

Abstract
Experimental studies suggest that the delivery of antiepileptic agents into the seizure focus might be of potential utility for the treatment of focal-onset epilepsies. Botulinum neurotoxin E (BoNT/E) causes a prolonged inhibition of neurotransmitter release after its specific cleavage of the synaptic protein synaptosomal-associated protein of 25 kDa (SNAP-25). Here, we show that BoNT/E injected into the rat hippocampus inhibits glutamate release and blocks spike activity of pyramidal neurons. BoNT/E effects persist for at least 3 weeks, as determined by immunodetection of cleaved SNAP-25 and loss of intact SNAP-25. The delivery of BoNT/E to the rat hippocampus dramatically reduces both focal and generalized kainic acid-induced seizures as documented by behavioral and electrographic analysis. BoNT/E treatment also prevents neuronal loss and long-term cognitive deficits associated with kainic acid seizures. Moreover, BoNT/E-injected rats require 50% more electrical stimulations to reach stage 5 of kindling, thus indicating a delayed epileptogenesis. We conclude that BoNT/E delivery to the hippocampus is both antiictal and antiepileptogenic in experimental models of epilepsy.
Keywords: neurotransmitter release, kainic acid, seizures, Morris water maze, neuronal death, hippocampal kindling
Introduction
A significant percentage of epileptic patients are resistant to conventional antiepileptic drugs (Engel, 1996; Regesta and Tanganelli, 1999). One therapeutic approach currently under investigation in animal models is the delivery of antiepileptic medication directly into the seizure focus in the brain (Kubek et al., 1998; Stein et al., 2000; Kohane et al., 2002; Tamargo et al., 2002). This strategy has the advantage of avoiding problems of whole-brain and systemic toxicity but requires that the action of the antiepileptic compound be long-lasting to prevent seizures for an extended period of time. Because excessive synaptic activity is the primary cause of seizures and seizure-induced brain damage (Meldrum, 1995; Holmes, 2002), drugs affecting the neurotransmitter release machinery in epileptogenic areas might be effective antiictal agents.
The seven botulinum neurotoxins (BoNTs; indicated with letters from A to G) are bacterial metalloproteases that act on peripheral cholinergic terminals and cause a long-lasting inhibition of neurotransmitter release (Schiavo et al., 2000). They act in the cytosol by cleaving core proteins of the neuroexocytosis apparatus named soluble N-ethylmaleimide-sensitive factor-attachment receptors (SNAREs) (Schiavo et al., 2000; Turton et al., 2002). BoNT/A and BoNT/E cleave synaptosomal-associated protein of 25 kDa (SNAP-25), BoNT/C cleaves both syntaxin and SNAP-25, and all other BoNTs act on vesicle-associated membrane protein/synaptobrevin (Rossetto et al., 2001). BoNT inhibition in peripheral cholinergic neurons is fully reversible after the degradation of the internalized toxin and the resynthesis of the cleaved proteins (Meunier et al., 2002). Data in the literature indicate that BoNTs block exocytosis also when directly applied to central neurons, with potencies and durations matching those observed for motor nerve terminals (Ashton and Dolly, 1988; Foran et al., 2003; Sutton et al., 2004). Remarkably, studies on hippocampal neurons in vitro demonstrate that BoNT/A and BoNT/E impair excitatory transmission with little or no effect on GABAergic synapses (Capogna et al., 1997; Verderio et al., 2004). This selective inhibition of glutamatergic versus GABAergic transmission by BoNT/E is attributed to the preferential localization of SNAP-25, the molecular target of BoNT/E, in synaptic terminals of excitatory hippocampal neurons (Verderio et al., 2004). Based on these findings, we explored the potential antiictal effects of in vivo administration of BoNT/E. We show that BoNT/E delivery to the rat hippocampus is anticonvulsant on acute seizures induced by kainic acid (KA). We also demonstrate that BoNT/E significantly retards the development of kindling epileptogenesis.
Materials and Methods
Animals. Long-Evans hooded rats bred in our animal facility were used in this study. Animals were housed with a 12 h light/dark cycle with food and water available ad libitum. All experimental procedures conformed to the European Communities Council Directive number 86/609/EEC.
BoNT/E injections. BoNT/E was obtained by Wako (Osaka, Japan) and was trypsin activated, purified, and tested as described previously (Schiavo and Montecucco, 1995). Its potency was evaluated with the mouse phrenic nerve-hemidiaphragm test. Two unilateral stereotaxic infusions of 1.5 μl of BoNT/E (50 nm) or vehicle (2% rat serum albumin in PBS) were made into the dorsal hippocampus under avertin anesthesia at postnatal day 35 (P35). P35 rats were chosen because they display a maximal sensitivity to KA-induced seizures (Ben-Ari, 1985; Stafstrom et al., 1993). Coordinates (in millimeters) from bregma were: nose bar, -2.5; for CA1, anteroposterior (AP), -2.4, lateral (L), +1.8, 2.1 below dura; for CA3, AP, -2.4, L, +3.3, H, 3 below dura.
Detection of cleaved SNAP-25. Cleaved SNAP-25 was detected by immunostaining and immunoblotting using a peptide-affinity purified polyclonal antibody raised against the BoNT/E truncated C-terminal peptide of SNAP-25 (CDMGNEIDTQNRQIDR). This antibody recognizes specifically cleaved SNAP-25 but not the whole protein. For immunohistochemistry, rats received hippocampal injections of BoNT/E (n = 10) or vehicle (n = 5) and were transcardially perfused with 4% paraformaldehyde 1-4 d later (Caleo et al., 2003). Brain coronal sections (40 μm thick; cut on a freezing microtome) were blocked with 10% normal goat serum in PBS and then incubated overnight with the anti-BoNT/E-cleaved SNAP-25 antibody diluted 1:300 in a PBS solution containing 1% serum and 0.3% Triton X-100. On the following day, sections were reacted with a biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) followed by avidin-biotin-peroxidase complex (ABC kit; Vector Laboratories) and diaminobenzidine reaction. For immunoblotting, rats (n = 13) received hippocampal injections of BoNT/E, and dorsal and ventral hippocampi (ipsilateral and contralateral to the injected side) were dissected after 1, 14, 21, and 35 d. Protein extracts (10 μg) were separated by electrophoresis and blotted, and filters were incubated with anti-BoNT/E-cleaved SNAP-25 (1:50 dilution) or anti-SNAP-25 (1:1000 dilution; Synaptic Systems, Goettingen, Germany) polyclonal antibodies, reacted with HRP-conjugated goat anti-rabbit secondary antibody (Bio-Rad, Hercules, CA), and developed by ECL (Amersham Biosciences, Amersham, UK). Filters were stripped and reprobed with anti-β-tubulin monoclonal antibody (1:500 dilution; Sigma, St. Louis, MO), which served as an internal standard for protein quantification.
Glutamate release. Glutamate release measurements were performed on superfused hippocampal synaptosomes from vehicle- and BoNT/E-treated rats (n = 7 per group), 1 d after intrahippocampal injection of 40 ng of kainic acid (see below for details). Preparation of synaptosomes was as described by Gobbi et al. (2002), and release of glutamate was assessed according to Di Stasi et al. (2002). Synaptosomes were depolarized by a 90 s pulse of 35 mm KCl. Glutamate overflow was measured by a Waters Associates (Milford, MA) Alliance HPLC analysis system. The analytical method involved automatic precolumn derivatization with o-phthalaldehyde followed by separation on C18 reverse-phase chromatography column and fluorimetric detection (Di Stasi et al., 2002).
Recordings of spike activity. Rats received unilateral injections of BoNT/E (n = 6) or vehicle (n = 3). Recordings of spike activity were performed in the injected hippocampus 1-2 d after BoNT/E or vehicle injection as described by Caleo et al. (2003). Animals were anesthetized with urethane (20% solution in saline; 0.7 ml/100 g of body weight, i.p.; Sigma) and placed in a stereotaxic frame. Body temperature was continuously monitored and maintained at 37°C by a thermostat-controlled electric blanket. After exposure of the cerebral surface, a micropipette (tip resistance, 2 MΩ) filled with 3 m NaCl was inserted into the brain to reach the dorsal hippocampus. Two to five penetrations per hemisphere were made to map spike activity in CA1 and CA3 sectors. Location of the recording sites was determined using histological controls (Caleo et al., 2003). Signals were amplified 25,000-fold, bandpass filtered (500-5000 Hz), and conveyed to a computer for storage and analysis.
EEG analysis after intrahippocampal KA. Rats were infused unilaterally into the left dorsal hippocampus with BoNT/E (n = 8) or vehicle (n = 8) as described above under avertin anesthesia. At the end of the infusion procedure, one screw electrode was placed over the parietal cortex ipsilateral to the injected hippocampus together with a ground lead over the nasal sinus. Two depth bipolar electrodes made of insulated nichrome wire (60 μm) were implanted bilaterally into the dorsal hippocampus (nose bar, -2.5 mm from bregma; AP, -2.4 mm from bregma; L, ±1.8 mm from bregma; 3.0 mm below dura), and a guide cannula was glued to the left side depth electrode and positioned on top of dura for the intrahippocampal injection of KA. Surface and depth electrodes were connected to a multipin socket and secured to the skull together with the injection cannula by acrylic dental cement. Two days after surgery, freely moving rats that were injected with BoNT/E or its vehicle received a unilateral injection of 40 ng of KA into the left hippocampus using a needle protruding for 3 mm below the guide cannula (Vezzani et al., 1999). The location of BoNT/E injection sites, KA infusion cannula, and recording electrodes is schematically shown in Figure 2, A and B.
Figure 2.

Antiictal effect of BoNT/E on EEG seizures induced by intrahippocampal KA. A, Three-dimensional drawing of the rat brain illustrating the location of BoNT/E injection (inj.) sites (open circles) and the placement of KA injection cannula and recording (rec.) electrodes. Position of the cortical electrode is indicated by an asterisk. The plane of the coronal section shown in B, corresponding to stereotaxic coordinate AP -2.4 mm from bregma (see Materials and Methods), is also indicated. B, Nissl-stained coronal section through the hippocampus indicating the sites of BoNT/E injection (open circles) and the position of the KA infusion cannula glued to the recording electrode (X). C, D, EEG tracing depicting seizure activity induced by unilateral injection of 40 ng of KA into the dorsal hippocampus of rats that received 2 d before the convulsant either vehicle (C) or BoNT/E (D). Traces on the left represent baseline recordings in the parietal cortex (CTX), right hippocampus (RHP), and left hippocampus (LHP). Traces on the right depict typical seizure episodes. Time elapsed from the KA injection is indicated. E-G, Quantification of EEG seizures in control (black circles) and BoNT/E-injected (open circles) animals (n = 8 per group). A significant reduction in the number and duration of EEG seizures was consistently detected in BoNT/E-treated rats. The onset time to the first seizure, the total number of seizures, and the total time spent in ictal activity are reported in E-G, respectively. Each point indicates one animal, and horizontal bars represent mean values ± SE.
To compare the effect of BoNT/E with that of a conventional antiepileptic drug, one group of rats (not previously treated with BoNT/E) received phenytoin (PHT; 50 mg/kg, i.p.; n = 7 rats) dissolved in propylene glycole, or vehicle (n = 7), 60 min before hippocampal delivery of 40 ng of KA.
EEG recordings on freely moving animals were performed using a four-channel EEG polygraph by an investigator who was unaware of the treatment of the animals. An initial 15-30 min recording was made to establish basal activity, and then EEG recordings were made continuously up to 4 h after KA administration. The KA dose (40 ng) was previously shown to induce EEG seizures recurring for ∼180 min in 100% of the rats without mortality (Vezzani et al., 1999). Our EEG analysis was based on visual inspection of tracings to detect and quantify ictal activity. EEG seizures (ictal episodes) were defined by the occurrence of discrete episodes consisting of the simultaneous occurrence of at least two of the following alterations in cortical and hippocampal leads of recording: high-frequency and/or multispike complexes and/or high-voltage synchronized spike or wave activity. The quantitative parameters chosen to quantify seizure activity were the latency to the first EEG seizure (onset), the total number of seizures occurring during the whole period of recording, and the total time spent in seizures that was reckoned by adding together the duration of all ictal episodes (Vezzani et al., 1999). During EEG seizures, the rats had the typical “frozen” appearance and apparently lost their reaction to external stimuli. “Wet dog shakes” were often observed at the end of seizure episodes. These behavioral sequels were not quantified in this study. Statistical analysis was performed by Student's t test and by two-way ANOVA followed by post hoc Tukey test.
Behavioral analysis after systemic KA. Behavioral but not EEG analysis of seizures was performed in rats systemically injected with KA.
Thirty rats received hippocampal injections of BoNT/E at P35. Control animals of the same age (n = 39) were injected with vehicle. One day after the injections, animals received a convulsive dose (8 mg/kg, i.p.) of KA (Ocean Produce International, Shelburne, Nova Scotia, Canada). Naive animals that only received KA at P36 (n = 20) were also used.
Rats were observed by an investigator unaware of the treatment. For each animal, behavior was scored every 5 min for a period of 4 h after KA administration, according to a previously defined seizure rating scale (Schauwecker and Steward, 1997; Bozzi et al., 2000): stage 0, normal behavior; stage 1, immobility; stage 2, stereotypies; stage 3, wet dog shakes, head bobbing; stage 4, sporadic clonus of forelimbs with rearing and falling; stage 5, generalized clonus with continuous rearing and falling (status epilepticus); stage 6, death. Statistical analysis was performed by two-way ANOVA followed by post hoc Tukey test.
In situ hybridization. Subsets of vehicle- and BoNT/E-injected rats (n = 5 per group) were randomly chosen among those used for the behavioral analysis of seizures after systemic KA (see above). Rats were killed 2.5 h after KA injection, and the brains were immediately frozen on dry ice. Coronal cryostat sections (20 μm thick) were collected on slides in serial order throughout the entire hippocampus and fixed in 4% paraformaldehyde. Nonradioactive in situ hybridization was performed according to standard protocols using a digoxigenin-labeled c-fos ribo-probe (Bozzi et al., 2000). Signal was detected by alkaline phosphatase-conjugated anti-digoxigenin antibody followed by alkaline phosphatase staining. To quantify the level of c-fos mRNA, digital images of three matching sections per animal (n = 5 rats per group) were analyzed using MCID-M4 software (Imaging Research, St. Catharines, Ontario, Canada). For the hippocampus, the profile of CA1, CA3, and dentate gyrus (DG) was outlined in each section, and signal intensity within each sector was measured. Background level was calculated in the corpus callosum of each section and subtracted from signal intensity values. The obtained values were averaged and used to calculate the average value per animal. These values (n = 5 per group) were used to calculate the mean ± SE for each group. Statistical analysis was performed by Student's t test.
Morris water maze. To evaluate the cognitive performance of rats during the time window of action of BoNT/E, eight animals that received BoNT/E at P35 were tested in the Morris water maze beginning from 3 to 7 d after treatment. These animals were compared with vehicle-injected animals of the same age (n = 8). A second group of rats (n = 5) injected with BoNT/E at P35 was allowed to recover for 5 weeks before the spatial learning test. Their performance was compared with that of age-matched rats (n = 10) that did not receive any treatment.
We also evaluated the cognitive performance of BoNT/E-injected and control rats treated with KA. Behavioral tests were begun on P70 (5 weeks after treatment) in both BoNT/E-treated (n = 13) and control (n = 26; n = 19 vehicle-injected and n = 7 naive) rats that received KA at P36.
Experiments were performed according to Mikati et al. (2001) and Cilio et al. (2001). Briefly, a circular tank (200 cm diameter) was filled with opaque water (22 ± 1°C), and a wooden platform (10 × 10 cm) was positioned in the center of one quadrant of the pool 2.5 cm below the water surface. On day 1 of testing, rats were placed in the pool for 60 s without the platform present to become habituated to the training environment. Rats were trained for 4 d (five trials per day) to locate and escape onto the submerged platform. The latency from immersion into the pool to escape onto the platform was recorded for each trial. On mounting the platform, rats were given a 30 s rest period. Rats that did not find the platform in 120 s were placed on the platform for 30 s. Rats experiencing a spontaneous seizure during testing were allowed to recover for 60 min before resumption of test. All experiments were conducted in a blinded manner. Statistical analysis was performed by two-way ANOVA followed by post hoc Tukey test.
Evaluation of neuronal loss. Hippocampal neuronal loss was evaluated in control (n = 22) and BoNT/E-infused rats (n = 11) treated with KA at the end of Morris water maze experiments (P77). Rats were perfused with 4% paraformaldehyde, and coronal sections throughout the dorsal hippocampus were processed in serial order for immunohistochemistry with mouse anti-neuronal-specific nuclear protein (NeuN) monoclonal antibody (1:500 dilution; Chemicon, Temecula, CA). Neuronal damage was scored in areas CA1 and CA3 of the hippocampus according to the following scale (Bozzi et al., 2000; Cilio et al., 2001): 0, no damage; 1, minimal damage (small spots of degeneration); 2, evident loss of pyramidal neurons; 3, complete disruption of hippocampal architecture. An average number of 10 sections per animal were analyzed by an investigator unaware of the treatment. A separate score was initially assigned to CA1 and CA3 regions of both sides of each section, and these values were used to calculate the mean damage score for each section. These values were averaged and the obtained damage scores for each animal were plotted. Statistical analysis was performed by the Mann-Whitney rank-sum test.
Rapid kindling. A total number of 11 rats at P35 were unilaterally infused in the right ventral hippocampus with BoNT/E (n = 5) or vehicle (n = 6) as described above, under avertin anesthesia (nose bar, -2.5 mm from bregma; CA1, AP, -4.7 mm from bregma; L, -5.5 mm from bregma; H, 6.0 mm below dura; CA3, AP, -4.7 mm from bregma; L, -5.0 mm from bregma; H, 5.0 mm below dura). After the end of the infusion procedure, twisted bipolar stimulating-recording electrodes were stereotaxically implanted bilaterally into the rat ventral hippocampal CA3 region (nose bar, -2.5 mm from bregma; AP, -4.7 mm from bregma; L, ±5.0 mm from bregma; 5.0 mm below dura), and bilateral screw electrodes were placed over the parietal cortex. The electrodes were connected to a multipin socket and secured to the skull with acrylic dental cement. Hippocampal kindling was started after a postoperative period of 2 d. Experiments were performed by an investigator unaware of the treatment. The rats were allowed to acclimatize in a Plexiglas cage, and an EEG recording was made for at least 15 min to assess basal activity. Rapid kindling was induced by delivering constant-current stimuli (50 Hz; 10 s trains of 400 μA; 1 ms bipolar square waves) unilaterally to the right ventral hippocampus through a bipolar electrode with a 5 min interval for 200 min (Kopp et al., 1999; Richichi et al., 2004), thus summing up to a total of 40 stimuli. Behavior was observed and scored according to a modified Racine's classification (Racine, 1972). Stage 2 was defined by unilateral forelimb retraction and mouth stereotypies, stage 3 by head nodding and dorsal muscle twitching, and stage 4-5 consisted of generalized clonic seizures without (stage 4) or with (stage 5) postural loss. The duration of the primary and secondary afterdischarges was measured in the stimulated hippocampus after each stimulation in every animal. Rat were considered fully kindled when they experienced at least three consecutive stage 4 or 5 seizures within the stimulation protocol. Twenty-four hours after the end of the stimulation period, rats received five additional electrical stimulations (retest day) as above to confirm kindling acquisition and maintenance. At the end of the stimulation protocol, kindled rats were killed, and the correct location of the stimulating electrode and the injection cannula was verified histologically in each animal. Statistical analysis was performed by two-way ANOVA followed by post hoc Tukey test and by Student's t test.
Results
Characterization of BoNT/E actions in the hippocampus
P35 rats received a unilateral injection of BoNT/E or vehicle into the hippocampus. Western blot for cleaved SNAP-25, performed 1 d after BoNT/E injection, demonstrated that SNAP-25 is efficiently proteolysed in vivo by BoNT/E (Fig. 1A). This effect was mainly restricted to the injected side, and cleaved SNAP-25 was almost undetectable in the contralateral hippocampus (Fig. 1A). Immunostaining for cleaved SNAP-25 confirmed the regional specificity of the BoNT/E effect (Fig. 1B). Anteroposterior spread of staining around the injection site was of ∼3 mm.
Figure 1.

Characterization of BoNT/E effects in the hippocampus. A, Representative immunoblotting for cleaved SNAP-25 (cl. S25) on protein extracts from the BoNT/E-injected (inj.) and contralateral, noninjected (not inj.) hippocampus of a P36 rat, 1 d after BoNT/E injection. β-tub, β-Tubulin (internal standard). B, Forebrain coronal section showing the hippocampus of a P36 rat, 1 d after unilateral BoNT/E injection. Immunostaining for cleaved SNAP-25 (dark labeling) is mainly restricted to the injected hippocampus (indicated by an asterisk). Scale bar, 3 mm. C, Effects of BoNT/E on Ca2+-dependent, 35 mm K+-evoked glutamate over flow from hippocampal synaptosomes prepared 3 d after toxin injection. Release is inhibited by ∼80% in BoNT/E-injected hippocampi. Data are expressed as mean ± SE (n = 7 animals per group; **p < 0.01; t test). D, Representative recordings of spontaneous spike activity from CA1 in vehicle- and BoNT/E-injected animals. E, Immunoblotting for cleaved SNAP-25 (cl. S25) on protein extracts from hippocampi of BoNT/E-treated rats at different times after BoNT/E injection. D, Dorsal hippocampus; V, ventral hippocampus. F, Immunoblotting for the intact SNAP-25 (S25) on protein extracts from hippocampi of BoNT/E-treated (B) and age-matched control (C) rats at different times after BoNT/E injection.
Ex vivo experiments on superfused hippocampal synaptosomes were performed to assess the effects of BoNT/E on glutamate overflow. The rats received BoNT/E (or vehicle) followed 2 d later by a single dose of intrahippocampal kainic acid (40 ng). Synaptosomes were prepared on the third day after BoNT/E. We found that the Ca2+-dependent fraction of potassium-induced glutamate release was markedly reduced by BoNT/E administration (n = 7 animals per group; Student's t test; p < 0.01) (Fig. 1C).
To characterize the effects of BoNT/E at the electrophysiological level, we performed in vivo recordings of spike activity from hippocampal pyramidal neurons in naive rats, 1-2 d after BoNT/E or vehicle injection. Spontaneous discharges of spikes of high amplitude were observed in the CA1 and CA3 sectors of naive rats. These discharges were potently inhibited by BoNT/E treatment, whereas injection of vehicle solution had no effect (Fig. 1D). The reduction in activity was specific to the BoNT/E-treated hippocampus and no effects were found in the contralateral, un-injected side. Thus, BoNT/E impairs excitatory transmission in the hippocampus, and the net electrophysiological effect is a silencing of spontaneous spike activity of pyramidal neurons.
To estimate the duration of the BoNT/E effects, we analyzed the time course of SNAP-25 cleavage after a single BoNT/E injection into the hippocampus. Cleaved SNAP-25 was detected in both the dorsal and ventral hippocampus 1 d after BoNT/E injection. The band was slightly reduced at 14 d, persisted up to 21 d, and was no longer detectable at 35 d (Fig. 1E). In keeping with the expression profile of cleaved SNAP-25, intact SNAP-25 was absent 1 d after BoNT/E, began to reappear at 14 d, and was completely replenished by 35 d (Fig. 1F). Thus, BoNT/E effects persist for at least 3 weeks in the injected hippocampus. Importantly, BoNT/E-injected animals never showed any sign of systemic intoxication (i.e., muscular paralysis) during this time window.
Effects of BoNT/E on EEG seizures induced by intrahippocampal KA
We investigated whether intracerebral BoNT/E application displays antiictal properties using as a model of acute electroencephalographic seizures triggered by intrahippocampal KA (Vezzani et al., 1999, 2002). Rats were given unilateral injections of BoNT/E or vehicle into the hippocampus, and seizures were induced 2 d later by focal KA application to the treated hippocampus. The location of BoNT/E injection sites, KA infusion cannula, and recording electrodes is schematically shown in Figure 2, A and B.
EEG seizures occurred simultaneously in all leads of recordings and recurred as discrete episodes for ∼180 min from their onset. Representative EEG tracings from vehicle- or BoNT/E-injected rats are shown in Figure 2, C and D. Quantification of seizure activity demonstrated that the onset time to seizures was delayed by fourfold in BoNT/E-treated animals (Student's t test; p < 0.001) (Fig. 2E). BoNT/E-injected rats showed a 2.6-fold reduction in the number of EEG seizures (Student's t test; p = 0.005) (Fig. 2F) and a fivefold decrease in the total time spent in seizure activity (Student's t test; p < 0.001) (Fig. 2G). The duration of individual ictal episodes was also reduced by the toxin (in minutes ± SE; vehicle, 2.4 ± 0.5; BoNT/E, 1.06 ± 0.1; Student's t test; p < 0.01).
We also compared the antiictal effects of BoNT/E with that of PHT, a standard anticonvulsant drug. PHT (50 mg/kg, i.p.; n = 7 rats) was administered 60 min before intrahippocampal KA. This dose of PHT was chosen based on previous studies (Ebert et al., 2000; N′Gouemo and Faingold, 2000) and was the highest dose that did not cause toxic effects in the animals (i.e., ataxia or sedation). PHT induced an average twofold delay in the onset of EEG seizures and a 2.5-fold reduction in the time spent in seizure activity (Student's t test; p < 0.01). PHT also reduced by 50% the number of EEG seizures (p < 0.01) (Fig. 3A-C). Thus, BoNT/E was significantly more effective than PHT in reducing KA-induced seizures (one-way ANOVA, p < 0.01; post hoc Tukey test, p < 0.01 for onset and time spent in seizures; one-way ANOVA, p < 0.01; post hoc Tukey test, p < 0.05 for number of seizures) (Fig. 3A-C).
Figure 3.

Comparison of the antiictal effects of PHT (50 mg/kg, i.p.; n = 7 rats) and intrahippocampal BoNT/E (n = 8 rats). Drugs were given 60 min (PHT) or 2 d (BoNT/E) before intrahippocampal KA. Graphs report the onset time to first the seizure (A), the total number of seizures (B), and the total time spent in ictal activity (C) expressed as percentage of the respective control (contr) values. Control animals (n = 15) received intrahippocampal KA and the respective vehicles. *p < 0.05; **p < 0.01; one way ANOVA followed by Tukey test. Error bars represent SE.
Unilateral intrahippocampal BoNT/E blocks behavioral seizures induced by systemic KA
We then assessed the behavioral response of BoNT/E-injected rats to systemic administration of KA. It is well established that parenteral KA induces limbic motor seizures originating in the hippocampus (Lothman and Collins, 1981; Ben-Ari, 1985). P35 rats received unilateral stereotaxic injections of BoNT/E (n = 30) or vehicle (n = 39) into the hippocampus. One day later, the animals received a single systemic injection of a convulsive dose of KA (8 mg/kg, i.p.). As a control, 20 uninjected rats were treated with the same dose of KA. KA treatment had a similar convulsant effect in both naive and vehicle-injected animals (two-way ANOVA; p > 0.05) (Fig. 4A). These rats showed initial immobility and staring followed by wet dog shakes, culminating in generalized clonic motor seizures with rearing and falling. Progression of clinical signs was dramatically different in BoNT/E-injected animals (Fig. 4A). Indeed, the trajectory in behavior score of BoNT/E-treated rats was dramatically different from that of control rats starting from 80 min after KA administration (two-way ANOVA, p < 0.001; post hoc Tukey test, BoNT/E vs vehicle and uninjected rats, p < 0.01). In Figure 4B, the results of the behavioral analysis are summarized as the maximum seizure rating scale value assigned to each animal during the 4 h of observation after KA administration. KA triggered typical limbic motor convulsions in 17 of 20 (85%) naive rats and 31 of 39 (79%) vehicle-injected rats. In contrast, the vast majority of the BoNT/E-injected rats showed only preconvulsive behaviors, and only 5 of 30 (16%) experienced limbic seizures after KA administration. ANOVA demonstrates that the anticonvulsant effect of BoNT/E is highly significant (one-way ANOVA, p < 0.001; post hoc Dunn's test, p < 0.01), whereas naive and vehicle-injected rats do not show significant differences (p > 0.05). Lethal toxicity induced by KA was also abolished by BoNT/E injection (Fig. 4B).
Figure 4.

BoNT/E prevents seizures induced by systemic KA. A, Progression of behavioral changes after systemic KA administration (8 mg/kg, i.p.) in the various treatment groups over a 4 h observation period (for details, see Materials and Methods). Data are mean seizure scores ± SE. B, Scatter plot reporting the maximum seizure score assigned to each experimental animal during a 4 h observation period after KA administration. The majority of the control animals reached status epilepticus (seizure stage 5), whereas only 1 of 30 BoNT/E-injected rats did so. **p < 0.01; one-way ANOVA followed by Dunn's test.
To further support these data, we performed activity mapping studies with c-fos mRNA in situ hybridization in a subset of rats randomly chosen among those used for behavioral analysis of seizures. A strong bilateral activation was observed in the hippocampus, thalamus, and cerebral cortex of control animals treated with KA (n = 5), whereas a dramatic decrease of c-fos induction was detected throughout the entire brain in BoNT/E-treated rats (n = 5) (Fig. 5A). Quantification of signal intensity revealed a strong reduction of c-fos mRNA within the injected hippocampus of BoNT/E-treated rats compared with the injected hippocampus of vehicle-treated animals. c-fos mRNA labeling in the uninjected contralateral hippocampus was also slightly, but not significantly, reduced in BoNT/E-treated rats (Fig. 5B). In keeping with the inhibition of seizure generalization, c-fos mRNA labeling was completely absent in the cerebral cortex of both hemispheres in BoNT/E-treated rats, except for a small area surrounding the injection track (Fig. 5A).
Figure 5.

BoNT/E markedly decreases KA-induced c-fos mRNA expression in the brain. A, In situ hybridization analysis of c-fos mRNA in coronal brain sections at the level of the dorsal hippocampus from a vehicle- and a BoNT/E-injected rat, 2.5 h after systemic KA. The vehicle-injected rat reached status epilepticus (stage 5), whereas the BoNT/E-injected animal showed only preconvulsive behaviors (stage 3). Asterisks indicate the injected sides. Scale bar, 3 mm. B, Quantification of KA-induced c-fos mRNA expression in the CA1, CA3, and DG sectors of the hippocampus from vehicle- (black columns) and BoNT/E- (white columns) treated rats. Separate quantifications were made for the hippocampus ipsilateral (ipsi) and contralateral (contra) to the injected side. Signal intensity is expressed as integrated optical density ± SE (n = 5 animals per group; *p < 0.05; Student's t test).
BoNT/E prevents spatial learning deficits induced by KA
We also determined whether BoNT/E prevents the cognitive deficits induced by KA by testing the animals in the Morris water maze, a well established hippocampus-dependent behavioral task. We first examined whether intrahippocampal injection of BoNT/E per se has an effect on cognitive performance by comparing toxin-injected rats with vehicle-injected rats. Clear deficits in acquisition of spatial learning were evident in rats tested 3-7 d after BoNT/E (i.e., during the time window of action of the toxin) (n = 8 animals per group; two-way ANOVA; p < 0.01) (Fig. 6A). However, in BoNT/E-injected animals (n = 5) allowed to recover for 5 weeks, a time at which the effect of BoNT/E is extinguished (Fig. 1E,F), we found a learning ability indistinguishable from that of control rats (n = 10) (two-way ANOVA; p = 0.38) (Fig. 6B). These long-term BoNT/E-injected rats also showed normal exploratory behavior in the open field (data not shown).
Figure 6.
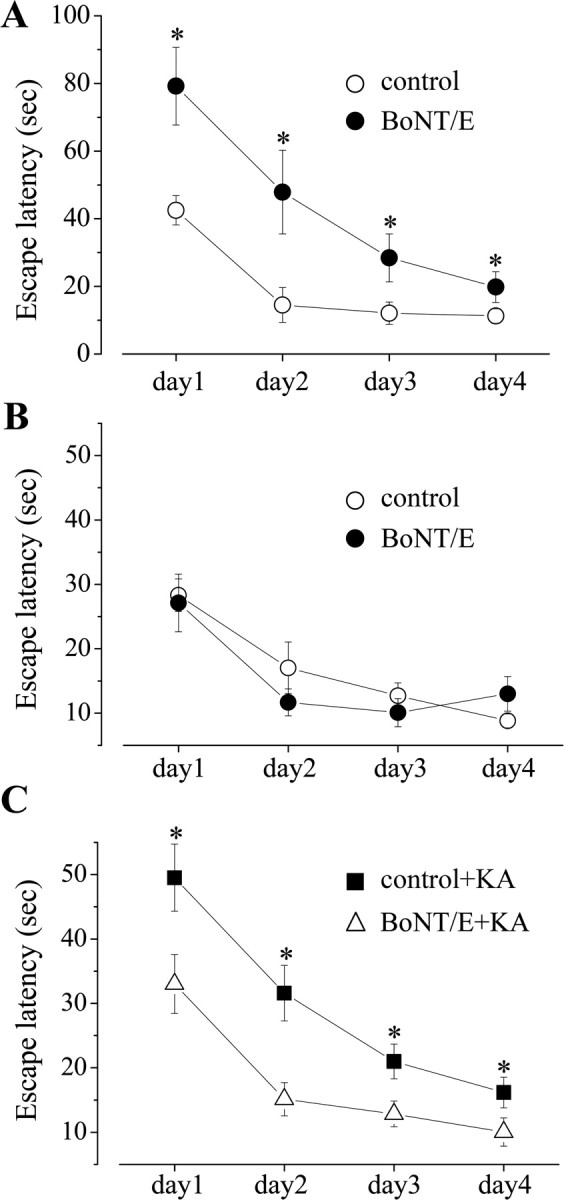
BoNT/E prevents spatial learning deficits induced by KA. A, Acquisition of place learning in the Morris water maze (MWM) for rats injected with BoNT/E into the hippocampus at P35 and tested 3-7 d later (BoNT/E; n = 8; black circles). Age-matched control rats were injected intrahippocampally with vehicle (control; n = 8; open circles). Time required to find the submerged platform (escape latency) is indicated in seconds and plotted as mean values ± SE across days of testing. Performance of vehicle-treated rats is better than that of BoNT/E-treated rats (two-way ANOVA; p < 0.001). B, Acquisition of place learning in the MWM for rats injected with BoNT/E into the hippocampus at P35 and tested 5 weeks later (BoNT/E; n = 5; black circles). Performance of naive, age-matched rats (control; n = 10) is shown by open circles and is superimposable to that of BoNT/E-treated rats. No statistically significant difference is observed between the two groups (two-way ANOVA; p = 0.38). C, Acquisition of place learning in the MWM for rats injected with BoNT/E at P35 and injected with KA at P36 (BoNT/E+KA; n = 13; open triangles). Performance of control plus KA rats (n = 26) is shown by black squares and includes data from both naive (n = 7) and vehicle-injected (n = 19) rats treated with KA at P36 because these groups did not differ. Behavioral testing began for all rats 5 weeks after KA. Control plus KA rats were slower to learn the task and never reached the performance of the BoNT/E-injected group. The asterisks in A and C indicate p < 0.01 (two-way ANOVA followed by post hoc Tukey test).
We next analyzed spatial learning in control (n = 26) and BoNT/E-injected (n = 13) animals treated with systemic KA at P36 and tested 5 weeks later. Control plus KA rats (Fig. 6C, black squares) performed poorly in the Morris water maze, consistent with previous studies (Stafstrom et al., 1993; Mikati et al., 2001). Performance of BoNT/E plus KA rats (Fig. 6C, open triangles) was significantly superior to that of control plus KA rats (two-way ANOVA; p < 0.001). Differences in performance could not be attributed to differences in swim speed, which was similar across the experimental groups (one-way ANOVA; p > 0.05) (data not shown). Importantly, BoNT/E plus KA rats showed spatial learning abilities comparable with that of age-matched naive rats (two-way ANOVA; p = 0.4) (Fig. 6, compare C, BoNT/E plus KA and B, control rats).
BoNT/E prevents neuronal loss induced by KA
Immunostaining for the neuronal marker NeuN was used to evaluate the extent of neuronal loss in the hippocampus of vehicle- and BoNT/E-injected rats treated with KA. Rats received intrahippocampal injections of vehicle or BoNT/E at P35, were treated with systemic KA at P36, and were killed at P77, after completion of the Morris water maze test. A significant preservation of hippocampal cells was found in BoNT/E-injected rats compared with abundant neuronal loss in CA1, CA3, and hilus in vehicle-injected rats treated with KA (Fig. 7A,B). Quantification of histological lesions in the dorsal hippocampus of control (black circles; n = 22) and BoNT/E-injected (open circles; n = 11) rats treated with KA is shown in Figure 7C.
Figure 7.
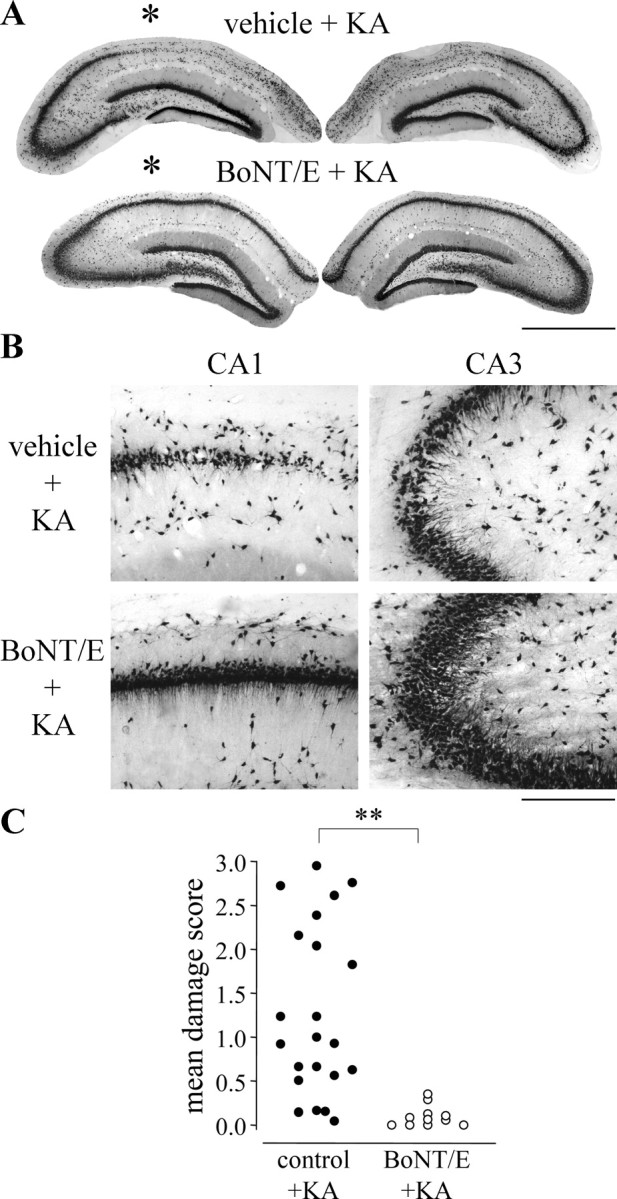
BoNT/E prevents hippocampal neuronal loss induced by KA. A, Representative coronal sections through the dorsal hippocampus of P77 rats that received vehicle (top) or BoNT/E (bottom) at P35 and KA at P36. Sections were immunostained for the neuronal marker NeuN. Note preservation of hippocampal neurons in the BoNT/E-injected rat, whereas widespread neuronal loss is evident in the vehicle-injected animal. The injected side is indicated by an asterisk. B, Representative pictures showing details of CA1 and CA3 regions of the hippocampus from vehicle- and BoNT/E-injected rats. In the vehicle-treated rat, restricted areas of degeneration and reduced thickness of pyramidal cell layer (damage score 1; see Materials and Methods) are evident in CA3, whereas a marked loss of pyramidal neurons (damage score 2) is observed in CA1. Preservation of hippocampal tissue (no damage, score 0) is detected in both CA1 and CA3 regions of the BoNT/E-treated animal. Scale bars: A, 1.2 mm; B, 200 μm. C, Quantification of histological lesions in the dorsal hippocampus of control (control+KA; black circles; n = 22) and BoNT/E-injected (BoNT/E+KA; open circles; n = 11) rats treated with KA. The control plus KA group pools data from both naive and vehicle-injected rats because these two groups did not differ. The extent of the loss of hippocampal pyramidal neurons was determined for each animal as the mean damage score value in CA1 and CA3 sectors of both hemispheres (for details, see Materials and Methods). Each point represents one animal. **p < 0.01; Mann-Whitney rank-sum test.
Effects of BoNT/E on kindling epileptogenesis
We studied whether BoNT/E, in addition to its antiictal effects, also displays antiepileptogenic-like actions using the rapid electrical kindling of the ventral hippocampus. As shown in Figure 8A, BoNT/E-injected animals showed a marked delay in the behavioral progression of kindling because they required significantly more stimulations to exhibit the first generalized motor seizure (stage 4-5 seizure; two-way ANOVA, p < 0.05; post hoc Tukey test, p < 0.01). The number of stage 4-5 seizures was also reduced (vehicle, 5.6 ± 0.5; BoNT/E, 1.7 ± 0.2; Student's t test; p < 0.01). BoNT/E treatment resulted in shorter duration of primary afterdischarge (AD) (cumulative AD; in minutes; mean ± SE; vehicle, 18.9 ± 1.1; BoNT/E, 11.9 ± 1.9; t test; p < 0.01) (Fig. 8B) and of secondary AD (vehicle, 10.2 ± 1.8; BoNT/E, 6.6 ± 1.2; t test; p < 0.05) (Fig. 8C). Both vehicle- and BoNT/E-injected group of rats showed stage 4-5 seizures 24 h after kindling acquisition, thus during the retest day.
Figure 8.

BoNT/E delays hippocampal kindling rate. A, Behavioral progression of seizures during kindling in vehicle- (black columns) and BoNT/E- (white columns) injected rats. BoNT/E-treated animals required more stimulations to reach stages (st.) 4 and 5 of kindling. Data represent mean ± SE (**p < 0.01; two-way ANOVA followed by post hoc Tukey test). Behavioral stages are scored according to Racine's classification (Vezzani et al., 2002). B, C, Duration of primary (B) and secondary (C) afterdischarge during kindling in BoNT/E- and vehicle-injected rats. Cumulative AD was reckoned in each rat by adding together the durations of the single AD induced in the hippocampus during kindling stimulations. Data represent the mean ± SE of cumulative AD. *p < 0.05; **p < 0.01; Student's t test.
Discussion
In the present study, we characterized the effects of intrahippocampal injections of BoNT/E and evaluated the anticonvulsant and antiepileptogenic properties of this treatment using models of acute seizures and kindling epileptogenesis in rats. We provide the first description of the effects of BoNT/E on CNS neurons in vivo. In addition, we present electrographic and behavioral evidence of a powerful anticonvulsant action of a prophylactic BoNT/E injection on both focal and generalized seizures induced by KA. Finally, we show that BoNT/E displays antiepileptogenic-like effects because it delays the development of hippocampal kindling.
Effects of BoNT/E in the hippocampus
BoNT/E interferes with neurotransmitter release in a very specific manner through its interaction with the SNARE protein SNAP-25. Previous studies in vitro (Bigalke et al., 1981; Ashton and Dolly, 1988; Verderio et al., 2004) have shown that BoNT/E dramatically impairs glutamatergic transmission but has little effect on GABA release. The low sensitivity of inhibitory transmission to BoNT/E is attributable to absence of SNAP-25 at GABAergic synapses, where it is replaced by its functional homolog SNAP-23, which is not cleaved by BoNT/E (Sadoul et al., 1997; Verderio et al., 2004).
Previous evidence has shown that treatment with BoNT/E prevents the occurrence of spontaneous EPSPs and greatly reduce the amplitude of evoked EPSPs in hippocampal slices (Capogna et al., 1997; Sutton et al., 2004). Consistent with this reduction of excitatory synaptic responses in vitro, our in vivo data indicate that BoNT/E potently inhibits the firing of pyramidal neurons in the hippocampus.
The effects of BoNT/E persist for ∼3 weeks after one single intrahippocampal injection, as determined by immunodetection of cleaved SNAP-25 and loss of intact SNAP-25. BoNT/E produced in the first week after treatment a marked deficit in the acquisition of spatial learning in the Morris water maze, a classic hippocampus-dependent task. This finding is in keeping with previous evidence showing that unilateral lesion of the hippocampus impairs performance in this task (van Praag et al., 1998). The normal Morris water maze performance showed by BoNT/E-treated rats 5 weeks after BoNT/E injection indicates that spatial learning deficits are transient in these animals. Similarly, reversible inhibition of neural activity via TTX infusion in the adult rat hippocampus has no subsequent deleterious effects on animal behavior (Lipska et al., 2002). However, we cannot exclude the possibility that more subtle aspects of hippocampal physiology, not investigated in our study, are permanently altered in BoNT/E-treated rats.
Antiictal and antiepileptogenic effects of BoNT/E
Our findings on both EEG and behavioral analysis of KA-induced seizures clearly show that intrahippocampal BoNT/E injection results in powerful anticonvulsant actions. The EEG data demonstrated that BoNT/E delays the onset and decreases the number and duration of EEG ictal episodes after focal delivery of KA. The behavioral analysis of seizures after systemic injection of KA showed that limbic status epilepticus was prevented by a unilateral intrahippocampal injection of BoNT/E, and this effect was associated with significant preservation of hippocampal neurons in BoNT/E-injected rats (Leite et al., 2002; Pitkanen, 2002). Mapping studies using c-fos as a functional marker of neuronal network activity demonstrated limited c-fos induction within the BoNT/E-injected hippocampus but a nearly normal activation of the contralateral one. Remarkably, c-fos mRNA labeling was absent from both cortices and thalamic nuclei of BoNT/E-treated rats, whereas a widespread bilateral activation of these areas was evident in vehicle-treated animals. These data indicate that the recruitment of hippocampi in both hemispheres is required for the progression from focal seizures to generalized motor convulsions.
Our data clearly show that a prophylactic administration of BoNT/E to the hippocampus can reduce focal seizures occurrence and their generalization. This treatment also prevents the neuronal loss and memory deficits that follow KA-induced seizures. Although our data establish a proof of principle of the effective antiictal activity of this toxin, it remains to be assessed whether BoNT/E is able to suppress spontaneous seizures and cognitive decline also when given after the establishment of chronic epilepsy.
The acute antiictal properties of BoNT/E were accompanied by a marked but transient spatial learning deficit as assessed in the Morris water maze task. This impairment is expected, given the acute effects of BoNT/E on hippocampal spiking activity and glutamate release, and obviously limits the possible clinical exploitation of the present findings. Indeed, although endorsed of a variety of side effects, current antiepileptic drugs spare a major amount of cognitive performance while preventing ictal activity. However, it has to be pointed out that the anticonvulsant efficacy of BoNT/E is superior to that of a conventional, systemically administered antiepileptic drug such as PHT. Another possible limitation to the use of intracerebral administration of BoNT/E comes from the demonstration that long-term suppression of glutamatergic activity can induce spontaneous withdrawal seizures (Tandon et al., 1996). Although this aspect awaits additional investigation, we never observed spontaneous behavioral seizures in BoNT/E-treated rats during daily cleaning and handling or during our behavioral testing.
We also evaluated the effects of BoNT/E on the development of hippocampal kindling. There is currently a lack of pharmacological treatments that can inhibit epileptogenesis and the progression of the disease (Pitkanen and Sutula, 2002; Stables et al., 2003), and kindling is a widely used model for screening potential antiepileptogenic treatments (Loscher, 2002; Stables et al., 2003). BoNT/E treatment retards the acquisition of stages 4 and 5 of kindling, indicating a delay in the development of epileptogenesis, likely because of its ability to inhibit the generalization of seizures at hippocampal onset. Reduction of primary AD is compatible with reduction of local hippocampal excitability and shortening of secondary AD indicates that reverberating activity within the limbic circuit is impaired (Racine, 1972; Nanobashvili et al., 2000; Vezzani et al., 2002). To validate the antiepileptogenic properties of BoNT/E, it will be important to determine whether administration of the toxin after electrically or chemically induced status epilepticus (i.e., during the latency period) can affect the onset or the incidence of the ensuing spontaneous seizures (Loscher, 2002; Stables et al., 2003).
In conclusion, we have shown that delivery of BoNT/E to the hippocampus is antiictal and potentially antiepileptogenic in experimental models of epilepsy. BoNTs have been widely exploited as a tool to investigate the molecular mechanisms controlling synaptic transmission at peripheral nerve terminals (Schiavo et al., 2000; Rossetto et al., 2001). Here, we provide evidence that BoNT/E injection into the brain results in significant inhibition of pathological hyper-activity such as seizures. Our data suggest that BoNT/E might be used as a novel tool to interfere with the mechanisms underlying seizure generation and epileptogenesis.
Footnotes
This work was supported by Fondazione Mariani (Grant R-04-38 to Y.B.), by Cofin (MM05192773-001), and by Fondo per gli Investimenti della Ricerca di Base (RBNE01RHZM 007). The financial support of Telethon-Italy (Grant GGP04086 to M.C. and Grant GP0272/01 to C.M.) is gratefully acknowledged. We thank C. Orsini, G. C. Cappagli, and C. Palla for excellent technical assistance.
Correspondence should be addressed to Matteo Caleo, Istituto di Neuroscienze, Consiglio Nazionale delle Ricerche, via G. Moruzzi 1, 56100 Pisa, Italy. E-mail: caleo@in.cnr.it.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251943-09$15.00/0
L.C. and Y.B. contributed equally to this work.
References
- Ashton AC, Dolly JO (1988) Characterization of the inhibitory action of botulinum neurotoxin type A on the release of several transmitters from rat cerebrocortical synaptosomes. J Neurochem 50: 1808-1816. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y (1985) Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14: 375-403. [DOI] [PubMed] [Google Scholar]
- Bigalke H, Heller I, Bizzini B, Habermann E (1981) Tetanus toxin and botulinum A toxin inhibit release and uptake of various transmitters, as studied with particulate preparations from rat brain and spinal cord. Naunyn Schmiedebergs Arch Pharmacol 316: 244-251. [DOI] [PubMed] [Google Scholar]
- Bozzi Y, Vallone D, Borrelli E (2000) Neuroprotective role of dopamine against hippocampal cell death. J Neurosci 20: 8643-8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M, Medini P, von Bartheld CS, Maffei L (2003) Provision of brain-derived neurotrophic factor via anterograde transport from the eye preserves the physiological responses of axotomized geniculate neurons. J Neurosci 23: 287-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, Gahwiler BH, Thompson SM (1997) Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci 17: 7190-7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilio MR, Bolanos AR, Liu Z, Schmid R, Yang Y, Stafstrom CE, Mikati MA, Holmes GL (2001) Anticonvulsant action and long-term effects of gabapentin in the immature brain. Neuropharmacology 40: 139-147. [DOI] [PubMed] [Google Scholar]
- Di Stasi AM, Mallozzi C, Macchia G, Maura G, Petrucci TC, Minetti M (2002) Peroxynitrite affects exocytosis and SNARE complex formation and induces tyrosine nitration of synaptic proteins. J Neurochem 82: 420-429. [DOI] [PubMed] [Google Scholar]
- Ebert U, Reissmuller E, Loscher W (2000) The new antiepileptic drugs lamotrigine and felbamate are effective in phenytoin-resistant kindled rats. Neuropharmacology 39: 1893-1903. [DOI] [PubMed] [Google Scholar]
- Engel J (1996) Introduction to temporal lobe epilepsy. Epilepsy Res 26: 141-150. [DOI] [PubMed] [Google Scholar]
- Foran PG, Mohammed N, Lisk GO, Nagwaney S, Lawrence GW, Johnson E, Smith L, Aoki KR, Dolly JO (2003) Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J Biol Chem 278: 1363-1371. [DOI] [PubMed] [Google Scholar]
- Gobbi M, Moia M, Pirona L, Ceglia I, Reyes-Parada M, Scorza C, Mennini T (2002) p-Methylthioamphetamine and 1-(m-chlorophenyl)piperazine, two non-neurotoxic 5-HT releasers in vivo, differ from neurotoxic amphetamine derivatives in their mode of action at 5-HT nerve endings in vitro. J Neurochem 82: 1435-1443. [DOI] [PubMed] [Google Scholar]
- Holmes GL (2002) Seizure-induced neuronal injury: animal data. Neurology 59 [Suppl 5]: S3-S6. [DOI] [PubMed] [Google Scholar]
- Kohane DS, Holmes GL, Chau Y, Zurakowski D, Langer R, Cha BH (2002) Effectiveness of muscimol-containing microparticles against pilocarpine-induced focal seizures. Epilepsia 43: 1462-1468. [DOI] [PubMed] [Google Scholar]
- Kopp J, Nanobashvili A, Kokaia Z, Lindvall O, Hokfelt T (1999) Differential regulation of mRNAs for neuropeptide Y and its receptor subtypes in widespread areas of the rat limbic system during kindling epileptogenesis. Brain Res Mol Brain Res 72: 17-29. [DOI] [PubMed] [Google Scholar]
- Kubek MJ, Liang D, Byrd KE, Domb AJ (1998) Prolonged seizure suppression by a single implantable polymeric-TRH microdisk preparation. Brain Res 809: 189-197. [DOI] [PubMed] [Google Scholar]
- Leite JP, Garcia-Cairasco N, Cavalheiro EA (2002) New insights from the use of pilocarpine and kainate models. Epilepsy Res 50: 93-103. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Halim ND, Segal PN, Weinberger DR (2002) Effects of reversible inactivation of the neonatal ventral hippocampus on behavior in the adult rat. J Neurosci 22: 2835-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W (2002) Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res 50: 105-123. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Collins RC (1981) Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res 218: 299-318. [DOI] [PubMed] [Google Scholar]
- Meldrum BS (1995) Excitatory amino acid receptors and their role in epilepsy and cerebral ischemia. Ann NY Acad Sci 757: 492-505. [DOI] [PubMed] [Google Scholar]
- Meunier FA, Schiavo G, Molgo J (2002) Botulinum neurotoxins: from paralysis to recovery of functional neuromuscular transmission. J Physiol (Paris) 96: 105-113. [DOI] [PubMed] [Google Scholar]
- Mikati MA, Tarif S, Lteif L, Jawad MA (2001) Time sequence and types of memory deficits after experimental status epilepticus. Epilepsy Res 43: 97-101. [DOI] [PubMed] [Google Scholar]
- Nanobashvili A, Airaksinen MS, Kokaia M, Rossi J, Asztely F, Olofsdotter K, Mohapel P, Saarma M, Lindvall O, Kokaia Z (2000) Development and persistence of kindling epilepsy are impaired in mice lacking glial cell line-derived neurotrophic factor family receptor alpha 2. Proc Natl Acad Sci USA 97: 12312-12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N′Gouemo P, Faingold CL (2000) Phenytoin administration reveals a differential role of pontine reticular formation and periaqueductal gray neurons in generation of the convulsive behaviors of audiogenic seizures. Brain Res 859: 311-317. [DOI] [PubMed] [Google Scholar]
- Pitkanen A (2002) Efficacy of current antiepileptics to prevent neurodegeneration in epilepsy models. Epilepsy Res 50: 141-160. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Sutula TP (2002) Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol 1: 173-181. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281-294. [DOI] [PubMed] [Google Scholar]
- Regesta G, Tanganelli P (1999) Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 34: 109-122. [DOI] [PubMed] [Google Scholar]
- Richichi C, Lin EJ, Stefanin D, Colella D, Ravizza T, Grignaschi G, Veglianese P, Sperk G, During MJ, Vezzani A (2004) Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci 24: 3051-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto O, Seveso M, Caccin P, Schiavo G, Montecucco C (2001) Tetanus and botulinum neurotoxins: turning bad guys into good by research. Toxicon 39: 27-41. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Berger A, Niemann H, Weller U, Roche PA, Klip A, Trimble WS, Regazzi R, Catsicas S, Halban PA (1997) SNAP-23 is not cleaved by botulinum neurotoxin E and can replace SNAP-25 in the process of insulin secretion. J Biol Chem 272: 33023-33027. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O (1997) Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 94: 4103-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Montecucco C (1995) Tetanus and botulism neurotoxins: isolation and assay. Methods Enzymol 248: 643-652. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C (2000) Neurotoxins affecting neuroexocytosis. Physiol Rev 80: 717-766. [DOI] [PubMed] [Google Scholar]
- Stables JP, Bertram E, Dudek FE, Holmes G, Mathern G, Pitkanen A, White HS (2003) Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia 44: 1472-1478. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL (1993) Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia 34: 420-432. [DOI] [PubMed] [Google Scholar]
- Stein AG, Eder HG, Blum DE, Drachev A, Fisher RS (2000) An automated drug delivery system for focal epilepsy. Epilepsy Res 39: 103-114. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM (2004) Regulation of dendritic protein synthesis by miniature synaptic events. Science 304: 1979-1983. [DOI] [PubMed] [Google Scholar]
- Tamargo RJ, Rossell LA, Kossoff EH, Tyler BM, Ewend MG, Aryanpur JJ (2002) The intracerebral administration of phenytoin using controlled-release polymers reduces experimental seizures in rats. Epilepsy Res 48: 145-155. [DOI] [PubMed] [Google Scholar]
- Tandon P, Liu Z, Stafstrom CE, Sarkisian M, Werner SJ, Mikati M, Yang Y, Holmes GL (1996) Long-term effects of excitatory amino acid antagonists NBQX and MK-801 on the developing brain. Brain Res Dev Brain Res 95: 256-262. [DOI] [PubMed] [Google Scholar]
- Turton K, Chaddock JA, Acharya KR (2002) Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem Sci 27: 552-558. [DOI] [PubMed] [Google Scholar]
- van Praag H, Qu PM, Elliott RC, Wu H, Dreyfus CF, Black IB (1998) Unilateral hippocampal lesions in newborn and adult rats: effects on spatial memory and BDNF gene expression. Behav Brain Res 92: 21-30. [DOI] [PubMed] [Google Scholar]
- Verderio C, Pozzi D, Pravettoni E, Inverardi F, Schenk U, Coco S, Proux-Gillardeaux V, Galli T, Rossetto O, Frassoni C, Matteoli M (2004) SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 41: 599-610. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG (1999) Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci 19: 5054-5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Michalkiewicz M, Michalkiewicz T, Moneta D, Ravizza T, Richichi C, Aliprandi M, Mule F, Pirona L, Gobbi M, Schwarzer C, Sperk G (2002) Seizure susceptibility and epileptogenesis are decreased in transgenic rats overexpressing neuropeptide Y. Neuroscience 110: 237-243. [DOI] [PubMed] [Google Scholar]