

Abstract
The sequential enzymatic actions of β-APP cleaving enzyme 1 (BACE1), presenilins (PS), and other proteins of the γ-secretase complex liberate β-amyloid (Aβ) peptides from larger integral membrane proteins, termed β-amyloid precursor proteins (APPs). Relatively little is known about the normal function(s) of APP or the neuronal compartment(s) in which APP undergoes proteolytic processing. Recent studies have been interpreted as consistent with the idea that APP serves as a kinesin-1 cargo receptor and that PS and BACE1 are associated with the APP-resident membranous cargos that undergo rapid axonal transport. In this report, derived from a collaboration among several independent laboratories, we examined the potential associations of APP and kinesin-1 using glutathione S-transferase pull-down and coimmunoprecipitation assays. In addition, we assessed the trafficking of membrane proteins in the sciatic nerves of transgenic mice with heterozygous or homozygous deletions of APP. In contrast to previous reports, we were unable to find evidence for direct interactions between APP and kinesin-1. Furthermore, the transport of kinesin-1 and tyrosine kinase receptors, previously reported to require APP, was unchanged in axons of APP-deficient mice. Finally, we show that two components of the APP proteolytic machinery, i.e., PS1 and BACE1, are not cotransported with APP in the sciatic nerves of mice. These findings suggest that the hypothesis that APP serves as a kinesin-1 receptor and that the proteolytic processing machinery responsible for generating Aβ is transported in the same vesicular compartment in axons of peripheral nerves requires revision.
Keywords: APP, presenilin, kinesin-1, axonal transport, Alzheimer's disease, proteolysis
Introduction
β-amyloid (Aβ) peptides, the principal components of parenchymal amyloid deposits in Alzheimer's disease (AD), are derived from membrane glycoproteins known as amyloid precursor proteins (APPs) (Price and Sisodia, 1998; Selkoe, 2001). Aβ peptides are liberated from APP by the sequential activities of a β-APP cleaving enzyme 1 (BACE1) aspartyl protease and a proteolytic complex, termed “γ-secretase” (for review see, Selkoe, 2001) consisting of presenilin (PS), nicastrin, PEN-2 (presenilin enhancer), and APH-1 (anterior pharynx defective) (De Strooper, 2003).
APP is transported in the fast component of anterograde transport in both peripheral and central axons (Koo et al., 1990; Sisodia et al., 1993; Buxbaum et al., 1998; Kaether et al., 2000), and membrane-tethered APP derivatives accumulate at terminal fields in the CNS (Buxbaum et al., 1998). Several lines of evidence indicate that Aβ is released at nerve terminals in the CNS (Lazarov et al., 2002; Sheng et al., 2002), but the neuronal compartments in which γ-secretase processing occurs have not been identified. The findings that APP interacts with the tandem repeat (TR) domains of kinesin-1 light chain (KLC) and that anterograde transport of APP is impaired in the sciatic nerves of KLC1-deficient mice, led to the hypothesis that APP is a kinesin-1 cargo receptor (Kamal et al., 2000). More recently, it has been reported that PS1 is rapidly transported in a membranous axonal organelle that also contains BACE1 and APP (Kamal et al., 2001). Aβ peptides seemed to be present in sciatic nerves, and the accumulation of these peptides at the proximal stump of ligated nerve led these authors to suggest that the concerted action of BACE1 and PS-γ secretase could mediate proteolysis of APP within axonally transported vesicles during transit along peripheral nerves (Kamal et al., 2001). In view of the substantial implications of these proposals for understanding the functions of APP and the transport of the secretases in both controls and cases of AD, we sought to reexamine these issues.
This report brings together data from six laboratories that examine the hypothesis that APP serves as a kinesin-1 cargo receptor and that the proteolytic machinery responsible for generating Aβ from APP are cotransported within APP-containing cargos. We show that recombinant fusion proteins containing various regions of either KLC1 or the cytoplasmic domains of APP can interact, but in a manner that is inconsistent with a specific association. Moreover, endogenous APP did not cofractionate with kinesin-1 in membrane fractions or detergent homogenates prepared from mouse brain. Significantly, we demonstrate that axonal transport of kinesin-1 and tyrosine kinase (Trk) receptors are not altered in the absence of APP in APP-/- mice. These studies do not support the hypothesis that APP is a kinesin-1 cargo receptor. Finally, biochemical and immunocytochemical assays show that neither PS1 nor BACE1 is cotransported with APP in sciatic nerves of mice. These observations, coupled with our inability to detect BACE1 and Aβ in sciatic nerves, suggests that in the PNS it is unlikely that there exists a coordinated axonal transport of the proteolytic processing machinery responsible for generating Aβ peptides.
Materials and Methods
Cloning of green fluorescent protein and glutathione S-transferase fusion proteins. Green fluorescent protein (GFP) fusion proteins were generated by subcloning of the enhanced GFP (EGFP) cDNA (Invitrogen, Gaithersburg, MD) in frame with the cDNA for human APP695 constructs into pcDNA3.1. Constructs included APP intracellular domain (AID; amino acids 649-695) and mutant AID missing the basolateral sorting signal (AIDΔBaSS; amino acids 652-656), the PEER motif domain (AIDΔPEER; amino acids 669-679), or the NPTY motif domain (AIDΔNPTY; amino acids 680-691). KLC1 cDNA (a kind gift from Dr. Lachman) and KLC1 deletion mutants were cloned in frame into pGEX-4T-2 (Amersham Biosciences, Arlington Heights, IL) glutathione S-transferase (GST-KLC). Deletion constructs included the following: C-terminal deletions (amino acids 1-500, KLC-CC/TPR; amino acids 1-363, KLC-ΔCT), the C terminus with all tandem repeats (amino acids 199-444; KLC-TR), the N terminus with the coiled coil domain (amino acids 1-181; KLC-CC), and an N-terminal deletion (amino acids 362-567; KLC-ΔNT). GST-APP/amyloid precursor-like protein (APLP) fusion proteins were generated by cloning the intracellular domain of APP (AID; amino acids 649-695), APLP1 (ALID1; amino acids 604-650), and APLP2 (ALID2; amino acids 717-763) in frame into pGEX4T-1 (Amersham Biosciences).
Expression and purification of GST fusion proteins in Escherichia coli. Expression of GST and GST fusion proteins was performed in 200 ml of BL21 E. coli cultures (DE3 strain; Novagen, Madison, WI) by addition of 1 mm isopropyl-β-d-thiogalactopyranoside at OD 0.3. After 2 h of induction, bacteria were lysed by incubating for 30 min with 1 mg/ml lysozyme plus 1 mm PMSF in PBS, pH 7.4, and sonicated with a Branson Sonifier 250 (24 times 30 pulses; Branson Ultrasonic, Danbury, CT). Triton X-100 was added to a final concentration of 1%, and cell lysates were centrifuged for 30 min at 20,000 × g at 4°C. Supernatant containing soluble GST or GST fusion proteins was incubated with 0.5 ml of glutathione-Sepharose beads (Amersham Biosciences) for 1 h at room temperature. Insoluble GST fusion proteins were solubilized in 6 ml of 6 m guanidinium HCl, 100 mm Tris, pH 8, and 50 mm DTT and sonicated again (two times 20 s on ice). Solubilized GST fusion proteins were incubated overnight at 4°C on a magnetic stirrer. Refolding of GST fusion proteins was performed by pulse renaturation in 1 m arginine, 100 mm Tris, pH 8, 5 mm DTT, and 2 mm EDTA starting with 0.1 mg/ml protein and a final concentration after the last pulse of <1.5 m guanidinium HCl. The refolded GST fusion proteins were dialyzed in PBS, pH 7.4, at 4°C and centrifuged at 40,000 × g for 30 min. The supernatant was incubated with 1 ml 50% glutathione-Sepharose beads for 1 h at room temperature. Loaded beads were washed three times with PBS, pH 7.4, and stored at 4°C as a 50% slurry. Protein concentration was determined using the Bio-Rad (Hercules, CA) protein assay.
Coupled in vitro transcription-translation. The in vitro transcriptiontranslation was performed using in vitro TNT 7 Quick Coupled Transcription-Translation System (Promega, Madison, WI) with 1 μg of pCDNA3.1-GFP, -GFP-AID, -GFP-AIDΔBaSS, -GFP-AIDΔPEER, or -GFP-AIDΔNPTY, GFP-ALID1, GFP-ALID2, Fe65, Numb, and PAT1a DNA with 250 μCi of [35S]methionine for labeling according to the manufacturer's instructions.
GST binding assay. An aliquot (8 μl) of in vitro-translated GFP or GFP-AID fusion proteins was incubated with 20 μl of glutathione-Sepharose beads (Amersham Biosciences) coupled with GST or GST fusion proteins for 1 h at room temperature in 750 μl of buffer H [50 mm Tris, pH 6.8, 50 mm KCl, 100 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, 1% (w/v) BSA, 0.1% (w/v) Triton X-100, 5 mm DTT]. Protein-bound beads were washed three times in buffer H, and bound proteins were analyzed by SDS-PAGE (12% Tris-glycine gels). Gels were subsequently dried and exposed for autoradiography (MS films; Amersham Biosciences) overnight.
Antibodies, immunoprecipitation, and Western blot analyses. For Western blot or immunoprecipitation-Western blot, the following antibodies were used as described previously: anti-APP N-terminus monoclonal antibody (mAb) 22C11 (Weidemann et al., 1989; Hilbich et al., 1993), anti-APP C-terminal polyclonal serum [CT15 (Sisodia et al., 1993), AB5352; Chemicon, Temecula, CA]; JF, a rabbit polyclonal serum provided by Joachim Herz (University of Texas Southwestern, Dallas, TX), mouse anti-α-tubulin (Sigma, St. Louis, MO), anti-β-tubulin (TUB2.1; Sigma), anti-PP2A (sc-6110; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-JIP3/Sunday Driver (SYD) SN1 serum (a gift from L. S. Goldstein, University of California San Diego, La Jolla, CA), antisynapsin I (MCA1224G; Serotec, Indianapolis, IN), rabbit anti-mouse PS1-N-terminal fragments (PS1-NTF) (Thinakaran et al., 1997), antikinesin light chain antibodies [63-90 (Stenoien and Brady, 1997)], L1, L2, and KLC-ALL (Pfister et al., 1989), anti-kinesin heavy chain H2 and SVK4 (Brady et al., 1990), anti-dynein (mAb 1618; Chemicon), antisynaptophysin (P38, SY38 clone; Boehringer Mannheim, Mannheim, Germany), H-93 (Santa Cruz Biotechnology), anti-low-molecular-weight neurofilament (anti-NFL) (Tu et al., 1995), anti-Trk (C14, s.c.-11; Santa Cruz Biotechnology), and anti-BACE1 antibodies [gifts from Drs. Seidah (Marcinkiewicz and Seidah, 2000), Xu (Shi et al., 2003), and Wong (Cai et al., 2001)]. For Aβ immunoprecipitation-Western blot, 26D6 mAb raised against aa 1-16 of Aβ was used (Lamb et al., 1999). In Figure 3A, full-length APP was detected in the sciatic nerve by immunoprecipitation using rabbit anti-APP C-terminal 5685 antibody, followed by Western blot analysis using goat anti-APP N′-terminal Karen antibody (Lee et al., 2005). The following antibodies were used for immunohistochemistry: CT15, 6E10 mAb raised against aa 1-17 of human Aβ, and affinity-purified rabbit anti-mouse PS1-NTF (Kim et al., 2001). PS1-NTF glutathione S-transferase fusion protein was used as a competition peptide for antibody-specificity assay (Kim et al., 2000). Nerve extracts were subject to Tris-glycine or Tris-tricine SDS-PAGE. Proteins were detected as described previously (Kim et al., 2000). Immunoprecipitation of Aβ and APP fragments from nerve extracts was performed as described previously (Kim et al., 2001). Briefly, protein extracts were suspended with an equal volume of immunoprecipitation (IP) buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholic acid, 0.5% NP-40) in the presence of a protease inhibitor mixture (Sigma) and 100 mm PMSF. The samples were boiled for 3 min and precleared with immobilized protein A-agarose (Pierce, Rockford, IL) for 30 min at 4°C. Samples were then incubated with antibodies overnight at 4°C. Complexes were collected using immobilized protein A- or G-agarose and washed with IP buffer for 30 min. Complexes were then loaded on 1 m sucrose gradient and centrifuged at 9000 × g, and the pellets were resuspended in sample buffer and boiled for 5 min before running. Stably transfected N2a cells used as positive controls in these experiments have been described previously (Thinakaran et al., 1996). Each one of the presented experiments was repeated at least three times.
Figure 3.

Expression and anterograde axonal transport of kinesin-1 and Trk receptor are APP independent. A, Protein extracts of sciatic nerves from wild-type mice (APP WT), mice with either homozygous or heterozygous APP depletion (Hm APP KO and Ht APP KO, respectively), and transgenic mice harboring FAD-linked APPswe (Tg2576 mice). Immunoblots for KHC, dynein, synaptophysin (Syp), neurofilament (NF), and β-tubulin show no differences at steady-state levels between the different genotypes, indicating that fast, slow, anterograde, and retrograde axonal transport are not affected by APP expression. B, Protein extracts prepared from ligated sciatic nerve of Ht APP KO, Hm APP KO, and APP WT mice. APP accumulation is evident in the proximal ligature of sciatic nerve of APP WT mice (22C11 panel, lane 8); reduced accumulation is observed in the proximal ligature of Ht APP KO mouse sciatic nerve (22C11 panel, lane 5). APP expression cannot be detected in the sciatic nerve of APP KO mice (22C11 panel, lanes 1-3). The accumulation level of Trk receptor (Trk panel), kinesin heavy chain (H2 antibodies, KHC panel), kinesin light chain (63-90 antibodies, KLC panel), and PS1 (PS1-NTF antibodies, PS1-NTF panel) at the proximal stump of the ligation site is comparable in sciatic nerve of APP-ablated and wild-type nontransgenic mice (compare lanes 2, 5, 8). I, Intact sciatic nerve; P, proximal stump of ligated sciatic nerve; D, distal stump of ligated sciatic nerve. C, Kinesin-1 expression in longitudinal sections of ligated sciatic nerve as detected by immunolabeling using anti-KHC H2 antibodies. Note kinesin-1 accumulation toward the proximal stump of the ligature.
Immunohistochemistry. Mouse sciatic nerves were frozen in dry icecold isopropanol and embedded in Tissue-Tek optimal cutting temperature compound (Sakura, Tokyo, Japan). Cryostat sections (10 μm) were fixed with 3.7% paraformaldehyde solution for 30 min at room temperature. Sections were washed and then blocked and permeabilized with 0.25% Triton X-100 -5% donkey serum in Tris buffer solution for 2 h at room temperature. Sections were incubated with primary antibodies for 2 h at room temperature, washed, and incubated with FITC or cyanine 3-conjugated secondary antibodies for 1 h. Sections were washed and mounted with glycerol-based mounting medium (polyvinyl alcohol mounting medium with antifading 1,4-diazabicyclo-[2.2.2]octane; Sigma). For specificity verification, αPS1-NTF antibodies were incubated with PS1-NTF glutathione S-transferase fusion protein for 2 h at 4°C before their introduction to cryostat sections. Images were taken using an Olympus (Melville, NY) Fluoview confocal laser scanning microscope equipped with Fluoview 2.1 software.
Transgenic mice. The background of wild-type nontransgenic mice is [C3H/HeJ × C57BL/6J F3] × C57BL/6J n1. Mice expressing a chimeric mousehuman APP695 harboring a human Aβ domain and mutations (K595N, M596L) linked to Swedish familial Alzheimer's disease (FAD) pedigrees (APPswe) have been described previously (Borchelt et al., 1996, 1997; Lee et al., 1997). The background strains for APPswe are [C3H/HeJ × C57BL/6J F3] × C57BL/6J n1. Tg2576 mice have been described previously (Hsiao et al., 1996). BACE1-deficient mice have been described previously (Cai et al., 2001). APP-deficient (KO) mice (Zheng et al., 1995) were obtained from Dr. Hui Zheng (Baylor Medical School, Houston, TX). Mice used in this study were 5 months of age.
Sciatic nerve ligation. Mice were anesthetized using a mixture of ketamine (75 mg/kg) and xylazine (4 mg/kg). The skin on the lateral surface of the right thigh was incised, and a section was made directly through the biceps femoris muscle. The sciatic nerve was exposed and doubly ligated using 4-0 silk sutures (Ethicon, New Brunswick, NJ). The skin was clipped with wound clips. Six or 24 h later, nerve segments (up to 0.5 cm) proximal and distal to the ligature were collected and frozen on dry ice. The contralateral nerve was kept intact. Corresponding contralateral nerve and cortex tissue were collected as well.
Sciatic nerve-crush injury. Mice were deeply anesthetized using isofluorane, and the left sciatic nerve was exposed and crushed for 20 s by pressing the nerve between Dumont blunt-tipped jeweler's forceps. The crush site was ∼2 cm distal to the L4/L5 articulation. Muscle was sutured using 5-0 polysorb suture (Ethicon), and the skin was clipped. Twentyfour hours later, sciatic nerve segments proximal and distal to the crush, as well as intact contralateral sciatic nerve segments, were dissected and frozen on dry ice.
Protein extraction from nerve. Sciatic nerve segments were homogenized in extraction buffer [50 mm Tris, pH 7.2, 150 mm NaCl, 5 mm EDTA, protease inhibitor mixture (Sigma), and 100 mm PMSF] using a glass-made microhomogenizer. After the addition of 1% SDS, the homogenate was centrifuged for 10 min. The protein concentration of the supernatant was determined using BCA protein assay (Pierce), and extract samples of equal amounts of proteins were analyzed by Western blot (see above).
Vesicle fractionation. Vesicle fractions were performed as described previously (Morfini et al., 2001, 2002) using adult mouse brains. Brains were homogenized in 10 ml of buffer [300 mm sucrose, 10 mm HEPES, pH 7.4, 5 mm EDTA, and 2% mammalian protease inhibitor mixture (Sigma)] and then centrifuged at 12,500 × gmax to eliminate cell debris, nuclei, and most mitochondria. Pellets from subsequent centrifugation steps generated three vesicle fractions: V0 (39,800 gmax), V1 (120,000 gmax), and V2 (260,000 gmax). The final supernatant was termed cytosol. For immunoblots, equal amounts of protein from V0, V1, V2, and cytosol fractions were used.
Immunoprecipitation of APP and kinesin-1 from mouse brain. Immunoprecipitations were performed as described by Kamal et al. (2000) and Rahman et al. (1999). Mouse brain was homogenized in 3 ml of NP-40 homogenization buffer (NB; 1% NP-40, 150 mm NaCl, 50 mm Tris, pH 8.0). Brain lysate was spun at 35,000 rpm for 10 min (TLA100.3 rotor; 66,000 × gmax; Beckman Coulter, Fullerton, CA), and the supernatant was collected. Protein A-Sepharose beads (Bio-Rad) were preblocked with 5% bovine serum albumin in NB for 1 h at 4°C and washed with NB before use. One hundred microliters of brain extract were brought to 800 μl with NB and precleared by incubating with 20 μl of protein A-Sepharose beads (Bio-Rad) for 1 h at 4°C. The solution was spun at 3000 rpm for 2 min in a microcentrifuge, and the supernatant was collected. The precleared brain lysate was then incubated with 5 μg each of affinity-purified monoclonal antibodies [SUK4, H2, 63-90, L1, L2, 22C11, normal mouse or rabbit IgG (Jackson ImmunoResearch, West Grove, PA)] or 5 μl of polyclonal anti-APP C terminus (AB5352; Chemicon) or antibodies directed against the last 13 residues of APP (JH; a generous gift from J. Herz, University of Texas Southwestern Medical Center, Dallas, TX). Lysates were incubated with rotation for 2 h at 4°C before adding 20 μl of protein A-Sepharose for an additional 1 h at 4°C. The solution was centrifuged as above, and the pellet and supernatant aliquots were saved. The pellet was washed three times in 1 ml of NB and finally washed one time in 500 μl of 50 mm Tris, pH 6.8. Equivalent amounts of immunoprecipitate were loaded on a 4-16% SDS-PAGE and processed for Western blotting as described above.
Results
APP does not bind directly to kinesin-1
Previous studies reported that the cytoplasmic domain of APP exhibits high affinity (∼16-18 nm) with KLC1 and KLC2 and that this interaction occurs with the KLC TR domains (Kamal et al., 2000). These data, together with the demonstration of the recovery of APP using antibodies raised against kinesin-1 heavy chain (KHC) or KLC and a reduction in levels of steady state or axonally transported APP in nerves from KLC1-deficient mice, led to the proposal that APP may serve as a kinesin-1 cargo receptor (Kamal et al., 2000).
To examine the hypothesis that APP interacts directly with kinesin-1, we performed three series of experiments. First, to evaluate the interactions between AID and KLC1, as well as to map potential interaction sites, various recombinant KLC1 and APP fusion proteins were used for in vitro binding assays (Fig. 1). Bacterially expressed GST-KLC constructs (Fig. 1A) were immobilized on glutathione-Sepharose beads and incubated with in vitro-translated, [35S]methionine-labeled GFP and GFP-AID fusion proteins (Fig. 1B). As described previously (Kamal et al., 2000), GST-KLC constructs containing the full set of TRs (GST-KLC-CC/TR) pulled down GFP-AID. Surprisingly, GST-KLC constructs containing only a subset of these repeats (GST-KLC-TR) pulled down similar levels of GFP-AID (Fig. 1B). GST fusion proteins carrying the entire TR domain bound with similar strength as observed for GFP-AID to GFP fused to the intracellular domain of APLP1 (GFP-ALID1) or APLP2 (GFP-ALID2) (Fig. 1C), indicating that the differences in the primary sequence of AID, ALID1, and ALID2 have no influence on the binding to KLC. Furthermore, all of these constructs also pulled down the highly soluble protein GFP, whereas GST and GST fused to a mutant KLC1 lacking the TRs (GST-KLC-CC) failed to do so. This observation suggested that the binding to GFP-AID was unique to TR-containing constructs but raised the possibility that this was caused by a nonspecific hydrophobic interaction. Consistent with this, binding of GFP to GST-KLC was elevated in the presence of high salt (500 mm NaCl) and reduced by higher concentrations of detergent (data not shown). Similarly, immobilized GST-KLC also pulled down recombinant Fe65 (Fig. 1F), a protein that is known to bind to AID (Kesavapany et al., 2002) but was not expected to interact directly with KLC1. Similarly, GST-KLC also pulled down PAT1a, a variant of PAT1 and mutant PAT1a missing either the N-terminal or C-terminal half (S.K., personal communication). These data indicate that GST-KLC1 constructs containing TRs interact with proteins as different as GFP, Fe65, and PAT1a and the APP, APLP1, and APLP2 intracellular domains. Thus, the TR domains in GST-KLC1 exhibit nonspecific binding to various polypeptides with suitable hydrophobic patches. This might be explained by misfolding of KLC1 in E. coli; therefore, we used GST-AID in a second set of GST pull-down analyses and tested for binding of in vitro-translated KLC1 (Fig. 1G) or recombinant KLC1 expressed in COS-7 cells (data not shown). Neither the in vitro-translated KLC1 nor the heterologously expressed KLC1 bound to GST or GST-AID, whereas other known interaction partners of APP, such as Fe65 or Numb, were specifically retained on beads loaded with GST-AID (Fig. 1G). Together, these data show that bacterially expressed KLC1 has nonspecific binding properties and that heterologously expressed or in vitro-translated KLC1, which has fewer nonspecific binding characteristics, does not bind to the APP intracellular domain.
Figure 1.

Nonspecific binding of AID and GST-KLC1 fusion proteins containing TRs. A, Schematic representation of GST-KLC1 deletion mutants used in GSTpull-down assays. For details, see Materials and Methods. B, Pull-down assays show that all GST-KLC constructs containing two or more TRs bind to in vitro-translated GFP-AID constructs and with lower affinity to the GFP-only control. Neither GST alone nor GST-KLC-CC (both lacking TRs) pulled down GFP-AID or GFP. The fact that TR-containing constructs pulled down both GFP-AID and to a lower extent the highly soluble protein GFP suggests that these are nonspecific protein-protein interactions. C, In addition to GFP-AID, both GFP-ALID1 and GFP-ALID2 were retained to a very similar extent on GST-KLCΔCT. Identical results were obtained with GST-KLC-TR or GST-KLC-CC/TR beads (data not shown). D, Schematic representation of GFP-APP intracellular domain deletion mutants used in GST pull downs (see Materials and Methods). E, GST-KLC-CC/TR binds with similar affinities to GFP-AID and GFP-AID constructs with deletions of the BaSS (G-AIDΔB), PEER (G-AIDΔP), or NPTY (G-AIDΔN) domains of APP. These represent domains that have been shown previously to affect trafficking of APP and map to the domain proposed by Kamal et al. (2000) to bind TR domain in KLC1. F, Beads containing GST-KLC with TR domains also pull down recombinant Fe65 with high affinity, again suggesting nonspecific binding of TR domains to proteins in this assay system. G, GST pull downs with GST fused to the intracellular domain of APP demonstrated binding to the known APP interacting proteins Fe65 and Numb, whereas no binding of KLC1 was observed under identical conditions.
Next, we prepared vesicle fractions from mouse brain under conditions that preserve kinesin-1 association with membranes (Tsai et al., 2000; Morfini et al., 2002) and examined the distribution of KHC, KLC, and APP in these fractions by Western blot analysis. We detected KHC and KLC in all vesicle fractions (Fig. 2A, lanes V0, V1, V2) and in the cytosol (Fig. 2A, lane Cyt). Each fraction had a characteristic stoichiometry and kinesin-1 (KLC and KHC) isoform composition. In contrast, APP, detected with an N-terminal antibody that detects both full-length and α- and/or β-secretase-generated soluble ectodomain derivatives, is associated primarily with V0 and V1 fractions with only trace amounts in V2 and low levels in the cytosol. V2 did contain high levels of KHC, KLC1, and KLC2, and the membrane-associated proteins synaptophysin (P38) and synapsin I. Some proteins such as the catalytic subunit of protein phosphatase 2A and PKB (data not shown) were enriched in the cytosolic fraction. This suggests that kinesin-1 and APP do not fully cofractionate in rodent brain, as would be expected if APP acted as the only cargo receptor for kinesin-1.
Figure 2.

Kinesin-1 and APP do not cofractionate or coimmunoprecipitate from brain lysate. A, Subcellular fractionation of mouse brain was performed as described previously (Morfini et al., 2002) to generate three vesicle fractions (V0, V1, and V2) and a cytosolic fraction (Cyt). Under the conditions of fractionation, KHCs and KLCs are found in all fractions, but KLCs exhibit differences in stoichiometry and isoform composition. In contrast, APP immunoreactivity is associated primarily with V0 and V1 fractions, with only trace amounts in V2 and low levels in the cytosol. V2 contains high levels of KHC, KLC1, KLC2, synaptophysin (P38), and synapsin I. This suggests that kinesin-1 and APP do not cofractionate in mouse brain. B, Immunoprecipitates of kinesin-1 from mouse brain show that Coomassie blue-stained levels of kinesin-1 heavy chain are obtained (CB), but no band corresponding to APP was seen. Immunoblots of whole-brain lysate show that kinesin-1 heavy chain (H2) is readily resolved from APP (22C11) in this gel system. These are distinguishable even when a mixture of both antibodies (H2+22C11) is used for immunoblotting. C, Immunoprecipitation of kinesin-1 from mouse brain using antibodies against either kinesin-1 heavy chains (SUK4 or H2) or light chains (63-90, L1, L2, or KLC-All) precipitated both KHC and KLC subunits (Pfister et al., 1989; Stenoien and Brady, 1997). Nevertheless, APP could not be detected in immunoprecipitates using kinesin-1 antibodies. Brain lysate (L) was a positive control. Negative controls, i.e., protein G beads without primary antibodies (B) or with normal mouse IgG show immunoprecipitation specificity. Similarly, kinesin-1 subunits were not detectable in immunoprecipitates with APP antibodies [Ct-APP and JH; see KHC (H2) for kinesin-1 heavy chain, KLC (63-90) for kinesin-1 light chain]. Antigens in immunoprecipitates were visualized using specific antibodies against kinesin-1 subunits (H2 or 63-90) or APP (22C11 or Ct-APP). D, Immunoblot analysis showed detectable amounts of the scaffolding protein JIP3/SYD in kinesin-1 immunoprecipitates (H2 IPP). Conversely, anti-JIP3 antibodies (SYd IPP) coprecipitate small but detectable amounts of KHC. Immunoprecipitations with normal mouse (NM IPP) or normal rabbit (NR IPP) IgGs were used as negative controls. Note the relative levels of JIP3/SYD compared with original protein input. E, Immunodepletion of kinesin-1 and APP from mouse brain lysate exhibits different depletion patterns. Immunodepletion of KHC from lysate with H2 comparing IPs and corresponding supernatant samples shows a gradual reduction in the level of both KHC (H2) and KLC (63-90) in the pellet (lanes 1-4) and the supernatant (lanes 13-16) after depletion cycles. In contrast, no change in APP level could be detected (Ct-APP, lanes 1-4 and 13-16, respectively). Similarly, a reduction in APP level was observed after depletion using APP-specific antibodies AB5352 (Ct-APP, pellet, lanes 9-12; supernatant, lanes 21-24), but no change in the level of kinesin-1 could be detected (KHC, lanes 9-12, 21-24; KLC, lanes 9-12, 21-24).
We then asked whether APP could be immunoisolated with antibodies directed against KHC or KLC. Immunoprecipitation of kinesin-1 from mouse brain with H2 antibodies, raised against KHC, show that Coomassie blue (CB)-stained levels of KHC were obtained, but no polypeptide with a molecular weight corresponding to APP was visible (Fig. 2B). Because immunoblots of whole-brain lysates readily resolved kinesin-1 heavy chain (H2) and APP (22C11) in this gel system (Fig. 2B), this could not result from either overlapping bands or detection sensitivity. Furthermore, APP and KHC were distinguishable even when a mixture of both antibodies (H2 plus 22C11) was used for immunoblotting. To determine whether this was unique to specific antibodies, we used a battery of antibodies raised against distinct epitopes on KHC (Fig. 2C, lanes SUK4 and H2) or KLC (Fig. 2C, lanes 63-90 to KLC-ALL). We prepared detergent-solubilized membranes from mouse brain and performed immunoprecipitations as described by Kamal et al. (2000). APP could not be coimmunoprecipitated at detectable levels with any of these antibodies (Fig. 2C, lanes SUK4 to KLC-ALL). Similarly, KHC or KLC could not be coimmunoprecipitated using three antibodies raised against APP (Fig. 2C, lanes 22C11 to JH). Notably, the 22C11 antibody did not recover detectable amounts of APP in immunoprecipitation, because the epitope between amino acids 66 and 81 is masked under the nondenaturing conditions used herein [(Hilbich et al., 1993); S.S.S., data not shown]; however, two antibodies to the APP C terminus (Ct-APP and JH) efficiently precipitated APP.
To enhance the ability to detect low-level interactions of APP with kinesin-1, we performed multiple rounds of immunoprecipitation to deplete the target proteins. In this case, depletion of the bulk of KHC from the lysate and corresponding supernatant samples (Fig. 2E, KHC panel, lanes 1-4) results in a parallel depletion in levels of KLC (Fig. 2E, KLC panel, lanes 1-4), but no apparent change in levels of APP in either pellet or supernatant fractions (Fig. 2E, Ct-APP panel, lanes 1-4 and 13-16, respectively). Similarly, APP could be significantly depleted from brain lysates in multiple cycles with an APP C-terminal antibody (Fig. 2E, Ct-APP panel, lanes 9-12); however, no changes in either KHC (Fig. 2E, KHC panel, lanes 9-12) or KLC (Fig. 2E, KLC panel, lanes 9-12) levels were detectable after four cycles of APP immunodepletion. No KHC was detectable in any of the IPs with AB5352, although the methods should detect as little as 1% of the original kinesin-1 (see below). No evidence for a direct interaction of kinesin-1 with APP was found in these immunohistochemical studies.
To evaluate whether our buffer conditions maintain the association of kinesin-1 and a known kinesin-1-associated protein, we asked whether JIP3/SYD, a scaffolding protein for the c-Jun N-terminal kinase (JNK) group of kinases (Bowman et al., 2000) would coprecipitate with kinesin-1 in our experiments (Fig. 2D). We used buffer conditions similar to those used by Kamal et al. (2000, 2001) to reveal the association of APP and kinesin-1. Significantly, antikinesin-1 H2 antibodies coprecipitated detectable levels of the scaffolding protein JIP3/SYD (Fig. 2D, lane H2 IPP), and this interaction was further confirmed by coprecipitation of kinesin-1 using anti-JIP3 antibodies (Fig. 2D, lane SYD IPP). Semiquantitative analysis (data not shown) indicates that ∼1% of total JIP3 from brain lysate cofractionates with kinesin-1 (Fig. 2D, input lanes), in agreement with the previous report [see Materials and Methods in Bowman et al. (2000)]. In summary, no interaction between APP and kinesin-1 was detected in mouse brain membranes under conditions that efficiently coisolate KHC, KLC, and JIP3/SYD.
Axonal transport of kinesin-1 and Trk receptors is independent of APP expression
To confirm findings reporting decreased steady-state levels and axonal transport of KLC and selected cargo molecules in the sciatic nerve of APP knock-out (KO) mice (Kamal et al., 2001), we assessed the levels of kinesin-1 in protein extracts prepared from the sciatic nerves of mice with homozygous (Fig. 3A, Hm APP KO, lanes 1, 2) or heterozygous (Fig. 3A, Ht APP KO, lanes 3, 4) deletions of APP and compared the levels with kinesin-1 in nerves of wild-type mice (Fig. 3A, APP WT, lanes 5, 6). We found no change in the steady-state levels of dynein and synaptophysin in nerve segments for any of the different genotypes (Fig. 3A, Dynein and Syp panels, respectively). Specifically, we did not find a reduction in the steady-state levels of kinesin-1 heavy chains in nerves of APP-deficient mice compared with nerves from APP WT animals (Fig. 3A, KHC panel, compare lanes 1-4, 5 and 6). We then asked whether axonal transport of KHC, KLC, TrkA, and PS1 were dependent on APP expression, as reported previously (Kamal et al., 2000, 2001). For this purpose, we ligated sciatic nerves from wild-type mice (Fig. 3B, lanes 7-9) and mice with heterozygous (Fig. 3B, lanes 4-6) or homozygous (Fig. 3B, lanes 1-3) deletion of APP. Axonally transported material was allowed to accumulate for 24 h, and Western blot analysis was performed on detergent homogenates prepared from ∼0.3 cm segments proximal or distal to the ligature. The levels of both KHC and KLC in proximal segments from ligated nerves were essentially the same for nerves from animals of the three different genotypes (Fig. 3B, KHC and KLC panels, compare lanes 2, 5, 8). We also observed no difference in levels of Trk receptors (Fig. 3B, Trk panel, lanes 2, 5, 8) that accumulate in the proximal segment of ligated nerves from these animals. Because accumulation of kinesin-1 at the proximal side of the ligature, as detected by Western blot, seemed to be less robust than the accumulation of APP or Trk (Fig. 3B), we examined kinesin-1 expression in longitudinal sections of ligated sciatic nerve of wild-type mice (Fig. 3C). Consistent with previous reports (Li et al., 1999), immunostaining using anti-KHC H2 antibodies confirmed accumulation of KHC toward the proximal stump of the ligature and reduced kinesin-1 expression distally (Fig. 3C). Thus, we conclude that axonal transport of both kinesin-1 and Trk occurs in an APP-independent manner. Our lack of evidence for a direct association between kinesin-1 and APP in brain and the absence of any effects of APP deficiency on axonal transport of kinesin-1 and Trk suggest that APP does not serve as a kinesin-1 cargo receptor.
Examination of PS1 expression in the samples in Figure 3B revealed that steady-state levels of PS1 expression in unligated nerves of mice were the same for all three genotypes (Fig. 3B, PS1-NTF panel, compare lanes 1, 4, 7). In addition, we did not observe an accumulation of PS1 at the segment proximal to the ligature of sciatic nerves prepared from mice of any of these genotypes (Fig. 3B, PS1-NTF panel, lanes 2, 5, 8). These findings suggested that levels of PS1 in sciatic nerve were not APP dependent and that the rapidly transported axonal vesicles that carry APP are unlikely to transport PS1. These new findings led us to investigate the proposal that PS1 is cotransported with APP in fast axonal transport in the sciatic nerve.
Analysis of PS1 transport in mouse sciatic nerve
Previous studies (Kamal et al., 2001; Papp et al., 2002) reported that PS1 is subject to rapid anterograde transport in mouse peripheral nerves. Moreover, Kamal et al. (2001) proposed that PS1 is cotransported with APP in the same vesicular compartment, in which APP gets cleaved to yield Aβ peptides. In light of this, we examined the axonal transport of PS1 and the production of Aβ peptides in nontransgenic mice. We placed two ligatures in the sciatic nerve just distal to the convergence of L4/L5 sensory fibers and motor afferents for 6 h and then prepared detergent-solubilized extracts from 0.3 cm segments proximal and distal to the ligatures. Western blot analysis using the APP C-terminal-specific antibody, CT-15, revealed that only highly glycosylated forms of full-length APP accumulate in the segment proximal to the ligature (Fig. 4A, CT15 panel, lane 5; compare with unligated contralateral nerve segment, lane 1, and with other segments, lanes 2-4) over this time frame, as described previously (Koo et al., 1990; Sisodia et al., 1993). The levels of underglycosylated APP remained unchanged in all segments and are a reflection of the APP expressed by cells intrinsic to the nerve, including Schwann cells and fibroblasts. We showed previously that these APP species are principally the APP-751 variant (Sisodia et al., 1993). In contrast, using the PS1-specific PS1-NTF antibody (Thinakaran et al., 1997), we were unable to detect any increase in the levels of the N-terminal, endoproteolytic derivative of mouse PS1 (PS1-NTF) in the proximal segment compared with the levels contained in other isolated segments (Fig. 4A, PS1-NTF panel).
Figure 4.
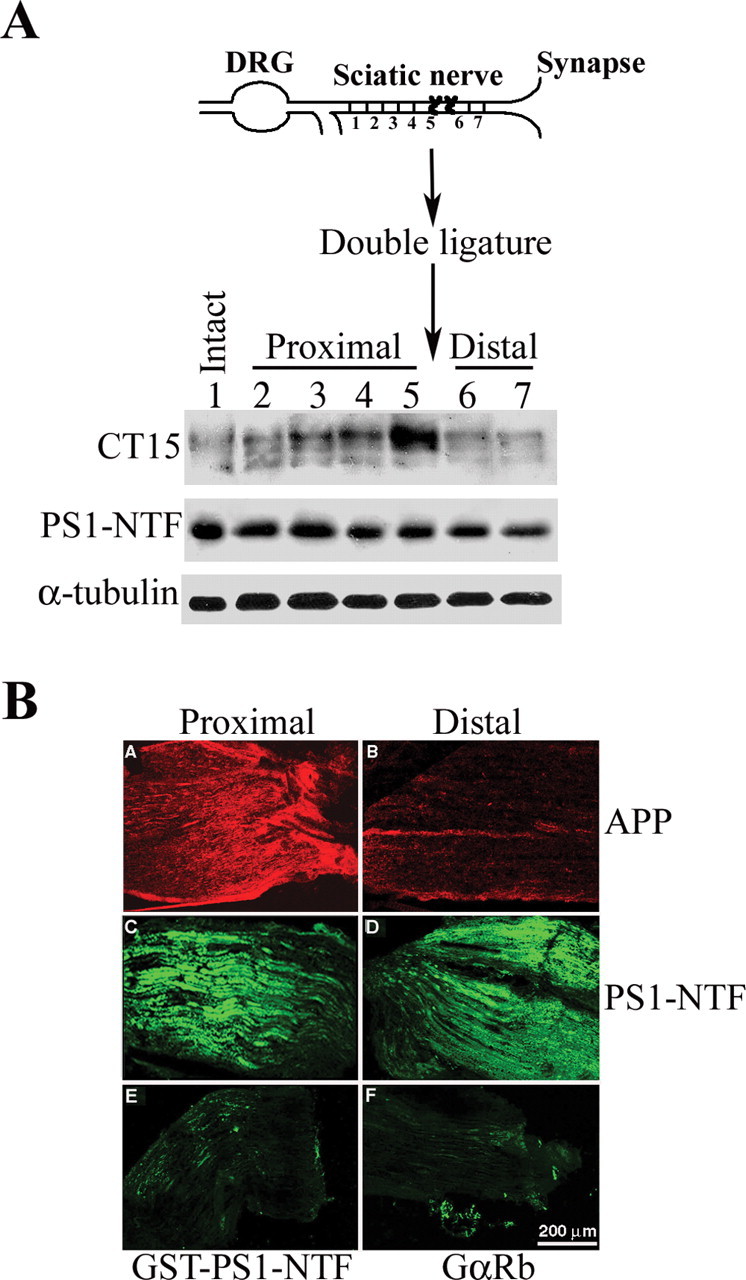
PS1 is not cotransported with APP in the same vesicular compartment by fast axonal transport. A, Top, Schematic diagram of sciatic nerve double ligation and nerve segments examined for protein expression. Bottom, Expression level of APP and PS1 in nerve segments at varying distances from the site of ligation in sciatic nerve of nontransgenic mice. APP expression level increases toward the site of ligation, whereas it decreases away from the distal ligature (CT15 panel, lanes 1-5). In contrast, PS1 expression level is comparable in intact sciatic nerve and nerve segments at varying distances from the site of ligation (PS1-NTF panel, lanes 1-7). B, APP and PS1 immunoreactivity in cryostat sections of the proximal and distal ends of ligated sciatic nerve. A, B, APP is detected using CT15 antibodies. Note the accumulation of APP proximal to the ligature (A), whereas weak immunoreactivity is observed at the distal stump (B). C, D, PS1 immunoreactivity as detected by affinity-purified αPS1-NTF antibodies; C, proximal stump; D, distal stump. Note that neither accumulation at the proximal stump nor reduced immunoreactivity at the distal stump can be detected. E, Specificity of immunostaining for PS1 was confirmed using αPS1-NTF antibodies preincubated with GST-PS1-NTF (see Materials and Methods). F, Immunostaining of mouse sciatic nerve cryostat sections using secondary antibodies only (fluorescein-conjugated goat anti-rabbit). Scale bar, 200 μm.
We considered the possibility that lack of PS1-NTF accumulation in the proximal segment of ligated sciatic nerve, as observed by Western blot analysis, may be a consequence of dissecting segments that were too wide, thus masking low levels of PS1-NTF that might have accumulated immediately proximal to the ligature. To examine this issue, we placed ligatures in the sciatic nerve of nontransgenic mice and analyzed the distribution of APP and PS1 in longitudinal, cryostat sections of the nerve by immunolabeling and visualization by indirect immunofluorescence microscopy (Fig. 4B). Confirming the Western blot analysis (Fig. 4A), we obtained high levels of CT-15 immunoreactivity in the segment proximal to the ligature and markedly lower levels in the distal ligature in nerve sections of nontransgenic mice (Fig. 4B, panels A and B, respectively). Parallel analysis with an affinity-purified preparation of the PS1-NTF antibody revealed similar levels of accumulated PS1 in both proximal and distal segments (Fig. 4B, panels C, D), with no apparent elevation in PS1 immunoreactivity immediately proximal to the ligature. PS1-NTF immunoreactivity in nerve segments was fully abolished by preincubation of the antibody with the immunogen, a GST fusion protein harboring the N-terminal 80 aa of PS1 (Fig. 4C, panel E). This data, and the demonstration that secondary antibodies alone (Fig. 4C, panel F) did not bind to nerve antigens, led us to conclude that the immunofluorescence signal observed using our PS1-NTF antibody is specific.
Together, these data indicate that APP and PS1 are not cotransported in the nerve. Thus, the transport kinetics of these two proteins is not consistent with the proposal that PS1 is anterogradely transported in a membrane compartment in which APP serves as a kinesin-1 receptor (Kamal et al., 2001).
Aβ and BACE1 are not cotransported with APP in the same vesicular compartment
To examine the proposal that Aβ peptides are generated within axonally transported vesicles carried by the fast anterograde component, we immunoprecipitated Aβ from detergent extracts of sciatic nerves of nontransgenic mice. Aβ was not detected using Western blot analysis or sandwich ELISA approaches (data not shown). Because Kamal et al. (2001) reported that both Aβ40 and Aβ42 peptides were readily detected in the transport component in nerves of nontransgenic mice, we attempted to enhance detection of Aβ in the sciatic nerve using antibody 26D6, raised against amino acids 1-16 of Aβ (Lamb et al., 1999), to immunoprecipitate Aβ from detergent extracts prepared from sciatic nerves of APPswe mice. Aβ was not detected in the proximal segment of ligated nerve from the transgenic mice (Fig. 5A, lane 5), although the ∼12 kDa APP C-terminal fragments (CTFs) were readily evident and accumulated in this segment. APP CTFs were likely to have been generated by β-secretase processing of full-length APPswe in the trans-Golgi network (Haass et al., 1995). To determine whether our inability to detect Aβ from nerve was a reflection of one or more technical issues, we used N2a cells that stably express APPswe and secrete abundant levels of Aβ peptides (Thinakaran et al., 1996). In addition, a segment of nerve in immunoprecipitation buffer was added to conditioned medium from homogenized APPswe N2a cells. This preparation was subjected to immunoprecipitation and Western blot analysis with 26D6 antibody, as before (Fig. 5A). In parallel, an aliquot of the conditioned medium from APPswe N2a cells, or detergent homogenate from the cortex of APPswe mice with an equivalent amount of protein as the sciatic nerve extracts, was analyzed by immunoprecipitation and Western blots. In cortices of APPswe mice, we detected ∼12 kDa APP-CTFs and very low levels of ∼4 kDa Aβ peptides (Fig. 5A, lane 3) (see long exposure). Identical levels of soluble Aβ peptides were immunoprecipitated from medium of APPswe cells and from the nerve homogenate that was “spiked” with conditioned medium from APPswe N2a cells (Fig. 5A, lanes 1, 2). Thus, the absence of detectable levels of Aβ peptides in the proximal segment of nerve from APPswe mice is not caused by inhibitors present in the nerve preparation or other technical limitations of the assay.
Figure 5.

Expression of BACE1 and Aβ is undetectable in mouse sciatic nerve. A, Western blot analysis of Aβ expression in protein extracts of stably transfected APPswe N2a cells (lane 1), APPswe N2a cells and sciatic nerve extract (lane 2), cortex (ctx) of transgenic mice harboring APPswe (lane 3), and ligated sciatic nerve of transgenic mice harboring APPswe (lanes 4-6). I, Intact contralateral nerve; P, segment proximal to the ligature; D, segment distal to the ligature. Accumulation of C-terminal fragments is shown, but no Aβ could be detected at the proximal stump of sciatic nerve of the FAD-linked APPswe variant (lanes 4-6). C-terminal fragments, as well as low levels of Aβ, could be detected in protein extract from the cortex of these mice using an amount of protein equivalent to that of the sciatic nerve extracts (lane 3; note “Aβ long exposure”). Aβ could be detected in N2a cells stably transfected with APPswe. Addition of nerve extract to N2a cells yielded the same level of Aβ as N2a alone (lanes 1 and 2, respectively). B, No specific band corresponding to BACE1 could be detected in protein extracts of ligated mouse sciatic nerve after crush injury. SN-P, Sciatic nerve segment proximal to the crush site; SN-D, sciatic nerve segment distal to the crush site. A band similar to the one observed in nontransgenic mice proximal and distal to the crush injury (BACE1 panel, lanes 2 and 3, respectively) was observed in protein extract of BACE1 KO sciatic nerve (BACE1 panel, lanes 5 and 6, respectively). To confirm our ability to detect BACE1, we examined BACE1 expression in the hippocampus of these mice. BACE1 was detectable in the hippocampus (Hipp) of nontransgenic mice (BACE1 panel, lane 1) but not in the hippocampus of BACE1 KO mice (BACE1 panel, lane 4). APP accumulated at the proximal segment of the crushed injury in the sciatic nerve of both the nontransgenic mice (APP panel, lane 2) and the BACE1 KO mice (APP panel, lane 5), confirming the blockade of fast anterograde axonal transport after the crush injury. As expected, increased expression of APP was observed in the hippocampus of BACE1 knock-out mice compared with nontransgenic mice (APP panel, compare lanes 4 and 1, respectively).
Finally, we examined the hypothesis that BACE1 is also transported in the same membranous compartment as APP. We performed Western blot and immunoprecipitation analyses using three independent antibodies (Marcinkiewicz and Seidah, 2000; Cai et al., 2001; Shi et al., 2003). With our antibodies (Cai et al., 2001), BACE1 was readily detectable as an ∼70 kDa species in protein extracts from hippocampus of nontransgenic mice (Fig. 5B, lane 1), whereas the immunoreactive band was absent in the protein extract from the hippocampus of BACE1-deficient mice (Fig. 5B, lane 4). In contrast, we were unable to detect BACE1 in detergent homogenates from sciatic nerve extracts from mice (Fig. 5B, compare lanes 2, 3, 5, 6). An immunoreactive band of the same size as BACE1 was detected in sciatic nerve extract of BACE1 KO mice, suggesting a nonspecific binding interaction of this polypeptide with the antibody in the sciatic nerve (Fig. 5B, lanes 5, 6). Moreover, there was no increase in levels of immunoreactive bands in the proximal segment compared with the distal segment of ligated nerves (Fig. 5B, lanes 2, 3). Notably, we observed elevated levels of mature, fully glycosylated APP in the hippocampus from BACE1-deficient mice (Fig. 5B, lane 4), as would be expected if BACE1 is inactivated.
Together, the absence of detectable BACE1 and Aβ in peripheral nerves is inconsistent with a model that proposes cotransport of the processing machinery that generates Aβ peptides in compartments within the axons. Thus, we conclude that in peripheral nerves APP is not coresident with components of the processing machinery within anterogradely transported membrane vesicles in peripheral axons. Furthermore, the absence of any detectable effects on the transport of known kinesin-1 cargos or of kinesin-1 itself in peripheral nerve in the absence of detectable APP is inconsistent with a role for APP as a receptor for kinesin-1.
Discussion
The unequivocal demonstrations that APP moves in fast anterograde transport in mammalian axons (Koo et al., 1990; Buxbaum et al., 1998) and the compelling demonstration that APP is proteolytically processed at nerve terminals have spurred efforts to examine the role played by APP in anterograde movement of membranous cargos and the molecular apparatus involved in APP processing in neurons. It has been reported that the cytoplasmic domain of APP binds with high affinity to the TR domains of KLC and that an APP-kinesin-1 complex is associated in vivo (Kamal et al., 2000). These findings, together with the observation that APP transport is impaired in mice with functionally ablated KLC1 alleles, led to the hypothesis that APP serves as a kinesin-1 cargo receptor (Kamal et al., 2000). In addition, it was suggested that PS1, a component of the γ-secretase machinery responsible for intramembranous processing of APP, is cotransported in the same vesicle cargo with APP (Kamal et al., 2001). Moreover, these studies suggested that BACE1 is rapidly transported anterogradely in APP-containing vesicles within nerves and that Aβ40 and Aβ42 peptides are generated during transport (Kamal et al., 2001). These findings, if replicated, would not only be of value in identifying an important functional role for APP, but also suggest a paradigm shift in our understanding of the pathogenesis of AD. In this report, we performed a series of experiments to test the hypothesis that APP serves as a kinesin-1 cargo receptor and that the molecular components involved in APP processing are cotransported in the sciatic nerves of wild-type and APPswe mice. Our findings are summarized below.
In contrast to the studies of Kamal et al. (2000), we were not able to demonstrate a direct interaction between APP and either KHC or KLC in vitro and in vivo. Because APP was reported to bind directly to the TR domains in KLCs with stoichiometry of two APP/KLC, we first examined possible direct interactions between these molecules using in vitro binding assays with recombinant KLC and APP fusion proteins (Kamal et al., 2000). Although we noted binding of the APP-ICD fusion protein to the KLC1 fusion protein, this may not reflect a physiologically relevant interaction. A wide variety of structurally unrelated proteins have been reported to interact with KLC through these domains, including Drosophila kinesin-1 associated protein YETI (Wisniewski et al., 2003), JIP1 and JIP2 (Matsuda et al., 2001; Verhey et al., 2001), Sunday Driver (Bowman et al., 2000), surface proteins in vaccinia virus (Ward and Moss, 2004), and the chaperone hsc70 (Tsai et al., 2000), as well as various unpublished interactions. The evidence thus suggests that TRs may be involved in protein-protein interactions, but TRs are poor candidates for determining cargo specificity. TR domain sequences are homologous in both KLC1 and KLC2 and highly conserved at the amino acid level (Tsai et al., 2000). Nonetheless, KLC1 and KLC2 appear to be associated with different subsets of membrane-bound organelles (Leopold et al., 1992; Elluru et al., 1995; Stenoien and Brady, 1997; Rahman et al., 1999). Hydrophobic stretches in TRs would tend to drive relatively nonspecific binding with other proteins containing suitable hydrophobic patches. This feature is a potential source of artifact for in vitro binding assays, making confirmation of putative interactions with in vivo assays critical. In our study, a series of in vivo assays with endogenous kinesin-1 and APP indicate that there is not a direct interaction between these two proteins. APP and kinesin-1 did not cofractionate under conditions in which kinesin-1 is retained on membrane fractions. In particular, there was no match between the distribution of KLC1 and APP, the proposed interacting partners (Kamal et al., 2000, 2001). Similarly, APP and kinesin-1 did not coimmunoprecipitate under nondenaturing conditions from lysates of mouse brain with any of a wide range of specific antibodies to different epitopes on KLC, KHC, and APP. In these experiments, KHC and KLC remained associated, indicating that high-affinity protein complexes were preserved.
Although Kamal et al. (2000, 2001) suggested that APP is required for kinesin-1-mediated axonal transport, we did not observe differences in the steady-state levels of KHC and KLC in sciatic nerves of APP-deficient mice compared with wild-type mice, nor were there differences in levels of kinesin-1 that accumulated in the proximal segment of ligated sciatic nerves from APP-deficient and wild-type animals. Consistent with this study, we showed that axonal transport of Trk also occurs in an APP-independent manner. The lack of effects of APP deficiency on axonal transport of kinesin-1 and Trk, and the lack of a direct association between kinesin-1 and APP in brain, leads us to conclude that APP is an unlikely candidate for a kinesin-1 cargo receptor, at least in peripheral nerves. In support of these findings, we (E. B. Lee and V. M.-Y. Lee) have shown that overexpression of BACE1 leads to a reduction in levels of axonally transported APP in the sciatic nerve, but without impairment in the transport of synaptophysin and kinesin-1 (Lee et al., 2005).
Clearly, APP is present in one or more of the membrane cargo vesicles moved by kinesin-1, but it is not required for the transport of those vesicles and does not attach directly to the kinesin-1 motor. This observation is consistent with the fact that APP KO mice are viable and phenotypically near normal (Zheng et al., 1995), whereas knock-outs of specific KHC (Tanaka et al., 1998; Kanai et al., 2000) or KLC1 (Rahman et al., 1999) genes are embryonic lethal, with neurological defects; however, it is still conceivable that APP and kinesin-1 could interact indirectly with a connection mediated by a linker protein. For example, a member of the JNK-signaling scaffold protein family, JIP1b, was reported to enhance the “interaction” of APP and kinesin-1 in vitro; however, these data are based only on in vitro analyses. Considering the tendency of KLC to bind nonspecifically to hydrophobic stretches, these data need clarification and further support by in vivo analysis.
Although Kamal et al. (2001) suggested that PS1 and APP are cotransported in rapid anterograde transport in sciatic nerve, our results do not support this hypothesis. For example, the steady-state levels of PS1 in unligated and ligated sciatic nerves of mice with functionally ablated APP genes are not different from those measured in nerves from wild-type mice, thus arguing against cotransport of APP and PS1 in the same vesicle component. Although it is conceivable that only small amounts of PS1 exit the cell body at a given time and any signal might be masked by the abundant levels of PS1 in the proximal segment that is contributed by cells intrinsic to the nerve itself, we consider this unlikely, because our immunohistochemical studies did not reveal accumulation of PS1 immediately proximal to the ligature during the intervals examined.
Additionally, we were not able to detect BACE1 or Aβ in sciatic nerves, nor were we able to detect accumulation of these polypeptides in the proximal segment of ligated nerves. Thus, our results do not support the hypothesis that components of the APP proteolytic processing machinery that are responsible for generating Aβ and Aβ itself are cotransported with APP in the fast transport component in peripheral nerves.
Many hypotheses have been generated to explain the functions or pathological roles of APP, PS1, BACE1, and other components of the complex that produce Aβ. For many years, the complexity of the disease and the absence of good animal models for AD made it difficult to evaluate the validity of these proposals, but currently available models and methodologies now may be used to test these hypotheses. The idea that all of the components for producing Aβ were localized in a single compartment had an attractive simplicity, but it was not consistent with what is known about the biology of Aβ and amyloid. The relative levels of the different components, their subcellular localization, and their varied biological roles are all inconsistent with a single compartment. Our test of the one-compartment model (Kamal et al., 2001) indicated that APP, PS1, and BACE1 exist primarily in different compartments in peripheral nerves. It may be that secretases are transported in these axons, but the levels (in contrast to APP) are so low that they are not detectable. More significantly, the levels of the secretases do not appear sufficient to generate Aβ that could deposit at sensory terminals or neuromuscular junctions. The situation may be different in CNS neurons, including basal forebrain cholinergic and entorhinal cortex neurons. In these cells, levels of APP and critical secretases appear to be sufficient to generate Aβ levels that lead to the formation of oligomeric species at synapses, leading to synaptic dysfunction and Aβ aggregation. In neurons, the various components of the APP processing complex must be specifically assembled at some point and transported before encountering membrane-tethered APP-CTFs to generate Aβ. Recognition of this requirement is an important step forward for our understanding of pathogenesis in Alzheimer's disease and possibly for the development of new therapeutics for targeting the formation and potential toxic effects of Aβ peptides.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant AG-021494, the Ellison Medical Foundation, and the Ruth Broad BioMedical Research Foundation (S.S.S.); NIH Grants NS23868, NS23320, NS41170, NS43408 (S.T.B.), NS41438 (V.E.K.), NS41438, and NS45150 (P.C.W.), AG11542 (V.M.-Y.L.), and AG05146; the Juvenile Diabetes Foundation (S.T.B.); the Deutsche Forschungsgemeinschaft (Ki819/1-1, SPP 1085/2); the Alzheimer Forschung Initiative Eingetragener Verein (S.K.); the Adler Foundation; and the Bristol-Myers Squibb Foundation (P.C.W.). S.S.S. and O.L. thank Dr. Seong-Hun Kim for the affinity-purified anti-PS1 antibodies. S.T.B. and G.A.M. acknowledge Huichuan Reyna for assistance with immunoblots and Joachim Herz for providing the JH antibody against APP. S.K. and A.S. thank Dr. Lachman for providing the hKLC1 cDNA.
Correspondence should be addressed to Sangram S. Sisodia, Department of Neurobiology, Pharmacology, and Physiology, The University of Chicago, Chicago, IL 60637. E-mail: ssisodia@drugs.bsd.uchicago.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252386-10$15.00/0
O.L. and G.A.M. contributed equally to this work.
References
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS (1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 17: 1005-1013. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939-945. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS (2000) Kinesin-dependent axonal transport is mediated by the Sunday Driver (SYD) protein. Cell 103: 583-594. [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS (1990) A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA 87: 1061-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS (1998) Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci 18: 9629-9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC (2001) BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci 4: 233-234. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) APH-1, PEN-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38: 9-12. [DOI] [PubMed] [Google Scholar]
- Elluru RG, Bloom GS, Brady ST (1995) Fast axonal transport of kinesin in the rat visual system: functionality of kinesin heavy chain isoforms. Mol Biol Cell 6: 21-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ (1995) The Swedish mutation causes early-onset Alzheimer's disease by β-secretase cleavage within the secretory pathway. Nat Med 1: 1291-1296. [DOI] [PubMed] [Google Scholar]
- Hilbich C, Monning U, Grund C, Masters CL, Beyreuther K (1993) Amyloid-like properties of peptides flanking the epitope of amyloid precursor protein-specific monoclonal antibody 22C11. J Biol Chem 268: 26571-26577. [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG (2000) Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell 11: 1213-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28: 449-459. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414: 643-648. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N (2000) KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci 20: 6374-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavapany S, Banner SJ, Lau KF, Shaw CE, Miller CC, Cooper JD, McLoughlin DM (2002) Expression of the Fe65 adapter protein in adult and developing mouse brain. Neuroscience 115: 951-960. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lah JJ, Thinakaran G, Levey A, Sisodia SS (2000) Subcellular localization of presenilins: association with a unique membrane pool in cultured cells. Neurobiol Dis 7: 99-117. [DOI] [PubMed] [Google Scholar]
- Kim SH, Leem JY, Lah JJ, Slunt HH, Levey AI, Thinakaran G, Sisodia SS (2001) Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J Biol Chem 276: 43343-43350. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 87: 1561-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb BT, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ (1999) Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci 2: 695-697. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS (2002) Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 22: 9785-9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM (2005) BACE overexpression alters the subcellular processing of APP and inhibits Aβ deposition in vivo. J Cell Biol 168: 291-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS (1997) Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med 3: 756-760. [DOI] [PubMed] [Google Scholar]
- Leopold PL, McDowall AW, Pfister KK, Bloom GS, Brady ST (1992) Association of kinesin with characterized membrane-bounded organelles. Cell Motil Cytoskeleton 23: 19-33. [DOI] [PubMed] [Google Scholar]
- Li JY, Pfister KK, Brady S, Dahlstrom A (1999) Axonal transport and distribution of immunologically distinct kinesin heavy chains in rat neurons. J Neurosci Res 58: 226-241. [PubMed] [Google Scholar]
- Marcinkiewicz M, Seidah NG (2000) Coordinated expression of β-amyloid precursor protein and the putative β-secretase BACE and α-secretase ADAM10 in mouse and human brain. J Neurochem 75: 2133-2143. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I (2001) c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci 21: 6597-6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Tsai MY, Szebenyi G, Brady ST (2001) Approaches to study interactions between kinesin motors and membranes. Methods Mol Biol 164: 147-162. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST (2002) Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 21: 281-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp H, Pakaski M, Kasa P (2002) Presenilin-1 and the amyloid precursor protein are transported bidirectionally in the sciatic nerve of adult rat. Neurochem Int 41: 429-435. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien DL, Brady ST, Bloom GS (1989) Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol 108: 1453-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci 21: 479-505. [DOI] [PubMed] [Google Scholar]
- Rahman A, Kamal A, Roberts EA, Goldstein LS (1999) Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J Cell Biol 146: 1277-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741-766. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE (2002) Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Aβ amyloidosis. J Neurosci 22: 9794-9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XP, Tugusheva K, Bruce JE, Lucka A, Wu GX, Chen-Dodson E, Price E, Li Y, Xu M, Huang Q, Sardana MK, Hazuda DJ (2003) β-secretase cleavage at amino acid residue 34 in the amyloid β peptide is dependent upon γ-secretase activity. J Biol Chem 278: 21286-21294. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL (1993) Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci 13: 3136-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST (1997) Immunochemical analysis of kinesin light chain function. Mol Biol Cell 8: 675-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N (1998) Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93: 1147-1158. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS (1996) Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “β-secretase” site occurs in the Golgi apparatus. J Biol Chem 271: 9390-9397. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS (1997) Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem 272: 28415-28422. [DOI] [PubMed] [Google Scholar]
- Tsai MY, Morfini G, Szebenyi G, Brady ST (2000) Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol Biol Cell 11: 2161-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM (1995) Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol 129: 1629-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol 152: 959-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B (2004) Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J Virol 78: 2486-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K (1989) Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell 57: 115-126. [DOI] [PubMed] [Google Scholar]
- Wisniewski TP, Tanzi CL, Gindhart JG (2003) The Drosophila kinesin-I associated protein YETI binds both kinesin subunits. Biol Cell 95: 595-602. [DOI] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisodia SS, Chen HY, Van der Ploeg LH (1995) β-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81: 525-531. [DOI] [PubMed] [Google Scholar]