure. Neuropharmacology 42: 1039–1042. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Schmidt WJ (2003) Glutamatergic mechanisms in addiction. Mol Psychiatry 8: 373–382. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587. [DOI] [PubMed] [Google Scholar]
- Vandergriff J, Rasmussen K (1999) The selective mGlu2/3 receptor agonist LY354740 attenuates morphine-withdrawal-induced activation of locus coeruleus neurons and behavioral signs of morphine withdrawal. Neuropharmacology 38: 217–222. [DOI] [PubMed] [Google Scholar]
- Vasquez C, Lewis DL (1999) The CB1 cannabinoid receptor can sequester G-proteins, making them unavailable to couple to other receptors. J Neurosci 19: 9271–9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O (2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81: 299–343. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW (2002) Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther 303: 608–615. [DOI] [PubMed] [Google Scholar]