

Abstract
Injury to peripheral nerves often results in a persistent neuropathic pain condition that is characterized by spontaneous pain, allodynia, and hyperalgesia. Nerve injury is accompanied by a local inflammatory reaction in which nerve-associated and immune cells release several pronociceptive mediators. Kinin B1 receptors are rarely expressed in nontraumatized tissues, but they can be expressed after tissue injury. Because B1 receptors mediate chronic inflammatory painful processes, we studied their participation in neuropathic pain using receptor gene-deleted mice. In the absence of neuropathy, we found no difference in the paw-withdrawal responses to thermal or mechanical stimulation between B1 receptor knock-out mice and 129/J wild-type mice. Partial ligation of the sciatic nerve in the wild-type mouse produced a profound and long-lasting decrease in thermal and mechanical thresholds in the paw ipsilateral to nerve lesion. Threshold changed neither in the sham-operated animals nor in the paw contralateral to lesion. Ablation of the gene for the B1 receptor resulted in a significant reduction in early stages of mechanical allodynia and thermal hyperalgesia. Furthermore, systemic treatment with the B1 selective receptor antagonist des-Arg9-[Leu8]-bradykinin reduced the established mechanical allodynia observed 7-28 d after nerve lesion in wild-type mice. Partial sciatic nerve ligation induced an upregulation in B1 receptor mRNA in ipsilateral paw, sciatic nerve, and spinal cord of wild-type mice. Together, kinin B1 receptor activation seems to be essential to neuropathic pain development, suggesting that an oral-selective B1 receptor antagonist might have therapeutic potential in the management of chronic pain.
Keywords: neuropathic pain, allodynia, hyperalgesia, B1 receptor, kinin, bradykinin
Introduction
Injury to a peripheral nerve in humans often results in a persistent neuropathic pain condition that is characterized by spontaneous pain, allodynia (pain responses to non-noxious stimuli), and hyperalgesia (exaggerated pain responses to noxious stimuli) (Malmberg and Basbaum, 1998). This type of chronic pain differs substantially from acute pain not only in terms of the persistence of pain but also with regard to the maladaptive changes, such as neuroplasticity, that have been described at various levels of the nervous system (Besson, 1999). Thus, the available analgesic drugs often have limited therapeutic value in the management of chronic pain and they may, in fact, represent a risk to the patient because of their common side effects (Woolf and Mannion, 1999). Therefore, the development of safe and efficacious drugs to treat chronic pain is an urgent priority.
Nerve injury is accompanied by a local inflammatory reaction in which nerve-associated and immune cells release several pronociceptive mediators such as cytokines, eicosanoids, and kinins (Tracey and Walker, 1995; Bennett, 1999). Of note, increased serum bradykinin levels have been found in patients with neuropathic pain (Blair et al., 1998).
Kinins are peptides formed in plasma and peripheral tissues in response to the activation of a class of enzymes, denoted “kallikreins,” on kininogen substrates. Kinins are involved in a wide range of physiological mechanisms, including control of blood pressure, smooth-muscle contraction or relaxation, vascular permeability, and pain transmission. Furthermore, kinins are implicated in pathological states such as arthritis, pancreatitis, and asthma (for review, see Calixto et al., 2000, 2004). The actions of kinins are mediated through the stimulation of two subtypes of G-protein-coupled receptors, denoted B1 and B2. The kinin B1 receptors exhibit higher affinity for the carboxypeptidase metabolites of kinins, des-Arg9-bradykinin and des-Arg10-kallidin. Usually, the B1 receptors are hardly expressed in nontraumatized tissues, but they can be expressed under certain conditions, such as those after tissue injury and infection (for review, see Marceau et al., 1998). In contrast, the B2 receptors for which bradykinin and kallidin exhibit great affinity are usually constitutively expressed and widely distributed throughout central and peripheral tissues (for review, see Calixto et al., 2000, 2004).
Once formed in the periphery, kinins activate Aδ and C fibers in sensory nerves producing pain, hyperalgesia, or allodynia in both humans and experimental animals. In addition, kinins may cause the release of other mediators such as neurokinins, calcitonin gene-related peptide, nitric oxide, and arachidonic acid metabolites, which also account for primarily their proinflammatory and nociceptive properties (for review, see Calixto et al., 2000, 2004) (Dray and Perkins, 1997).
Recently, the use of both B1 and B2 knock-out mice has led to a better understanding of the role played by kinins in physiological and pathological processes (Borkowski et al., 1995; Pesquero et al., 2000). For example, the deletion of the B1 receptor gene significantly decreases acute and chronic inflammatory nociception (Pesquero et al., 2000; Ferreira et al., 2001, 2002). In the present study, we examine the contribution of the kinin B1 receptor to the chronic nociception produced by peripheral nerve injury using knock-out mice, selective drugs, and the measurement of mRNA levels.
Materials and Methods
Animals. Experiments were conducted using male and female wild-type 129/J mice and kinin B1 receptor knock-out mice (20-30 g; 129/J background) kept at controlled room temperature (22 ± 2°C) under a 12 h light/dark cycle (lights on at 6:00 A.M.) and 60-90% humidity. The animals were obtained from the Department of Biophysics at the Federal University of São Paulo (Brazil). Deletion of the entire coding sequence for the kinin B1 receptor was achieved as described previously (Pesquero et al., 2000). The experiments were performed in accordance with current guidelines for the care of laboratory animals and ethical guidelines for the investigation of pain in conscious animals (Zimmermann, 1983). The number of animals and intensity of noxious stimuli used were the minimum necessary to demonstrate the consistent effects of drug treatments or genetic manipulation.
Partial sciatic nerve ligation. For the induction of chronic neuropathy, male and female mice were anesthetized by intraperitoneal injection of 7% chloral hydrate (0.6 ml/kg; Vetec, Rio de Janeiro, Brazil). A partial ligation of the right sciatic nerve was made by tying one-third to one-half of the dorsal portion of the sciatic nerve, using a similar procedure to that described for rats by Seltzer et al. (1990) and for mice by Malmberg and Basbaum (1998). In sham-operated mice, the nerve was exposed without ligation.
Measurement of thermal hyperalgesia. Thermal hyperalgesia was measured using the paw-withdrawal latency according to the method described by Hargreaves et al. (1988), with minor modifications. After challenge, hyperalgesia was measured at several time points after nerve injury (1-42 d), as described below. Thermal baseline measures were obtained from nonoperated animals 1 d before nerve injury. Mice were placed in clear plastic chambers (7 × 9 × 11 cm) on an elevated surface and allowed to acclimatize to their environment for 1.5 h before testing. The heat stimulus was directed to the plantar surface of each hindpaw in the area immediately proximal to the toes. The infrared intensity was adjusted to obtain basal paw-withdrawal latencies of ∼11 s. An automatic 20 s cutoff was used to prevent tissue damage.
Measurement of mechanical allodynia. Mechanical nociceptive thresholds in mice were measured as the withdrawal response frequency to application of von Frey hairs (Stoelting, Chicago, IL). Six hairs with forces of 0.07, 0.16, 0.6, 1, 2, and 4 g were applied 10 times each to the plantar surface of each hindpaw following an alternating sequence and in ascending order of force. The monofilament was applied at intervals of 2 s to slightly different loci on the plantar surface of both hindpaws. A positive withdrawal response was considered valid only when the hindpaw was completely removed from the platform. The frequency of positive responses was calculated after 10 applications of the filament. The frequency of response was measured before and 1-42 d after nerve injury. Mechanical baseline measures were obtained from nonoperated animals 1 d before nerve injury. Mice were placed individually in clear Plexiglas boxes (9 × 7 × 11 cm) on elevated wire mesh platforms to allow access to the ventral surface of the hindpaws. Animals were acclimatized to the testing chambers, and the static mechanical withdrawal threshold was determined before and after nerve injury. The involvement of the B1 receptor in mechanical allodynia was tested by using the selective antagonist of the B1 receptor des-Arg9-[Leu8]-bradykinin (150 nmol/kg, s.c.; Sigma, St. Louis, MO) (Ferreira et al., 2001).
Quantitative real-time PCR. The expression of B1 receptor mRNA was measured using a quantitative real-time PCR according to the method described previously (Argañaraz et al., 2004). Several days after nerve lesion, mice (n = 3-6 for each group) were killed, and the plantar skin of the right hindpaw, right sciatic nerve, dorsal portion of spinal cord, and whole cerebral cortex were isolated, dissected, and frozen in liquid nitrogen and stored at -80°C. Thawed tissue was homogenized in 0.3-1 ml of TRIzol reagent (Invitrogen, Gaithersburg, MD), and total RNA was isolated according to the instructions of the manufacturer. Before cDNA synthesis, RNA samples were pretreated with DNase I (Invitrogen) to avoid genomic DNA contamination. Reverse transcription was performed using 2 μg of total pure RNA, 50 ng of random hexamer primers, and 200 U of Maloney murine leukemia virus reverse transcriptase (Invitrogen), as described by the manufacturers. Samples were submitted to a20 μl reaction using TaqMan Amplification system with an ABI PRISM 7000 Sequence Detection system (Applied Biosystems, Foster City, CA). Multiplex reactions were performed with 600 ng of cDNA for kinin B1 receptor and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) amplification. Oligonucleotide primers and fluorogenic probe sets for Taq-Man real-time PCR were designed for kinin B1 receptor using Assays-by-Design service (Applied Biosystems) to meet all TaqMan design guidelines. The probes were synthesized with the reporter dye 6-carboxyfluorescein (6-FAM) covalently linked at the 5′-end, and the quencher dye 6-carboxy-tetramethyl-rhodamine was linked to the 3′- end of the probe. For GAPDH amplification, commercial TaqMan rodent GAPDH control reagents (Applied Biosystems) were used. Differently from kinin B1 receptor probe, the GAPDH probe was VIC-labeled, allowing us to use it for multiplex detection. Each reaction was performed with 10 μl of Master Mix (Applied Biosystems), 1 μl of a mix containing two primers (18 μm each) and a probe (5 μm) specific to mRNA of kinin B1 receptor (probe B1:5′-CACAGGAACCCAGACAG-3′, forward primer: 5′-CCATACAAAACCCCAGCTGAA-3′, reverse primer: 5′-CTTTGGTTAGAAGGCTGTAGCTTCA-3′), and 1 μl of each GAPDH primer and the VIC-labeled probe (10 μm each). The cycle conditions were as follows: 50°C for 2 min and then 95°C for 10 min, followed by 50 cycles of 95°C for 15 s (melting step), and 60°C for 1 min (anneal/extend step). Both FAM and VIC correspondent fluorescences were acquired at the end of each extend phase. The PCR cycle, when a given fluorescence threshold is crossed by the amplification curve, was considered our first parameter to analyze mRNA expression and named Ct. ΔCt values were calculated by subtracting GAPDH Ct from kinin B1 receptor Ct to obtain the 2-ΔCt parameter, which represents relative B1 receptor/GAPDH expression.
Measurement of overt nociception. The procedure used was similar to that described previously (Ferreira et al., 2004). Twenty microliters of des-Arg9-bradykinin solution (10 nmol/paw; Sigma) were injected intraplantarly under the surface of the right hindpaw 7 d after sham surgery or partial sciatic nerve lesion in wild-type mice. Separate groups of animals received an intraplantar injection of vehicle (PBS). Animals were placed individually in chambers (transparent glass cylinders of 20 cm diameter) and were adapted for 20 min before algogen or vehicle injection. After challenge, mice were observed individually for 10 min. The amount of time spent licking the injected paw was measured with a chronometer and was considered as indicative of overt nociception.
Skin temperature measurement. Apart from nociceptive hypersensitivity, sciatic nerve lesions may cause abnormal cutaneous temperature regulation. Thus, the skin temperature of the ipsilateral and contralateral paw was measured 7 d after surgery using a surface radiation thermometer (Pro Check, Taipei, Taiwan) as described previously (Ferreira et al., 2004).
Data analysis. The results are presented as means ± SEM of four to six animals. The statistical significance of differences between groups was analyzed by means of Student's t test or ANOVA followed by Student-Newman-Keuls test when appropriate. p values <0.05 were considered indicative of significance.
Results
Partial ligation of the sciatic nerve in the wild-type mouse produced a profound and prolonged decrease in thermal and mechanical nociceptive thresholds observed in the paw ipsilateral to the nerve lesion (Figs. 1, 2). Neither threshold changed in the sham-operated animals or in the paw contralateral to the lesion (Figs. 1, 2). We found a significant reduction in the paw-withdrawal latency to the heat stimulus as early as 1 d after nerve injury that was stable until 21 d compared with sham-operated wild-type animals (Fig. 1A). At 28 d after the nerve injury, the paw-withdrawal latencies to thermal stimulation returned to baseline values (Fig. 1A).
Figure 1.
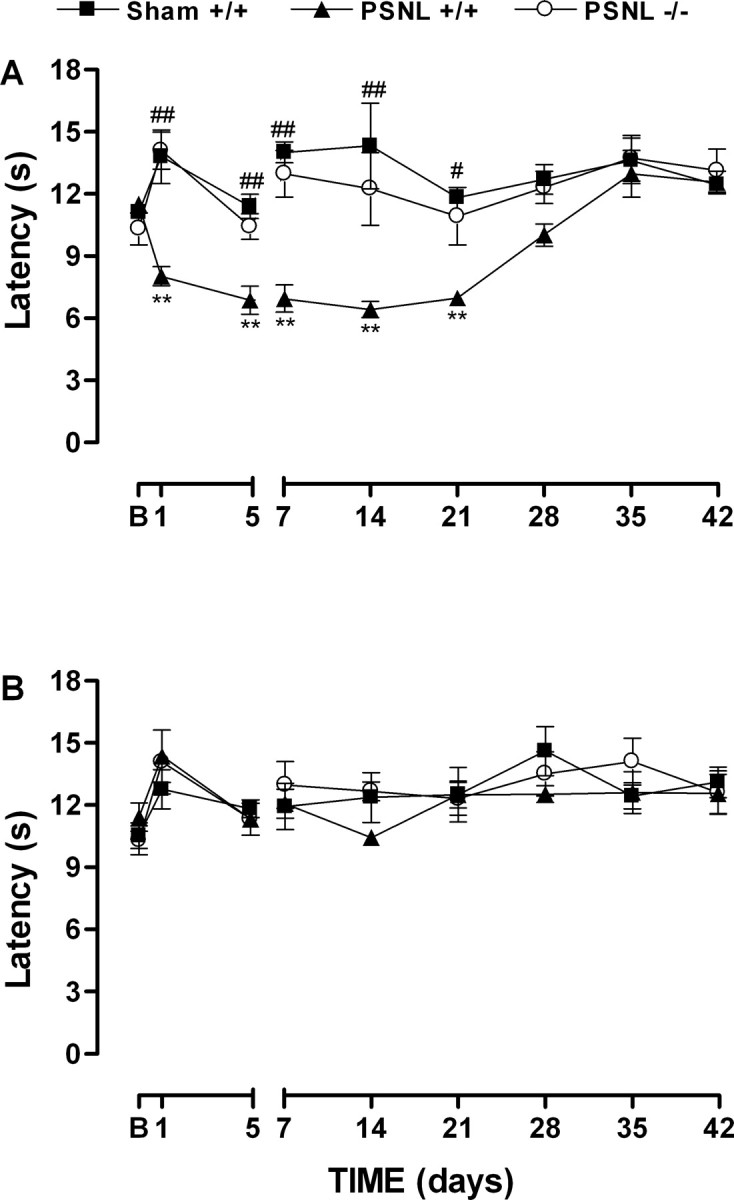
Time-dependent thermal hyperalgesia in the ipsilateral paw (A) but not in contralateral paw (B) induced by partial sciatic nerve lesion (PSNL) in wild-type (+/+) or B1 receptor knock-out (-/-) mice. Data represent the latencies of the response to thermal stimuli. Each point represents the mean ± SEM of four to six mice. In some cases, the error bars are hidden within the symbols. *p < 0.05 or **p < 0.01 denotes the significance level when compared with the wild-type sham-operated group. #p < 0.05 or ##p < 0.01 denotes the significance level when compared with the wild-type PSNL group (one-way ANOVA followed by Student-Newman-Keuls test).
Figure 2.
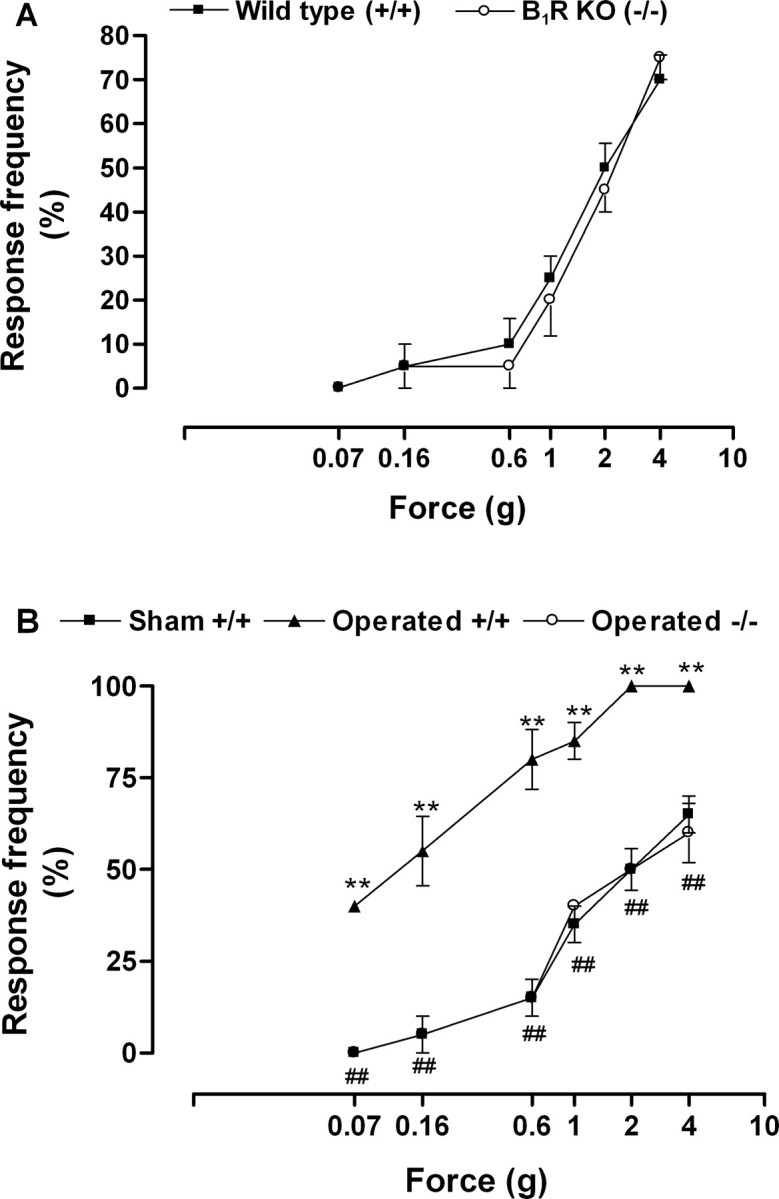
A, Mechanical sensitivity to von Frey hairs stimulation in wild-type (+/+) or B1 receptor knock-out (KO) (-/-) mice before nerve injury. B, Increased mechanical sensitivity in the ipsilateral paw observed 7 d after partial sciatic nerve lesion (PSNL) in wild-type mice (+/+) but not in B1 receptor knock-out mice (-/-). Data represent the response frequency to mechanical stimuli. Each point represents the mean ± SEM of four to six mice. In some cases, the error bars are hidden within the symbols. *p < 0.05 or **p < 0.01 denotes the significance level when compared with the wild-type sham-operated group. #p < 0.05 or ##p < 0.01 denotes the significance level when compared with the wild-type PSNL group (one-way ANOVA followed by Student-Newman-Keuls test).
In the absence of neuropathy, we found no difference in the paw-withdrawal responses to thermal stimulation between B1 receptor knock-out mice and wild-type mice (10.3 ± 0.7 and 10.5 ± 0.6 s, respectively). Ablation of the gene for the B1 receptor caused a significant reduction in thermal hyperalgesia produced by nerve injury (Fig. 1A). This anti-hyperalgesic response was observed from 1 to 21 d after lesion.
Before nerve injury, wild-type mice showed an increase in the frequency of responses to mechanical stimulation with von Frey hairs of higher forces (1-4 g) but little change in the responses to weaker von Frey hairs (0.07-0.6 g) (Fig. 2A). Moreover, B1 receptor knock-out mice displayed a similar pattern of response to high and weak mechanical stimulation (Fig. 2A). Thus, von Frey hairs from 0.07 to 0.6 g were considered innocuous stimuli for both wild-type and B1 receptor knock-out mice.
Mechanical allodynia produced by nerve injury was characterized by a pronounced and long-lasting increase in response frequency to innocuous von Frey hairs stimulation in the paw ipsilateral to the lesion (Fig. 2B). In contrast to thermal hyperalgesia, mechanical allodynia developed at day 1, reached a maximum at day 7 after nerve ligation, and remained increased for >42 d (Fig. 3A). B1 receptor gene deletion completely reversed mechanical allodynia from 1 to 28 d after nerve injury (Fig. 3A). However, this anti-allodynic effect became only partial 35 d after lesion and disappeared at day 42 (Fig. 3B). Moreover, we were not able to detect mechanical allodynia in the contralateral paw (data not shown).
Figure 3.
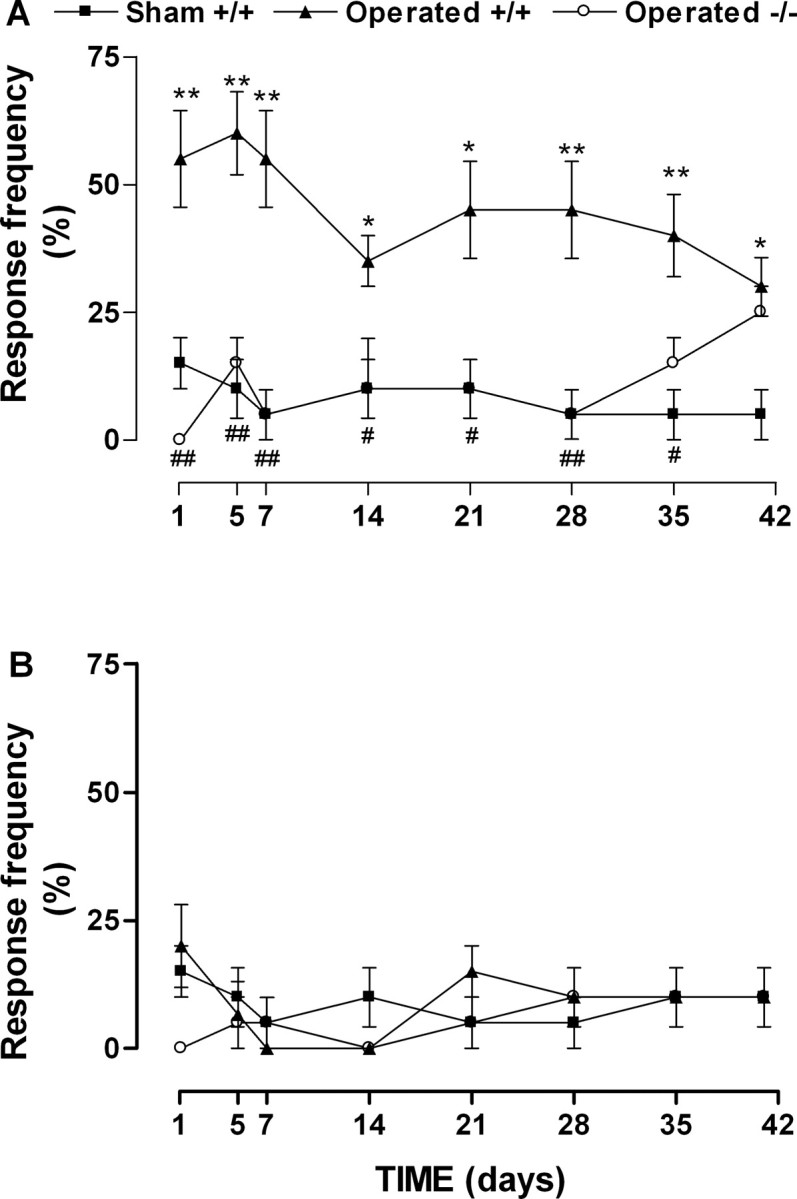
Time-dependent mechanical allodynia in ipsilateral paw (A) but not in contralateralpaw (B) induced by partial sciatic nerve lesion in wild-type (+/+) or B1 receptor knock-out (-/-) mice. Data represent the frequency response to 0.16 g von Frey hair stimulation. Each point represents the mean ± SEM of four to six mice. In some cases, the error bars are hidden within the symbols. *p < 0.05 or **p < 0.01 denotes the significance level when compared with the wild-type sham-operated group. #p < 0.05 or ##p < 0.01 denotes the significance level when compared with the wild-type partial sciatic nerve lesion group (one-way ANOVA followed by Student-Newman-Keuls test).
To further confirm the participation of the B1 receptor in neuropathic pain, a separate group of wild-type mice was treated with the selective B1 receptor antagonist des-Arg9-[Leu8]-bradykinin (150 nmol/kg, s.c.). Des-Arg9-[Leu8]-bradykinin administration to wild-type mice 7 d after partial sciatic nerve ligation, when the maximal pain hypersensitivity is already installed, also greatly reduced the mechanical allodynia (Fig. 4). The antinociceptive effect of des-Arg9-[Leu8]-bradykinin was shortlasting, maximal 1 h after treatment (inhibition of 69.6 ± 5.9%). In agreement with that after gene deletion, this dose of antagonist did not alter the frequency responses of mechanical stimulation of sham-operated animals (50 ± 5.7 and 45 ± 5.0% of frequency responses against 2.0 g of stimulation when assessed 7 d after injury). As occurred for the gene lacking, the treatment with des-Arg9-[Leu8]-bradykinin was also capable of reducing mechanical allodynia when the antagonist was administered 14 and 28 d, but not 42 d, after nerve injury (Fig. 4B).
Figure 4.

Antinociception produced by treatment with the selective B1 receptor antagonist des-Arg9-[Leu8]-bradykinin (DALBK; 150 nmol/kg) in mechanical allodynia in the ipsilateral paw observed after partial sciatic nerve lesion in wild-type mice. A, Time course of the antiallodynic effect 7 d after surgery. B, Anti-allodynic effect of DALBK 7-42 d after nerve injury when administered 1 h before mechanical allodynia measurement. Data represent the frequency response to 0.16 g von Frey hair (VFH) stimulation. Each point represents the mean ± SEM of five to six mice. *p < 0.05 or **p < 0.01 denotes the significance level when compared with PBS-treated mice. ##p < 0.01 denotes the significance level when compared with baseline (B) value without hair stimulation (one-way ANOVA followed by Student-Newman-Keuls test). The point 0 on the x-axis represents the measured mechanical allodynia immediately before drug treatment.
Next, the expression of B1 receptor mRNA was quantified by real-time reverse transcription (RT)-PCR in tissues of mice after sciatic nerve lesion or sham operation. Basal expression of B1 receptor mRNA was detected in plantar hindpaw skin, sciatic nerve, spinal cord, and cerebral cortex of wild-type mice (Fig. 5). However, 7 d after nerve lesion, we observed an increased of B1 expression in ipsilateral paw skin, right sciatic nerve, and spinal cord obtained from operated mice (Fig. 5). No expression of B1 receptor mRNA could be detected in B1 receptor gene-deficient mice (results not shown). Moreover, the increase of expression of B1 receptor mRNA was also detected in paw skin of injured wild-type mice 14, 28, and 42 d after nerve injury (441 ± 216, 241 ± 127, 849 ± 103% of increase over sham-operated animals, respectively). These results suggested that the increase in mRNA appears to mainly relate with the development and the maintenance of early stages of neuropathic pain but not with the maintenance of its late stage. In fact, intraplantar injection of the selective B1 receptor agonist des-Arg9-bradykinin produced overt nociception in ligated but not in, when assessed, sham-operated wild-type mice 7 d after surgery (Fig. 6A).
Figure 5.

Levels of expression of kinin B1 receptor mRNA in the paw skin (A), sciatic nerve (B), spinal cord (C), and cerebral cortex (D) 7 d after sham surgery or partial sciatic nerve ligation (PSNL) in wild-type mice (+/+) assessed by real-time RT-PCR assay. All data have been normalized for levels of GAPDH expression within the same sample. Each bar represents the mean ± SEM of three to six mice. *p < 0.05 denotes the significance level when compared with the sham-operated group (Student's t test).
Figure 6.
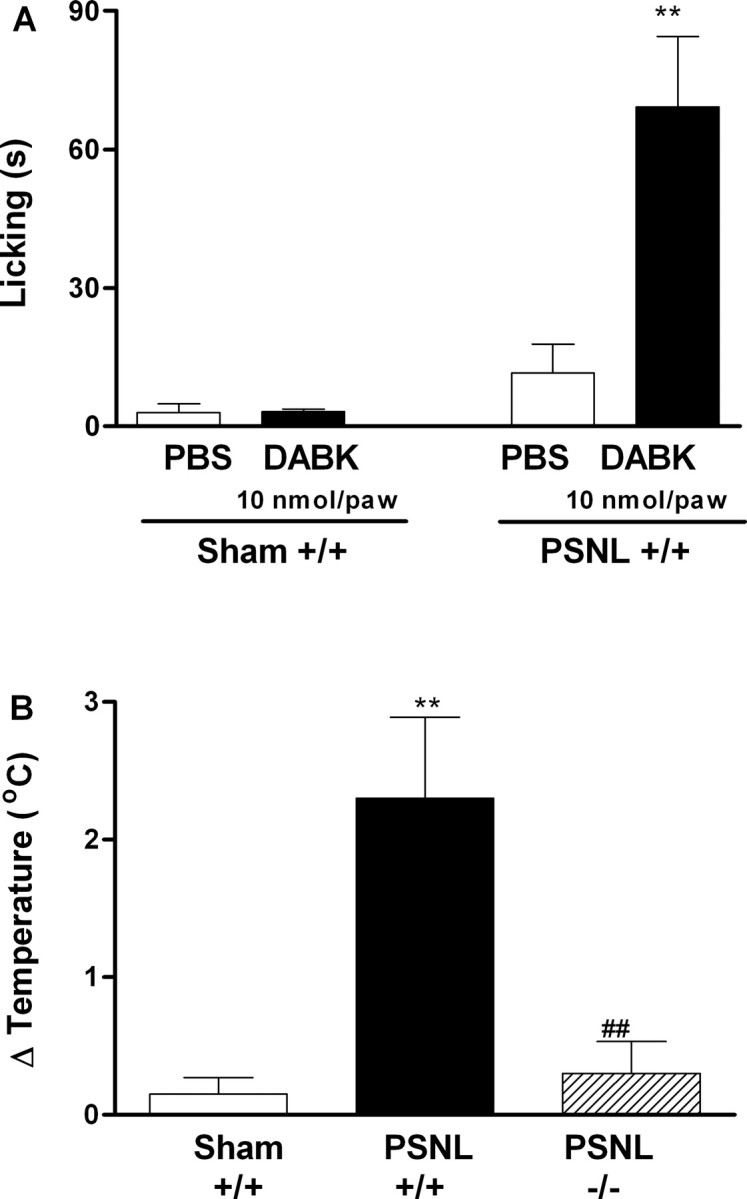
A, Overt nociception produced by intraplantar injection of des-Arg9-bradykinin (DABK; 10 nmol per paw) in wild-type mice (+/+) 7 d after partial sciatic nerve ligation (PSNL). B, Difference in temperature [Δ Temperature (°C)] between ipsilateral and contralateral hindpaw surface observed 7 d after unilateral sciatic nerve lesion. Each point represents the mean ± SEM of four to six mice. **p < 0.01 denotes the significance level when compared with wild-type sham-operated group. ##p < 0.01 denotes the significance level when compared with wild-type PSNL group (one-way ANOVA followed by Student-Newman-Keuls test).
In addition to nociceptive hypersensitivity, other symptoms similar to clinical features of human neuropathies may occur after partial sciatic nerve ligation in mice, including abnormal cutaneous temperature regulation. Accordingly, we observed a significant increase in the skin surface temperature of the ipsilateral paw 7 d after nerve injury in wild-type mice (Fig. 6B). Notably, the B1 receptor gene deletion abolished this cutaneous heating (Fig. 6B). However, we were not able to detect significant modifications in skin temperature from 14 to 42 d after nerve injury either in operated or in sham-operated animals (results not shown).
Discussion
Painful neuropathies may result from nerve injury as well as the effects of drugs, diseases, toxins, and metabolic disorders (Woolf and Mannion, 1999). Because of the as yet poor understanding of the mechanisms underlying these syndromes, therapy does not provide satisfactory pain relief for many patients. Consequently, these patients suffer from chronic intractable pain (Seltzer, 1995).
Several studies have demonstrated the participation of kinins and their receptors in neuropathic pain induction. Increased levels of B1 and B2 receptor mRNA or protein have been found in dorsal root ganglia (DRGs) after sciatic nerve constriction in rats and mice (Petersen et al., 1998; Eckert et al., 1999; Levy and Zochodne, 2000; Yamaguchi-Sase et al., 2003; Rashid et al., 2004). Of note, the systemic administration of B1 or B2 receptor antagonists has been found to reduce thermal hyperalgesia and mechanical allodynia produced by sciatic nerve constriction in rats (Levy and Zochodne, 2000; Yamaguchi-Sase et al., 2003; Gougat et al., 2004). Plasma seems to be the main source of endogenous kinins after nerve injury, and there is recent evidence demonstrated that neuropathic pain is reduced in mutant plasma kininogen-deficient B/N-Katholiek rats when compared with normal B/N-Kitasato rats (Yamaguchi-Sase et al., 2003).
The present work extended these previous observations by demonstrating that gene deletion or pharmacological inhibition of the B1 receptor in mice practically abolished the nociceptive hypersensitivity produced by nerve injury. This effect appeared as early as 1 d after lesion, and it was found significant until 28 d after the surgery, suggesting that the B1 receptor is critically involved in both the development and the early maintenance of neuropathic pain symptoms. In contrast, thermal hyperalgesia was not observed, and mechanical allodynia was reduced only in the later stages of nerve injury (35-42 d after surgery), despite the detection of increased levels of B1 receptor mRNA. Interestingly, at this time, the mechanical allodynia was reinstalled in B1 receptor knock-out mice, and the B1 receptor antagonist was not capable of reducing allodynia. Because regeneration occurs after constrictive injury to the sciatic nerve (Myers et al., 1996), it is quite possible that under this circumstance, B1 receptor activity is not relevant to the production of neuropathic pain and probably other mediators substitute for the nociceptive action of kinins.
Pain is produced by the stimulation of small-diameter primary afferent fibers that innervate regions of the head and body and arise from cell bodies in the trigeminal ganglion and DRG, respectively (Julius and Basbaum, 2001). B1 receptor mRNA and protein are constitutively expressed in mouse, rat, and monkey DRG (Seabrook et al., 1997; Levy and Zochodne, 2000; Ma et al., 2000; Wotherspoon and Winter, 2000; Shughrue et al., 2003; Yamaguchi-Sase et al., 2003; Rashid et al., 2004). B1 receptors are predominantly expressed by small-diameter DRG neurons colocalized with isolectin B4 and calcitonin gene-related peptide that are contained in C and Aδ fibers (Ma, 2001). Moreover, the B1 receptor is expressed in both peripheral and spinal terminals of primary afferent fibers (Wotherspoon and Winter, 2000; Ma and Heavens, 2001; Shughrue et al., 2003). B1 receptors are newly expressed 7 d after partial sciatic nerve injury in mice mainly in non-neuronal satellite cells and in large myelinated DRG neurons (Rashid et al., 2004). Because there is evidence that large A fibers mediate the mechanical allodynia in rats with partial sciatic nerve lesion (Shir and Seltzer, 1990) and B1 receptor knock-out mice have reduced allodynia, it seems that this novel expression of B1 receptors is potentially related to the production of the persistent mechanical allodynia observed in the early stages of neuropathy.
In the present study, we have shown that B1 receptor mRNA was normally expressed in some tissues important for the detection, transmission, and modulation of pain, including plantar paw skin, sciatic nerve, spinal cord, and cerebral cortex. Moreover, the involvement of B1 receptors in neuropathy was further confirmed by the upregulation of B1 receptor mRNA several days after sciatic nerve injury. It has been well demonstrated that several stimuli are able to upregulate B1 receptor, including proinflammatory cytokines, mitogen-activated protein kinases (MAPK), and nuclear factor κB (NFκB) (for review, see Calixto et al., 2000, 2004). We can suggest that similar mechanisms might be involved in B1 receptor upregulation in the present study, because proinflammatory cytokines are produced, and MAPK and NFκB are activated after sciatic nerve injury (Ma and Bisby, 1998; Okamoto et al., 2001; Ma and Quirion, 2002). We also observed an increase in levels of B1 receptor mRNA in samples of ipsilateral paw skin and sciatic nerve 7 d after injury, a finding that could suggest a role for B1 receptors in the abnormal perception of noxious and innocuous stimuli seen in early stages neuropathy. This upregulation seems to be functional, because the intraplantar injection of the selective B1 receptor agonist des-Arg9-bradykinin produced overt nociception in nerve-injured, but not in sham-operated, wild-type mice. These results reinforce the recent data obtained by Rashid et al. (2004), showing that intraplantar administration of des-Arg10-kallidin was able to induce both nociceptive reflex and activation of ERK (extracellular signal-regulated kinase) in DRG neurons in ligated, but not sham-operated, mice.
B1 receptors are also found in the CNS, which contains all of the components of the kallikrein-kinin system and is also involved in nociceptive processing (Couture and Lindsey, 2000; Ferreira et al., 2002). B1 receptors have been identified in the superficial layers of the dorsal horn confined mainly to the terminals of primary sensory nerve fibers (Couture and Lindsey, 2000; Wotherspoon and Winter, 2000). Using an in vitro spinal cord preparation, Pesquero et al. (2000) demonstrated that B1 receptor stimulation increases the C-fiber component, but not the Aβ-fiber component, of the ventral root potential produced by electrical excitation of the dorsal root of naive mice. This indicates that the B1 receptor functions specifically in nociceptive synaptic pathways and appears to be involved in some forms of central sensitization. In fact, intrathecal injection of B1 receptor antagonists reduces the inflammatory phase of formalin-induced pain and chronic inflammatory pain caused by Complete Freund's Adjuvant in mice and rats (Ferreira et al., 2002; Fox et al., 2003). Moreover, the use-dependent facilitation of spinal cord neuron firing (wind-up) was significantly reduced (∼50%) in B1 receptor knock-out mice when compared with the wild-type littermates (Pesquero et al., 2000). We have shown that B1 receptor mRNA is upregulated in dorsal spinal cord after partial sciatic nerve lesion, further suggesting a role for spinal B1 receptors in neuropathy. Because the development of spinal sensitization is an important consequence of nerve injury (Sah et al., 2003), these data indicate that the nociceptive impairment observed in B1 receptor knock-out mice might be attributed to, at least in part, a deficit in the pathological plasticity of the spinal neurons.
Subsets of dorsal horn neurons that project axons and transmit pain messages to higher brain structures are involved in the somatic, affective, and autonomic responses to pain (Hunt and Mantyh, 2001). In this respect, we have shown that B1 receptor mRNA is constitutively expressed in the cerebral cortex of mice. This result is in line with literature showing basal B1 receptor expression in rat somatosensory cortex (Ongali et al., 2003; Shughrue et al., 2003). However, the function of cortical B1 receptors still remains obscure.
Besides thermal and mechanical hypersensitivity, animals subjected to sciatic nerve injury exhibit other signs similar to clinical features of human painful neuropathies, including abnormal sympathetic activity, abnormal growth of hair, and cutaneous temperature regulation (Wakisaka et al., 1994). Similar to our observations in mice, the ipsilateral plantar surface in rats was warmer than that of the contralateral paw during the first week after loose ligation of sciatic nerve, thereafter becoming cooler (Wakisaka et al., 1991, 1994). It has been reported that early heating of the paw surface is dependent on sympathetic vasoconstriction (Wakisaka et al., 1994). Furthermore, partial nerve injury-induced pain is mediated by sympathetic activity (Shir and Seltzer, 1991; Malmberg and Basbaum, 1998). Interestingly, functional B1 receptors are expressed in sympathetic neurons, because their activation by agonists is able to depolarize superior cervical ganglia neurons in vitro (Seabrook et al., 1995, 1997). In addition, postganglionic sympathetic terminals are involved in B1 receptor agonist-induced hyperalgesia (Khasar et al., 1995). However, the participation of sympathetic fibers in nociception mediated by B1 receptors activation during neuropathy still needs to be determined.
Besides being caused by nerve injury, painful neuropathy may also develop in diabetes (Woolf and Mannion, 1999; Sah et al., 2003). It has been reported recently that thermal hyperalgesia in diabetic mice was blocked by the systemic treatment with selective B1 receptor antagonists (Gabra and Sirois, 2002, 2003a,b). Moreover, intrathecal administration of a B1 receptor agonist produces thermal hyperalgesia in hyperglycemic rats (Couture et al., 2001). Thus, the activation of B1 receptors is a critical step in the production of neuropathic pain, and B1 receptor blockade is able to not only prevent the development of nociception but also reduce an established painful condition. Of interest are the results showing that oral treatment with the newly synthesized nonpeptide B1 receptor antagonist SSR240612 [(2R)-2-[((3R)-3-(1,3-benzodioxol-5-yl)-3-[[(6-methoxy-2-naphthyl)sulfonyl]amino] propanoyl)amino]-3-(4-[[2R,6S)-2,6-dimethylpiperidinyl]methyl] phenyl)-N-isopropyl-N-methylpropanamide hydrochloride] was able to reduce the thermal hyperalgesia produced by sciatic nerve injury in rats (Gougat et al., 2004). These findings support the notion that the development of oral-selective B1 receptor antagonists might be expected to have clinical therapeutic potential in the management of neuropathic pain.
Footnotes
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Financiadora de Estudos e Projetos, the Programa de Apoio aos Núcleos de Excelência, and the Fundação de Ciência e Tecnologia do Estado de Santa Catarina (Brazil). J.F., A.B., and M.A.S.M. are PhD students receiving grants from CNPq and Fundação de Amparo à Pesquisa do Estado de São Paulo (Brazil).
Correspondence should be addressed to João B. Calixto, Department of Pharmacology, Universidade Federal de Santa Catarina, Campus Universitário, Trindade, Bloco D, Caixa Postel 476, 88049-900 Florianópolis, Santa Catarina, Brazil. E-mail: calixto@farmaco.ufsc.br or calixto3@terra.com.br.
J. Ferreira's present address: Department of Chemistry, Universidade Federal de Santa Maria, 97105-900, Santa Maria, Rio Grande do Sul, Brazil. E-mail: ferreiraj99@bol.com.br.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252405-08$15.00/0
References
- Argañaraz GA, Silva Jr JA, Perosa SR, Pessoa LG, Carvalho FF, Bascands JL, Bader M, Trindade ES, Amado D, Cavalheiro EA, Pesquero JB, Naffah-Mazzacoratti MG (2004) The synthesis and distribution of the kinin B1 and B2 receptors are modified in the hippocampus of rats submitted to pilocarpine model of epilepsy. Brain Res 1006: 114-125. [DOI] [PubMed] [Google Scholar]
- Bennett GJ (1999) Does a neuroimmune interaction contribute to the genesis of painful peripheral neuropathies? Proc Natl Acad Sci USA 96: 7737-7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson JM (1999) The neurobiology of pain. Lancet 353: 1610-1615. [DOI] [PubMed] [Google Scholar]
- Blair SJ, Chinthagada M, Hoppenstehdt D, Kijowski R, Fareed J (1998) Role of neuropeptides in pathogenesis of reflex sympathetic dystrophy. Acta Orthop Bel 64: 448-451. [PubMed] [Google Scholar]
- Borkowski JA, Ranson RW, Seabrook GR, Trumbauer M, Chen H, Hill RG, Strader CD, Hess JF (1995) Targeted disruption of a B2 bradykinin receptor in mice eliminates bradykinin action in smooth muscle and neurons. J Biol Chem 270: 13706-13710. [DOI] [PubMed] [Google Scholar]
- Calixto JB, Cabrini DA, Ferreira J, Campos MM (2000) Kinins in pain and inflammation. Pain 87: 1-5. [DOI] [PubMed] [Google Scholar]
- Calixto JB, Medeiros R, Fernandes ES, Ferreira J, Cabrini DA, Campos MM (2004) Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes. Br J Pharmacol 143: 803-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture R, Lindsey CJ (2000) Brain kallikrein-kinin system: from receptors to neuronal pathways and physiological functions. In: Handbook of chemical anatomy: peptide receptors (Quirion R, Björklund A, Hökfeld T, ed), pp 241-298. Amsterdam: Elsevier.
- Couture R, Harrisson M, Vianna RM, Cloutier F (2001) Kinin receptors in pain and inflammation. Eur J Pharmacol 429: 161-176. [DOI] [PubMed] [Google Scholar]
- Dray A, Perkins MN (1997) Kinins and pain. In: The kinin system (Farmer SG, ed), pp 157-172. San Diego: Academic.
- Eckert A, Segond von Banchet G, Sopper S, Petersen M (1999) Spatiotemporal pattern of induction of bradykinin receptors and inflammation in rat dorsal root ganglia after unilateral nerve ligation. Pain 83: 487-497. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Campos MM, Pesquero JB, Araujo RC, Bader M, Calixto JB (2001) Evidence for the participation of kinins in Freund's adjuvant-induced inflammatory and nociceptive responses in kinin B1 and B2 receptor knockout mice. Neuropharmacology 41: 1006-1012. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Campos MM, Araujo R, Bader M, Pesquero JB, Calixto JB (2002) The use of kinin B1 and B2 receptor knockout mice and selective antagonists to characterize the nociceptive responses caused by kinins at the spinal level. Neuropharmacology 43: 1188-1197. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Silva GL, Calixto JB (2004) Involvement of vanilloid receptors in the overt nociception induced by B2 kinin receptor activation in mice. Br J Pharmacol 141: 787-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Wotherspoon G, McNair K, Hudson L, Patel S, Gentry C, Winter J (2003) Regulation and function of spinal and peripheral neuronal B1 bradykinin receptors in inflammatory mechanical hyperalgesia. Pain 104: 683-691. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Sirois P (2002) Role of bradykinin B1 receptors in diabetes-induced hyperalgesia in streptozotocin-treated mice. Eur J Pharmacol 457: 115-124. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Sirois P (2003a) Kinin B1 receptor antagonists inhibit diabetes-induced hyperalgesia in mice. Neuropeptides 37: 36-44. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Sirois P (2003b) Beneficial effect of chronic treatment with the selective bradykinin B1 receptor antagonists, R-715 and R-954, in attenuating streptozotocin-diabetic thermal hyperalgesia in mice. Peptides 24: 1131-1139. [DOI] [PubMed] [Google Scholar]
- Gougat J, Ferrari B, Sarran L, Planchenault C, Poncelet M, Maruani J, Alonso R, Cudennec A, Croci T, Guagnini F, Urban-Szabo K, Martinolle JJ, Soubrie P, Finance O, Le Fur G (2004) SSR240612 [(2R)-2-[((3R)-3-(1,3-benzodioxol-5-yl)-3-[[(6-methoxy-2-naphthyl)sulfonyl]amino]propanoyl)amino]-3-(4-[[2R,6S)-2,6-dimethylpiperidinyl]methyl]phenyl)-N-isopropyl-N-methylpropanamide hydrochloride], a new non-peptide antagonist of the bradykinin B1 receptor: biochemical and pharmacological characterization. J Pharmacol Exp Ther 309: 661-669. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method to measure thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Mantyh PW (2001) The molecular dynamics of pain control. Nat Rev Neurosci 2: 83-91. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413: 203-210. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao FJ, Levine JD (1995) Inflammation modulates the contribution of receptor-subtypes to bradykinin-induced hyperalgesia in the rat. Neuroscience 69: 685-690. [DOI] [PubMed] [Google Scholar]
- Levy D, Zochodne DW (2000) Increased mRNA expression of the B1 and B2 bradykinin receptors and antinociceptive effects of their antagonists in an animal model of neuropathic pain. Pain 86: 265-271. [DOI] [PubMed] [Google Scholar]
- Ma Q-P (2001) The expression of bradykinin B1 receptors on primary sensory neurones that give rise to small calibre sciatic nerve fibres in rats. Neuroscience 107: 665-673. [DOI] [PubMed] [Google Scholar]
- Ma Q-P, Heavens R (2001) Basal expression of bradykinin B1 receptor in the spinal cord in humans and rats. NeuroReport 12: 2311-2314. [DOI] [PubMed] [Google Scholar]
- Ma Q-P, Hill R, Sirinathsinghji D (2000) Basal expression of bradykinin B1 receptor in peripheral sensory ganglia in the rat. NeuroReport 18: 4003-4005. [DOI] [PubMed] [Google Scholar]
- Ma W, Bisby MA (1998) Increased activation of nuclear factor κB in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res 797: 243-254. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R (2002) Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 99: 175-184. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI (1998) Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain 76: 215-222. [DOI] [PubMed] [Google Scholar]
- Marceau F, Hess JF, Bachvarov DR (1998) The B1 receptors for kinins. Pharmacol Rev 50: 357-386. [PubMed] [Google Scholar]
- Myers RR, Heckman HM, Rodriguez M (1996) Reduced hyperalgesia in nerve-injured WLD mice: relationship to nerve fiber phagocytosis, axonal degeneration, and regeneration in normal mice. Exp Neurol 141: 94-101. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA (2001) Pro- and anti-inflammatory cytokine gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain. Exp Neurol 169: 386-391. [DOI] [PubMed] [Google Scholar]
- Ongali B, Campos MM, Bregola G, Rodi D, Regoli D, Thibault G, Simonato M, Couture R (2003) Autoradiographic analysis of rat brain kinin B1 and B2 receptors: normal distribution and alterations induced by epilepsy. J Comp Neurol 461: 506-519. [DOI] [PubMed] [Google Scholar]
- Pesquero JB, Araujo RC, Heppenstall PA, Stucky CL, Silva Jr JA, Walther T, Oliveira SM, Pesquero JL, Paiva AC, Calixto JB, Lewin GR, Bader M (2000) Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc Natl Acad Sci USA 97: 8140-8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M, Eckert AS, Segond von Banchet G, Heppelmann B, Klusch A, Kniffki KD (1998) Plasticity in the expression of bradykinin binding sites in sensory neurons after mechanical nerve injury. Neuroscience 83: 949-959. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Inoue M, Matsumoto M, Ueda H (2004) Switching of bradykinin-mediated nociception following partial sciatic nerve injury in mice. J Pharmacol Exp Ther 308: 1158-1164. [DOI] [PubMed] [Google Scholar]
- Sah DW, Ossipov MH, Porreca F (2003) Neurotrophic factors as novel therapeutics for neuropathic pain. Nat Rev Drug Discov 2: 460-472. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Bowery BJ, Hill RG (1995) Bradykinin receptors in mouse and rat isolated superior cervical ganglia. Br J Pharmacol 115: 368-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabrook GR, Bowery BJ, Heavens R, Brown N, Ford H, Sirinathsinghji DJS, Borkowski JA, Hess JF, Strader CD, Hill RG (1997) Expression of B1 and B2 bradykinin receptor mRNA and their function roles in sympathetic ganglia and sensory root ganglia neurones from wild-type and B2 receptor knockout mice. Neuropharmacology 36: 1009-1017. [DOI] [PubMed] [Google Scholar]
- Seltzer Z (1995) The relevance of animal neuropathy models for chronic pain in humans. Semin Neurosci 7: 211-219. [Google Scholar]
- Seltzer Z, Dubner R, Shir Y (1990) A novel behavioural model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43: 205-218. [DOI] [PubMed] [Google Scholar]
- Shir Y, Seltzer Z (1990) A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neurosci Lett 115: 62-67. [DOI] [PubMed] [Google Scholar]
- Shir Y, Seltzer Z (1991) Effects of sympathectomy in a model of causalgiform pain provided by partial sciatic nerve injury in rats. Pain 45: 309-320. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Ky B, Austin CP (2003) Localization of B1 bradykinin receptor mRNA in the primate brain and spinal cord: an in situ hybridization study. J Comp Neurol 465: 372-384. [DOI] [PubMed] [Google Scholar]
- Tracey DJ, Walker JS (1995) Pain due to nerve damage: are inflammatory mediators involved? Inflamm Res 44: 407-411. [DOI] [PubMed] [Google Scholar]
- Wakisaka S, Kajander KC, Bennett GJ (1991) Abnormal skin temperature and abnormal sympathetic vasomotor innervation in an experimental painful peripheral neuropathy. Pain 46: 299-313. [DOI] [PubMed] [Google Scholar]
- Wakisaka S, Shibata M, Takikita S, Yoshiya I, Kurisu K (1994) Effects of sympathectomy on the cutaneous temperature abnormalities in rats with chronic constriction injury of the sciatic nerve. Neurosci Lett 173: 5-8. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353: 1959-1964. [DOI] [PubMed] [Google Scholar]
- Wotherspoon G, Winter J (2000) Bradykinin B1 receptor is constitutively expressed in the rat sensory nervous system. Neurosci Lett 294: 175-178. [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Sase S, Hayashi I, Okamoto H, Nara Y, Matsuzaki S, Hoka S, Majima M (2003) Amelioration of hyperalgesia by kinin receptor antagonists or kininogen deficiency in chronic constriction nerve injury in rats. Inflamm Res 52: 164-169. [DOI] [PubMed] [Google Scholar]
- Zimmermann M (1983) Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109-110. [DOI] [PubMed] [Google Scholar]