

Abstract
Type B coxsackieviruses (CVB) frequently infect the CNS and, together with other enteroviruses, are the most common cause of viral meningitis in humans. Newborn infants are particularly vulnerable, and CVB also can infect the fetus, leading to mortality, or to neurodevelopmental defects in surviving infants. Using a mouse model of neonatal CVB infection, we previously demonstrated that coxsackievirus B3 (CVB3) could infect neuronal progenitor cells in the subventricular zone (SVZ). Here we extend these findings, and we show that CVB3 targets actively proliferating (bromodeoxyuridine+, Ki67+) cells in the SVZ, including type B and type A stem cells. However, infected cells exiting the SVZ have lost their proliferative capacity, in contrast to their uninfected companions. Despite being proliferation deficient, the infected neuronal precursors could migrate along the rostral migratory stream and radial glia, to reach their final destinations in the olfactory bulb or cerebral cortex. Furthermore, infection did not prevent cell differentiation, as determined by cellular morphology and the expression of maturation markers. These data lead us to propose a model of CVB infection of the developing CNS, which may explain the neurodevelopmental defects that result from fetal infection.
Keywords: migration, neuropathology, proliferation, infection, stem cells, coxsackievirus
Introduction
Enteroviral infections of the CNS can lead to aseptic meningitis and encephalitis, which are serious, and sometimes fatal, diseases. The host CNS seems to be particularly susceptible during early development; enteroviruses can infect the fetus, with serious consequences, including neurodevelopmental defects (Gear and Measroch, 1973; Daley et al., 1998; Sauerbrei et al., 2000; Euscher et al., 2001), and this vulnerability is retained in newborns (Ratzan, 1985; Hsueh et al., 2000). Coxsackieviruses, members of the enterovirus genus and picornavirus family, are the single most common cause of enteroviral CNS infection in neonates, and we have recently developed an animal model system that allows us to evaluate coxsackievirus B3 (CVB3) infection and pathogenesis in the neonatal CNS (Feuer et al., 2003). Using this model, we have shown that neuronal progenitor cells are susceptible to CVB3 infection. These cells could support viral protein expression during their migration and differentiation into mature (postmitotic) neurons, some of which underwent caspase-3-induced cell death, resulting in lesions in the hippocampus, olfactory bulb, and cortex (Feuer et al., 2003).
We wanted to expand these initial observations by answering three related questions. First, does CVB3 preferentially target proliferating cells in the neonatal CNS? Such a scenario would support our in vitro data, which showed that CVB3 productively infects dividing cells and can remain latent within quiescent cells (Feuer et al., 2002, 2004). To answer this question, we investigated the relationship between viral protein expression and cell division; the latter was examined using antibodies against markers of previous cell division [the nucleoside analog bromodeoxyuridine (BrDU)] and active cell proliferation [the nuclear antigen Ki67 (Kiel 67 antigen)]. Second, are all neonatal neuronal progenitor cells susceptible to CVB3 infection? Our previous study identified infection of type B stem cells and maturing neuronal precursors. Here, we expand our analysis to include a complete visualization of the anterior and posterior subventricular zones (SVZs) and the rostral migratory stream (RMS), allowing us to identify additional infected progenitor cell types. Third, do CVB-infected progenitor cells exploit the host migratory machinery, allowing the virus to spread through the CNS? We identified radial glia, which serve as the scaffolding for the migration of neuronal precursors, and we evaluated their association with virus-infected cells.
Materials and Methods
Recombinant enhanced green fluorescent protein-CVB. The generation of enhanced green fluorescent protein (eGFP)-CVB3, a recombinant CVB3 expressing eGFP has been described previously (Feuer et al., 2002). Viral stocks were prepared on HeLa cells and diluted in DMEM (Invitrogen, Gaithersburg, MD) before inoculation.
Mice, viral inoculations, and BrDU administration. BALB/c mice were obtained from The Scripps Research Institute animal facilities, and breeding pairs were observed daily, to ensure that pups were identified within 24 h of birth. These 1-d-old pups were infected intracranially with 2 × 106 pfu of eGFP-CVB3. The procedure for intracranial inoculation of 1-d-old pups has been described previously (Feuer et al., 2003). In some experiments, at the time of inoculation with virus, pups also were injected intraperitoneally with BrDU (500 μg/g body weight). One or 2 d after infection, pups were killed by hypothermia/CO2 intoxication, followed by immediate decapitation. The brain was fixed by immersion in 10% neutral-buffered Formalin for >4 h and paraffin embedded.
Immunofluorescence staining. eGFP expression was observed in unstained sagittal or transverse paraffin-embedded sections overlaid with PBS. For immunofluorescence studies, paraffin-embedded sections (3 μm thickness) were deparaffinized with three washes in xylene and serial washes in 100, 90, and 70% ethanol, followed by a final wash in PBS. The detection of Ki67, RC2 (a marker for radial glia), polysialylated neural cell adhesion molecule (PSA-NCAM), nestin, and neuronal nuclear antigen (NeuN) required high-temperature antigen unmasking in 0.01 m citrate buffer, pH 6.0. For BrDU staining, deparaffinized sections were treated with 4.2N HCl for 15 min, washed three times with PBS, and blocked with 10% normal goat serum (NGS) for 30 min. Both antigen unmasking and HCl treatment caused denaturation of eGFP, preventing its detection by normal means; in these cases, eGFP was detected by immunofluorescence, as described below. Immunofluorescence staining procedures have been described previously (Feuer et al., 2003). Briefly, sections were blocked with 10% NGS for 30 min and incubated overnight with primary antibody at 4°C. All antibody dilutions were made in 2% NGS. Class III β-tubulin was detected using a rabbit primary antibody (Covance Research Products, Cumberland, VA), diluted 1:500. The secondary antibody was a biotinylated goat anti-rabbit IgG [heavy and light chains (H+L)] (Vector Laboratories, Burlingame, CA) diluted 1:500 in 2% NGS and incubated on sections for 30 min. BrDU was detected using a rat anti-BrDU primary antibody (Accurate Chemicals, Westbury, NY) at 1:100 and a biotinylated goat anti-rat IgG (H+L) secondary antibody (Vector Laboratories) diluted 1:200. Immunological detection of eGFP was performed using rabbit anti-GFP (Molecular Probes, Eugene OR), neat or diluted 1:20, and incubated at 4°C overnight, followed by 30 min incubation with anti-rabbit FITC secondary antibody (Rockland Immunochemicals, Gilbertsville, PA) used at 1:100. When using primary antibodies derived from mouse [anti-NeuN at 1:1000 (Chemicon, Temecula, CA); anti-nestin at 1:50 (Chemicon); prediluted anti-Ki67 used neat (Novocastra Laboratories, Newcastle, UK); anti-RC2 concentrate diluted 1:50 (Developmental Studies Hybridoma Bank, Iowa City, IA); and anti-PSA-NCAM at 1:50 (PharMingen, San Diego, CA)], the Mouse on Mouse kit (Vector Laboratories) was used as described by the manufacturer. After staining with secondary antibodies, all sections were washed twice with PBS and incubated for 30 min with either a streptavidin-rhodamine red complex (Jackson ImmunoResearch, West Grove, PA) or a streptavidin-7-amino-4-methyl-coumarin-3-acetic acid blue complex (Vector Laboratories) diluted 1:500 in 2% NGS. Specificity controls for immunostaining included sections stained in the absence of primary antibody or in the presence of rabbit IgG control antibody at 0.1 μg/ml (Vector Laboratories) and staining of uninfected neonatal brains. For detection of DNA/nuclei using 4′,6-diamidino-2-phenylindole (DAPI), sections were incubated for 2 min in a 300 nm solution of DAPI dilactate (Molecular Probes) in PBS and then washed once with PBS. Sections were overlaid with Vectashield mounting medium (Vector Laboratories) and observed by fluorescence microscopy (Axiovert 200 inverted microscope; Zeiss, Oberkochen, Germany) for eGFP (green), the indicated cellular marker (red or blue), and DAPI nuclear staining (blue). Green, red, and blue channel images were merged using Axio-Vision software (Zeiss).
In situ hybridization. Our in situ hybridization procedure has been described previously (Feuer et al., 2003). Briefly, a 33P-labeled anti-sense RNA (421 bases) probe for the 5′ untranslated region of CVB3 was generated using the MAXIscript in vitro transcription kit (Ambion, Austin, TX), as described by the manufacturer. In situ hybridization procedures were performed using the mRNAlocater in situ hybridization kit (Ambion), as described by the manufacturer. The radiolabeled probe (107 cpm) was applied to deparaffinized sections, and the sample was sealed in a humidified chamber and incubated at 46°C for 18 h. After washing and RNase A treatment, slides were immersed in photographic emulsion, held at 4°C for 6 d, and then developed. Finally, slides were stained with hematoxylin (1 min) and eosin (1.5 min) (H&E) and mounted with Cytoseal (VWR Scientific, West Chester, PA).
Quantitation of Ki67+ cells in the SVZ. The numbers of Ki67+ cells present in the SVZs of infected and uninfected mice were calculated as follows. For each fluorescent image, a black and white picture was generated using NIH ImageJ (public domain software), and these black and white images were analyzed using ImageJ to enumerate the signals (which represent Ki67+ cells). ImageJ settings were as follows: threshold range, 30-255; and pixel size range, 25-5000. These numerical values were graphed for mice at 1, 2, and 5 d after infection and for uninfected mice of the same ages.
Results
Coxsackievirus distribution in the neonatal CNS coincides with regions that contain BrDU+ cells
Our laboratory has described the construction of a recombinant CVB3 expressing eGFP (Feuer et al., 2002) and its use to characterize the distribution of CVB3 within the CNS of neonatal mice (Feuer et al., 2003). Soon after infection, viral protein expression was observed in the choroid plexus and in neuronal progenitor cells near the lateral ventricles. These progenitor cells were identified by their expression of nestin (Lendahl et al., 1990), a neuroepithelial stem cell intermediate filament protein that is present in the majority of neural stem cells in the mouse CNS (Mignone et al., 2004). Our goal in the present study was to evaluate the relationship between viral infection/protein expression and cellular proliferation/migration. Cells that had proliferated during the period of observation were identified by inoculating the mice with the nucleoside analog BrDU, which is incorporated into DNA during mitosis, and can be detected using immunohistochemistry (Wynford-Thomas and Williams, 1986). We inoculated 1-d-old neonatal mice with BrDU, and they were infected with eGFP-CVB3. One or 2 d later, pups were killed, and sagittal brain sections were obtained. Viral RNA was identified by in situ hybridization, and cells that had divided were located using antibodies against BrDU (Fig. 1). Viral RNA was present in the olfactory bulb (OB), the RMS, the SVZ of the lateral ventricle, and adjacent to the fourth ventricle (Fig. 1A). BrDU staining (Fig. 1B) identified mitotically active cells in the same areas; the regional colocalization between viral RNA and BrDU+ cells was striking. Higher-power magnification of the OB shows that viral RNA is distributed in relatively discrete layers within that structure (Fig. 1C), and the viral RNA distribution coincides with that of viral gene expression, as revealed by eGFP signal (Fig. 1D). Our subsequent studies evaluated in greater detail the relationship between CVB3 infection and cell status in the RMS, OB, SVZ, retrosplenial cortex (RSC), and fourth ventricle.
Figure 1.
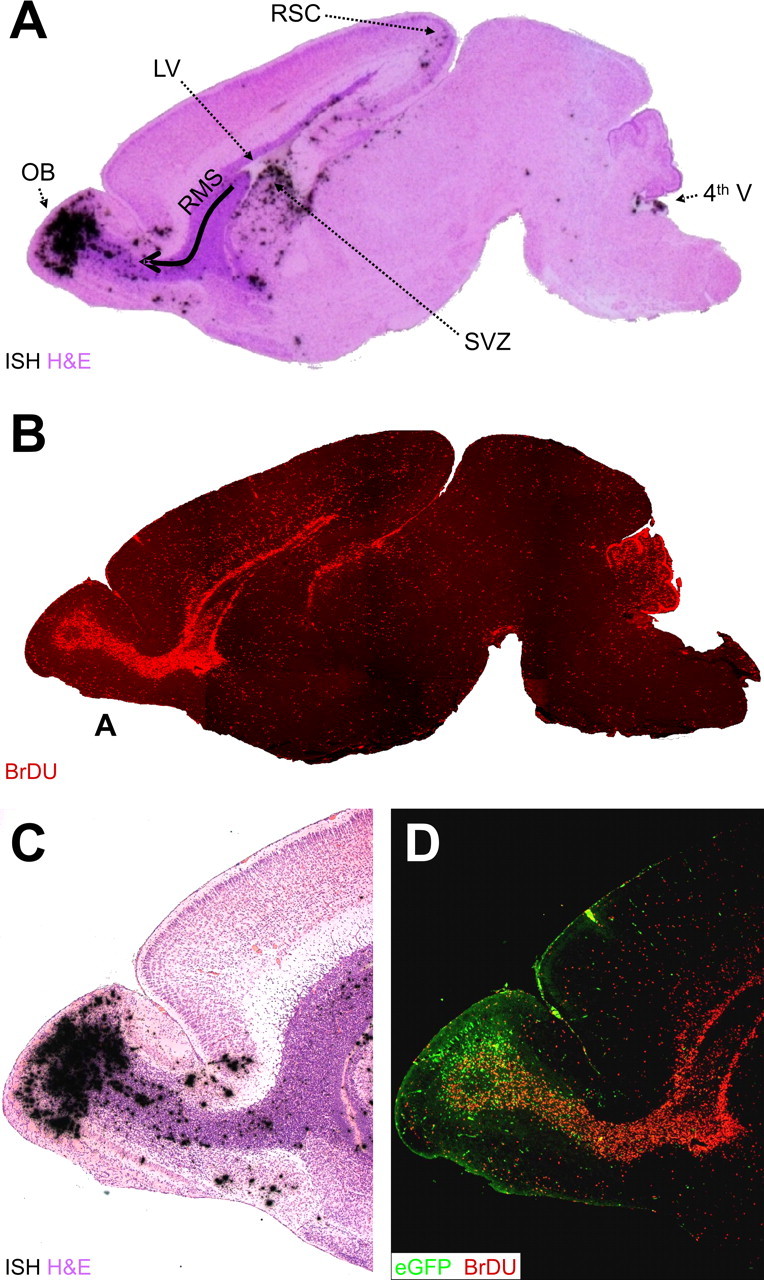
Distribution of CVB and proliferating cells in the neonatal CNS. We treated 1-d-old pups with BrDU (intraperitoneally), and we infected them with eGFP-CVB3 (2 × 106 pfu, i.c.). After 2 d, the brain was harvested and fixed in 10% neutral-buffered Formalin. Sagittal sections were deparaffinized (see Materials and Methods) and probed for viral RNA by in situ hybridization or immunostained using antibody against BrDU. A, H&E-stained sagittal section, close to the midline, with viral RNA revealed by in situ hybridization (black dots). Viral RNA is present in the OB, the RMS (curved arrow), the ventral SVZ below the lateral ventricle (LV), the RSC, and in the region of the fourth ventricle (4th V). B, A similar sagittal section (composite of 5× images) showing BrDU staining (red) that highlights the RMS, as well as the cerebellar cortex. C, Higher-power image (5×) showing the layered distribution of viral RNA in the OB and scattered throughout the RMS. D, Viral protein expression is shown (5×) by eGFP (green), and cells that have divided are indicated by BrDU staining (red).
Viral protein expression is observed in migratory neurons within the OB and the RMS
First, we investigated the relationship between CVB3 and cell status in the RMS in mice infected with eGFP-CVB. BrDU staining identifies cells that have undergone mitosis but does not provide information on the proliferative status of cells at the time of harvest. For this purpose, we evaluated the expression of Ki67, a well characterized nuclear antigen that is expressed during all stages of the cell cycle in dividing cells but is absent from quiescent (G0) or nondividing cells (MacCallum and Hall, 2000). The RMS contains migrating progenitor and immature neuronal cells, which begin their tangential migration near the lateral ventricle and end their journey in the OB (Luskin and Coskun, 2002). H&E staining shows the characteristic dark granular pattern of the RMS, leading toward the OB (Fig. 2A, arrow shows direction of cell migration in the RMS). Viral protein (eGFP) is most abundantly expressed in the OB (discussed below), but eGFP+ cells also are present within the RMS in close proximity to proliferating (Ki67+) cells (Fig. 2B). We have shown that, in tissue culture, CVB3 protein expression is tightly associated with the cell cycle; cells infected with eGFP-CVB and held at G0 or G2/M expressed very little eGFP, but protein expression resumed when the cells were permitted to cycle (Feuer et al., 2002). Therefore, we anticipated that most eGFP+ cells in the RMS would be cycling, and thus would express Ki67. However, contrary to our expectations, we found that eGFP+ and Ki67+ cells in the RMS were adjacent but distinct; few coexpressing (yellow) cells were identified (Fig. 2C). A similar relationship was observed when an adjacent section was evaluated for the presence of BrDU (Fig. 2D); once again, very few double-positive cells were visible. However, many of the eGFP+ cells showed leading or lagging axonal progressions (Fig. 2C,D, notched arrows), which are characteristic of migratory neurons. Nestin staining identified neuronal progenitor cells in the RMS and OB (Fig. 2E), and a higher-power magnification allowed a number of eGFP+ nestin+ cells to be identified, consistent with viral protein expression in some migrating neuronal precursors (Fig. 2F, notched arrows); most of the eGFP+ cells also were nestin+. We conclude that CVB3 is present in migratory neurons in the RMS but that these cells rarely show signs of active proliferation (Ki67) or recent division (BrDU); however, most of the infected cells express an early neuronal marker and display the morphological hallmarks of migratory neurons.
Figure 2.
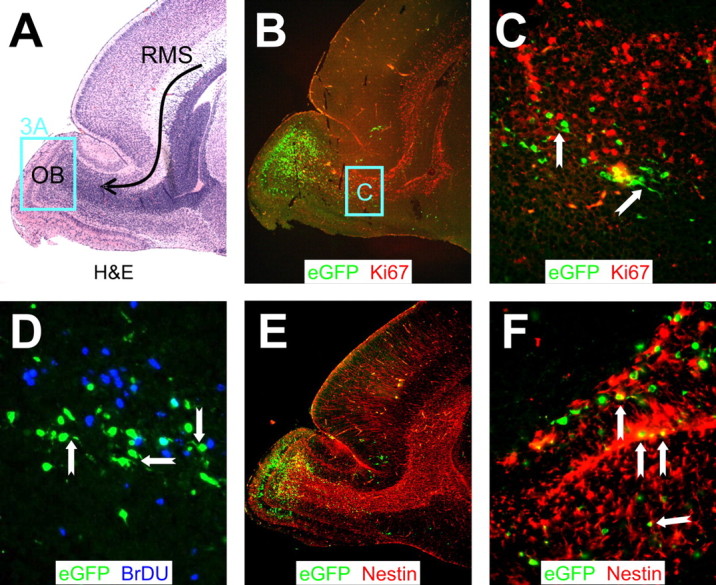
CVB3 protein expression in migratory neuroblasts within proliferating regions of the RMS. Sagittal sections of the same infected brain presented in Figure 1 were immunostained with antibodies against Ki67, BrDU, and nestin. A, H&E staining. The curved black arrow represents the RMS, which contains neuroblasts migrating from the SVZ to the OB. The distinct layers of the olfactory bulb were apparent, and a higher magnification of this area (represented by the cyan box) is evaluated in Figure 3A. B, Proliferating cells in the RMS were detected using an antibody against Ki67. Infected cells (eGFP+) were observed within proliferating (Ki67+; red) regions extending from the anterior SVZ into the olfactory bulb. C, Higher magnification (B, cyan box) revealed infected cells in close proximity to, yet distinct from, proliferating cells. Many infected cells in the RMS exhibited morphological similarities to migratory neuroblasts, with characteristic leading or lagging axonal progressions (notched arrows). D, Similarly, infected cells in the RMS were observed in close proximity to, yet distinct from, BrDU+ (blue) cells. Notched arrows indicate infected migratory neuroblasts. E, Nestin+ (red) staining was seen throughout the RMS. F, Higher magnification of E demonstrated direct colocalization of nestin with infected migratory neuroblasts. A, B, E, 5× objective; C, 20× objective with an additional twofold computer-generated magnification; D, 20× objective with an additional threefold computer-generated magnification; F, 40× objective.
Viral protein expression in OB: cell types and association with cell proliferation and maturation
The OB contains several distinct layers of neurons, visible in low-power section in Figure 2A, from which the indicated region is presented at higher power in Figure 3A. The glomerular layer (G) lies immediately below the cribriform plate and contains spheroidal glomerular cells (which receive input signals from olfactory receptors) and periglomerular interneurons. Glomerular cell signals are transferred to neurons in the mitral layer (M) via mitral cell primary dendrites, which span the external plexiform layer that separates the G and M layers. Mitral cell output is modified by interactions with granule interneurons, which can be anatomically and functionally divided into two layers; a superficial layer (grs) and a deep layer (grd). The majority of CNS neurons appear to be nonrenewing, but the OB is a striking exception. Throughout the lifespan of the host, the two populations of interneurons in the olfactory bulb (periglomerular cells and granule cells) are removed and replaced by cells that originate in the SVZ and migrate to their final destination via the RMS (Luskin and Coskun, 2002; Murase and Horwitz, 2002), in which these abundant neuronal precursors are revealed by nestin staining (Fig. 2E). Once in the OB, the cells complete their differentiation, becoming mature neurons, which can be identified by their expression of the neuronal nuclear antigen NeuN (Mullen et al., 1992). A discrete layer of NeuN+ cells is clearly visible in the OB (Fig. 3B), and a higher-power magnification identified cells in grs as the source of the NeuN signal (Fig. 3C). Virus protein (eGFP) was present in many cells in grd and also in mitral neurons [which, as reported previously (Mullen et al., 1992), lack NeuN staining, despite apparently being postmitotic mature neurons]. An infected mitral cell appears to retain its morphological characteristics, with a single primary dendrite ascending through the external plexiform layer, toward the glomerular layer (Fig. 3D). Together with our observation of eGFP in migrating cells, it appears that viral protein expression in neuronal progenitors, and even in mature neurons, does not invariably lead to the loss of cell morphology or function.
Figure 3.
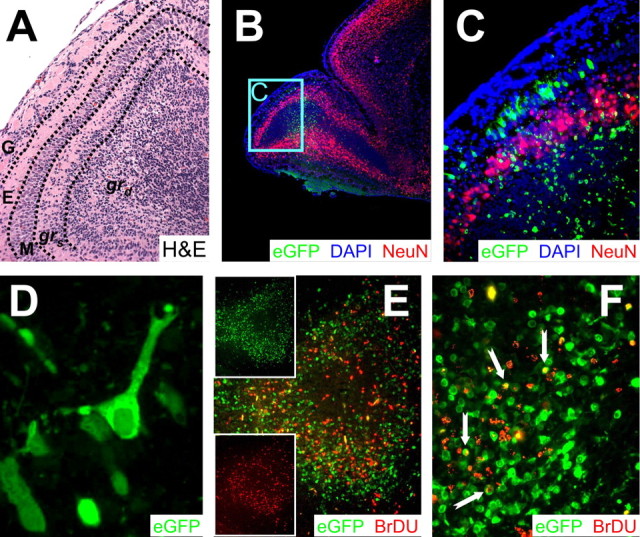
CVB3 protein expression and markers of cellular proliferation and maturation in the OB. All samples represent brains from mice that were infected at 1 d after birth and killed 2 d after infection. A, High magnification of the OB region shown in Figure 2A. The various layers of the OB, described in detail in Results, are separated by dashed black lines. B, Infected cells positive for NeuN were identified in the grs cell layer of the OB (5× magnification). The majority of NeuN staining (red) included a distinct halo of cells extending within the grs layer of the olfactory bulb and continuing throughout the cortex. Very little NeuN staining was identified within the RMS. DAPI stain (blue), which identified the nucleus of all cells, helped to define tissue section morphology. DAPI signal merged with NeuN generated a magenta color illustrating the nuclear localization of NeuN antigen. C, Higher magnification (B, cyan box) of the olfactory bulb demonstrated infection in the mitral cell layer and in granule cells in both the grs and grd layers. Infected cells in the mitral cell layer were NeuN-. However, infected granule cells were NeuN+ in the grs cell layer and NeuN- in the grd cell layer of the OB. The periglomerular cell layer was spared from infection. D, Many infected cells in the mitral cell layer had characteristic mitral cell morphology with a single projection positioned toward the periglomerular cell layer. E, Regional colocalization of virus protein expression (eGFP+) and cellular proliferation (BrDU+; red) was observed in the granule cell layer of the olfactory bulb. Individual channels for eGFP or BrDU (insets) demonstrated a close association of infected and dividing cells. F, Higher magnification of the olfactory bulb revealed some BrDU+ infected cells (yellow; notched arrows). A, C, E, 20× objective; B, 5× objective; D, 100.8× objective; F, 63× objective.
Studies of viral protein expression (eGFP) and cell proliferation (BrDU) in the OB at 2 d after infection revealed a very strong regional colocalization (Fig. 3E). Furthermore, many BrDU+ cells in the granule cell layer of the olfactory bulb also expressed high levels of viral protein (Fig. 3F, arrows), but the great majority of infected cells had no detectable levels of BrDU. Few BrDU+ infected cells were observed later during infection (data not shown), possibly because of the loss of these cells through caspase 3- or siva-mediated apoptosis (Henke et al., 2000; Feuer et al., 2003).
CVB3 can infect type A and type B stem cells in the SVZ
The SVZ serves as the source of neuronal progenitor cells; cells from the anterior SVZ travel in the RMS to the OB, and cells originating in the posterior SVZ migrate to form the cerebral cortex. These neuronal progenitors are themselves derived from stem cells in the SVZ. The primary stem cell lineage appears to be the type B cell, characterized by cytoplasmic extensions that protrude through the ependymal cell layer, toward the ventricle. Type B stem cells can divide and expand into, first, type C cells, and subsequently into type A cells, which are neuroblasts that migrate as chains extending from the SVZ (Doetsch et al., 1999) (for review, see Doetsch, 2003). All three cell types are thought to be capable of division. We showed previously that CVB3 could infect type B cells in the SVZ (Feuer et al., 2003), but we evaluated neither the possible infection of type C or type A cells, nor the proliferative status of the infected cells. To begin to address these issues, we focused on the anterior SVZ, using transverse sections. Figure 4A shows virus-infected (eGFP+) cells in the SVZ, near the lateral ventricle; the apposed ventricular ependymal layers are indicated by dashed white lines. At higher magnification (Fig. 4B), clusters of infected cells are apparent; this clustering, together with the anatomical location close to, but not contiguous with, the ependyma, is consistent with both type C and type A stem cells. Nestin staining demonstrated the expected high frequency of neuronal progenitor cells in the SVZ (Fig. 4C), and a high magnification of a cluster of eGFP+ cells (Fig. 4D) revealed that the surrounding cellular processes contained this intermediate filament, consistent with type C and type A cells, which are nestin+ (Doetsch et al., 1997). BrDU costaining indicated that, in several cases, these virus-infected cells had recently divided (Fig. 4E,F, arrow). The nature of these cells was determined by staining for PSA-NCAM, a marker that is absent from type C cells but is present in type A cells (Doetsch, 2003). As shown in Figure 4G, several of the clustered virus-infected cells expressed PSA-NCAM, identifying them as type A cells (white notched arrows); however, many of the PSA-NCAM+ cells were uninfected (cyannotched arrow). Finally, our evaluation of BrDU incorporation also revealed infected (eGFP+) cells with a morphology characteristic of the type B stem cell lineage; representative examples are shown in Figure 4, H and I, with their cytosolic processes extending through the ependymal cell layer (apposing ependyma are shown as a white dashed line). In summary, these data suggest that CVB3 can infect primordial progenitor cells (type B) in the CNS, some of which have recently divided. Furthermore, the virus can be detected in progeny type A cells, which also show signs of cell division. However, as with the RMS and OB, only a minority of cells showed markers both of infection (eGFP) and of previous cell division (BrDU).
Figure 4.

CVB3 infects type A, as well as type B, stem cells. Transverse paraffin-embedded sections were deparaffinized and immunostained using antibodies against nestin, BrDU, or PSA-NCAM. The ependymal cell layer is represented by the dashed white line. A, Many clusters of infected cells (eGFP+) were observed within the subventricular zone. B, Higher magnification (A, cyan box) revealed the dense clusters of infected cells (notched arrows) morphologically similar to type A neuronal progenitor cells within the SVZ. C, As expected, nestin staining (red) was observed near the lateral ventricle and within the SVZ. D, Higher magnification (C, cyan box) demonstrated that these clusters of infected cells were nestin+, suggesting that these cells share immunological similarities with type A or type C neuronal progenitor cells. E, BrDU staining (red) indicated that many cells near the SVZ had recently divided. F, Higher magnification (E, cyan box) showed direct colocalization between infected and BrDU+ cells (arrows), indicating that these clusters of cells had recently divided. G, PSA-NCAM staining (red) allowed us to distinguish type A and type C neuronal progenitor cells in the SVZ. Clusters of infected cells (notched white arrows) near the SVZ were found to be PSA-NCAM+, which suggests that type A neuronal progenitor cells are susceptible to infection; uninfected clusters also were present (notched cyan arrow). H, Merged images revealed colocalization of nuclear BrDU+ staining (red) and infected cells (cytoplasmic eGFP) near the lateral ventricle. An infected cell (100× with an additional twofold computer-generated magnification) labeled with BrDU (notched arrow) is visible protruding through the ependymal cell layer into the ventricle, having morphological similarities to a type B cell. I, Two infected cells labeled with BrDU are shown deeper within the subventricular zone. A, C, E, 20× objective; B, 100× objective; D, F, H, I, 100× objective with an additional twofold computer-generated magnification; G, 20× objective with an additional threefold computer-generated magnification.
Many CVB-infected cells leave the SVZ, but these cells show no evidence of proliferation
Nestin+ cells are abundant around the SVZ, and the nestin signal extends in long processes that radiate from the SVZ (Figs. 4C, 5A); nuclei (Fig. 5A, blue) can be seen studding these long chains, which comprise migrating neuroblasts (see Fig. 7). The developmental status of the cells changes soon after they leave the immediate region of the SVZ; staining for neuron-specific class III β-tubulin, a marker of immature neurons (Moody et al., 1989), is absent from the cells within the SVZ but is abundant in cells lying outside this structure (Fig. 5B). Infected cells are readily detected both within the SVZ and in migrating cells, and proliferating (Ki67+) cells also are numerous (Fig. 5C). However, at higher magnification, clearly the Ki67 and eGFP signals are entirely separate (Fig. 5D); the addition of DAPI staining to identify cell nuclei (Fig. 5E) confirms this point and also shows that, in this region, (1) a large proportion, ∼30%, of the cells are infected (green cytosol, blue nuclei), (2) a similar proportion of cells are proliferating (pink nuclei) but are uninfected, and (3) a minority of cells are neither infected nor proliferating (blue nuclei, no cytosolic staining).
Figure 5.

Proliferation and infection are mutually exclusive in cells exiting the SVZ. Transverse paraffin-embedded sections from an infected brain 1 d after infection were immunostained using antibodies against nestin, β-tubulin, and Ki67. Dashed lines represent the ependymal cell layer of the lateral ventricle. A, Nestin staining (red) colocalized with the majority of infected (eGFP+) cells in the SVZ. B, The majority of β-tubulin expression (blue) was detected farther away from the SVZ, although a few infected cells were observed within the β-tubulin staining region. C, Infected cells regionally colocalized with proliferating cells expressing Ki67 (red) in the subventricular zone near the lateral ventricle. D, Higher magnification (C, cyan box) revealed infected cells adjacent to, yet distinct from, Ki67+ cells. Almost all Ki67+ cells were eGFP-, and nearly all eGFP+ cells were Ki67-. E, DAPI (blue) nuclear counterstain allowed visualization of all cells within the section and illustrated the lack of Ki67/eGFP colocalization. The nuclear localization of Ki67 antigen and cytosolic distribution of eGFP were evident after merging the red, blue, and green channels. A-C, 20× objective; D, E, 100.8× objective.
Figure 7.
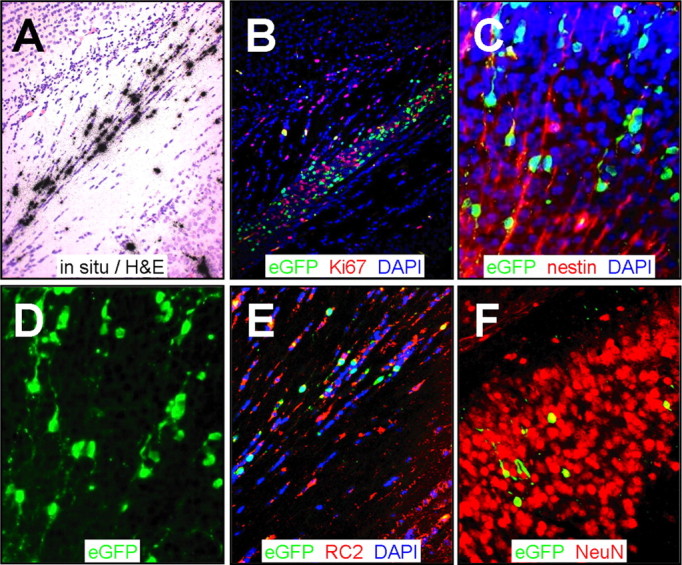
Virus-infected cells are arrayed as chains, contiguous with radial glia. Paraffin-embedded sections were obtained from the brain 2 d after infection and stained using antibodies against Ki67, nestin, and RC2. Viral infection was evaluated by in situ hybridization (CVB3 5′ untranslated region probe) or by viral protein expression (eGFP). A, In situ hybridization identified infected cells near the retrosplenial cortex in longitudinal arrays. B, Proliferating cells were identified in close proximity to infected cells (eGFP+) in this region. C, Sections stained for nestin and counterstained with DAPI (blue) revealed infected nestin+ cells that appeared to be migratory neuroblasts associated with radial glia. D, Single-channel (eGFP) image of C highlighted the chain-like distribution of infected cells with long extensions entering into the cortex, reminiscent of radial glial cells. E, RC2 staining (red) identified many infected radial glial cells contiguous with the retrosplenial cortex. F, Many infected NeuN+ cells were observed deeper within the cortex. A, B, 20× objective; C, D, 20× objective with an additional approximately threefold computer-generated magnification; E, F, 20× objective with an additional approximately two-fold computer-generated magnification.
Reduced numbers of proliferating (Ki67+) cells in the SVZ of CVB3-infected neonatal mice
Next, we quantitated the effect of CVB3 infection on the numbers of proliferating (Ki67+) cells in the CNS. We infected 1-d-old mice with eGFP-CVB3, and they were killed 1, 2, or 5 d later. Uninfected mice of the same ages were killed for comparative analyses. SVZ sections were stained to detect Ki67+ cells, and the fluorescent images revealed a severe depletion of Ki67+ cells by 5 d after infection (Fig. 6A). These images were captured, and the Ki67+ signals were quantitated using ImageJ software as described in Materials and Methods. As shown in Figure 6B, CVB3 infection resulted in a ∼75% reduction in the number of proliferating cells.
Figure 6.
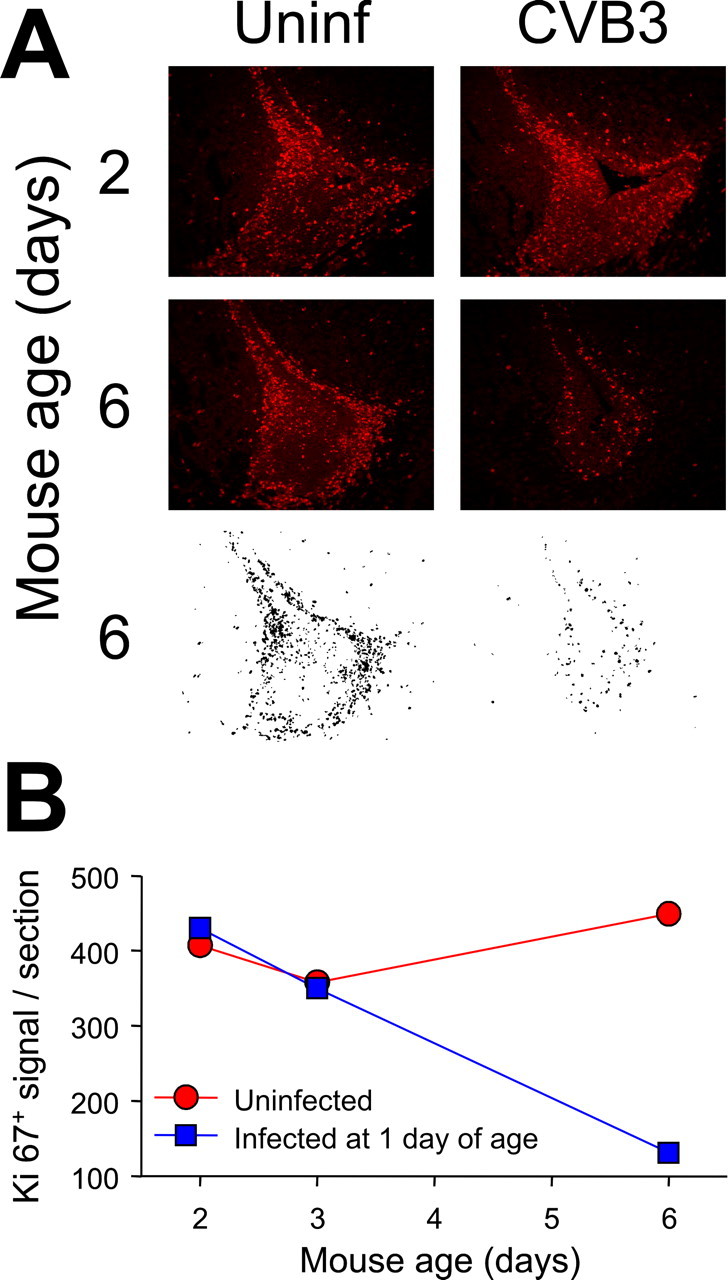
Quantitative analysis of Ki67+ cells in the SVZ. A, We infected 1-d-old pups with eGFP-CVB3 (2 × 106 pfu, i.c.), and they were killed 1, 2, or 5 d later. Uninfected pups of the same ages were evaluated for comparative purposes. Sections of the SVZ were stained with an antibody against Ki67 (red) and observed by fluorescence microscopy. To enumerate Ki67+ cells, black and white images were generated using ImageJ, as described in Materials and Methods; examples are shown for the 6-d-old mice. B, The number of Ki67+ cells per section are shown at each time point for uninfected mice (red) and infected mice (blue).
CVB3-infected cells migrate along radial glia associated with the SVZ
The cortex is generated during embryogenesis by the migration of neuronal progenitor cells, which form long chains of cells, tethered to radial glia that form a scaffolding for the migrating cells (Noctor et al., 2002). Such neurogenesis also has been observed in the adult cortex, and it is now hypothesized that radial glial cells may persist after birth and continue to give rise to new neurons (Alvarez-Buylla et al., 2001; Noctor et al., 2001). These chains were readily visualized in our neonatal animals (Fig. 5A), allowing us to investigate whether infected cells might move along this route, toward the cortex. Viral RNA was detected by in situ hybridization in longitudinal arrays of cells near the posterior SVZ (Fig. 7A) in a region containing many proliferating (Ki67+) cells (Fig. 7B). The migrating cells were nestin+ (Fig. 7C), and the chain-like orientation of infected cells was highlighted by eGFP expression (Fig. 7C,D). Staining with RC2, a monoclonal antibody specific for radial glia (Misson et al., 1988), identifies the radial scaffold, and the contiguity of both uninfected cells (DAPI-stained nuclei) and infected cells (eGFP+) with this scaffold is apparent (Fig. 7E). After reaching the cerebral cortex, the cells completed their differentiation and expressed the NeuN marker; in all cases, virus-infected cells also were NeuN+ (Fig. 7F, yellow cells). These data suggest that CVB3-infected neuronal precursor cells can, despite the infection, migrate along the radial glia, toward the developing cerebral cortex, and can complete their differentiation into mature neurons.
Infection of neuronal progenitors and stem cells in regions adjacent to the fourth ventricle
The source of viral RNA signal around the fourth ventricle also was analyzed. A near-midline sagittal section of this region (Fig. 8A,B) revealed scattered virus infection (eGFP+), as well as numerous neuronal progenitor cells (nestin+). At higher power, eGFP+ radial processes were clearly visible (Fig. 8C) and were distributed similarly to nestin (Fig. 8D). The eGFP signal was directly overlapping with nestin, as demonstrated by the yellow signal that is present in the composite figure (Fig. 8E); we conclude that rCVB infects neuronal progenitor cells in the area of the fourth ventricle. Ki67 staining of the periventricular region revealed numerous proliferating cells, as well as several eGFP+ cells in the ependymal layer (Fig. 8F). Some of these virus-infected cells also were BrDU+ (Fig. 8G,H), and some had the morphology of type B stem cells (Fig. 8I). These observations indicate that the patterns identified in the OB, SVZ, and RSC are maintained throughout the neonatal CNS: eGFP-CVB preferentially infects very early precursor cells but appears not to disturb the capacity of the cells to migrate and differentiate.
Figure 8.
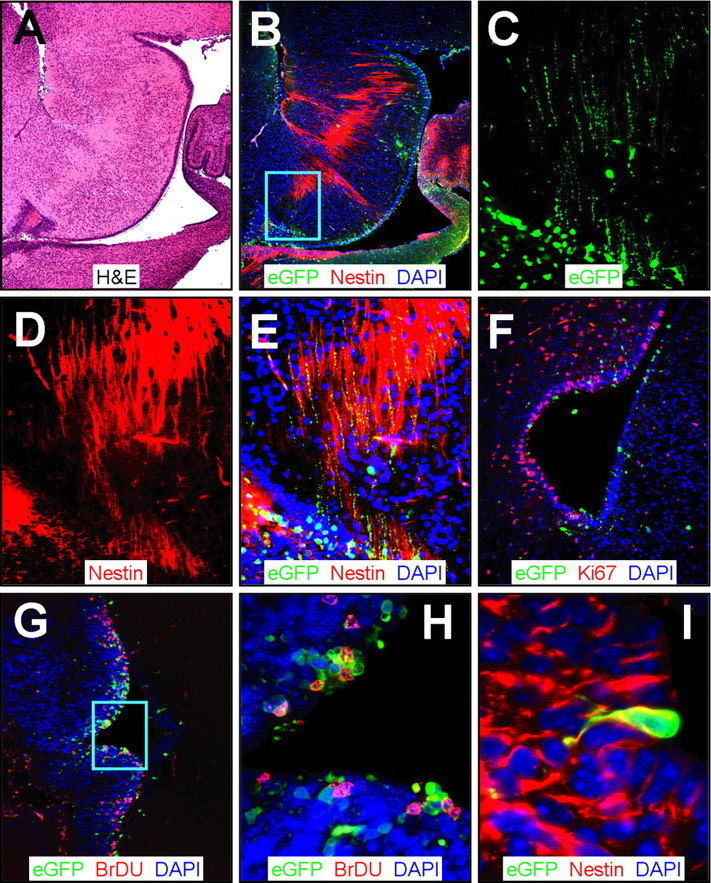
CVB3 infection adjacent to the fourth ventricle. Sagittal (A-E) and transverse (F-I) paraffin sections of the posterior brain of a newborn mouse at 1 d after infection were immunostained using antibodies against nestin, Ki67, and BrDU. A, Low-magnification H&E-stained sagittal section identified features of the cerebellum, the fourth ventricle, and the ependymal cell layer. B, Infected cells (eGFP+) were evident near the ependymal cell layer in the area below the fourth ventricle and in the region of the dorsal tegmental nucleus. Nestin+ staining (red) was observed stretching across the ependymal cell layer into the surrounding parenchyma. The cyan box represents the area expanded in C-E. C, Higher magnification revealed infected (eGFP+) cells with long cellular processes. D, Similarly, nestin staining demonstrated long extensions of neuronal progenitor cells found near the fourth ventricle. E, Direct colocalization of nestin and infected axonal processes was evident (yellow), indicating that neuronal progenitor cells in the fourth ventricle were susceptible to infection. F, Ki67 staining (red) showed many proliferating cells near the ependymal cell layer and infected regions of the fourth ventricle. G, Similarly, many BrDU cells (red) were found closely associated with infected cells near the ependymal cell layer. H, Higher magnification (G, cyan box) demonstrated direct colocalization of many infected cells with BrDU staining. I, Many nestin+ (red) infected cells protruded through the ependymal cell layer in the fourth ventricle, consistent with infected type B stem cells, as seen near the lateral ventricle (Fig. 4).
Discussion
Our previous studies of CVB3 infection of the neonatal CNS showed that several cell types expressed viral protein: type B stem cells, β-tubulin+ immature neurons, and NeuN+ mature neurons. We hypothesized that this might represent a progression, in which progenitor cells were infected early and harbored the infection, while continuing to migrate and differentiate into mature neurons. Herein, we confirm and extend those findings by examining viral infection and host cell status in several regions of the CNS that generate, contain, or are destinations for proliferating and/or migratory cells.
Several lines of evidence suggest that the status of the host cell plays a key role in determining the outcome of CVB infection. Recent studies from several laboratories, including our own, indicate that cellular activation and proliferation may be required for productive coxsackievirus infection in tissue culture cells (Feuer et al., 2002; Luo et al., 2002), and viral binding to target cells has been found to stimulate cellular activation via the ERK1/2 (extracellular signal-regulated kinase) pathway (presumably through receptor signaling), which may in turn provide a better cellular setting for viral replication (Luo et al., 2002). In vivo studies correlate with these findings. Our laboratory has shown that CVB3 undergoes a burst of replication in B cells trafficking through splenic germinal centers, which are sites of active B cell proliferation (Mena et al., 1999), and both coxsackievirus-induced myocarditis and productive infection of T cells are dependent on the expression of p56lck kinase, a signal transduction molecule involved in cellular activation (Liu et al., 2000). Thus, proliferating or highly activated cells appear to favor productive CVB infection, and we proposed (Feuer et al., 2003) that this preference may contribute to the extreme susceptibility of newborns to coxsackievirus infection and viral-induced pathology (Kaplan et al., 1983; Modlin, 1988).
Many cells in the developing CNS express high levels of the major CVB receptor protein mCAR (murine coxsackievirus-adenovirus receptor) (Honda et al., 2000; Hotta et al., 2003) and thus represent possible targets for infection. However, the cerebellum is a notable exception, and this may explain the absence of viral signal in this region (Fig. 1A), despite the presence of numerous proliferating cells (Fig. 1B). We hypothesize that, although proliferating neuronal progenitor cells are found within the cerebellum, the lack of mCAR expression prevents infection of these cells. In contrast to the cerebellum, the SVZ, RMS, OB, cortex, and fourth ventricle are highly susceptible to CVB3 infection (Fig. 1). All of these areas represent sites of neonatal neurogenesis; neuronal progenitor cells (nestin+, β-tubulin-) and immature neurons (nestin+, β-tubulin+) were readily detected, as expected. Many infected neuronal precursors were identified within the RMS (Fig. 2F). Infected mature neurons also were relatively abundant, as shown by our analyses of the OB, in which infected mitral neurons were identified (Fig. 3C). These cells, although mature neurons, do not express the NeuN marker (Mullen et al., 1992) but can be identified by their distinctive morphology, which appears to be maintained despite CVB3 infection (Fig. 3D). NeuN staining revealed a very distinct layer of NeuN+ cells (Fig. 3B), and, in the OB, this signal was derived mainly from grs; cells in grd were NeuN- (Fig. 3C). These findings may reflect those reported in another study, in which cells in the OB granule layer also were subdivided into two populations based on their ability to become dopaminergic neurons (Baker et al., 2001). Cells in the deep layer (grd) did not express dopaminergic markers. In contrast, cells in grs were dopaminergic and were thought to rapidly cross the mitral and external plexiform layers, to reach their final destination in the glomerular layer as periglomerular neurons. This population may correspond to the NeuN+ cells that we see in grs, and one would predict that resulting periglomerular neurons might also be NeuN+. However, in our study, we failed to identify NeuN+ neurons within the glomerular layer, as has been reported previously in rats carrying lesions in the RMS (Fukushima et al., 2002). Several possible scenarios may explain the absence of NeuN+ cells in the periglomerular layer: (1) periglomerular progenitor neurons may be more sensitive to the apoptotic effects of infection compared with granule cells, and these cells may die before reaching the periglomerular cell layer; or (2) infection of neuronal progenitor cells may influence the differentiation of these cells, thereby causing all migratory neuroblasts to differentiate preferentially into granule cells, which reside in grd.
To determine whether CVB3 targets proliferating cells in the neonatal CNS, we used antibodies against Ki67 and BrDU to visualize the complete RMS, as well as other regions of the brain. Proliferating (Ki67+ or BrDU+) cells were abundant and overlapped with sites of infection, as expected. The tissue surrounding the cerebral ventricles contains four main cell types. Immediately apposed to the ventricular space is a layer of ciliated epithelium, comprising ependymal cells. These are surrounded by the SVZ, which contains the remaining three cell types, all of which are actively proliferating to varying degrees (Doetsch et al., 1999; Temple, 2001). Type B stem cells, many of which have a stretched morphology, contact the ventricle with cytoplasmic processes that penetrate the ependymal layer. These cells give rise to type C stem cells which, in turn, rapidly divide to generate type A cells, or neuroblasts (Doetsch, 2003). We have shown previously that type B cells can be infected with CVB3 (Feuer et al., 2003); here, we confirm this finding, and we show also that these cells express markers of cell division in both the SVZ (Fig. 4H,I) and the equivalent region adjacent to the fourth ventricle (Fig. 8G-I). In addition, we observed clusters of infected cells within the SVZ that lay close to, but not contiguous with, the lateral ventricle (Fig. 4A,B). Many of these infected cells also were BrDU+ (Fig. 4E,F), consistent with their highly proliferative phenotype, and some of them were PSA-NCAM+ (Fig. 4G), indicative of type A stem cells. The susceptibility of both type B and type A cells to CVB infection renders it highly probable that the intermediary type C cells also may be infected. Type C cells cannot be positively identified using marker proteins, but eGFP+ PSA-NCAM- cells are present (Fig. 4G) and may represent this cell type. Thus, the proliferating cells that represent the roots of the neurogenic tree appear to be highly susceptible to CVB3 infection, but, to our surprise, these are the only CNS cells in which infection and proliferation appear to readily coexist; contrary to our expectations, in most regions of the CNS, we found it difficult to identify cells that showed signs of both infection (eGFP) and proliferation (Ki67 or BrDU). The low frequency of infected proliferating cells was exemplified by the data in Figure 5, which revealed that cells exiting the SVZ can be eGFP+ or Ki67+ but not both; infected (GFP+) cells exiting the SVZ can no longer proliferate, as judged by their lack of Ki67 expression. The consequences of CVB3 infection on precursor cell abundance are shown quantitatively in Figure 6, which demonstrates that the number of Ki67+ cells in the SVZ of CVB3-infected neonatal mice was substantially reduced (by ∼75%) by 5 d after infection.
Despite the apparent inhibitory effect on cell proliferation, CVB3 infection appears to disrupt neither migration nor maturation of these neuronal precursors. Virus-infected cells were abundant in the RMS and retained the morphological characteristics of migrating cells (nestin+, with leading and trailing axonal processes) (Fig. 2C, D, F). Furthermore, infected cells with similar migratory morphology were plentiful in the region leading to the cerebral cortex (Fig. 7C,D), and these cells were closely associated with radial glia (Fig. 7E), strongly suggesting that they use the normal physiological scaffold to guide them to their ultimate destination.
Our working model is presented in diagrammatic form in Figure 9. We propose that CVB3 preferentially infects dividing SVZ type B cells without necessarily interrupting cell division. Consequently, infected type C cells are produced, and these, in turn, generate infected type A progeny neuroblasts, which can exit the SVZ. However, in contrast to their uninfected counterparts, and presumably as a consequence of viral infection, these infected neuronal precursors can no longer proliferate (and, therefore, are Ki67-) (Fig. 9, dark gray circles). Relevant to this idea, others have shown that CVB3 infection of HeLa cells can arrest the cell cycle at the G1/S phase (Luo et al., 2003). The immune system is involved in CVB3-induced myocardial disease (Gebhard et al., 1998), and we considered its possible involvement in the depletion of progenitor cells reported in this paper. However, we think it unlikely that the adaptive immune system is involved because, unlike myocarditis, the CNS events are observed at very early times after infection (1-2 d), when the adaptive immune response is very weak, and the effects observed in RAG (recombination-activating gene) knock-out mice are indistinguishable from those described herein (data not shown). Our data show that these proliferation-defective infected cells can still migrate along the RMS or radial glia and also retain their capacity to differentiate into mature neurons (black circles). Figure 9 also makes clear one prediction of our model: the combination of diminished cellular proliferation after exiting the SVZ and virus-induced lysis of mature cells implies that the final destinations of those infected progenitor cells (e.g., the OB or cerebral cortex) should be depleted of mature neurons, as we observed in the glomerular layer (Fig. 3) and as has been reported in a transgenic model of echovirus infection (Hughes et al., 2003). We are currently investigating the magnitude of this depletion throughout the neonatal CNS; if extensive, it could contribute to the neurodevelopmental deficits that have been identified in survivors of fetal CVB infections (Euscher et al., 2001; Genen et al., 2004).
Figure 9.

Model for CVB3 dissemination in the neonatal CNS. A graphical representation of CVB3 infection of the neonatal CNS. The SVZ of uninfected mice (left column) contains three populations of neuronal progenitor cells (white circles). Type B cells give rise to type C cells, which eventually produce migratory neuroblasts (type A cells). All progenitor cells are known to proliferate extensively. Neuronal progenitor cells migrate to the OB or cortex through the RMS or radialglia (RG) and differentiate into mature neurons (hatched circles). In an infected mouse (right column), CVB3 infects some of the type B and A progenitor cells, and type C cells also may be susceptible to infection. CVB3 inhibits the proliferation of neuronal progenitor cells exiting the SVZ (dark gray circles) as judged by Ki67 expression (quantitated in Fig. 6); these nondividing progenitor cells nevertheless retained their ability to migrate and to differentiate into infected mature neurons (black circles). However, many of these infected neurons may eventually die as a result of caspase-3-mediated apoptosis, and this, together with the reduction in Ki67+ cells exiting the SVZ, may lead to a marked reduction in mature neurons (right side of infected mouse column) and thereby to the neurodevelopmental defects associated with CVB infections.
Footnotes
This work was supported by National Institutes of Health Grant R-01 AI-42314 (to J.L.W.) and National Multiple Sclerosis Society advanced postdoctoral fellowship Grant FA 1551-A-1 (to R.F.). This is manuscript number 16571-NP from Scripps Research Institute. We are grateful to Annette Lord for excellent secretarial support and to the histology laboratory of The Scripps Research Institute for skilled technical assistance.
Correspondence should be addressed to J. Lindsay Whitton, Department of Neuropharmacology, CVN-9, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail: lwhitton@scripps.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252434-11$15.00/0
References
- Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD (2001) A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci 2: 287-293. [DOI] [PubMed] [Google Scholar]
- Baker H, Liu N, Chun HS, Saino S, Berlin R, Volpe B, Son JH (2001) Phenotypic differentiation during migration of dopaminergic progenitor cells to the olfactory bulb. J Neurosci 21: 8505-8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley AJ, Isaacs D, Dwyer DE, Gilbert GL (1998) A cluster of cases of neonatal coxsackievirus B meningitis and myocarditis. J Paediatr Child Health 34: 196-198. [DOI] [PubMed] [Google Scholar]
- Doetsch F (2003) A niche for adult neural stem cells. Curr Opin Genet Dev 13: 543-550. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17: 5046-5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97: 703-716. [DOI] [PubMed] [Google Scholar]
- Euscher E, Davis J, Holzman I, Nuovo GJ (2001) Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol 98: 1019-1026. [DOI] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Slifka MK, Whitton JL (2002) Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 76: 4430-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL (2003) Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol 163: 1379-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL (2004) Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med Microbiol Immunol (Berl) 193: 83-90. [DOI] [PubMed] [Google Scholar]
- Fukushima N, Yokouchi K, Kawagishi K, Moriizumi T (2002) Differential neurogenesis and gliogenesis by local and migrating neural stem cells in the olfactory bulb. Neurosci Res 44: 467-473. [DOI] [PubMed] [Google Scholar]
- Gear JH, Measroch V (1973) Coxsackievirus infections of the newborn. Prog Med Virol 15: 42-62. [PubMed] [Google Scholar]
- Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL (1998) Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 153: 417-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genen L, Nuovo GJ, Krilov L, Davis JM (2004) Correlation of in situ detection of infectious agents in the placenta with neonatal outcome. J Pediatr 144: 316-320. [DOI] [PubMed] [Google Scholar]
- Henke A, Launhardt H, Klement K, Stelzner A, Zell R, Munder T (2000) Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J Virol 74: 4284-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R (2000) The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res Mol Brain Res 77: 19-28. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Honda T, Naito M, Kuwano R (2003) Developmental distribution of coxsackie virus and adenovirus receptor localized in the nervous system. Brain Res Dev Brain Res 143: 1-13. [DOI] [PubMed] [Google Scholar]
- Hsueh C, Jung SM, Shih SR, Kuo TT, Shieh WJ, Zaki S, Lin TY, Chang LY, Ning HC, Yen DC (2000) Acute encephalomyelitis during an outbreak of enterovirus type 71 infection in Taiwan: report of an autopsy case with pathologic, immunofluorescence, and molecular studies. Mod Pathol 13: 1200-1205. [DOI] [PubMed] [Google Scholar]
- Hughes SA, Thaker HM, Racaniello VR (2003) Transgenic mouse model for echovirus myocarditis and paralysis. Proc Natl Acad Sci USA 100: 15906-15911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Klein SW, McPhee J, Harper RG (1983) Group B coxsackievirus infections in infants younger than three months of age: a serious childhood illness. Rev Infect Dis 5: 1019-1032. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD (1990) CNS stem cells express a new class of intermediate filament protein. Cell 60: 585-595. [DOI] [PubMed] [Google Scholar]
- Liu P, Aitken K, Kong YY, Opavsky MA, Martino T, Dawood F, Wen WH, Kozieradzki I, Bachmaier K, Straus D, Mak TW, Penninger JM (2000) The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat Med 6: 429-434. [DOI] [PubMed] [Google Scholar]
- Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM (2002) Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J Virol 76: 3365-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Zhang J, Dastvan F, Yanagawa B, Reidy MA, Zhang HM, Yang D, Wilson JE, McManus BM (2003) Ubiquitin-dependent proteolysis of cyclin D1 is associated with coxsackievirus-induced cell growth arrest. J Virol 77: 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luskin MB, Coskun V (2002) The progenitor cells of the embryonic telencephalon and the neonatal anterior subventricular zone differentially regulate their cell cycle. Chem Senses 27: 577-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DE, Hall PA (2000) The biochemical characterization of the DNA binding activity of pKi67. J Pathol 191: 286-298. [DOI] [PubMed] [Google Scholar]
- Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard JR, Whitton JL (1999) The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol 155: 1205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G (2004) Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol 469: 311-324. [DOI] [PubMed] [Google Scholar]
- Misson JP, Edwards MA, Yamamoto M, Caviness Jr VS (1988) Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res 44: 95-108. [DOI] [PubMed] [Google Scholar]
- Modlin JF (1988) Perinatal echovirus and group B coxsackievirus infections. Clin Perinatol 15: 233-246. [PubMed] [Google Scholar]
- Moody SA, Quigg MS, Frankfurter A (1989) Development of the peripheral trigeminal system in the chick revealed by an isotype-specific anti-beta-tubulin monoclonal antibody. J Comp Neurol 279: 567-580. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201-211. [DOI] [PubMed] [Google Scholar]
- Murase S, Horwitz AF (2002) Deleted in colorectal carcinoma and differentially expressed integrins mediate the directional migration of neural precursors in the rostral migratory stream. J Neurosci 22: 3568-3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR (2001) Neurons derived from radial glial cells establish radial units in neocortex. Nature 409: 714-720. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR (2002) Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci 22: 3161-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzan KR (1985) Viral meningitis. Med Clin North Am 69: 399-413. [DOI] [PubMed] [Google Scholar]
- Sauerbrei A, Gluck B, Jung K, Bittrich H, Wutzler P (2000) Congenital skin lesions caused by intrauterine infection with coxsackievirus B3. Infection 28: 326-328. [DOI] [PubMed] [Google Scholar]
- Temple S (2001) The development of neural stem cells. Nature 414: 112-117. [DOI] [PubMed] [Google Scholar]
- Wynford-Thomas D, Williams ED (1986) Use of bromodeoxyuridine for cell kinetic studies in intact animals. Cell Tissue Kinet 19: 179-182. [DOI] [PubMed] [Google Scholar]