

Abstract
Dopamine receptor agonist and NMDA receptor antagonist activation of the mesolimbic dopamine system increases locomotion and disrupts prepulse inhibition of the acoustic startle response (PPI), paradigms frequently used to study both the pharmacology of antipsychotic drugs and drugs of abuse. In rats, virally mediated overexpression of the neurotensin 1 (NT1) receptor in the nucleus accumbens antagonized d-amphetamine- and dizocilpine-induced PPI disruption, hyperlocomotion, and d-amphetamine-induced rearing. The NT receptor antagonist SR 142948A [2-[[5-(2,6-dimethoxyphenyl)-1-(4-N-(3-dimethylaminopropyl)-N-methylcarbamoyl)-2-isopropylphenyl)-1H-pyrazole-3-carbonyl]amino] adamantane-2-carboxylic acid, hydrochloride] blocked inhibition of dizocilpine-induced hyperlocomotion mediated by overexpression of the NT1 receptor. Together, these results suggest that increased nucleus accumbens NT neurotransmission, via the NT1 receptor, can decrease the effects of activation of the mesolimbic dopamine system and disruption of the glutamatergic input from limbic cortices, resembling the action of the atypical antipsychotic drug clozapine. In contrast to clozapine, virally mediated overexpression of the NT1 receptor in the nucleus accumbens had prolonged protective effects (up to 4 weeks after viral injection) without perturbing baseline PPI and locomotor behaviors. These data further confirm the NT1 receptor as the receptor mediating the antistimulant- and antipsychotic-like properties of NT and provide rationale for the development of NT1 receptor agonists as novel antipsychotic drugs. In addition, the NT1 receptor vector might be a valuable tool for understanding the mechanism of action of antipsychotic drugs and drugs of abuse and may have potential therapeutic applications.
Keywords: schizophrenia, prepulse inhibition, locomotion, amphetamine, dizocilpine, viral vector
Introduction
Dopamine (DA) receptor agonists and NMDA receptor antagonists are well known drugs of abuse as well as psychotomimetic drugs. In animal models of drug abuse, both classes of compounds stimulate place preference, increase locomotor activity, and possess high addictive potential (Cami and Farre, 2003). Additionally, DA receptor agonists and NMDA receptor antagonists disrupt sensorimotor gating in rats and humans (Braff et al., 2001) and induce psychotic-like symptoms in humans (Lieberman et al., 1987; Krystal et al., 1994). A deficit in sensorimotor gating is thought to be one of the primary deficits in schizophrenia leading to cognitive fragmentation (Venables, 1966). Sensorimotor gating is operationally measured by prepulse inhibition of the acoustic startle (PPI) (Swerdlow et al., 1994). Another well studied behavioral paradigm, drug-induced hyperlocomotion, is based on the increased arousal associated with activation of the mesolimbic DA system and is an animal model used both in drug abuse and schizophrenia research. The ability to counteract drug-induced hyperlocomotion and disruption of PPI are tests widely used to detect compounds with antipsychotic properties (Arnt, 1995).
The tridecapeptide neurotensin (NT) is implicated in reward, drug addiction, and motor control (Caceda et al., 2003; Kinkead and Nemeroff, 2004). NT antagonizes DA action at the level of the nucleus accumbens (NAcc), most likely via the NT1 receptor. In addition, there is considerable evidence that NT may act as an endogenous antipsychotic and be involved in the pathophysiology of schizophrenia and the mechanism of action of antipsychotic drugs (Binder et al., 2001b; Kinkead and Nemeroff, 2004). Our group has shown that pharmacological (Binder et al., 2002), environmental (Binder et al., 2001a), and genetic (Kinkead et al., 2005) disruption of NT neurotransmission is associated with sensorimotor gating deficits. Examination of the effects of microinjections of NT on PPI (Feifel et al., 1997) and locomotion (Ervin et al., 1981; Steinberg et al., 1994) suggests that the locus of the proposed antipsychotic-like action of NT is the NAcc. It has been proposed that the antistimulant- and/or antipsychotic-like properties of NT are mediated via the NT1 receptor (Steinberg et al., 1994; Casti et al., 2004). The NT1 receptor is a G-protein-coupled receptor linked to protein kinase A and PKC (Shi and Bunney, 1992) and is located in discrete forebrain areas, including the ventral tegmental area, substantia nigra, and less abundantly in the NAcc and caudate (Binder et al., 2001b).
Previous studies investigating the antipsychotic and antistimulant effects of NT were limited by the inherent anatomical nonspecificity of systemically administered NT receptor ligands and knock-out animals and by the development of receptor downregulation after repeated administration of NT receptor agonists (Kinkead and Nemeroff, 2004). For these reasons, a viral vector overexpressing the NT1 receptor was developed. The following studies were designed to investigate the effects of NT1 receptor overexpression in the NAcc on d-amphetamine- and dizocilpine-induced behaviors. It was hypothesized that NT1 receptor overexpression in the NAcc would increase NT neurotransmission and antagonize the stimulant effects of both d-amphetamine and dizocilpine, with implications for the development of novel treatments for schizophrenia and drug abuse.
Materials and Methods
Construction of the viral vector. In collaboration with J. Jacob (Yerkes National Primate Research Center, Atlanta, GA), a lentiviral vector composed of three plasmids was obtained. The first plasmid (pCMO2) encodes the viral backbone and carries the green fluorescent protein (GFP) gene and the transgene. The second plasmid (pΔ8.9) carries most of the HIV genome, except for the deleted accessory proteins. The third plasmid (pVSV-G) encodes a pseudotyped vesicular stomatitis virus capsule that confers extended tropism for any mammalian cell.
A BamHI site was added at the 5′ end and an EcoRI site was added at the 3′ end of the internal ribosome entry site (IRES) sequence of the pIRES2-EGFP (Clontech, Mountain View, CA) by high-fidelity PCR. A similar procedure was used to add an EcoRI site at the 5′ end and a BamHI site at the 3′ end of the NT1 receptor sequence of the pBlueScript-NT1 receptor plasmid (K. Tanaka, Osaka University, Osaka, Japan). Subsequently, both PCR products were double-digested with EcoRI and BamHI (Promega, Madison, WI) and ligated with a BamHI cut pCMO2 lentivirus backbone. This BamHI site is located in the immediate 3′ vicinity of the GFP sequence in pCMO2, as shown in Figure 1. Escherichia coli competent cells were then transformed with the ligation mix, and clones were screened for fragments of known length after digestion with EcoRI and BamHI. The same digestions confirmed the appropriate orientation of the insert. The sequence of the transgene was confirmed at the DNA Sequencing Facility at the University of Pennsylvania (Philadelphia, PA).
Figure 1.
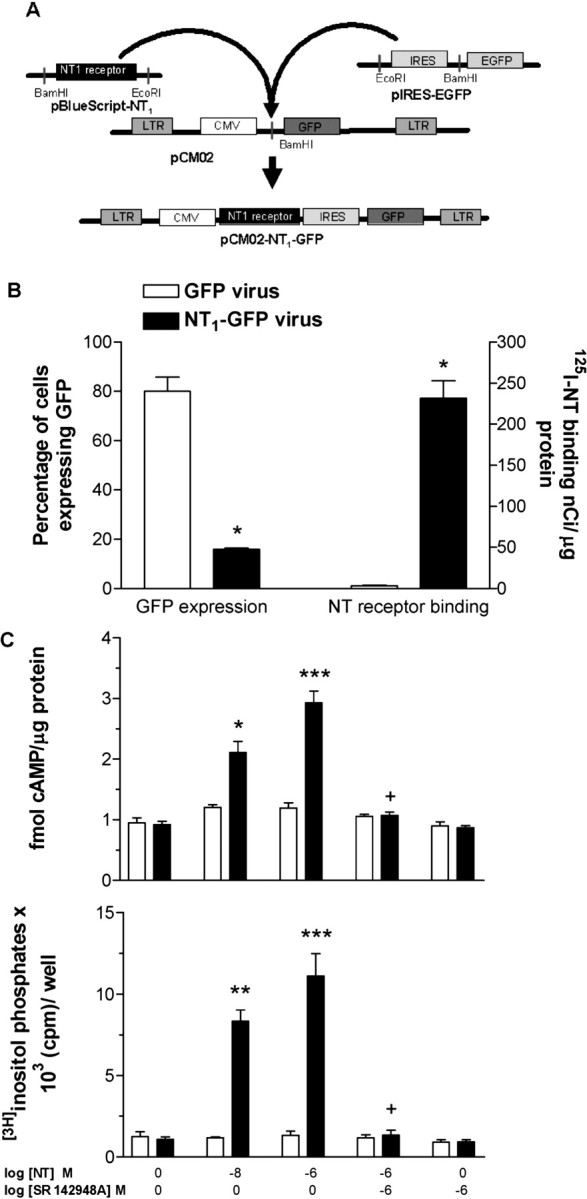
Construction of the NT1-GFP viral vector. A, Strategy for subcloning the NT1 receptor gene and IRES sequence into the pCMO2 backbone to produce the NT1-GFP vector. B, HEK 293 cells infected with the NT1-GFP vector increased NT1 receptor binding and expression of GFP, whereas cells infected with the GFP vector did not express the receptor but expressed GFP fivefold greater than cells infected with the NT1-GFP vector. Results are mean ± SEM of triplicate experiments. *p < 0.001 compared with cells infected with the GFP vector (Student's t test). C, Demonstration of coupling of the virally overexpressed NT1 receptor to cAMP and IP3 formation in HEK 293 cells. Data are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with cells infected with the GFP virus within the same treatment group; +p < 0.001 compared with other NT1-GFP groups that received NT but not SR 142948A. The vehicle for SR 142948A was DMSO (final concentration, 0.1%) in saline. prot, Protein.
Next, the viral particles were packed with a triple transfection of human embryonic kidney (HEK) 293 cells with pΔ8.9, pVSV-G, and either of the two backbones: pCMO2 (GFP) or pCMO2 (NT1-GFP). The supernatant was changed after 24 h. Forty-eight hours after transfection, the supernatant containing viral particles was collected, centrifuged, and filtered. Concentration of the viral particles was achieved by ultracentrifugation of the supernatant at 100,000 × g for 90 min at 4°C. The pellet was resuspended in PBS and 1% BSA in 1:200 of the original volume [unless noted otherwise, all of the chemicals were purchased from Sigma-Aldrich (St. Louis, MO)]. After a second ultracentrifugation at 14,000 × g for 90 min at 4°C, the pellet was resuspended in 1:1000 of the original volume in Tris-buffered saline. Viral titers were calculated by serial dilution and quantification of the p24 antigen (Retro-Tek HIV-1 p24 Antigen ELISA; Zeptometrix Corporation, Buffalo, NY). Final viral concentrations were 108 to 109 viral units/ml.
In vitro testing of the NT1 vector. To test in vitro activity of the virus, 106–7 viral units/million cells was applied to HEK 293 cells. After 48 h, the expression of GFP and NT1 was assessed by flow cytometry and [125I]-NT binding assay, respectively.
[125I]-NT binding assay. An approximate total of 36 × 106 HEK 293 cells were harvested 2 d after infection with either GFP or NT1-GFP virus. Cells were homogenized in 50 mm Tris-HCl and 5 mm EDTA buffer, pH 7.4 (Polytron; Brinkmann Instruments, Westbury, NY) and centrifuged at 17,000 × g for 30 min, and the pellet was resuspended in 50 mm Tris-HCl buffer, pH 7.4. An aliquot was removed for protein quantification, and the remaining sample was incubated with 1 nm [125I]-Tyr3-NT (specific activity, 2200 Ci/mmol; PerkinElmer, Boston, MA), with or without 1 μm nonradioactive NT for 30 min at room temperature. The reaction was stopped with ice-cold 50 mm Tris-HCl buffer, pH 7.4, containing 0.2% BSA, followed by rapid filtration under reduced pressure through glass filters presoaked in ice-cold 50 mm Tris-HCl buffer, pH 7.4, with 0.3% polyethylenimine. Radioactivity was counted on an Clinigamma model 1274 (LKB Instruments, Gaithersburg, MD) with 50% counting efficiency. Counts per minute were converted to nanocuries using a data sheet from the vendor. Protein content was quantified with the BCA protein assay kit (Pierce, Rockford, IL) using a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA). Results were finally expressed as nanocurie per microgram of protein.
Flow cytometry. Two days after infection with either GFP or GFP-NT1 virus, HEK 293 cells were harvested by incubation in EDTA, washed with PBS, and resuspended in 2.0 ml of PBS. Cells were then analyzed for GFP fluorescence using a FACSCalibur flow cytometer. After gating on viable cells, data were analyzed using CellQuest software (Becton Dickinson, Mountain View, CA). Cells were analyzed at typical rates of 100–300 cells/s with 20,000 gated events. The laser excitation wavelength was 488 nm, and the photomultiplier bandpass filter for GFP was FL1, 525 nm. Plots of events against fluorescent intensity were used. Results are expressed as mean ± SEM of the percentage of cells expressing GFP fluorescence.
cAMP formation. Confluent HEK 293 cells in 12-well plates were incubated in controlled salt solution (in mm: 120 NaCl, 5 KCl, 0.8 MgCl2, 1.8 CaCl2, and 15 glucose) containing 1.0 mm 3-isobutylmethyl xanthine, pH 7.5, for 60 min at 37°C. Triplicates were run for each concentration in every experiment, and the experiments were replicated three or four times. The reaction was stopped with the addition of ice-cold Ro 20-1724 [4-[(3-butoxy-4-methoxyphenyl)methyl]-2-imidazolidinone] (Tocris Cookson, Ellisville, MO). The plates were incubated on ice under constant movement for 10 min and scraped. cAMP was quantitated using a commercial ELISA kit (Amersham Biosciences, Piscataway, NJ). Protein concentrations were determined using a commercial kit (Protein BCA kit; Pierce). Final cAMP concentrations were expressed as femtomoles of cAMP per microgram of protein.
PI hydrolysis. Confluent HEK 293 cells in 12-well plates were incubated with 4 μCi/ml [3H]inositol (American Radiolabeled Chemicals, St. Louis, MO) in incomplete DMEM overnight at 37°C. The cells were washed with incomplete DMEM supplemented with 10 mm Li CaCl2 and 25 mm HEPES, pH 8.0, for 20 min at 37°C. NT was dissolved in double-distilled water, and SR 142948A [2-[[5-(2,6-dimethoxyphenyl)-1-(4-N-(3-dimethylaminopropyl)-N-methylcarbamoyl)-2-isopropylphenyl)-1H-pyrazole-3-carbonyl]amino] adamantane-2-carboxylic acid, hydrochloride] was dissolved in DMSO (final concentration, 0.1%) and distilled water. The assay was initiated by the addition of NT (0, 10–8, and 10–6m final concentration) or the NT receptor antagonist SR 142948A (10–6 m final concentration). The reaction was terminated by the addition of ice-cold 1 mm formic acid, and plates were incubated on ice for 2 h. Next, 0.7 m ammonium hydroxide was added to each well, and supernatant was harvested and pipetted onto 1 ml Dowex columns (1-X8, 100–200 mesh in the formate form; Dow Chemical, Midland, MI). The columns were washed with distilled water to remove free [3H]inositol and 2 × 6 ml of 60 mm ammonium formate/5 mm disodium tetraborate to remove [3H]glycerophosphoinositol. [3H]inositol 1-phosphate was eluted with 8 ml of 200 mm ammonium formate/100 mm formic acid, and the radioactivity was determined by liquid scintillation spectrometry. Results are expressed as [3H]inositol phosphate × 103 (cpm)/well.
Animals and housing. All animals were housed in an environmentally controlled animal facility with food and water available ad libitum and with a regular light cycle (lights on 7:00 A.M. to 7:00 P.M.). All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee in compliance with National Institutes of Health (http://grants.nih.gov/grants/olaw/olaw.htm) recommendations based on National Research Council guidelines (Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research, Washington, DC: National Academies Press, 2003). Adult male Long–Evans rats (Harlan Sprague Dawley, Indianapolis, IN) were used for the anatomical and behavioral studies.
Surgical procedures. All surgical procedures were performed under aseptic conditions. Rats were anesthetized using an anesthetic mixture containing ketamine, acepromazine, and xylazine (Lipman et al., 1990). Animal heads were fixed in a stereotaxic frame, and holes were drilled in the skull. One or 2 μl of the virus (GFP or NT1-GFP) stock were injected in the caudate putamen [anteroposterior (AP), 0.2; mediolateral (ML), ±3.1; dorsoventral (DV), –6.0] at the rate of 0.2 μl/min for the time course experiment and at 1.0 μl at the same rate into the NAcc (AP, 2.0; ML, ±1.5; DV, –7.0) for the behavioral experiments. After 5 min, the cannula was removed, the holes were filled with dental cement, and the incision was sutured. After surgery, animals in all groups were single housed until the finalization of experiments. Rats in the control and clozapine groups did not undergo any surgical manipulation.
Drug administration. Different sets of rats were used for PPI and locomotion experiments. Between weeks 2 and 4 after viral injections, rats were tested in the following way. Nonoperated animals received either clozapine (20 mg/kg dissolved in sodium tartrate and two drops of Tween 20, i.p.; n = 16) or vehicle (n = 48; for control, NT1-GFP, and GFP groups). Twenty minutes later, one-half of the animals in each of the four groups (n = 8) received saline or d-amphetamine (2 mg/kg, s.c., dissolved in saline). Ten minutes later, animals were placed either in the PPI boxes or the activity chambers. After 7–10 d, the same experiment was performed using dizocilpine (0.1 mg/kg, s.c., dissolved in saline) counterbalancing for animals that received d-amphetamine and saline.
PPI. PPI refers to the ability of a weak auditory stimulus to transiently inhibit the response to a closely following strong stimulus and is a well characterized operational measure of sensorimotor gating (Swerdlow et al., 1994). PPI was performed between 11:00 A.M. and 4:00 P.M. in a startle chamber (San Diego Instruments, San Diego, CA). Startle amplitude was measured by converting the vibrations of a Plexiglas cylinder (resting platform) caused by the rat startle response into analog signals by a piezoelectric unit. These signals were then digitized, represented as arbitrary startle units, and stored in a personal computer. The testing session began with 5 min of acclimatization to the startle chamber in the presence of 65 dB background white noise. Testing consisted of nine 120 dB pulses alone and 18 pulses preceded (100 ms) by a prepulse 4, 8, or 12 dB above background. Pulses were presented in a pseudorandom order with an average of 15 s between trials. The percentage of PPI (%PPI) was calculated using the following formula: %PPI = 100–(startle amplitude with prepulse × 100/startle amplitude with pulse alone). The latency to response peak was determined as the point of maximal amplitude that occurred within 100 ms from the startle stimulus. Peak latency facilitation was calculated using the following formula: peak pulse alone latency-peak prepulse/pulse latency.
Locomotion assessment. Activity measurements were evaluated by placing rats in an open field consisting of a clear Plexiglas box (40 × 40 × 39 cm) with a black floor in standard room light. Activity was recorded at 5 min intervals for 2 h and quantified by a computer-operated tracking system of 16 photo beams per side (TruScan System; Coulbourn Instruments, Allentown, PA). The distance traveled in each 5 min interval was measured as the total of all vectored x--y-coordinate changes. In addition, data were collected for the number of entries in the vertical planes to reflect two-leg rearing.
In the experiments with the NT receptor antagonist, SR 142948A was dissolved in DMSO (final concentration, 0.1%) and saline and administered intraperitoneally at 100 μg/kg 1 h before dizocilpine or saline.
Receptor binding autoradiography. Animals were deeply anesthetized with Euthanasia 5 solution and decapitated, and the brains were removed immediately, frozen on dry ice, and stored at –70°C. Fresh frozen brains were sliced on a cryostat in 20 μm sections, mounted on Superfrost slides (Fisher Scientific, Pittsburgh, PA), and stored at 70°C until use. At the time of the assay, slides were gradually brought to room temperature and desiccated. Sections were fixed for 2 min in 0.1% paraformaldehyde, pH 7.4, at 4°C and preincubated for 15 min at room temperature in incubation buffer (50 mm Tris base, 10 mm MgCl2, 2 mm EGTA, and 0.1% BSA, pH 7.4). Incubation with 0.2 nm [125I]-NT (DuPont NEN, Boston, MA) was performed in incubation buffer for 60 min at room temperature. Nonspecific binding was determined in the presence of 1 μm unlabeled NT. Sections were washed twice for 5 min at 4°C in 50 mm Tris and 0.1% BSA, rinsed twice in ice-cold water, and quickly dried under a constant stream of cool air. Dried sections were exposed to Biomax MR film (Eastman Kodak, Rochester, NY) for 3 d.
Film quantification. Quantitative receptor autoradiography was performed by computerized densitometry [Imaging Research (AIS), St. Catherines, Ontario, Canada]. Optical densities were calibrated with coexposed 14C standards revealing brain substance-like quench coefficients for iodinated isotopes (Amersham Biosciences) and converted to nanocuries per milligram of protein. Measurements were obtained from a rectangular area in the caudate at +0.2 mm relative to bregma. To establish the increase in NT receptor binding by the vector above baseline levels, measurements from the side that received the GFP vector or vehicle were subtracted from those of the contralateral side that received injection of the NT1-GFP vector. The calculation of the longitudinal spread of the NT1 receptor overexpression was performed by counting the number of consecutive sections (one every 100 μm) with [125I]-NT binding 10% higher than the control side. This number minus one was then multiplied by 100 μm.
Calculation of the Kd of the virally overexpressed NT1 receptor in the rat caudate. A saturation experiment was performed by incubating different sets of slides (n = 12) with increasing concentrations of [125I]-NT: 0.01, 0.1, and 1.0 nm. Measurements from the side that received the vehicle were subtracted from those of the contralateral side that received injection of the NT1-GFP vector. The dissociation constant (Kd) was calculated using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego CA).
Data analysis. Full descriptive statistics were computed for each variable within each group. Data from in vitro NT receptor binding and GFP expression were analyzed with the Student's t test. Multiple two-way ANOVAs were used to analyze data from a second-messenger response to NT in NT1-GFP-infected HEK 293 cells. Data from the d-amphetamine and dizocilpine challenge on locomotion were analyzed with two-way ANOVA (drug by treatment). Three-way ANOVA (antagonist pretreatment-by-drug-by-viral injection) was used to analyze the effects of d-amphetamine and dizocilpine on PPI and the effects of NT receptor antagonist pretreatment on dizocilpine-induced hyperlocomotion. Separate two-way ANOVAs at each prepulse intensity were performed. Post hoc comparisons were calculated with Tukey's test. Significance was set at p < 0.05.
Results
Construction of a lentiviral vector expressing the NT1 receptor
A lentiviral vector was chosen because of the crucial capability of integration of the transgene into the genome of nondividing cells and the related properties of long-term expression of the transgene, very low toxicity, and immune host reaction, as well as the ability to infect neuronal cells (Blomer et al., 1997). A bicistronic vector carrying GFP as a reporter gene and the NT1 receptor were constructed based on the lentiviral system described by Naldini et al. (1996) (Fig. 1A). HEK 293 cells (normally devoid of NT receptors and GFP) were infected with either the NT1-GFP or GFP vector. Cells infected with the NT1-GFP vector exhibited a marked increase in NT receptor binding and expression of GFP (Fig. 1B). In contrast, although cells infected with the GFP vector did not have increased NT receptor binding, they had fivefold higher GFP expression than those infected with the NT1-GFP vector, a finding in agreement with previous studies demonstrating stronger expression of a transgene when in the immediate vicinity of a promoter rather than after an IRES sequence (Martinez-Salas, 1999). Functional coupling of the virally overexpressed NT1 receptor to Gs and Gq proteins was then confirmed in vitro by examination of NT-induced cAMP and IP3 formation in NT1-GFP- and GFP-infected HEK 293 cells (Fig. 1C). There was a significant effect of SR 142948A (cAMP: F(1,51) = 8.7, p < 0.01; IP3: F(1,37) = 7.7, p < 0.001), NT (cAMP: F(2,51) = 11.5, p < 0.001; IP3: F(2,37) = 6.1, p < 0.01), virus (cAMP: F(1,51) = 14.6, p < 0.001; IP3: F(1,37) = 5.7, p < 0.001), an SR 142948A-by-virus interaction (cAMP: F(1,51) = 5.7, p < 0.05; IP3: F(1,37) = 6.6, p < 0.05), and an NT-by-virus interaction (cAMP: F(2,51) = 5.6, p < 0.01; IP3: F(2,37) = 5.7, p < 0.01). NT significantly increased cAMP and IP3 formation in NT1-GFP vector-infected cells compared with GFP vector-infected cells. The effect of NT was completely blocked by pretreatment with the NT receptor antagonist SR 142948A. These results are in agreement with previous studies demonstrating functional coupling of the NT receptor to both Gs and Gq proteins (Hermans and Maloteaux, 1998).
Effects of NT1 receptor overexpression in the NAcc on PPI
The initial characterization of virally mediated NT1 receptor overexpression in vivo was performed in the rat caudate. NT receptor binding was significantly increased at sustained levels from 2 weeks up to 4 months after microinjection of the NT1-GFP vector in the rat striatum (Kd = 0.37 nm) and was restricted to an area of ∼1 mm from the site of injection. Additional histological examination (Nissl staining and glial fibrillary acidic protein immunohistochemistry) showed no apparent signs of toxicity or proliferative vascular changes at the injection sites. This absence of significant tissue damage is in agreement with previous reports using lentiviral vectors (Blomer et al., 1997).
The effects of virally mediated overexpression of the NT1 receptor in the NAcc on d-amphetamine- and dizocilpine-induced disruption of PPI were then examined. In preliminary experiments, it was demonstrated that there was no significant difference in baseline PPI or d-amphetamine or dizocilpine effects on PPI in sham or vehicle-injected animals compared with the GFP group; therefore, all subsequent experiments were run with a nonsurgical and a GFP virus group as controls. Sixty-four male Long–Evans rats were tested for PPI, ranked according to the average PPI response, and distributed to four groups (n = 16): control, clozapine, GFP virus, and NT1-GFP virus. Rats from the virus groups received 1 μl of either GFP or NT1-GFP virus (10–8 to 10–9 viral units/ml) bilaterally in the NAcc. Animals in the control and clozapine groups did not undergo any surgical procedure. One week after surgery (a time point when there is no measurable increase in NT receptor binding; data not shown), all rats were again tested for PPI to exclude possible confounding behavioral effects related to the surgical procedure, and no differences were observed between groups (data not shown). Between the second and fourth weeks after surgeries, a period in which previous experiments have shown consistent and sustained NT1 receptor overexpression, the effects of d-amphetamine (2.0 mg/kg, s.c.) or dizocilpine (0.1 mg/kg, s.c.) on PPI were examined. After completion of behavioral experiments, the animals were killed, and NT1 receptor overexpression was confirmed with [125I]-NT autoradiography. NT binding in the NAcc in the animals receiving the NT1-GFP virus was significantly increased compared with animals receiving the GFP virus (Fig. 2A). Additionally, placement of the injection cannula was confirmed. In three animals, the cannula was placed outside the NAcc, and they were excluded from additional analysis. These animals were in the group of the five with highest disruption of PPI by d-amphetamine and dizocilpine, the values of which were significantly different from the rest of the NT1-GFP virus-injected animals (data not shown).
Figure 2.
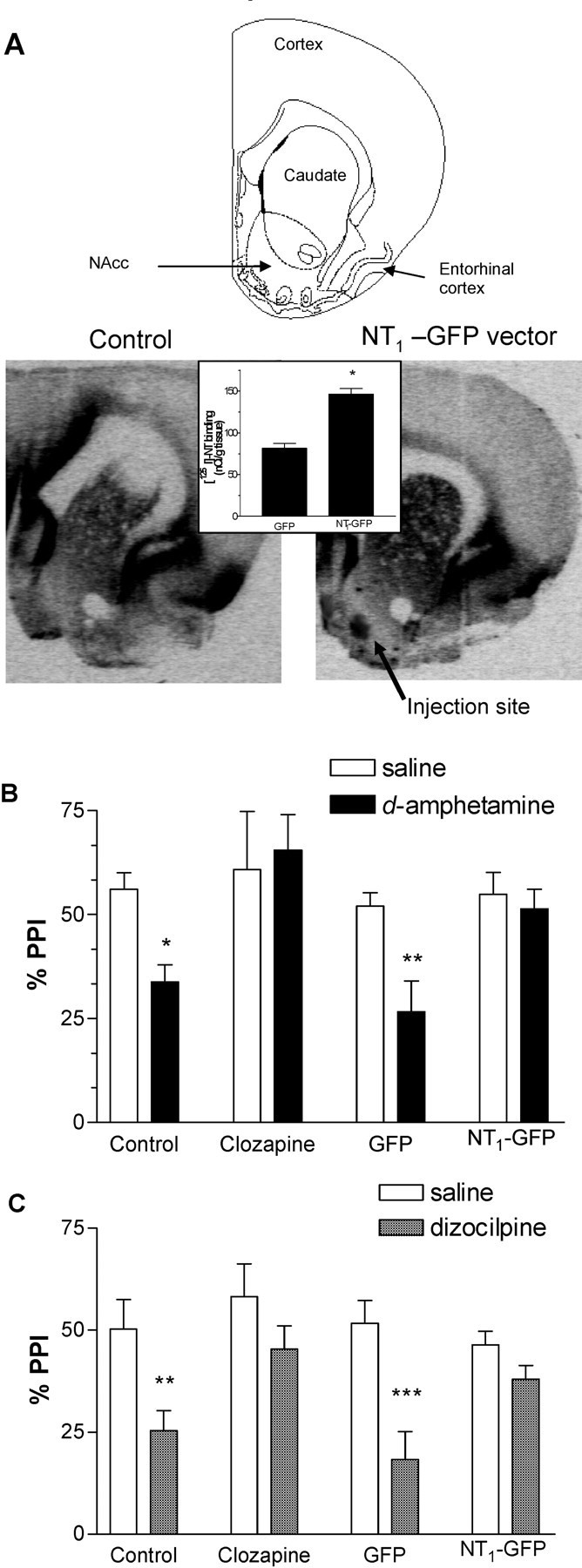
NT1-GFP vector injection in the NAcc did not modify baseline PPI but had clozapine-like effects against d-amphetamine- and dizocilpine-induced PPI. A, NT receptor binding in the rat NAcc assessed by [125I]-NT binding autoradiography in nonsurgical controls (left) or 4 weeks after injection of the NT1-GFP vector (right) (bregma, +2 mm). Inset, Comparison of [125I]-NT binding in the NAcc in animals that were given injections of the GFP and the NT1-GFP viruses 3 weeks after injection (n = 17–20). Data are mean ± SEM. *p < 0.001 (Student's t test). B, Protection of PPI from d-amphetamine (2 mg/kg)-induced disruption at 4 dB prepulse intensity. C, Protection of PPI from dizocilpine (0.1 mg/kg)-induced disruption at 4 dB prepulse intensity. Data are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with saline within each group (Tukey's honestly significant difference).
There were no significant effects of group (control, clozapine, NT1-GFP, or GFP) on pulse-alone startle amplitude, basal PPI, peak latency facilitation, or startle habituation (data not shown). d-Amphetamine did not modify pulse-alone startle amplitude in any group, but dizocilpine significantly increased prepulse startle amplitude in the groups receiving clozapine and the NT1-GFP virus (data not shown). Two-way ANOVA indicated a significant effect of group (F(3,63) = 2.9; p < 0.05) and dizocilpine (F(1,63) = 2.9; p < 0.05). Analysis of the effects of d-amphetamine on PPI by three-way ANOVA indicated significant effects of prepulse intensity (F(2,345) = 19.8; p < 0.001), d-amphetamine (F(1,345) = 76.5; p < 0.001), group (F(3,345) = 10.0; p < 0.001), and a d-amphetamine-by-group interaction (F(3,345) = 7.5; p < 0.001). Based on previous literature demonstrating that d-amphetamine only disrupts PPI at low prepulse intensities, we ran separate two-way ANOVAs at each prepulse intensity. There was a significant effect of d-amphetamine at a 4 dB prepulse (F(1,60) = 12.0; p = 0.001) but not at 8 and 12 dB prepulse intensity, an effect reported previously and thought to be strain specific (Swerdlow et al., 2005). Post hoc analysis revealed that at the 4 dB prepulse intensity, d-amphetamine (2 mg/kg) disrupted PPI in the control and GFP groups (p < 0.05). d-Amphetamine did not significantly disrupt PPI in the NT1-GFP or clozapine groups. Analysis of the effect of dizocilpine on PPI by three-way ANOVA indicated significant effects of prepulse intensity (F(2,294) = 34.6; p < 0.001), dizocilpine (F(1,294) = 21.6; p < 0.001), and group (F(3,294) = 6.1; p < 0.001). Post hoc analysis revealed that dizocilpine disrupted PPI in the animals receiving saline or the GFP vector (p < 0.01) but not in animals receiving clozapine (20 mg/kg, i.p.) or the NT1-GFP vector at all prepulse intensities. The effects of d-amphetamine and dizocilpine on PPI at the 4 dB prepulse intensity are shown in Figure 2. Together, these results demonstrate that NT1 receptor overexpression in the NAcc did not modify baseline PPI but antagonized the PPI disrupting effects of both d-amphetamine and dizocilpine, resembling the effects of the atypical antipsychotic drug clozapine.
Effects of NT1 receptor overexpression in the NAcc on locomotor activity
In another set of animals, the effects of NT1 receptor overexpression in the NAcc on d-amphetamine- and dizocilpine-induced hyperlocomotion (Fig. 3) and rearing (Fig. 4) were examined between 2 and 4 weeks after viral injections. There were no significant effects of group (control, clozapine, NT1-GFP, or GFP) on baseline locomotion and rearing. Analysis of the effect of d-amphetamine on locomotion and rearing by two-way ANOVA indicated a significant effect of group (locomotion: F(3,60) = 7.3, p < 0.001; rearing: F(3,60) = 11.3, p < 0.001), d-amphetamine (locomotion: F(1,60) = 198.6, p < 0.001; rearing: F(1,60) = 67.5, p < 0.001), and a group-by-d-amphetamine interaction (locomotion: F(3,60) = 2.8, p = 0.05; rearing: F(3,60) = 5.1, p = 0.004). Post hoc analysis revealed that d-amphetamine (2 mg/kg)-induced rearing, but not hyperlocomotion, was significantly reduced in animals receiving clozapine or the NT1-GFP virus. To elucidate whether the lack of effect of NT1-GFP virus injections on d-amphetamine-induced hyperlocomotion was quantitative (dose of d-amphetamine too high) versus qualitative (effect of d-amphetamine would not be blocked regardless of dose), a similar experiment was performed in the same animals 3 weeks after viral injection with a lower dose of d-amphetamine (1.0 mg/kg). Two-way ANOVA indicated a significant effect of group (locomotion: F(3,51) = 19.9, p < 0.001; rearing: F(3,51) = 13.6, p < 0.001), d-amphetamine (locomotion: F(1,51) = 69.9, p < 0.001; rearing: F(1,51) = 12.6, p < 0.001), and a group-by-d-amphetamine interaction (locomotion: F(3,51) = 3.7; p < 0.05). Post hoc analysis revealed that d-amphetamine (1.0 mg/kg) significantly increased locomotion and rearing in the control and GFP groups, whereas it (1.0 mg/kg) had significantly less effect on locomotion and rearing in animals receiving clozapine or the NT1-GFP vector. Analysis of the effect of dizocilpine on locomotion and rearing by two-way ANOVA indicated a significant effect of group (locomotion: F(3,60) = 3.6, p = 0.02; rearing: F(3,60) = 5.6, p = 0.002), dizocilpine (locomotion: F(1,60) = 122.9, p < 0.001; rearing: F(1,60) = 39.4, p < 0.001), and a group-by-dizocilpine interaction (rearing: F(3,60) = 5.4; p = 0.003). In contrast to d-amphetamine (2.0 mg/kg), whereas dizocilpine increased locomotion in the control and GFP groups, rearing was significantly decreased in all groups. The hyperlocomotor effect of dizocilpine was significantly reduced compared with the control group in the NT1-GFP and clozapine groups. These results indicate that NT1 receptor overexpression in the NAcc did not alter baseline locomotor behavior but antagonized d-amphetamine (in a dose-dependent manner) and dizocilpine effects on locomotor activity.
Figure 3.

NT1-GFP vector injection in the NAcc did not modify baseline locomotion but had clozapine-like effects against d-amphetamine- and dizocilpine-induced hyperlocomotion. A, B, NT1 receptor overexpression in the NAcc decreased d-amphetamine-induced hyperlocomotion only at the lower dose (1 mg/kg). C, NT1 receptor overexpression in the NAcc antagonized dizocilpine-induced hyperlocomotion. Data are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with saline within each group; +p < 0.05, ++p < 0.01, and +++p < 0.001 compared with d-amphetamine/dizocilpine in the control group (Tukey's honestly significant difference).
Figure 4.

NT1-GFP vector injection in the NAcc did not modify baseline rearing but had clozapine-like effects against d-amphetamine-induced rearing. A, B, NT1 receptor overexpression in the NAcc decreased d-amphetamine (1.0 and 2.0 mg/kg)-induced increase in rearing. C, Dizocilpine decreased rearing in all groups. Data are mean ± SEM. *p < 0.01 and **p < 0.001 compared with saline within each group; +p < 0.05, ++p < 0.01, and +++p < 0.001 compared with d-amphetamine/dizocilpine in the control group; #p < 0.05 and ##p < 0.001 compared with saline in the control group (Tukey's honestly significant difference).
Blockade of the behavioral effects of NT1 receptor overexpression in the NAcc with an NT receptor antagonist
To determine whether the observed behavioral effects of increased NT1 receptor expression in the NAcc were a direct consequence of increased NT neurotransmission or secondary to alterations in other neurotransmitter systems as suggested previously (Tanganelli et al., 1994), the ability of the NT receptor antagonist SR 142948A to block the behavioral effects of NT1 receptor overexpression on dizocilpine-induced hyperlocomotion was examined. We chose drug-induced locomotion and not PPI to test for direct NT neurotransmission, because SR 142948A by itself counteracts d-amphetamine- and dizocilpine-induced PPI disruption (our manuscript in preparation). Three-way ANOVA indicated significant effects of SR 142948A (F(1,138) = 9.2; p = 0.003), dizocilpine (F(1,138) = 66.9; p < 0.001), and an SR 142948A-by-dizocilpine interaction (F(1,138) = 5.4; p < 0.05). Post hoc analysis revealed that in rats with NT1 receptor overexpression in the NAcc, SR 142948A had no effect on baseline locomotion but completely abolished the protective effect against dizocilpine-induced hyperlocomotion (p < 0.01) (Fig. 5). This experiment shows that the protective effect of NT1 receptor overexpression in the NAcc was directly dependent on NT neurotransmission.
Figure 5.

NT receptor antagonist SR 142948A (SR) (100 μg/kg) prevents the protective effect of NT1-GFP vector administration on dizocilpine-induced hyperlocomotion. Data are mean ± SEM. *p < 0.01 and **p < 0.001 compared with saline within each group; +p < 0.05 compared with the GFP–vehicle–dizocilpine and NT1–GFP–SR–dizocilpine groups. The vehicle for SR 142948A was DMSO (final concentration, 0.1%) in saline.
Discussion
Viral vectors have been used to study social behavior and other models of psychiatric conditions (Carlezon et al., 1997; Clark et al., 2002; Lim et al., 2004). However, to the best of our knowledge, this is the first report of the use of viral vector technology in relation to the mechanism of action of antipsychotic drugs or other psychotherapeutic agents. The strategy of overexpressing the NT1 receptor rather than the NT peptide itself was chosen for several reasons. First, the NT1 receptor is downregulated in response to sustained exposure to NT or NT receptor agonists (Hermans and Maloteaux, 1998), potentially leading to the development of tolerance (Hertel et al., 2001, 2002; Boules et al., 2003). Second, overexpression of the NT1 receptor allows for specific examination of NT1 receptor-mediated NT neurotransmission, avoiding the potentially confounding behavioral effects of NT at other NT receptor subtypes. Third, because NT is first expressed as part of a large propeptide containing NT and a related peptide, neuromedin N, it is critical to include the nearby necessary sequences for the correct intracellular trafficking of the peptide. Finally, virally mediated overexpression of neurotransmitter receptors has been successful in previous experiments (Carlezon et al., 1997; Lim et al., 2004).
Our results demonstrate that virally mediated increase in NT neurotransmission in the NAcc did not modify baseline PPI or locomotion; however, it antagonized the behavioral effects of excitation of the mesolimbic DA system and disruption of limbic cortical input into the NAcc induced by a DA receptor agonist and an NMDA receptor antagonist, respectively. Additionally, in the animals in which the viral injection did not reach the NAcc and were excluded, both psychotomimetic drugs still worked. These findings confirm the NAcc as a brain region mediating the antistimulant properties of NT, in concurrence with the effects of intra-NAcc injection of NT (Ervin et al., 1981; Steinberg et al., 1994; Feifel et al., 1997). In addition, unlike studies demonstrating tolerance to the effects of chronic administration of NT receptor agonists (Hertel et al., 2001, 2002; Boules et al., 2003), increased NT neurotransmission via virally mediated increase in NT1 receptor expression antagonized the behavioral effects of d-amphetamine and dizocilpine for several weeks and after repeated challenge with psychotomimetic drugs, indicating the sustained behavioral efficacy of this approach. Although it was possible that the effects of increased NT1 receptor expression in the NAcc were not directly attributable to increased NT neurotransmission, but attributable to secondary changes in other neurotransmitter systems (Tanganelli et al., 1994), systemic administration of the NT receptor antagonist SR 142948A blocked the protective effect of NT1 receptor overexpression on dizocilpine-induced hyperlocomotion. Therefore, the behavioral effects of overexpression of the NT1 receptor are most likely as a result of the enhanced effect of NT release in the presence of additional receptors, and not attributable to compensatory changes downstream of the overexpressed NT receptors. Future experiments are necessary to address whether this phenomenon is generalizable to hyperlocomotion induced by other substances such as DA receptor agonists.
Psychostimulants act at different levels of the mesolimbic system; however, it is believed that the common mechanism of action of acute psychostimulant administration (as well as several other drugs of abuse) is inhibition of medium spiny GABAergic neurons in the NAcc leading to a reduction in its functional output (Cami and Farre, 2003; Chao and Nestler, 2004). DA receptor agonists and NMDA receptor antagonists act through different, not yet fully understood, pharmacological mechanisms (Millan et al., 1999). Mesolimbic DA and glutamatergic (prefrontal cortex, hippocampus, and amygdala) inputs converge in the NAcc and in several cases on the same medium spiny cell (O'Donnell and Grace, 1995). Hippocampal and amygdala projections into the NAcc are antagonistic (Pennartz et al., 1994), and DA can facilitate or attenuate cortical inputs (Brady and O'Donnell, 2004; Hjelmstad, 2004). Furthermore, recent experiments suggest that DA and glutamate might be released from the same midbrain cells (Chuhma et al., 2004). It is believed that acute administration of most drugs of abuse mimic a functional over activation of the mesolimbic DA system, leading to hyperpolarization of NAcc spiny cells and reduction of their GABAergic output, mainly to the ventral pallidum (VP). However, downstream from the NAcc, the circuits activated by d-amphetamine and dizocilpine seem to diverge. Whereas the behavioral effects of d-amphetamine and other DA receptor agonists are mediated by reduction in the inhibitory tone of the NAcc–VP projections and an increase in VP functional output (Swerdlow et al., 1990), the behavioral actions of dizocilpine are independent of the VP and less well understood (Kretschmer and Koch, 1998; Mele et al., 1998; Kretschmer, 2000).
Although NMDA receptor antagonists induce DA release in the NAcc (Karreman et al., 1996; Millan et al., 1999), evidence suggests that the primary site for dizocilpine behavioral effects are NMDA receptors in the amygdala and the ventral hippocampus (but not the NAcc) (Bakshi and Geyer, 1998) and non-NMDA receptors in the NAcc (Hauber and Andersen, 1993; Bubser et al., 1995). NT is known to antagonize D2 receptor responses via receptor–receptor interactions (Fuxe and Agnati, 1985) and convergence of second-messenger systems (Binder et al., 2001b). However, it is tempting to speculate that the antipsychotic-like properties of NT in the NAcc could be related to modulation of neurotransmitter systems other than DA, including serotonin, GABA, glutamate, or adenosine. It is possible that the net result of increased NT neurotransmission is to decrease the hyperpolarizing effects of acute administration of DA receptor agonists and NMDA receptor antagonists on accumbal medium spiny cells, preserving the functional output of the nucleus and maintaining an inhibitory tone over the VP and other brain regions.
Increased NT1 receptor in the NAcc antagonized the behavioral effects of two different types of psychotomimetic drugs: d-amphetamine and dizocilpine, a property shared with clozapine and most atypical antipsychotic drugs but not with haloperidol (Geyer and Ellenbroek, 2003). This supports the view that NT neurotransmission in the NAcc might mediate the therapeutic action of atypical antipsychotic drugs and directly supports the role of the NT1 receptor in this action. However, in contrast to clozapine, increasing NT1 receptors in the NAcc did not modify baseline PPI or locomotion. Therefore, manipulation of NT neurotransmission may protect sensorimotor gating and locomotion from pharmacological disruptions without perturbing these behaviors in normal or baseline conditions, with the potential for the development of novel antipsychotic treatment with decreased side-effect potential.
PPI is altered in schizophrenic patients and their relatives and is normalized after antipsychotic drug treatment (Swerdlow et al., 1994; Braff et al., 2001). In rodents, pharmacological agents (DA receptor agonists, NMDA receptor antagonists, 5-HT2A receptor agonists), neonatal lesions, and environmental manipulations disrupt PPI (Koch, 1999; Swerdlow et al., 2001). PPI and locomotion are two behaviors regulated by the interaction of the mesolimbic DA system and the cortical–NAcc–VP circuit (Pennartz et al., 1994; Swerdlow et al., 2001). Psychostimulants, via increased DA in the NAcc, are thought to disrupt PPI and induce hyperlocomotion. However, despite these commonalities, there is a dissociation of the neuronal substrates regulating locomotion and sensorimotor gating as indicated by the differential time course of d-amphetamine effects in these behaviors (Sills, 1999), differential susceptibility to DAergic and adrenergic regulation in the NAcc (Schwienbacher et al., 2002), and differential sensitivity to DA receptor agonists (larger doses required to disrupt PPI than locomotor activity) (Geyer et al., 2001). Consistent with these observations was the observation that NT1 receptor overexpression in the NAcc completely blocked d-amphetamine- and dizocilpine-induced PPI deficits, while only partially antagonizing the effects of these drugs on locomotor activity.
Our results provide additional support for the development of NT1 receptor agonists as novel antipsychotic drugs and clinically relevant compounds for the treatment of drug abuse. Not only does NT neurotransmission mediate the acute activating effects of psychostimulants (Betancur et al., 1998), but also the development of drug-induced behavioral sensitization (Horger et al., 1994; Rompré and Perron, 2000; Costa et al., 2001; Panayi et al., 2002).
Additional studies examining the antipsychotic properties of increased NT1 receptor overexpression in the NAcc in environmental (isolation rearing) and genetic (NT1 receptor knock-out mice or Brattleboro rats) animal models demonstrating deficits in sensorimotor gating, as well as exploring the effects of increasing NT1 receptor expression in other brain regions, are warranted. In addition, viral vector overexpression of the NT1 receptor may be used to specifically address the role of NT in the pathology of drug abuse including development of behavioral sensitization, self-administration, and reinstatement, with potential implications for novel therapeutic approaches in this field. Utilization of the NT1 receptor lentiviral vector has the potential to provide further insight into the mechanism of action of antipsychotic drugs and may have potential therapeutic applications for schizophrenia, drug abuse, and other psychiatric disorders.
Footnotes
This work was supported by National Institutes of Health Grant MH39415.
Correspondence should be addressed to Dr. Charles B. Nemeroff, Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine, Suite 4000 Woodruff Memorial Research Building, 101 Woodruff Circle, Atlanta, GA 30322. E-mail: cnemero@emory.edu.
DOI:10.1523/JNEUROSCI.4282-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511748-09$15.00/0
References
- Arnt J (1995) Differential effects of classical and newer antipsychotics on the hypermotility induced by two dose levels of d-amphetamine. Eur J Pharmacol 283: 55–62. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Geyer MA (1998) Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci 18: 8394–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C, Cabrera R, de Kloet ER, Pélaprat D, Rostène W (1998) Role of endogenous neurotensin in the behavioral and neuroendocrine effects of cocaine. Neuropsychopharmacology 19: 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, Kinkead B, Owens MJ, Kilts CD, Nemeroff CB (2001a) Enhanced neurotensin neurotransmission is involved in the clinically relevant behavioral effects of antipsychotic drugs: evidence from animal models of sensorimotor gating. J Neurosci 21: 601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, Kinkead B, Owens MJ, Nemeroff CB (2001b) Neurotensin and dopamine interactions. Pharmacol Rev 53: 453–486. [PubMed] [Google Scholar]
- Binder EB, Gross RE, Nemeroff CB, Kilts CD (2002) Effects of neurotensin receptor antagonism on latent inhibition in Sprague-Dawley rats. Psychopharmacology 161: 288–295. [DOI] [PubMed] [Google Scholar]
- Blomer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH (1997) Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol 71: 6641–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boules M, McMahon B, Wang R, Warrington L, Stewart J, Yerbury S, Fauq A, McCormick D, Richelson E (2003) Selective tolerance to the hypothermic and anticataleptic effects of a neurotensin analog that crosses the blood-brain barrier. Brain Res 987: 39–48. [DOI] [PubMed] [Google Scholar]
- Brady AM, O'Donnell P (2004) Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo J Neurosci 24: 1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR (2001) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology 156: 234–258. [DOI] [PubMed] [Google Scholar]
- Bubser M, Tzschentke T, Hauber W (1995) Behavioural and neurochemical interactions of the AMPA antagonist GYKI 52466 and the noncompetitive NMDA antagonist dizocilpine in rats. J Neural Transm Gen Sect 101: 115–126. [DOI] [PubMed] [Google Scholar]
- Caceda R, Kinkead B, Nemeroff CB (2003) Do neurotensin receptor agonists represent a novel class of antipsychotic drugs? Semin Clin NeuropsychiatryNeuropsychiatry 8: 94–108. [DOI] [PubMed] [Google Scholar]
- Cami J, Farre M (2003) Drug addiction. N Engl J Med 349: 975–986. [DOI] [PubMed] [Google Scholar]
- Carlezon Jr WA, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, Nestler EJ (1997) Sensitization to morphine induced by viral-mediated gene transfer. Science 277: 812–814. [DOI] [PubMed] [Google Scholar]
- Casti P, Marchese G, Casu G, Ruiu S, Pani L (2004) Blockade of neurotensin receptors affects differently hypo-locomotion and catalepsy induced by haloperidol in mice. Neuropharmacology 47: 128–135. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ (2004) Molecular neurobiology of drug addiction. Annu Rev Med 55: 113–132. [DOI] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S (2004) Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci 24: 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MS, Sexton TJ, McClain M, Root D, Kohen R, Neumaier JF (2002) Overexpression of 5-HT1B receptor in dorsal raphe nucleus using herpes simplex virus gene transfer increases anxiety behavior after inescapable stress. J Neurosci 22: 4550–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa FG, Frussa-Filho R, Felicio LF (2001) The neurotensin receptor antagonist, SR48692, attenuates the expression of amphetamine-induced behavioural sensitisation in mice. Eur J Pharmacol 428: 97–103. [DOI] [PubMed] [Google Scholar]
- Ervin GN, Birkemo LS, Nemeroff CB, Prange Jr AJ (1981) Neurotensin blocks certain amphetamine-induced behaviours. Nature 291: 73–76. [DOI] [PubMed] [Google Scholar]
- Feifel D, Minor KL, Dulawa S, Swerdlow NR (1997) The effects of intra-accumbens neurotensin on sensorimotor gating. Brain Res 760: 80–84. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Agnati LF (1985) Receptor-receptor interactions in the central nervous system. A new integrative mechanism in synapses. Med Res Rev 5: 441–482. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Ellenbroek B (2003) Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry 27: 1071–1079. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR (2001) Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology 156: 117–154. [DOI] [PubMed] [Google Scholar]
- Hauber W, Andersen R (1993) The non-NMDA glutamate receptor antagonist GYKI 52466 counteracts locomotor stimulation and anticataleptic activity induced by the NMDA antagonist dizocilpine. Naunyn Schmiedebergs Arch Pharmacol 348: 486–490. [DOI] [PubMed] [Google Scholar]
- Hermans E, Maloteaux JM (1998) Mechanisms of regulation of neurotensin receptors. Pharmacol Ther 79: 89–104. [DOI] [PubMed] [Google Scholar]
- Hertel P, Byskov L, Didriksen M, Arnt J (2001) Induction of tolerance to the suppressant effect of the neurotensin analogue NT69L on amphetamine-induced hyperactivity. Eur J Pharmacol 422: 77–81. [DOI] [PubMed] [Google Scholar]
- Hertel P, Olsen CK, Arnt J (2002) Repeated administration of the neurotensin analogue NT69L induces tolerance to its suppressant effect on conditioned avoidance behaviour. Eur J Pharmacol 439: 107–111. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO (2004) Dopamine excites nucleus accumbens neurons through the differential modulation of glutamate and GABA release. J Neurosci 24: 8621–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horger BA, Taylor JR, Elsworth JD, Roth RH (1994) Preexposure to, but not cotreatment with, the neurotensin receptor antagonist SR 48692 delays the development of cocaine sensitization. Neuropsychopharmacology 11: 215–222. [DOI] [PubMed] [Google Scholar]
- Karreman M, Westerink BH, Moghaddam B (1996) Excitatory amino acid receptors in the ventral tegmental area regulate dopamine release in the ventral striatum. J Neurochem 67: 601–607. [DOI] [PubMed] [Google Scholar]
- Kinkead B, Nemeroff CB (2004) Neurotensin, schizophrenia and antipsychotic drug actions. In: Disorders of synaptic plasticity (Smythies J, ed), pp 328–342. London: Elsevier.
- Kinkead B, Dobner PR, Egnatashvili V, Murray T, Deitemeyer N, Nemeroff CB (2005) Neurotensin-deficient mice have deficits in prepulse inhibition: restoration by clozapine but not haloperidol, olanzapine or quetiapine. J Pharmacol Exp Ther 315: 256–264. [DOI] [PubMed] [Google Scholar]
- Koch M (1999) The neurobiology of startle. Prog Neurobiol 59: 107–128. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD (2000) NMDA receptor antagonist-induced dopamine release in the ventral pallidum does not correlate with motor activation. Brain Res 859: 147–156. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD, Koch M (1998) The ventral pallidum mediates disruption of prepulse inhibition of the acoustic startle response induced by dopamine agonists, but not by NMDA antagonists. Brain Res 798: 204–210. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers Jr MB, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Kane JM, Alvir J (1987) Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology 91: 415–433. [DOI] [PubMed] [Google Scholar]
- Lim MM, Wang Z, Olazabal DE, Ren X, Terwilliger EF, Young LJ (2004) Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 429: 754–757. [DOI] [PubMed] [Google Scholar]
- Lipman NS, Marini RP, Erdman SE (1990) A comparison of ketamine/xylazine and ketamine/xylazine/acepromazine anesthesia in the rabbit. Lab Anim Sci 40: 395–398. [PubMed] [Google Scholar]
- Martinez-Salas E (1999) Internal ribosome entry site biology and its use in expression vectors. Curr Opin Biotechnol 10: 458–464. [DOI] [PubMed] [Google Scholar]
- Mele A, Thomas DN, Pert A (1998) Different neural mechanisms underlie dizocilpine maleate- and dopamine agonist-induced locomotor activity. Neuroscience 82: 43–58. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Brocco M, Gobert A, Joly F, Bervoets K, Rivet J, Newman-Tancredi A, Audinot V, Maurel S (1999) Contrasting mechanisms of action and sensitivity to antipsychotics of phencyclidine versus amphetamine: importance of nucleus accumbens 5-HT2A sites for PCP-induced locomotion in the rat. Eur J Neurosci 11: 4419–4432. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1995) Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci 15: 3622–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayi F, Dorso E, Lambas-Senas L, Renaud B, Scarna H, Berod A (2002) Chronic blockade of neurotensin receptors strongly reduces sensitized, but not acute, behavioral response to d-amphetamine. Neuropsychopharmacology 26: 64–74. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Groenewegen HJ, Lopes da Silva FH (1994) The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol 42: 719–761. [DOI] [PubMed] [Google Scholar]
- Rompré PP, Perron S (2000) Evidence for a role of endogenous neurotensin in the initiation of amphetamine sensitization. Neuropharmacology 39: 1880–1892. [DOI] [PubMed] [Google Scholar]
- Schwienbacher I, Fendt M, Hauber W, Koch M (2002) Dopamine D1 receptors and adenosine A1 receptors in the rat nucleus accumbens regulate motor activity but not prepulse inhibition. Eur J Pharmacol 444: 161–169. [DOI] [PubMed] [Google Scholar]
- Shi WX, Bunney BS (1992) Actions of neurotensin: a review of the electrophysiological studies. Ann NY Acad Sci 668: 129–145. [DOI] [PubMed] [Google Scholar]
- Sills TL (1999) Amphetamine dose dependently disrupts prepulse inhibition of the acoustic startle response in rats within a narrow time window. Brain Res Bull 48: 445–448. [DOI] [PubMed] [Google Scholar]
- Steinberg R, Brun P, Fournier M, Souilhac J, Rodier D, Mons G, Terranova JP, Le Fur G, Soubrie P (1994) SR 48692, a non-peptide neurotensin receptor antagonist differentially affects neurotensin-induced behaviour and changes in dopaminergic transmission. Neuroscience 59: 921–929. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA (1990) GABAergic projection from nucleus accumbens to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res 532: 146–150. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Taaid N, Geyer MA (1994) Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry 51: 139–154. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL (2001) Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology 156: 194–215. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Kuczenski R, Goins JC, Crain SK, Ma LT, Bongiovanni MJ, Shoemaker JM (2005) Neurochemical analysis of rat strain differences in the startle gating-disruptive effects of dopamine agonists. Pharmacol Biochem Behav 80: 203–211. [DOI] [PubMed] [Google Scholar]
- Tanganelli S, O'Connor WT, Ferraro L, Bianchi C, Beani L, Ungerstedt U, Fuxe K (1994) Facilitation of GABA release by neurotensin is associated with a reduction of dopamine release in rat nucleus accumbens. Neuroscience 60: 649–657. [DOI] [PubMed] [Google Scholar]
- Venables PH (1966) Psychphysiological aspects of schizophrenia. Br J Med Psychol 39: 289–297. [Google Scholar]