

Abstract
Protein kinase Mζ (PKMζ), an autonomously active atypical PKC isoform, is both necessary and sufficient for enhanced synaptic transmission during long-term potentiation (LTP) maintenance. LTP, however, evolves through several temporal phases, which may be mediated by distinct molecular mechanisms of potentiation. Here, we determined the specific phase of LTP maintained by PKMζ. Using a selective, cell-permeable ζ-pseudosubstrate inhibitor at concentrations that block potentiation produced by postsynaptic perfusion of PKMζ, we inhibited PKMζ activity at various times after tetanization of Schaffer collateral/commissural-CA1 synapses. Inhibition of PKMζ did not affect baseline AMPA receptor-mediated synaptic transmission or an early phase of LTP. In contrast, the inhibitor reversed established LTP when applied 1, 3, or 5 h after tetanic stimulation. Control nontetanized pathways within the hippocampal slices were unaffected. An inactive scrambled version of the peptide had no effect on LTP. Thus, persistent, increased phosphorylation by PKMζ specifically maintains the late phase of LTP.
Keywords: PKMζ, PKCζ, long-term potentiation, memory, ZIP, αPKC
Introduction
Several protein kinases are important for long-term potentiation (LTP), a persistent form of synaptic plasticity thought to be a physiological substrate of memory (Bliss and Collingridge, 1993). Many studies have focused on four essential kinases: calcium/calmodulin-dependent protein kinase II (CaMKII), mitogen-activated protein (MAP) kinase, protein kinase A (PKA), and PKC (Sweatt, 1999). Each of these kinases is activated by post-translational mechanisms involving second messengers that convert the basal enzyme from an inactive to active conformation (Schwartz and Greenberg, 1987). Although some of the post-translational modifications triggered by LTP, such as CaMKII autophosphorylation (Lisman, 1994) and PKC oxidation (Klann et al., 1998), may be persistent, eventually these kinases either return to their inactive states or are replaced by their inactive precursors (Schwartz and Greenberg, 1987; Schwartz, 1993). Consistent with this transient mechanism of action, the activities of CaMKII (Otmakhov et al., 1997; Chen et al., 2001), MAP kinase (English and Sweatt, 1997), PKA (Frey et al., 1993; Matthies and Reymann, 1993; Huang and Kandel, 1994), and most forms of PKC (Malinow et al., 1989; Colley et al., 1990; Lopez-Molina et al., 1993) are critical within ∼30 min after tetanic stimulation, but not afterward.
PKC, however, is a family of ∼15 different isoforms, each with individual cellular functions (Newton, 2001). We found previously that, during LTP, a single PKC isozyme, called PKMζ (Sacktor et al., 1993), is activated by a mechanism fundamentally different from that of the kinases important for LTP induction (Osten et al., 1996; Hernandez et al., 2003). A typical full-length PKC isoform consists of an N-terminal regulatory domain and a C-terminal catalytic domain (Newton, 2001). The regulatory domain contains binding sites for second messengers and a pseudosubstrate sequence that inhibits the catalytic domain. Second messengers activate a full-length PKC by binding to the regulatory domain and causing a transient conformational change that releases the inhibition of the pseudosubstrate.
PKMζ, in contrast, consists of the independent catalytic domain of the atypical PKCζ isoform, and, lacking inhibition from the pseudosubstrate of a regulatory domain, is a persistently active enzyme. Although PKM is usually thought of as a cleavage product of full-length PKC (Kishimoto et al., 1983), we found that PKMζ is not formed in LTP by proteolysis but by gene expression of a brain-specific PKMζ mRNA, which is generated by an internal promoter within the PKCζ gene (Hernandez et al., 2003) and transported to synaptodendritic compartments of neurons (Muslimov et al., 2004). Tetanic stimulation induces protein synthesis from the PKMζ mRNA, persistently increasing the levels of the kinase during LTP maintenance (Sacktor et al., 1993; Osten et al., 1996; Hernandez et al., 2003).
Phosphorylation by PKMζ potently enhances AMPA receptor (AMPAR)-mediated synaptic transmission, and this enhancement fully occludes LTP (Ling et al., 2002). In addition, inhibition of PKMζ activity 1 h after tetanization reverses the maintenance of AMPAR-mediated synaptic potentiation (Ling et al., 2002). LTP, however, is not a unitary phenomenon but evolves through several temporal phases (Bliss and Collingridge, 1993; Sweatt, 1999). Protein synthesis inhibitors have been used to define an early protein synthesis-independent phase of potentiation lasting ∼1-3 h and a late protein synthesis-dependent phase (Stanton and Sarvey, 1984; Frey et al., 1988; Otani et al., 1989; Nguyen et al., 1994; Osten et al., 1996). Therefore, PKMζ could potentially mediate synaptic enhancement in the early phase, the transition from early to late, or the late phase of LTP. In this study, we determined the temporal window of LTP maintained by PKMζ activity by using the cell-permeable ζ-pseudosubstrate inhibitory peptide (ZIP), which acts on PKMζ by reconstituting the inhibition of the missing PKCζ regulatory domain (Laudanna et al., 1998). Applying this agent at various times during LTP, we found that PKMζ specifically maintains the late phase of LTP.
Materials and Methods
Animals. For all of the experiments, hippocampi of male Sprague Dawley rats were removed after decapitation under halothane-induced anesthesia, according to the standards of the Animal Use and Care Committee of the State University of New York Downstate Medical Center. Animals were 15-20 d of age, except for two animals that were >35 d of age used to examine the role of PKMζ at 5 h after tetanization. Because the results were equivalent to those obtained from the younger animals, the experiments were pooled in Figure 3E.
Figure 3.

Persistent PKMζ phosphorylation maintains the late phase of LTP at 3 and 5 h after tetanization. A, A 5 μm concentration of ZIP applied 3 h after tetanization reversed the potentiated pathway but had no effect on the control pathway (n = 6). The level of potentiation before drug exposure was significantly reduced by 1 h after the inhibitor was applied (p < 0.01), and, by 2 h, the tetanized pathway had returned to baseline. B, Infusion of 2.5 μm partially reversed potentiation after 2 h of drug perfusion (p < 0.05; n = 5). C, D, A 1 μm concentration of ZIP (C; n = 6) or a 5 μm concentration of the scrambled inactive peptide (D; n = 6) had no effect on either tetanized or nontetanized pathways. E, A 5 μm concentration of ZIP applied 5 h after tetanization reversed the potentiated pathway but had no effect on the control pathway (n = 4). F, A 5 μm concentration of scrambled inactive peptide had no effect (n = 4).
Hippocampal slice preparation, stimulation, and recording. Transverse hippocampal slices (450 μm) were prepared as described previously (Sacktor et al., 1993). Hippocampi were dissected, bathed in cold dissecting buffer, and sliced with a McIlwain tissue slicer. The dissecting buffer contained (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 11 glucose, 10 MgCl2, and 0.5 CaCl2 and was bubbled with 95% O2/5% CO2, bringing the solution to pH 7.4. After 30 min, the slices were placed in an interface recording chamber (total volume, 2.1 ml; Fine Science Tools, Foster City, CA), prewarmed to 31.5 ± 0.5°C, and perfused at 0.175 ml/min with the saline solution (1.2 mm MgCl2 and 1.7 mm CaCl2).
A bipolar platinum/iridium (80/20%) electrode (Frederick Haer Company, Bowdoinham, ME) was placed in CA1 stratum radiatum to evoke field EPSPs from Schaffer collateral/commissural-CA1 synapses. Field EPSPs were recorded using glass microelectrodes with a resistance of 3-8 MΩ filled with the recording saline and positioned 200 μm from the stimulating electrodes. Current intensity of test stimuli (20-40 μA; duration, 0.1 ms) was set to produce one-third of maximal EPSPs (1.5-2.0 mV). The frequency of test stimulation was every 15 s. Baseline EPSPs were recorded for at least 30 min to ensure stability of the response. Data were collected and analyzed using Superscope (GW Instruments, Somerville, MA). The slope of the field EPSP was measured between 10 and 50% of the initial phase of the EPSP response. The tetanization protocol was four 100 Hz, 1 s trains, set at 75% of the maximal EPSP response (15-30 μA above the current intensity of test stimuli), delivered at 5 min intertrain intervals (Scharf et al., 2002). Data were analyzed with Excel (Microsoft, Redmond, WA) and StatView (version 4.1; SAS Institute, Cary, NC). Values are presented as mean ± SEM; within-group differences were determined by paired t test, and between-group comparisons were determined by unpaired t test.
In all of the LTP experiments, a second stimulation pathway in the radiatum that received only test pulse stimulation served as a control. To ensure that the pathways were separate, a paired-pulse facilitation protocol was used to demonstrate that no facilitation occurred at a 50 ms pulse interval. The test pulses of the two pathways were set to alternate the delivery of the pulses every 7.5 s, resulting in each pathway receiving a pulse every 15 s.
The myristoylated ζ-pseudosubstrate peptide (myr-SIYRRGAR-RWRKL-OH) and its corresponding scrambled control peptide (myr-RLYRKRIWRSAGR-OH; both from Biosource International, Camarillo, CA) (Laudanna et al., 1998) were dissolved in an aqueous stock concentration of 10 mm, stored at -20°C, and diluted in the physiological saline immediately before use at the designated concentrations. Anisomycin and emetine was applied at a final bath concentration of 25 and 20 μm, respectively, in 0.01% DMSO. LTP in the presence of 0.01% DMSO was indistinguishable from that without the drug presented in Figure 1 A (data not shown).
Figure 1.
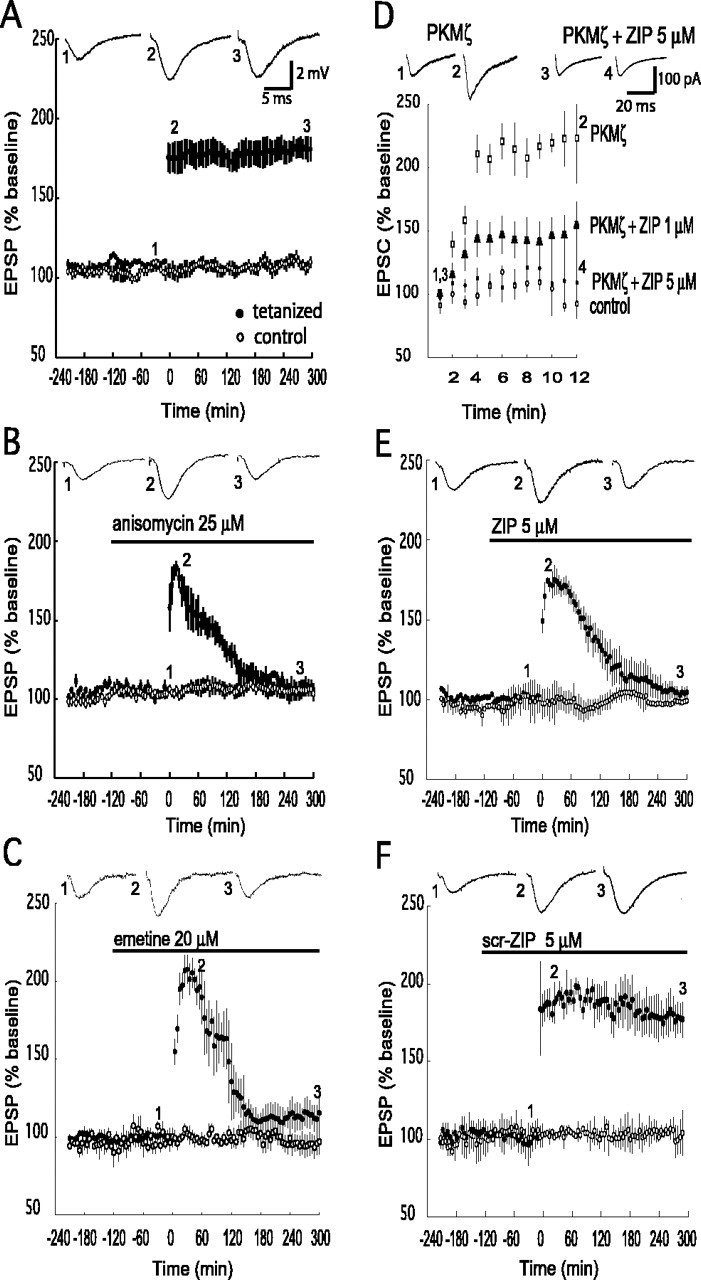
Phosphorylation by PKMζ is critical for the late phase, but not the early phase, of LTP. EPSPs were monitored in two independent pathways within the same slice (filled circles, tetanized pathway; open circles, control nontetanized pathway). Representative EPSP traces from the times indicated are shown. Tetanic stimulation occurs at time 0. A, Tetanization resulted in stable potentiation for 5 h (n = 6). B, C, Anisomycin (B; 25 μm) or emetine (C; 20 μm) infused into the recording chamber 2 h before tetanization blocks the late phase of LTP (filled circles). The potentiated pathway returned to baseline by 3 h (n = 4). Compared with the initial posttetanization response, a significant reduction in potentiation occurred after 2 h (p<0.05). No effects were observed on control pathways for either drug. D, Bath application of myristoylated ZIP blocked potentiation of AMPAR EPSCs produced by postsynaptic perfusion of PKMζ through a whole-cell recording pipette. Whole-cell recordings showed potentiation of evoked AMPAR responses by PKMζ (open squares; p < 0.05 for values between 1 and 12 min of recording). Bath application of 5 μm ZIP showed strong inhibition of potentiation (filled circles); 1 μm showed partial inhibition (filled triangles). Control baseline recordings without PKMζ in the pipette or inhibitor are shown (open circles); n = 4 for all of the experiments. E, Application of ZIP had no effect on baseline or early LTP (5 μm; n = 5) but prevented the late phase, with the potentiated pathway returning to baseline in 3 h. We observed a significant reduction of the initial potentiation by 2 h after tetanization (p < 0.05). No effect was observed on the nontetanized pathway. F, Bath application of scr-ZIP (5 μm; n = 5) had no effect on LTP or the nontetanized pathway.
Whole-cell recording obtained from CA1 pyramidal cells visualized in hippocampal slices were performed as described previously (Ling et al., 2002). The recording buffer contained (in mm) 124 NaCl, 5 KCl, 26 NaHCO3, 1.6 MgCl2, 4 CaCl2, and 10 glucose, pH 7.3, bubbled with 95% O2/5% CO2, and was perfused at 4.8 ml/min in an ∼1.5 ml submerged chamber at 32-34°C. Recording pipettes had tip resistances of 3-5 MΩ and contained 130 mm Cs-methanesulfonate, 10 mm NaCl, 10 mm HEPES, 2 mm EGTA, 2 mm Na-ATP, and 0.5 mm Na-GTP, pH adjusted to 7.30 with KOH, with or without 18 nm baculovirus/Sf9-cell recombinantly expressed PKMζ (0.43 pmol·min-1·μl-1 phosphotransferase activity) (Ling et al., 2002). ZIP was applied at varying concentrations to the bath before cell break-in, and experiments with ZIP were interleaved with experiments without the drug. Synaptic events were evoked by extracellular stimulation with an electrode placed in stratum radiatum every 15 s and measured at peak amplitude. Signals were recorded under voltage clamp with a Warner PC-501A amplifier (Warner Instruments, Hamden, CT). Brief voltage steps (5 mV; 10 ms) from cell holding potential (-75 mV) were applied to monitor cell access resistance, capacitance, and input resistance. Cells were accepted for study if the input resistance was >100 MΩ and the access resistance was 8-12 MΩ. If cell access resistance changed significantly during recording (>20%), the data were discarded. Data signals were digitized at 100 kHz by a 16-bit digitizer (Digidata 1322A; Axon Instruments, Union City, CA) and then stored and analyzed with pClamp software (Axon Instruments) on an IBM-compatible Pentium 4 microcomputer.
Results
Inhibition of PKMζ phosphorylation blocks the late phase, but not the early phase, of LTP
We used four strong tetanic trains separated by 5 min, a standard protocol for producing late LTP (Scharf et al., 2002) (Fig. 1A). We determined the temporal window of the protein synthesis-independent phase for this protocol by bath applying the protein synthesis inhibitors anisomycin (25 μm) (Fig. 1B) (Scharf et al., 2002) and emetine (20 μm) (Fig. 1C) (Raymond et al., 2000) for 2 h before tetanization and continuously thereafter. Consistent with previous studies (Stanton and Sarvey, 1984; Otani et al., 1989; Nguyen et al., 1994; Osten et al., 1996; Raymond et al., 2000; Scharf et al., 2002), the early, protein synthesis-independent phase was stable for ∼30 min and then declined to baseline by 3 h after tetanus.
To examine the role of PKMζ in LTP produced by this tetanization protocol, we used the cell-permeable atypical PKC-selective inhibitor ZIP (Laudanna et al., 1998; Ling et al., 2002). We assessed the efficacy of bath applications of ZIP in hippocampal slices by directly examining the ability of the drug to inhibit PKMζ-mediated synaptic potentiation (Fig. 1D). We obtained whole-cell recordings of evoked AMPAR responses from CA1 pyramidal cells with PKMζ in the recording pipette, as described previously (Ling et al., 2002). Confirming our previous results, in the absence of the inhibitor, postsynaptic perfusion of PKMζ caused a robust potentiation of AMPAR responses (224 ± 36 at 12 min of recording, baseline at 1 min set at 100%, compared with 92 ± 4% in control recordings without PKMζ). Bath application of 5 μm ZIP strongly inhibited the ability of perfused PKMζ to potentiate synaptic AMPAR responses (responses were 109 ± 28 of baseline after 12 min of recording). A 1 μm concentration of ZIP caused a partial reduction of PKMζ-mediated potentiation (responses were 154 ± 18 of baseline after 12 min), indicating that 5 μm is close to the lowest effective dose for complete blockade of PKMζ-mediated synaptic potentiation.
We therefore examined the role of PKMζ in LTP by applying 5 μm ZIP before tetanization (Fig. 1E). Application of the agent had no effect on baseline synaptic transmission. In the presence of ZIP, tetanization produced synaptic potentiation that was stable for ∼30 min and then declined to baseline by 3 h (Fig. 1E, filled circles). The decline in responses in the presence of ZIP was specific to potentiated synapses, because the drug had no effect on an independent control pathway simultaneously recorded within each slice (Fig. 1E, open circles). An inactive version of the ZIP peptide with a scrambled amino acid sequence (scr-ZIP) did not affect baseline synaptic transmission or LTP (Fig. 1F). Thus, PKMζ phosphorylation did not appear to contribute to an early phase of LTP (similar in kinetics to the protein synthesis-independent phase) but was critical for the late phase of LTP.
Persistent phosphorylation by PKMζ establishes late LTP
The blockade of late LTP by the PKMζ inhibitor could be because only early transient PKMζ activity is necessary to induce the late phase, as has been described for PKA (Chetkovich et al., 1991; Frey et al., 1993; Matthies and Reymann, 1993; Huang and Kandel, 1994; Nguyen et al., 1994; Abel et al., 1997), or because persistent PKMζ activity is required to generate the late phase. To examine these alternatives, we applied 5 μm ZIP 1 h after tetanization (Fig. 2A, filled circles). ZIP reversed LTP maintenance but had no effect on a control nontetanized pathway (Fig. 2A, open circles). The kinetics of the decline in potentiation with 5 μm was similar to that observed previously with 1 μm (Ling et al., 2002), suggesting that these are saturating doses for LTP reversal at 1 h after tetanization. To further characterize the reversal of this intermediate phase of LTP, we progressively lowered the dose of ZIP to obtain the range of its IC50 value (Fig. 2B-D). Doses of 0.25-0.5 μm had partial effects, whereas 0.1 μm was ineffective. Thus, persistent PKMζ activity is required to establish the late phase of LTP.
Figure 2.

Persistent PKMζ phosphorylation is critical for an intermediate phase of LTP. A, Infusion of 5 μm ZIP reversed potentiation when applied 1 h after tetanization (filled circles). By 3 h after tetanization, the level of EPSP responses was significantly reduced compared with preapplication levels (p < 0.01) and was not significantly different from the nontetanized pathway (open circles). B, Infusion of 0.5 μm ZIP produced a significant reduction of the initial potentiation by 3 h after tetanization (p < 0.05); the level of potentiation at that time was significantly higher than the EPSPs from the nontetanized pathway (p<0.02). By 5 h after LTP induction, the two pathways were not significantly different. C, Infusion of 0.25 μm ZIP significantly reduced the initial potentiation by 3 h after tetanization (p < 0.02) but by 5 h had not completely reversed the EPSP potentiation to the level of the nontetanized pathway (p < 0.05). D, Infusion of 0.1 μm had no effect. No effect of the inhibitor was observed in the control pathways in any experiment; n = 5-6 for all experiments.
Persistent phosphorylation by PKMζ maintains late LTP
PKMζ inhibition at 1 h after tetanus might reverse LTP maintenance because PKMζ phosphorylation is necessary only for the transition from early to late LTP or because the persistent activity of PKMζ is critical for maintaining the late phase itself. To distinguish between these possibilities, we began the application of 5 μm ZIP 3 h after tetanus (Fig. 3A). The PKMζ inhibitor completely reversed the late phase of LTP at 3 h (Fig. 3A, filled circles). No effect on the nontetanized pathway was observed (Fig. 3A, open circles). To characterize the reversal of late-phase LTP, we determined the IC50 range for ZIP at 3 h after tetanization. A dose of 2.5 μm reduced the level of potentiation after 2 h of drug perfusion (Fig. 3B), and 1.0 μm had no apparent effect (Fig. 3C); therefore, the IC50 value of ZIP for reversing the late phase was >1 and ≤2.5 μm. A 5 μm concentration of the inactive, scrambled version of the inhibitor had no effect (Fig. 3D).
To examine further the critical requirement of persistent PKMζ activity in late LTP maintenance, we determined the effect of the inhibitor when applied 5 h after tetanization. A 5 μm concentration of ZIP completely reversed LTP at this time point, with no effect on a nontetanized control pathway within the slices (Fig. 3E). A 5 μm concentration of scrambled peptide had no effect on LTP maintenance (Fig. 3F). Thus, the persistent activity of PKMζ maintains synaptic potentiation during late LTP.
Discussion
These results demonstrate a unique role for persistent PKMζ activity in the maintenance of the late phase of LTP. We used ZIP, a cell-permeable, selective peptide inhibitor of PKMζ to block the constitutive activity of PKMζ. We determined that bath application of 5 μm ZIP onto hippocampal slices was close to the lowest dose that fully inhibits the ability of the kinase to potentiate AMPAR responses. We then found that this dose, although it does not block early LTP, reverses late LTP when applied 1, 3, and 5 h after tetanization, without affecting control nontetanized pathways. Although we cannot exclude the possibility that ZIP may also be affecting another kinase at this dose, other kinases known to be important for induction are unlikely to be inhibited [e.g., ZIP is ∼60 times less potent on CaMKII than on PKMζ (Ling et al., 2002)].
To our knowledge, no other molecule has been shown to selectively maintain potentiated synapses in the late phase of LTP. For example, despite its persistent autophosphorylation, CaMKII activity appears to have a specific effect on LTP for less than an ∼2 min period during induction (Otmakhov et al., 1997; Chen et al., 2001) or a general nonspecific effect on synaptic transmission (Waxham et al., 1993). MAP kinase (English and Sweatt, 1997) and conventional/novel PKCs (Malinow et al., 1989; Lopez-Molina et al., 1993) are necessary only within ∼30 min after tetanization and may be important for the induction of new protein synthesis (Kelleher et al., 2004). PKA, which regulates the induction of transcriptional changes in late LTP (Matthies and Reymann, 1993; Abel et al., 1997), as well as earlier changes (Otmakhova et al., 2000), is also necessary only for the first 30 min after a tetanus (Huang and Kandel, 1994). Thus, the function of PKMζ in LTP maintenance is fundamentally different from that of the kinases important in LTP induction. This is consistent with the unique molecular properties of PKMζ as an autonomously active kinase that is synthesized during LTP maintenance (Sacktor et al., 1993; Osten et al., 1996; Hernandez et al., 2003). Because the time window of new protein synthesis critical for LTP maintenance is relatively brief after tetanization (Otani et al., 1989; Frey and Morris, 1997), our results suggest that the persistence of newly synthesized PKMζ protein, rather than its continual synthesis, is the persistent molecular mechanism maintaining late LTP in hippocampal slices.
Synaptic potentiation in the early phase of LTP may be related to the activity of the other kinases implicated in induction, several of which can enhance synaptic transmission, including CaMKII (Lledo et al., 1995; Shirke and Malinow, 1997), conventional/novel PKC (Malenka et al., 1986; Hu et al., 1987), and PKA (Carroll et al., 1998). Other molecules, such as BDNF, may be involved in extending the duration of this early potentiation (Pang et al., 2004) and may interact with PKMζ. PKMζ-mediated potentiation itself first becomes apparent 1 h after tetanic stimulation, when the early phase begins to decline. Therefore, we cannot exclude the possibility that PKMζ-mediated potentiation might have begun earlier than 1 h but was masked by the potentiation produced by the other molecules important for early LTP. Interestingly, we observed previously a rapid block of potentiation by intracellular perfusion of a dominant-negative inhibitor of PKMζ soon after tetanization paired with postsynaptic depolarization in whole-cell recordings (Ling et al., 2002). A rapid potentiation by PKMζ may be observed under these conditions because intracellular dialysis during standard whole-cell recordings eliminates synaptic potentiation by the conventional/novel PKCs (Carroll et al., 1998).
Persistent PKMζ phosphorylation appears critical for both the transition from the early to late phase of LTP and the maintenance of the late phase. These functions of PKMζ may differ, however, because the IC50 value of ZIP increases between 1 and 3 h after tetanization, as late LTP becomes more established. Although comparisons between the rapid bath exchange/submerged chamber used for whole-cell recording and the slow exchange/interface chamber used for maintaining late LTP should be made cautiously (Ho et al., 2004), it appears that a partial inhibition of PKMζ activity is sufficient to block the transition from early to late LTP, whereas a more complete block is required to reverse established LTP. Because ZIP is a competitive inhibitor of protein substrate binding to PKMζ, establishing potentiation at synapses might require more PKMζ-substrate interactions than maintaining potentiation. Alternatively, there may be separate substrates involved at different stages of maintenance with distinct affinities for PKMζ. Interestingly, previous observations with H7 [1-(5-isoquinolinesulfonyl)-2-methylpiperazine], a nonspecific inhibitor of several kinases, including ζ, also showed a similar increase in the IC50 value with time during LTP maintenance (Colley et al., 1990).
The synaptic potentiation during late LTP is usually hypothesized to be attributable to a change in synaptic structure (Yuste and Bonhoeffer, 2001; Harris et al., 2003) (but see Lang et al., 2004); thus, the observation that an enzyme, albeit one in a unique, autonomously active form, is critical for maintaining late LTP, may be surprising. Recent work on synapses and spines, however, suggests that many of their components, including neurotransmitter receptors, scaffolding proteins, and actin (Matus et al., 2000; Smart and Halpain, 2000; Ehlers, 2003), are rapidly cycling into and out of these structures. Therefore, enzymes that organize the cytoskeleton and its binding proteins may be key to maintaining the structural changes that could contribute to LTP expression. Thus, the persistent increased PKMζ activity that mediates late LTP might play a master role in establishing and maintaining structural changes in the face of protein turnover (Ruiz-Canada et al., 2004).
Footnotes
This work was supported by National Institutes of Health Grants MH53576 and MH57068 (T.C.S.).
Correspondence should be addressed to Dr. Todd C. Sacktor, Departments of Physiology and Pharmacology, Box 29, State University of New York Downstate Medical Center, 450 Clarkson Avenue, Brooklyn, NY 11203. E-mail: tsacktor@downstate.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251979-06$15.00/0
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615-626. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Nicoll RA, Malenka RC (1998) Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol 80: 2797-2800. [DOI] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE (2001) Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol 85: 1368-1376. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Gray R, Johnston D, Sweatt JD (1991) N-methyl-d-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA 88: 6467-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley PA, Sheu FS, Routtenberg A (1990) Inhibition of protein kinase C blocks two components of LTP persistence, leaving initial potentiation intact. J Neurosci 10: 3353-3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231-242. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272: 19103-19106. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG (1997) Synaptic tagging and long-term potentiation. Nature 385: 533-536. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reyman KG, Matthies H (1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57-65. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang Y-Y, Kandel ER (1993) Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260: 1661-1664. [DOI] [PubMed] [Google Scholar]
- Harris KM, Fiala JC, Ostroff L (2003) Structural changes at dendritic spine synapses during long-term potentiation. Philos Trans R Soc Lond B Biol Sci 358: 745-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AI, Blace N, Crary JF, Serrano PA, Leitges M, Libien JM, Weinstein G, Tcherapanov A, Sacktor TC (2003) Protein kinase M zeta synthesis from a brain mRNA encoding an independent protein kinase C zeta catalytic domain. Implications for the molecular mechanism of memory. J Biol Chem 278: 40305-40316. [DOI] [PubMed] [Google Scholar]
- Ho OH, Delgado JY, O'Dell TJ (2004) Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in vitro. J Neurochem 91: 1344-1357. [DOI] [PubMed] [Google Scholar]
- Hu G-Y, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Anderson P, Greengard P (1987) Protein kinase C injection into hippocampal pyramidal cells elicits features of long-term potentiation. Nature 328: 426-429. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1994) Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem 1: 74-82. [PubMed] [Google Scholar]
- Kelleher III RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116: 467-479. [DOI] [PubMed] [Google Scholar]
- Kishimoto A, Kajikawa N, Shiota M, Nishizuka Y (1983) Proteolytic activation of calcium-activated, phospholipid-dependent protein kinase by calcium-dependent neutral protease. J Biol Chem 258: 1156-1164. [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD (1998) A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem 273: 4516-4522. [DOI] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS (2004) Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci USA 101: 16665-16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC (1998) Evidence of zeta protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. J Biol Chem 273: 30306-30315. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC (2002) Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat Neurosci 5: 295-296. [DOI] [PubMed] [Google Scholar]
- Lisman J (1994) The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci 17: 406-412. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA (1995) Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA 92: 11175-11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Molina L, Boddeke H, Muller D (1993) Blockade of long-term potentiation and of NMDA receptors by the protein kinase C antagonist calphostin C. Naunyn Schmiedebergs Arch Pharmacol 348: 1-6. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA (1986) Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321: 175-177. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862-866. [DOI] [PubMed] [Google Scholar]
- Matthies H, Reymann KG (1993) Protein kinase A inhibitors prevent the maintenance of hippocampal long-term potentiation. NeuroReport 4: 712-714. [DOI] [PubMed] [Google Scholar]
- Matus A, Brinkhaus H, Wagner U (2000) Actin dynamics in dendritic spines: a form of regulated plasticity at excitatory synapses. Hippocampus 10: 555-560. [DOI] [PubMed] [Google Scholar]
- Muslimov IA, Nimmrich V, Hernandez AI, Tcherepanov A, Sacktor TC, Tiedge H (2004) Dendritic transport and localization of protein kinase Mzeta mRNA: implications for molecular memory consolidation. J Biol Chem 279: 52613-52622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC (2001) Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101: 2353-2364. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994) Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265: 1104-1107. [DOI] [PubMed] [Google Scholar]
- Osten P, Valsamis L, Harris A, Sacktor TC (1996) Protein synthesis-dependent formation of protein kinase Mζ in LTP. J Neurosci 16: 2444-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Marshall CJ, Tate WP, Goddard GV, Abraham WC (1989) Maintenance of long-term potentiation in rat dentate gyrus requires protein synthesis but not messenger RNA synthesis immediately post-tetanization. Neuroscience 28: 519-526. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE (1997) Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci 17: 5357-5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE (2000) Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci 20: 4446-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306: 487-491. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Thompson VL, Tate WP, Abraham WC (2000) Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci 20: 969-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, Budnik V (2004) New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron 42: 567-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E (1993) Persistent activation of the ζ isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci USA 90: 8342-8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T (2002) Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol 87: 2770-2777. [DOI] [PubMed] [Google Scholar]
- Schwartz JH (1993) Cognitive kinases. Proc Natl Acad Sci USA 90: 8310-8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JH, Greenberg SM (1987) Molecular mechanisms for memory: second-messenger induced modifications of protein kinases in nerve cells. Annu Rev Neurosci 10: 459-476. [DOI] [PubMed] [Google Scholar]
- Shirke AM, Malinow R (1997) Mechanisms of potentiation by calcium-calmodulin kinase II of postsynaptic sensitivity in rat hippocampal CA1 neurons. J Neurophysiol 78: 2682-2692. [DOI] [PubMed] [Google Scholar]
- Smart FM, Halpain S (2000) Regulation of dendritic spine stability. Hippocampus 10: 542-554. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM (1984) Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci 4: 3080-3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (1999) Toward a molecular explanation for long-term potentiation. Learn Mem 6: 399-416. [DOI] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD (1993) Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res 609: 1-8. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001) Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071-1089. [DOI] [PubMed] [Google Scholar]