

Abstract
The glutamate receptor (GluR) agonist-binding site consists of amino acid residues in the extracellular S1 and S2 domains in the N-terminal and M3-M4 loop regions, respectively. In the present study, we sought to confirm that the conserved ligand-binding residues identified in the AMPA receptor S1S2 domains also participate in ligand binding of GluR6 kainate receptors. Amino acid substitutions were made in the GluR6 parent at R523, T690, and E738 to alter their potential interactions with ligand. Mutant receptors were expressed in human embryonic kidney 293 cells, confirmed by Western blot analysis, and tested by [3H]kainate binding and patch-clamp recording. Each of the binding site mutations was sufficient to reduce [3H]kainate binding to undetectable levels and eliminate functional responses to glutamate or kainate. As with our studies of other nonfunctional mutants (Fleck et al., 2003), immunocytochemical staining and cell-surface biotinylation studies showed that the mutant receptors were retained intracellularly and did not traffic to the cell surface. Endoglycosidase-H digests and colocalization with endoplasmic reticulum (ER) markers demonstrated that the mutant receptors are immaturely glycosylated and retained in the ER. Immunoprecipitation, native PAGE, and functional studies confirmed that the GluR6-binding site mutants are capable of multimeric assembly, indicating their retention in the ER does not result from a gross protein folding error. Together, these results confirm the role of R523, T690, and E738 directly in ligand binding to GluR6 and further support our previous report that nonfunctional GluRs are retained intracellularly by a functional checkpoint in ER quality control.
Keywords: glutamate receptor, kainate receptor, ER retention, binding site, mutant, trafficking
Introduction
Glutamate receptor (GluR) ion channels mediate excitatory synaptic transmission in the mammalian CNS. Ionotropic GluRs are divided into three discrete subtypes according to their pharmacological profiles: AMPA, kainate (KA), and NMDA receptor channels (for review, see Seeburg, 1993; Dingledine et al., 1999). Functional GluRs are multimeric proteins assembled from individual subunits, each between 900 and 1300 amino acids in length. AMPA receptors are composed of the subunits GluRA-D, also referred to as GluR1-4, respectively. KA receptors are composed of the subunits GluR5-7 and KA1-2 (Lerma, 2003). NMDA receptors include the NR1 subunit in conjunction with NR2A-D (McIlhinney et al., 2003; Prybylowski and Wenthold, 2004). The overall topology of the individual subunits is similar: an N-terminal extracellular segment followed by a transmembrane domain (M1), a reentrant pore-forming loop (M2), a second extracellular loop between transmembrane domains M3 and M4, and a C-terminal intracellular segment. Numerous studies indicate that two extracellular domains critical to agonist binding are located in the N-terminal segment and in the M3-M4 loops; these segments are referred to as S1 and S2, respectively (Stern-Bach et al., 1994; Leuschner and Hoch, 1999).
Atomic-level studies by x-ray crystallography have provided an elegant description of the AMPA receptor agonist-binding site based on a soluble GluR2 S1S2 construct examined in unbound or various ligand-bound conformations (Armstrong et al., 1998; Armstrong and Gouaux, 2000). Amino acid residues making ligand interactions in the GluR2 S1S2 construct include R485 in the S1 segment and T655 and E705 in the S2 segment. Not surprisingly, these residues are particularly well conserved among other GluR subunits, and their critical role in AMPA receptor function is confirmed by mutagenesis studies of full-length receptors (Uchino et al., 1992; Mano et al., 1996; Lampinen et al., 1998; Abele et al., 2000).
We previously examined a series of mutations in the GluR6 S2 domain to identify molecular differences between KA and AMPA receptors that confer unique functional and pharmacological properties (Fleck et al., 2003). The results of ours and other comparative studies (Smith and Howe, 2000; Bowie and Lange, 2002) suggest there may be structural and stoichiometric differences in KA and AMPA receptor gating that have yet to be resolved. In addition, we identified several nonfunctional mutations that interfered with GluR6 and GluR1 trafficking to the cell surface, raising new questions about the structural and functional constraints on GluR trafficking.
In the present study, we demonstrate that the residues identified in the GluR2 S1S2 crystal structures are, likewise, critical to ligand binding and function of GluR6 KA receptors. Moreover, the elimination of agonist binding interferes with cell-surface expression and causes accumulation of nonfunctional GluR6 receptors in the endoplasmic reticulum (ER), suggesting a functional binding site is required for proper GluR6 trafficking. Similar defects in GluR1 and other nonfunctional mutations suggest that both KA and AMPA receptor trafficking and cell-surface expression are regulated by a functional checkpoint in ER quality control.
Materials and Methods
Cell cultures and transfections. Human embryonic kidney 293 (HEK293) fibroblasts (CRL 1573; American Type Culture Collection, Manassas, VA) were cultured in minimal essential medium supplemented with 10% fetal bovine serum and 2 mm glutamine (Invitrogen, Gaithersburg, MD) and maintained at 37°C in a 5% CO2 environment. Cells were plated into 25 cm2 flasks and passaged twice weekly to fresh flasks or plated onto poly-d-lysine-coated dishes (35 mm). Primary rat astrocyte cultures were obtained from Dr. T. Lindsley (Albany Medical College) and harvested and maintained as described previously (Goslin et al., 1998). Transfections were done the following day using the LipofectaminePLUS reagents (Invitrogen) as described previously (Fleck et al., 2003). Astrocyte cultures in 60 mm2 dishes were transfected with 2.4 μl of Lipofectamine 2000 (Invitrogen) and 3 μg of cDNA. Cells were then incubated for 18-48 h before recording or 36 h before immunocytochemical and biochemical experiments. Plasmids encoding enhanced green fluorescent protein, enhanced yellow fluorescent protein (EYFP)-ER, and EYFP-Golgi were from Clontech (Palo Alto, CA). EYFP-ER encodes a calreticulin signal sequence, EYFP, and the KDEL sequence containing fusion protein. EYFP-Golgi plasmid encodes the N-terminal localization sequence of 1,4-galactosyltransferase fused to EYFP. Wild-type GluR6 (GluR6-wt) and GluR1-wt (flip variant) constructs were a gift from Dr. Mark L. Mayer (National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD) and Dr. Peter Seeburg (Max-Planck-Institute for Medical Research, Heidelberg, Germany). Mutations were introduced by QuikChange mutagenesis (Stratagene, La Jolla, CA) using PfuTurbo DNA polymerase (Stratagene) and custom primers (Sigma Genosys, Woodlands, TX). Mutant cDNAs were amplified in transformed DH5α cells (Invitrogen, Rockville, MD), purified using Qiafilter Maxiprep kits (Qiagen, Los Angeles, CA), selected by diagnostic restriction digests (New England Biolabs, Beverly, MA), and confirmed by automated sequencing (Center for Comparative Functional Genomics, State University of New York, Albany, NY).
Patch-clamp recording with rapid solution exchange. Cells were superfused continuously with standard extracellular solution containing 150 mm NaCl, 3 mm KCl, 5 mm HEPES, 1 mm MgCl2, 1.8 mm CaCl2, 10 mm glucose, and 0.1 mg/ml phenol red, pH 7.3. Recording microelectrodes were fabricated from thin-walled borosilicate glass capillary tubes (TW150F; World Precision Instruments, Sarasota, FL) having resistances of 2-4 MΩ when filled with an internal solution (IC) containing the following (in mm): 135 CsCl, 10 CsF, 10 HEPES, 5 EGTA, 1 MgCl2, and 0.5 CaCl2, pH 7.2, 295 mOsm. Whole-cell and outside-out patch recordings were performed in voltage clamp at a holding potential of -70 mV using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Ultrafast solution exchange was achieved using an LSS-3100 piezo-translator (Burleigh Instruments, Fishers, NY), as described previously (Fleck et al., 2003). Control and agonist solutions were driven simultaneously at a rate of 0.3 ml/min through the two parallel barrels of a theta tube. The rate of solution exchange was ≤100 μs as determined by the open-tip junction currents measured at the end of each experiment. For [Q]/[R] heteromeric assembly studies, current-voltage (I-V) relationships were generated at 10 mV voltage steps from -100 to +100 mV with IC containing 30 μm spermine. Whole-cell recording using a multivalve solution exchange system was performed as described previously (Fleck et al., 2003). The rate of solution exchange using this system was ∼2 ms, as determined from open-tip junction currents, but was further limited by cell diameter. Current signals were filtered at 1-5 kHz with an eight-pole Bessel filter (Cygnus Technologies, Delaware Watergap, PA), digitized at 2-20 kHz, and stored on a Macintosh PowerPC-G3 computer using an ITC-16 interface (Instrutech, Great Neck, NY) under the control of the data acquisition and analysis program Synapse (Synergy Research, Silver Spring, MD).
Radioligand binding. HEK293 cells were transfected in T75 flasks and grown for 36 h before washing three times in ice-cold Tris-buffered saline (TBS; 50 mm Tris and 154 mm NaCl, pH 7.4). Cells were lysed in hypotonic 10 mm Tris buffer supplemented with protease inhibitor mixture (Sigma) and suspended with a Teflon-glass homogenizer. Membrane fractions were obtained by centrifugation at 20,000× g for 20 min. Protein concentrations were determined by the BCA protein assay (Pierce, Rockford, IL). Membrane aliquots were stored overnight at -80°C and used the following day. Assays were done in triplicate using 60 μg of membrane protein homogenate resuspended in 50 mm Tris HCl buffer, pH 7.4, and incubated with up to 300 nm [3H]kainate (NEN, Boston, MA). Reactions were initiated by the addition of membranes and persisted for 1 h on ice. Bound and free radioligands were separated by rapid-filtration with ice-cold assay buffer over Whatman (Maidstone, UK) GF/B filters presoaked in 0.3% polyethyleneimine. Radioactivity was determined by liquid scintillation spectrometry in biodegradable scintillation fluid. Nonspecific binding was determined in the presence of 1 mm unlabeled glutamate and represented 10-12% of the total counts for GluR6-wt at a 100 nm concentration.
Immunocytochemistry and confocal microscopy. For N-GluR6 and C-GluR6 staining, cells were washed in TBS and fixed in TBS plus 4% formalin 36 h after transfection. Nonspecific antibody interactions were blocked with 2% goat serum in TBS. Cell-surface GluR6 staining was examined using chick anti-N-terminal GluR6 (Aves Laboratories, Tigard, OR), followed by goat anti-chick Alexa 488 (Molecular Probes, Eugene, OR), as described previously (Fleck et al., 2003). Samples were then permeabilized with 0.1% Triton X-100, and total GluR6 content was determined by staining with rabbit anti-C-terminal GluR6/7 (Upstate Biotechnology, Lake Placid, NY) and goat anti-rabbit Alexa 568 (Molecular Probes). For localization studies with EYFP markers, C-GluR6 was visualized with goat anti-rabbit Alexa 568. Coverslips were then fixed onto slides using Vectashield (Vector Laboratories, Burlingame, CA). Samples were examined through a 100× oil immersion lens on an Olympus IX71 microscope (Olympus, Tokyo, Japan), an OZ confocal-laser scanning fluorescent microscope (Noran Instruments, Middletown, WI), or a dual-photon Zeiss (Thornwood, NY) inverted LSM 510 Meta NLO laser-scanning confocal microscope using a Plan-Apochromat 63×/1.4 oil differential interference contrast lens. Confocal images were acquired and analyzed on a Silicon Graphics (Mountain View, CA) O2 computer, and all final images were generated with Adobe Photoshop (Adobe Systems, San Jose, CA). Antibody concentrations were optimized by evaluating nonspecific staining of nontransfected HEK293 cells.
Cell-surface biotinylation and Western blot analysis. At 36 h after transfection, HEK293 cells were washed three times in ice-cold borate buffer (100 mm NaCl and 10 mm borate, pH 8.8) and subsequently incubated on ice twice for 10 min with 50 μg/ml EZ-link-sulfo-NHS-Biotin (Pierce). Biotin was removed, the reaction was quenched by the addition of 1 m NH4Cl, and the cells were washed three times in ice-cold TBS. Membranes were then scraped in 1 ml of radioimmunoprecipitation assay (RIPA) buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 10 mm sodium phosphate, 2 mm EDTA, and 0.2% sodium vanadate) supplemented with protease inhibitor mixture (Sigma) and solubilized for 1 h at 4°C. Nonsolubilized material was removed by centrifugation at 16,000 × g for 10 min. The solubilized protein concentration was determined by BCA assay, and 200 μg of protein was incubated with 100 μl of washed UltraLink NeutrAvidin Immobilized beads (Pierce) for 4 h at 4°C with rotation. Beads were subsequently washed twice with RIPA buffer and twice with TBS. Proteins were eluted by boiling in Laemmli buffer, subjected to SDS-PAGE, and transferred to nitrocellulose. The quantity of immunoreactive biotinylated GluR6 was evaluated by Western blot analysis using anti-C-terminal GluR6/7 (Upstate Biotechnology) and goat anti-rabbit alkaline phosphatase (Bio-Rad, Hercules, CA). Bands were visualized using ImmunStar AP substrate (Bio-Rad) and ECL Hyperfilm (Amersham Biosciences, Piscataway, NJ). Densitometric measurements were made using the Kodak EDAS290 digital camera and Kodak 1D Analysis System (Eastman Kodak, Rochester, NY). GluR6 surface expression was determined as a ratio of biotinylated GluR6 to total GluR6. Nontransfected cells were biotinylated and NeutrAvidin precipitated in parallel as an additional control.
Glycosylation. Thirty-six hours after transfection, cells were washed in PBS. Cells were harvested in 50 mm Na-acetate, 10 mm EDTA, and 0.2% Triton X-100, pH 5.5, for endoglycosidase-H (EndoH) or 250 mm NaPO4 and 0.2% Triton X-100, pH 7.5, for PNGaseF and supplemented with protease inhibitor mixture for 1 h at 4°C. Samples were centrifuged at 3000 × g for 4 min, and the pellet was discarded. Samples were incubated at 37°C overnight for EndoH or for 3 h for PNGaseF, and samples were reduced with β-mercaptoethanol as per the manufacturer's instructions. After the addition of Laemmli sample buffer, proteins were separated by SDS-PAGE and transferred to nitrocellulose. Immunoreactive GluR6 were visualized using anti-C-terminus GluR6 antibodies (Upstate Biotechnology).
Immunoprecipitation. HEK293 cells were cotransfected with R6R1C and GluR6-wt, R523G, T690G, or E738G (1:1) in 25 cm2 flasks 36 h before use. Cells were washed in PBS and then harvested and solubilized in 50 mm Tris HCl, pH 7.2, 2 mm EDTA, and 0.2% Triton X-100, supplemented with protease inhibitor mixture. After 1 h, samples were centrifuged at 3000 × g for 5 min, and the pellet was discarded. Fifty micrograms of solubilized protein were incubated with 0.5 μg of anti-C-GluRA overnight at 4°C. Immunoreactive product was isolated by precipitation on MagnaBind Anti-Rabbit Beads (Pierce) after extensive washes. Precipitated proteins were separated by SDS-PAGE on 8% gels.
Nonreducing-PAGE. HEK293 cells were washed in ice-cold TBS, and proteins were solubilized in 50 mm Tris HCl, pH 7.2, 20 mm NaCl, 5 mm EDTA, and 1% Triton X-100 supplemented with protease inhibitor mixture. After centrifugation at 10,000 × g for 20 min, supernatants were diluted in native sample buffer and separated by PAGE under nonreducing conditions on precast 3-7.5% Tris-glycine gradient gels (Jule Biotechnologies, Milford, CT). Immunoreactive bands were detected with C-GluR6 antibodies as described above.
Antibodies and other materials. Chick polyclonal anti-N-GluR6 was raised against the peptide fragment SDNKDSFYVSLYPDFSS, corresponding to amino acids 139-155 in the extracellular N-terminal domain of GluR6, coupled with keyhole limpet hemocyanin (Fleck et al., 2003). Rabbit polyclonal anti-C-GluR6/7 and anti-C-GluRA antisera were obtained from Upstate Biotechnology; Alexa-conjugated goat anti-chicken and anti-rabbit secondary antibodies were obtained from Molecular Probes. PNGaseF was purchased from New England Biolabs. EndoH was obtained from Roche Diagnostics (Indianapolis, IN). Other reagents were purchased from Sigma or RBI-Sigma (Natick, MA).
Results
GluR6-binding site mutations
Crystal structure analysis has determined that the polar side chains of residues R485, T655, and E705 in the GluR2 S1S2-binding pocket (Fig. 1A) contribute the principal ionic bonds with agonist and competitive ligands (Armstrong et al., 1998; Armstrong and Gouaux, 2000). To determine whether the homologous residues in GluR6 are, likewise, involved in ligand binding and agonist-induced channel opening, we identified the equivalent GluR6 residues from linear sequence alignments between AMPA and KA receptor subunits (Fig. 1B) and tested various substitutions at these positions in subsequent experiments. The arginine (R485) residue in the GluR2 S1 segment was identified as R523 in GluR6. The threonine (T655) and glutamate (E705) residues in the GluR2 S2 segment were identified as T690 and E738, respectively, in GluR6. Site-directed mutagenesis was used to produce the GluR6 glycine mutants R523G, T690G, and E738G; corresponding alanine (A) mutants and the more conservative E738D mutant were also produced. Wild-type and mutant GluR6 receptors were expressed in HEK293 cells and examined by a variety of biochemical and functional measures.
Figure 1.

Linear sequence alignment of AMPA and KA receptors. A, GluR2 and equivalent GluR6 agonist-binding residues are shown making the principal ionic interactions with KA (cyan) (Armstrong et al., 1998). Oxygen (red) and nitrogen (blue) atoms and hydrogen bonds (green dashed lines) are shown. B, Linear amino acid sequence alignments of AMPA and KA subunits indicating the positions and conservation of putative ligand-binding residues (BS) examined by site-directed mutagenesis of GluR6.
[3H]Kainate binding
Radioligand binding studies were performed to examine the effects of amino acid substitutions at the putative agonist-binding residues. Membranes were isolated from HEK293 cells expressing recombinant GluR6-wt or R523G, T690G, E738G, or E738D mutant receptors. Saturation binding analysis for [3H]kainate binding at GluR6-wt yielded a KD of 36 nm and a Bmax of 3.8 pmol/mg protein (Fig. 2A), consistent with previous reports (Bettler et al., 1991; Sommer et al., 1992; Tygesen et al., 1994; Zhou et al., 1997; Keinanen et al., 1998). Binding to the mutant receptors was assessed at 100 nm [3H]kainate relative to GluR6-wt. Under these conditions, GluR6-wt specifically bound 19,100 dpm (nonspecific binding, 2200 dpm) of [3H]kainate. No specific [3H]kainate binding was detected for the R523G, T690G, or E738G mutant (Fig. 2B). Only trace-specific binding was detectable to the E738D mutant (specific binding, ≈500 dpm). Western blot analysis confirmed that the assayed membrane samples contained immunoreactive GluR6 proteins of expected molecular mass (114 kDa) at expression levels comparable with GluR6-wt (Fig. 2B, inset). As an additional control of the binding assay, we tested the nonfunctional D776T mutant, which we found previously is tonically desensitized and retained in the ER (Fleck et al., 2003). At the 100 nm concentration, specific [3H]kainate binding was 5300 dpm (∼28% of GluR6-wt). Saturation binding analysis revealed a KD of 48 nm for the D776T mutant, comparable with GluR6-wt. Thus, we conclude the R523G, T690G, or E738G mutation effectively eliminated KA binding in the nanomolar range to GluR6.
Figure 2.
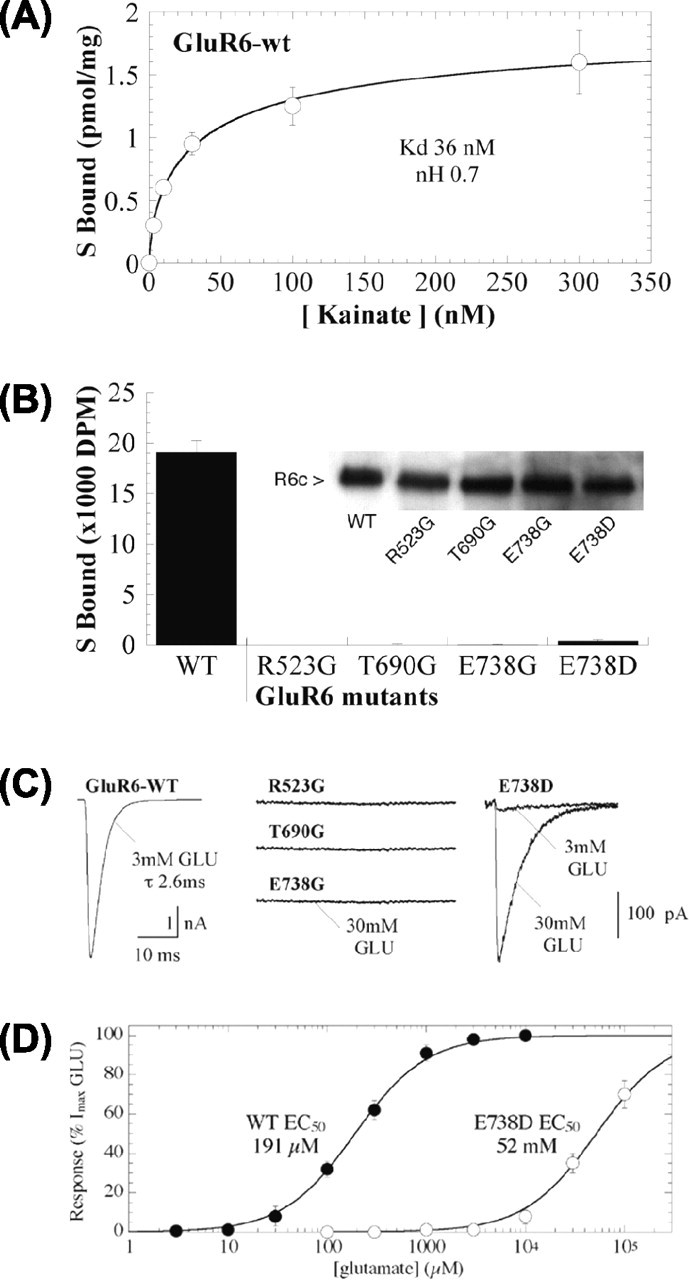
[3H]Kainate binding and functional assessment by patch-clamp recording of GluR6-binding site mutants. A, Saturation analysis of [3H]kainate binding to GluR6-wt expressed in HEK293 cells yielded a KD of 36 nm and a Bmax of 3.8 pmol/mg protein. Specific binding was determined in the presence (non specific) and absence (total) of 1 mm glutamate.B, Specific binding of 100 nm [3H]kainate to GluR6-wt, R523G, T690G, E738G, and E738D (60 μg of protein). Western blots confirmed similar amounts of C-GluR6 immunoreactivity in 25 μg of protein used for the binding assays (inset). C, Outside-out patches were pulled from HEK293 cells expressing GluR6-wt, R523G, T690G, or E738G, voltage clamped at -70 mV, and tested by ultrafast application of 3 or 30 mm glutamate (GLU). For GluR6-wt, 3 mm glutamate was saturating and elicited large peak currents (2.8 ± 0.4 nA). No currents were detected in patches from cells expressing R523G, T690G, or E738G. Application of 3 or 30 mm glutamate demonstrates that E738D receptors are functional, whereas 3 mm glutamate is no longer saturating for this mutant. D, Dose-response curve showing the rightward shift of the EC50 value (>100-fold) for the mutation E738D compared with GluR6-wt in peak response to glutamate.
Functional studies
Because the radioligand binding studies are limited to nanomolar agonist concentrations, we proceeded with functional analyses to determine the extent to which the mutations altered agonist-induced channel opening. Recombinant GluR6 mutant receptors were expressed in HEK293 cells and examined by outside-out patch-clamp recording with ultrafast application of 3 mm glutamate or 300 μm KA. Neither glutamatenor KA-evoked currents were observed for any of the glycine-substituted mutant receptors (R523G, T690G, or E738G) (Fig. 2C); peak current amplitudes were 2.8 ± 0.4 nA (n = 22) for GluR6-wt in parallel transfections (Fig. 2C). Indeed, we have been unable to measure any currents from these mutant receptors up to 100 mm glutamate in patch or whole-cell configurations or after treatment with 10 μm concanavalin A to enhance the measurement of whole-cell currents. These results suggest an absolute requirement for each of these three residues for ligand binding and GluR6 function. Additional studies were performed to clarify the extent of agonist interaction with the more conservative E738D mutant. In this case, 3 mm glutamate elicited small but measurable currents in patches from E738D-expressing cells (58 ± 17 pA; n = 7) (Fig. 2C, right), whereas application of 30 mm glutamate to the same patches stimulated much larger currents (841 ± 239 pA). Further dose-response analysis revealed that 30 mm glutamate was nonsaturating for E738D, but rather approached the EC50 value (52 mm) (Fig. 2D). Thus, the shortened side chain of the mutant E738D results in a dramatic shift in the EC50 value compared with GluR6-wt (EC50 = 200 μm) (Fleck et al., 2003).
Surface expression
Western blot analysis with antibodies raised against the GluR6 C-terminal domain indicated that the nonfunctional GluR6-binding site mutants are expressed as full-length, immunoreactive GluRs (Fig. 2B, inset). Nonetheless, we have previously reported that other nonfunctional mutants are retained intracellularly by an ER retention mechanism (Fleck et al., 2003). We therefore examined the cellular expression of the GluR6-binding site mutants by immunofluorescence and confocal microscopy. Transfected HEK293 cells expressing GluR6-wt or GluR6 mutants R523G, T690G, E738G, or E738D were fixed and stained with antibodies raised against an extracellular N-terminal domain to label receptors at the cell surface. Cells were then permeabilized and counterstained with antibodies to an intracellular C-terminal domain. GluR6-wt receptors were expressed robustly on the cell surface as indicated by intense N-GluR6 immunofluorescence; intracellular C-GluR6 labeling is also evident (Fig. 3A, top left). Nontransfected cells and controls omitting primary antibodies were uniformly negative (data not shown). The staining pattern of cells expressing the E738D mutant closely resembled that of GluR6-wt (Fig. 3A). In contrast, N-GluR6 labeling on the cell surface was entirely negative for the R523G, T690G, and E738G mutants, whereas C-GluR6 antibodies intensely labeled an intracellular compartment reminiscent of the ER (Fig. 3A). To confirm that the observed trafficking defect was not an artifact of recombinant expression in non-neural cells, we also examined the subcellular localization of GluR6-wt and T690G in transfected rat primary astrocytes. The lack of cell-surface expression and subcellular distribution pattern were reproduced in T690G-transfected astrocytes (Fig. 3B, right). Staining of a parallel control transfection demonstrated that the cell surface and intracellular localization of GluR6-wt (Fig. 3B, left) was similar in astrocytes to that observed in HEK293 cells (see supplemental Figs. S1 and S2, available at www.jneurosci.org as supplemental material). Thus, the failure of the GluR6-binding-deficient mutants to forward traffic to the cell surface is not restricted to HEK293 cells but also occurs in neural cells that can express native GluRs. These results indicate that, unlike GluR6-wt and E738D, but similarly to other nonfunctional mutants, the glycine-substituted-binding site mutants do not traffic to the cell surface and are therefore not amenable to electrophysiological studies.
Figure 3.

Cell-surface expression of the GluR6-binding site mutants. A, Confocal images of extracellular N-GluR6 (green) and intracellular C-GluR6 (red) immunofluorescence staining in HEK293 cells expressing GluR6-wt and GluR6-binding site mutants. N-GluR6 staining before cell membrane permeabilization demonstrates robust surface expression of GluR6-wt and E738D. In contrast, the absence of N-GluR6 staining indicates that R523G, T690G, and E738G are not present at the cell surface. C-GluR6 staining was evident for GluR6-wt and GluR6 mutants after permeabilization. Staining of nontransfected controls was negative. B, Primary cultures of rat astrocytes transfected with GluR6-wt or T690G were stained for N- and C-GluR6 as above. C, Biotinylated cell-surface proteins (50 μg) were precipitated with NeutrAvidin beads. Only trace amounts, if any, of R523G, T690G, and E738G were found at the cell surface. GluR6-wt and E738D were both isolated in the biotinylated fraction. The percentage of cell-surface expression of immunoreactive GluR6 was determined by densitometry using 10 μg of total protein. The blot is representative of three separate experiments. IB, Immunoblot; IP, immunoprecipitate. D, Biotinylation studies, using GluR1-wt as a control, revealed that a homologous GluR1-binding site mutant, R499G, also fails to reach the cell surface.
To quantify the extent of cell-surface expression of the GluR6-binding site mutants, we biotinylated the cell-surface proteins of transfected HEK293 cells and isolated the biotin-linked proteins by precipitation with NeutrAvidin-linked beads. The GluR6 content of the precipitates was evaluated by Western blotting with C-GluR6 antibodies. GluR6 cell-surface expression relative to total expression was determined by densitometric analysis. Consistent with immunofluorescence results, GluR6-wt and the E738D mutant were expressed in the biotinylated surface fraction at levels of 56 and 63% of totals, respectively, whereas R523G, T690G, and E738G were either not detected or present in very low abundance in the biotinylated surface fractions (Fig. 3C). These results support the immunofluorescence studies indicating that the glycine-substituted GluR6-binding site mutants do not traffic to the cell surface.
To determine whether AMPA receptor trafficking is regulated in a similar manner, we evaluated the effect of mutating R499 to G in the GluR1 AMPA receptor (equivalent to R523G in GluR6). Cell-surface biotinylation studies revealed that GluR1-wt receptors were expressed in the biotinylated surface fraction at levels of 19% of the totals, whereas R499G was not detected in the biotinylated surface fraction (Fig. 3D). Neither could any agonist-evoked responses be detected in functional studies with R499G (data not shown). These results suggest a general mechanism exists to prevent trafficking of both nonfunctional AMPA and KA receptors.
ER retention
Trafficking of multimeric proteins to the cell surface is tightly controlled. The ER is the site of synthesis and maturation of proteins destined for secretion or insertion into the plasma membrane, including receptors and ion channel proteins. Numerous quality control mechanisms exist to retain ER-resident proteins and immature, monomeric, or misfolded proteins, and thus to ensure that only properly folded and fully assembled oligomeric proteins gain exit from the ER into the secretory pathway (Deutsch, 2002, 2003; Ellgaard and Helenius, 2003; Wanamaker et al., 2003). The most commonly observed ER quality control mechanisms involve the association of newly synthesized proteins with ER chaperones and folding enzymes such as calnexin, calreticulin, BiP/GRP94, protein disulfide isomerase, ERp57, and ERp72 (Reddy and Corley, 1998; Ellgaard et al., 1999; Trombetta and Parodi, 2003). These chaperones assist in protein folding, maturation, and assembly and serve as retention factors for the immature proteins as they are being processed. We examined the intracellular localization of the binding site mutants to delineate where forward trafficking was disrupted. GluR6-wt, R523G, T690G, E738D, and E738G were individually cotransfected with the ER marker EYFP-ER in HEK293 cells. After fixation and permeabilization, immunoreactive GluR6 was detected with anti-C-terminal GluR6 antibodies. GluR6-wt and E738D exhibited their characteristic diffuse cellular distribution including prominent cell-surface expression (Fig. 4A, left panels). The staining patterns of the binding-deficient mutants were reticulated and colocalized exclusively with the EYFP-ER marker, suggesting that they do not forward traffic beyond the ER (Fig. 4A, right panels). In contrast, the absence of colocalization with a EYFP-Golgi marker indicated that the GluR6-binding site mutants do not proceed to the Golgi (Fig. 4B).
Figure 4.

ER retention and glycosylation state of GluR6-binding site mutants. A, Confocal images of intracellular C-GluR6 (red) immunofluorescence staining in HEK293 cells coexpressing the EYFP-ER marker and GluR6-wt or GluR6-binding site mutants. GluR6-wt and E738D demonstrate robust surface staining that is not colocalized with EYFP-ER. In contrast, R523G, T690G, and E738G are not present at the cell surface and are colocalized entirely with EYFP-ER. The merged images show the overlapping localization of C-GluR6 and EYFP-ER markers. Identical results were obtained with antibodies to other ER markers, calnexin and calreticulin. B, Representative confocal images of the GluR6-wt (top) or the GluR6-binding-deficient mutant R523G (bottom) cotransfected with the EYFP-Golgi marker. The merged image of immunofluorescence staining demonstrates the lack of colocalization of R523G (red) and the EYFP-Golgi marker (green). C, Triton X-100 (0.5%) solubilized lysates of HEK293 cells transfected with GluR6-wt or GluR6-binding site mutants were subjected to EndoH digest. Western blot analysis of control (Gly) and EndoH-digested (Cut) samples (15-30 μg of protein) reveals that R523G, T690G, and E738G are entirely EndoH sensitive. In contrast, the majority of GluR6-wt and E738D are EndoH insensitive. D, Parallel digests demonstrate PNGaseF and EndoH deglycoslate R523G, T690G, and E738G to the same extent, confirming their immature N-linked glycosylation state. The blots are representative of at least three separate experiments.
To confirm and quantify the ER retention of the GluR6-binding site mutants, we further examined their glycosylation state. GluR6 contains nine N-linked glycosylation sites (Everts et al., 1997). EndoH cleaves N-linked high-mannose-containing sugars on immature glycoproteins found in ER localized proteins. Further glycoprotein maturation to EndoH-resistant carbohydrates is achieved in the Golgi. Thus, glycoprotein sensitivity to EndoH serves as an indicator of subcellular localization. PN-GaseF, on the other hand, cleaves all Asn-glycans and thus serves as a control for full deglycosylation of Asn-linked glycoproteins. After transfection, we examined the extent of glycosylation of GluR6-wt and the binding site mutants in total cell proteins after membrane solubilization by treatment with the EndoH and PN-GaseF. Immunoblots with C-terminal GluR6 antibody indicate that monomeric, denatured GluR6-wt and E738D mutants are predominantly an ∼114 kDa band (Fig. 4C). Treatment of GluR6-wt and E738D with EndoH resulted in two bands of ∼111 and 94 kDa (Fig. 4C), whereas PNGaseF treatment yielded a single species of 94 kDa (Fig. 4D). The majority of GluR6-wt and E738D (84 and 85%, respectively) were insensitive to EndoH digestion, indicating that they are predominately mature glycoproteins. In contrast, treatment of R523G, T690G, and E738G with EndoH resulted in the complete shift of an immunoreactive ∼114 kDa band to a single band of 94 kDa (Fig. 4C). PNGase digest of GluR6-wt and all of the mutant GluR6-binding site proteins yielded a single band of 94 kDa representing the completely deglycosylated form of GluR6 (Fig. 4D). The equivalent deglycosylations by EndoH and PN-GaseF of R523G, T690G, and E738G reflect their immature glycosylation state and indicate that the GluR6-binding site mutants are not processed through the medial Golgi. Thus, immunofluorescence, biotinylation, and enzymatic studies all indicate the binding-deficient mutants are entirely retained in the ER.
Alanine mutants
As an additional test of the structural elements mediating ER retention of binding site mutant proteins, we tested two additional alanine mutants, R523A and E738A. Specifically, there was some concern that the added flexibility conferred by glycine substitutions might uniquely disrupt the local binding site structure to impact protein folding. This seems unlikely because adjacent substitutions of E524G or K525G are perfectly well tolerated (Fleck et al., 2003). Indeed, the R523A and E738A mutants mimicked the glycine substitutions in all respects (data not shown). N-GluR6 surface staining was uniformly negative, whereas C-GluR6 staining was intense and entirely consistent with ER localization. Likewise, biotinylation studies failed to resolve any immunoreactive R523A or E738A at the cell surface, whereas glycosidase digestion indicated these mutants were entirely EndoH-sensitive immature glycoproteins. Thus, ER retention is not unique to the glycine substitutions but occurs in other loss-of-function mutations as well.
GluR oligomerization
Numerous studies indicate that efficient and appropriate oligomerization is required for robust cell-surface expression of multimeric proteins (Deutsch, 2002; Ma and Jan, 2002; McIlhinney et al., 2003; Wanamaker et al., 2003). Indeed, one of the quality control functions of the ER is to retain unassembled subunits. The principal mechanism to ensure the proper oligomerization of multisubunit receptors and ion channels involves ER retention/retrieval via interactions with the coatamer protein (COPI) complex on recycling vesicles from Golgi (Letourneur et al., 1995). The best known ER retention/retrieval signals are the KDEL motif in many ER-resident proteins and dibasic di-lysine (e.g., KKXX) or arginine (RXR) motifs (Teasdale and Jackson, 1996; Zerangue et al., 1999, 2001). Oligomeric proteins assembled in the proper conformation escape ER retention by association and masking of their ER retention/retrieval motifs, whereas improper conformations leave these motifs exposed to COPI-mediated retrieval. Such mechanisms ensuring that only proper oligomeric forms are expressed on the cell surface were shown recently to regulate trafficking of GluR6 KA receptors (S. Yan et al., 2004), NMDA receptors (Okabe et al., 1999; Standley et al., 2000; Scott et al., 2001), nicotinic acetylcholine receptors (Keller et al., 2001), serotonin type-3 receptors (Boyd et al., 2002), K+ channels (Zerangue et al., 1999, 2001; Hough et al., 2000), Ca2+ channels (Bichet et al., 2000), and various G-protein-coupled receptors (Margeta-Mitrovic et al., 2000; Calver et al., 2001; Chan et al., 2001; Pagano et al., 2001; Grunewald et al., 2002).
To determine whether ER retention of the glycine-substituted mutants results from an inability to multimerize, we examined the ability of the GluR6-binding site mutants to assemble with a chimera consisting of the N-terminal domain of GluR6 through M4 and the intracellular C terminus of GluR1 (R6R1C). R6R1C serves as a useful marker because it contains all of the determinants critical to GluR6 self-assembly (Leuschner and Hoch, 1999; Ayalon and Stern-Bach, 2001) and also allows for isolation of immunodetectable heteromers. Specifically, the R6R1C was coexpressed with GluR6-wt, R523G, T690G, or E738G and immunoprecipitated with anti-C-GluR1 antibodies. The precipitated proteins were then probed by Western blot analysis using anti-C-GluR6 antibodies to detect proteins containing an intact GluR6 C terminus heteromerized with R6R1C. After cotransfection with R6R1C, Western blot analysis using C-terminus GluR6 antibody demonstrated that GluR6-wt, R523G, T690G, and E738G were all immunoprecipitated at similar levels by the C-GluR1 antibody (Fig. 5A, left). The reciprocal experiment in which proteins were immunoprecipitated with anti-C-GluR6 and detected with anti-C-GluR1 produced correspondingly appropriate results (data not shown).
Figure 5.
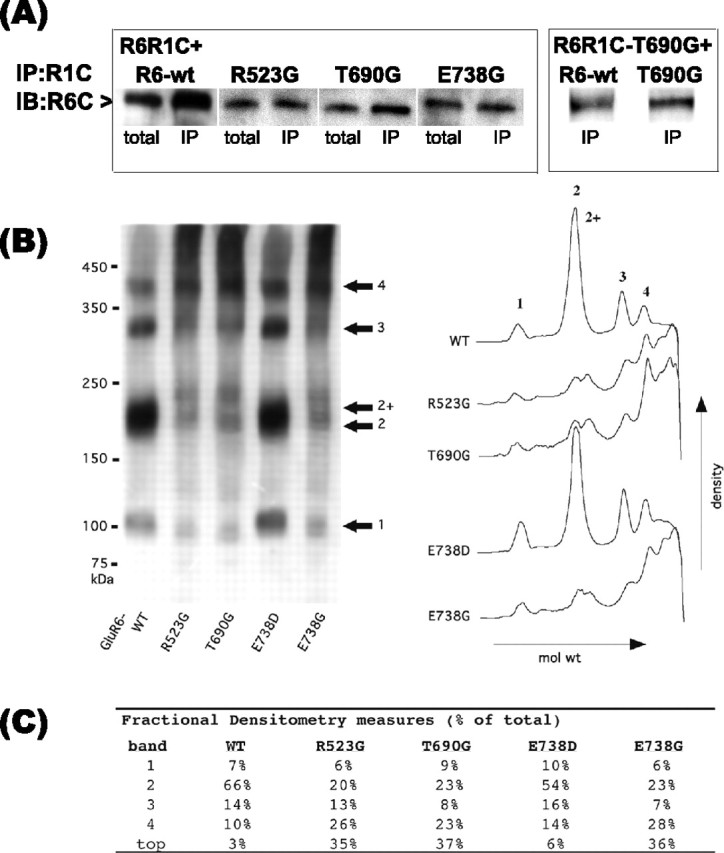
GluR6-binding site mutant oligomerization. A, Solubilized protein (50 μg) from cotransfection of R6R1C with GluR6-wt, R523G, T690G, or E738G (1:1) or R6R1C-T690G with GluR6-wt or T690G was immunoprecipitated (IP) with rabbit C-GluR1 antibodies (left). Washed precipitates subjected to Western blot analysis with C-GluR6 antibodies demonstrate that the GluR6-binding site mutants and GluR6-wt oligomerize with R6R1C. Control immunoprecipitates of R6R1C and GluR6-wt transfected alone were negative. The blot is representative of duplicate experiments. IB, Immunoblot. B, Native-PAGE densitometric analysis. Total cell proteins solubilized with 1% Triton X-100 were separated by PAGE on 3-7.5% Tris-glycine gradient gels under nonreducing conditions. C-GluR6-immunoreactive proteins segregated to four prominent bands having approximate molecular masses of 100, 200, 300, and 400 kDa, consistent with GluR6 monomers, dimers, trimers, and tetramers. B, C, Densitometric analysis indicating relative amounts of immunoreactive proteins of varying molecular weight. Relative intensity differences might reflect either a greater prevalence of large nonfunctional mutant receptor complexes (R523G, T690G and E738G) or indicate that the mutant receptor complexes are more stable than their functional counterparts. Relative band intensities are given combining the two putative dimeric forms.
The requirements for subunit association were further assessed by studies using the R6R1C construct also containing the T690G mutation R6R1C-T690G. Immunoprecipitation studies demonstrated that GluR6-wt and T690G assembled with R6R1C-T690G (Fig. 5A, right). These immunoprecipitation studies confirm that the C terminus of GluR6 is not required for GluR6 subunit association. Furthermore, the assembly of T690G with R6R1C-T690G demonstrates oligomeric association of two binding-deficient subunits, implying that oligomerization occurs when both participating subunits are binding deficient and nonfunctional. The immunoprecipitation studies with the R6R1C constructs do not distinguish between dimer and higher-order oligomer formation, although one might expect monomers and dimers would be preferentially retained in the ER. To test this, we further probed the assembly status of homomeric binding-deficient GluR6 mutants by native Western blot analysis. The high-molecular weight species detected under nonreducing conditions are consistent with the assembly of GluR6-wt and E738D into oligomers (Fig. 5B). Although we cannot exclude that the higher-molecular weight bands might reflect the interaction of GluR6 with other accessory proteins, the apparent molecular weights of the immunoreactive bands are multiples, suggesting the formation of GluR6 dimers, trimers, and tetramers. The nonfunctional binding-deficient mutants R523G, T690G, and E738G display a similar banding pattern, suggesting that they form these same higher-order oligomeric complexes. Two separate and distinct bands resolved around 200-240 kDa may represent two classes of GluR6 dimers bearing post-translational modifications or dimers associated with unknown accessory protein(s). The double dimer peaks existed for all constructs but were less well resolved for GluR6-wt and E738D. Densitometric analysis (Fig. 5B,C) indicated that ∼30% of total nonfunctional mutant proteins remained near the top of the gel, likely as aggregates or multiprotein complexes, whereas the putative dimeric forms were reduced by an equivalent amount compared with GluR6-wt or E738D. In light of the glycosylation and immunocytochemistry studies, it appears that the homomeric GluR6-binding-deficient subunits are fully assembled and retained in the ER regardless of their monomeric or oligomeric assembly status.
As an additional test, we next used a functional assay to determine whether the binding site mutants can contribute to functional oligomers, which traffic to the cell surface. Toward this end, we generated the M2-edited [R] forms of GluR6-wt, E738D, and E738G and coexpressed them with GluR6-wt[Q]. The Q/R editing site in M2 lines the ion channel pore conferring distinct ion selectivity and rectification properties on GluRs (Bowie and Mayer, 1995; Seeburg et al., 1998). Edited GluRs contain an arginine (R), whereas unedited GluRs contain glutamine (Q) at the analogous position (Q621 in GluR6). GluRs composed of unedited Q-containing subunits are Ca2+ permeable and subject to voltage-dependent block by polyamines such as spermine. In heteromers containing both Q and R isoforms, the R form dominates, the receptors become insensitive to spermine block, and the I-V relationship is therefore linearized. The R form thus serves as an electrophysiological tag in heteromerization studies. We coexpressed GluR6-wt[R], E738D[R] or E738G[R] with the Q-form of GluR6-wt (GluR6-wt[Q]). Patch-clamp recordings of homomeric GluR6-wt[Q], in the presence of 30 μm spermine in the internal solution, yielded the expected inwardly rectifying I-V relationship (Fig. 6A). Cotransfection of the Q and R forms of GluR6-wt (1:2) resulted in a nearly linear I-V relationship, indicative of the heteromerization of the Q and R forms of GluR6-wt. As a quantitative index of subunit heteromerization, a conductance ratio was determined from the ratio of current amplitudes measured at +60 and -60 mV. In this way, a linear I-V curve is represented by a value of 1, whereas inward or outward rectification yield values <1 or >1, respectively. The rectification ratio of homomeric GluR6-wt[Q] was 0.05 ± 0.01 (n = 10), indicating strong inward rectification. In contrast, the rectification ratio of GluR6-wt[Q] plus GluR6-wt[R] (1:2) was 0.87 ± 0.02 (n = 7), reflecting a nearly linear I-V curve after heteromerization of the [Q] and [R] forms of GluR6-wt (Fig. 6B, Table 1). In the same way, functional heteromeric receptors composed of GluR6-wt[Q] plus E738D (1:2) or GluR6-wt[Q] plus E738G[R] (1:2) were clearly evident from the linearization of the I-V curves and an increased rectification ratio of 0.55 ± 0.08 (n = 7) or 0.34 ± 0.09 (n = 12), respectively (Fig. 6B, Table 1). The reduction of voltage-dependent block indicates that E738D[R] and E738G[R] form functional heteromers with GluR6-wt[Q] and that these heteromers can traffic to the cell surface. Because biotinylation studies demonstrate that E738G[R] by itself, like E738G[Q], does not traffic to the cell surface (data not shown), these results indicate that assembly into a functional heteromer partially overcomes the ER retention of E738G.
Figure 6.
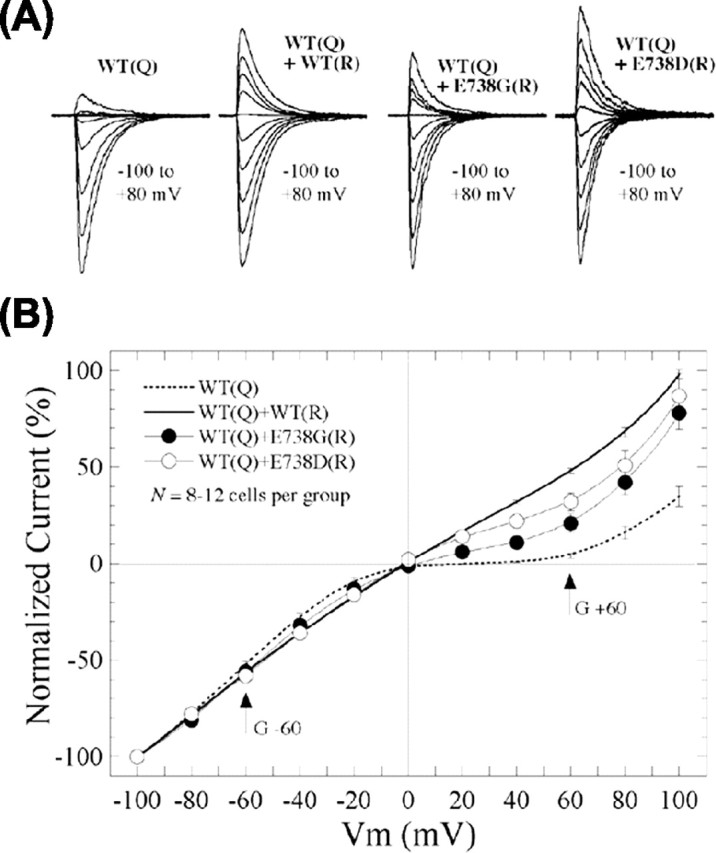
Coassembly rescues function and trafficking of binding site mutants. A, Representative glutamate-evoked currents at holding potentials from -100 to +80 mV in patches from HEK293 cells cotransfected with GluR6-wt[Q] and R-forms of GluR6-wt, E738G, or E738D (1:2). B, I-V plots were constructed from compiled data of homomeric GluR6-wt[Q] and GluR6-wt[Q] cotransfected with GluR6-wt, E738G, or E738D (n ≥ 7). Inward rectification of homomeric GluR6-wt[Q] was relieved to varying degrees by coexpression of GluR6-wt, E738G, or E738D, indicating the formation and appearance of functional oligomers at the cell surface.
Table 1.
Functional analysis of GluR6 [Q]/[R] oligomers
|
|
Rectification ratio |
IPeak (nA) |
τDesensitization (ms) |
n |
|---|---|---|---|---|
| GluR6-wt[Q] | 0.05 ± 0.01 | 3.3 ± 0.4 | 3.0 ± 0.2 | 10 |
| GluR6-wt[Q]/[R] | 0.87 ± 0.02 | 2.1 ± 0.4 | 3.1 ± 0.3 | 7 |
| GluR6-wt[Q]/E738D[R] | 0.55 ± 0.08 | 1.1 ± 0.2 | 2.8 ± 0.2 | 7 |
| GluR6-wt[Q]/E738G[R] |
0.34 ± 0.09 |
1.1 ± 0.3 |
2.8 ± 0.3 |
12 |
The rectification ratio is the current amplitude at + 60 mV/amplitude at −60 mV. Mean maximal current amplitudes (IPeak) and exponential time constants of desensitization (τDesensitization) are averages for 7-12 patches. HEK293 cells were cotransfected with [Q]/[R] at a 1:2 ratio.
Interestingly, the mean maximal current amplitudes and rectification ratios after coexpression of GluR6-wt[Q] with either E738D[R] or E738G[R] were reduced by approximately one-half compared with GluR6-wt[Q]/[R] (Table 1). Because the rates of desensitization (τdes) were unaltered (Table 1), the changes in amplitude and voltage-dependent block presumably reflect a combination of decreased efficiency in heteromer formation and reduced agonist efficacy when heteromers are formed from GluR6-wt[Q] and either E738D[R] or E738G[R]. Specifically, the presence of binding-deficient subunits within a functional heteromer would be expected to lower agonist-induced ion conductance with a resultant net decrease in macroscopic current amplitudes (Rosenmund et al., 1998; Swanson et al., 2002). Additionally, the intermediate sensitivity to voltage-dependent block (as evidenced by the intermediate rectification ratios and reduced spermine affinity) suggests either the E738D[R] and E738G[R] subunits do not coassemble with GluR6-wt[Q] as efficiently as GluR6-wt[R] or the unbound subunits contribute less to rectification (see additional information in supplemental Fig. S3, available at www.jneurosci.org as supplemental material). In either case, these results do clearly demonstrate that the mutant subunits are able to coassemble with wild-type subunits to form functional heteromers that traffic to the cell surface, indicating the restoration of function is sufficient for ER export and surface expression.
Discussion
The present study demonstrated that agonist binding and consequent agonist-induced function were essentially eliminated by point mutations of the putative ligand-binding residue R523, T690, or E738 in GluR6. Concomitantly, N-GluR6 surface staining and cell-surface biotinylation studies indicated that glycine or alanine substitutions at these residues eliminated cell-surface expression of the mutant GluRs in HEK293 cells and primary astrocytes. Immunofluorescence colocalization and glycosylation studies indicated that the nonfunctional mutants are retained in the ER. Coimmunoprecipitation and native PAGE experiments confirmed that the mutants form oligomeric complexes, and functional studies indicated that they could be rescued from ER retention by restoration of function by coassembly with wild-type subunits. Together with our previous results on other loss-of-function mutations (Fleck et al., 2003), these results suggest that ER quality control includes a “functional” checkpoint to regulate KA receptor trafficking.
GluR6-binding residues
Crystal structure studies of a soluble construct of the S1S2-binding domain of GluR2 determined that critical interactions are formed between the backbone and side chains of residues R485 in S1 and T655 and E705 in S2 with KA and water molecules (Armstrong et al., 1998; Armstrong and Gouaux, 2000). Of these, the R485 (S1) and E705 (S2) residues are perfectly conserved in all AMPA and KA subunits. The T655 (S2) residue is also perfectly conserved, except for a serine substitution in KA1. These same residues are also mostly conserved in NMDA subunits. As such, our report is hardly surprising but confirms that mutations to the homologous residues in GluR6 (R523, T690 and E738) interfere with agonist-receptor interactions. This indicates the agonist-binding pocket is not atypical for GluR6 despite various amino acid substitutions and alignment gaps throughout the S1S2 domain.
The residue homologous to GluR2-R485 is perfectly conserved in all AMPA, KA, and NMDA subunits and may be the principal residue to bind during the initial docking step of agonist association (Abele et al., 2000; Armstrong and Gouaux, 2000). Mutagenesis studies confirm the equivalent residue is critical for agonist-induced function in AMPA (Uchino et al., 1992), KA (Ayalon and Stern-Bach, 2001), and NMDA (Laube et al., 1997; Anson et al., 1998) receptors. The residue homologous to GluR2-T655 is also conserved in AMPA, KA, and NMDA subunits, except KA1, which substitutes a serine, and NR1, which substitutes a valine at this position and confers its altered selectivity for glycine (Furukawa and Gouaux, 2003). The T655 interaction with agonist is underscored by alanine mutagenesis studies of both AMPA and NR2 receptors (Anson et al., 1998; Lampinen et al., 2002). Likewise, the residue homologous to GluR2-E705 is perfectly conserved in all AMPA and KA subunits and is critical to agonist-receptor interactions in AMPA receptors (Mano et al., 1996; Lampinen et al., 1998; Abele et al., 2000). Additional S1S2 domain residues are more or less conserved in GluRs, which may facilitate ligand binding directly or influence the subtle structure of the binding pocket in different subtypes.
Various alterations in side chain polarity, charge, or length of the ligand-binding-site residues impact radioligand binding to the KA-binding protein (Paas et al., 1996; Wo et al., 1999) and AMPA receptors (Kawamoto et al., 1997; Lampinen et al., 1998, 2002; Abele et al., 2000; Jouppila et al., 2002). In the S1 domain, the conservative GluR4-R507K mutation eliminated high-affinity antagonist binding (Jouppila et al., 2002), and the equivalent GluR6-R523K mutation reportedly increased the EC50 value for glutamate activation by >50-fold. In the S2 domain, the GluR4-E727D mutation alters agonist binding, whereas neutralizing the charge, E727A, eliminated AMPA and high-affinity antagonist binding (Lampinen et al., 1998; Jouppila et al., 2002). Similarly, we found that the reduction in side chain length in the GluR6-E738D mutant increased the EC50 value for glutamate activation by >20-fold, whereas parallel studies found no detectable responses or binding to E738G.
Loss-of-function mutations
In previous studies (Fleck et al., 2003), we reported that functionally defective GluR6 mutant receptors and related GluR1 mutant receptors displayed reduced or no surface expression attributable to prominent ER retention. The cause of the trafficking errors was unclear. We suggested the ER retention might reflect a functional checkpoint in ER quality control but could not entirely exclude other possibilities. The present studies were therefore conceived in part to determine whether the ER retention phenotype was, in fact, the result of loss of function rather than any structural defects that might lead to a failure of oligomeric assembly. Glycine or alanine substitutions at the principal ligand-binding residues in GluR6 (R523, T690, and E738) reduced agonist binding and agonist-evoked currents to undetectable levels. As predicted, these nonfunctional receptors were almost entirely retained in the ER as determined by EndoH digests and immunofluorescence colocalization with ER markers. Virtually no receptor could be detected on the cell surface by N-terminal staining or surface biotinylation assays. These same loss-of-function mutations apparently did not prohibit assembly of oligomeric GluR6 receptors, and indeed they could be rescued by coexpression with GluR6-wt. In contrast, the E738D mutation, which reduced but did not eliminate glutamate binding, did not prevent ER export, mature glycosylation, or surface expression. Similar ER retention is reported for other nonfunctional mutations including GluR6-D776G/T (Fleck et al., 2003), GluR1-R499G, K500G, K507G (Fig. 3) (Fleck et al., 2003), KA2-T675A/E (Valluru et al., 2005), and GLR1-E770A in Caenorhabditis elegans (Grunwald and Kaplan, 2003) but not numerous other functional S1S2 mutants (Fleck et al., 2003), suggesting a general process exists in the ER to monitor the functional state of GluR channels in their fully assembled form.
Functional quality control
Mechanisms for ER retention generally involve chaperone interactions with structural domains that are exposed or buried in the process of protein folding and oligomerization. So how might GluR function be related to trafficking? Glutamate, the natural agonist, is prevalent intracellularly at 4-14 mm concentrations (Berger et al., 1977; Shank and Aprison, 1988), much of this associated with the ER (Meeker et al., 1989). Given the affinity for glutamate binding to GluRs is in the 50-250 nm range, it follows that nascent GluR subunits would rapidly isomerize to their glutamate-bound conformation, perhaps before assembly, and that oligomeric GluRs also would naturally exist in a bound conformation. Glutamate binding to the receptor causes at least two alternative conformational rearrangements, one associated with channel opening, the other with desensitization. Thus, GluR function could be monitored by the structural rearrangements associated with glutamate binding and gating. In this way, ER chaperone proteins might bind or release their cargo in a state-dependent manner, because a structural motif is either exposed or buried in the process of channel opening or desensitization. Alternatively, ion flux through the open channel might play a role in the process. Given intracellular conditions, the predominant ionic gradient between ER and cytosol should be Ca2+, which is sequestered in the ER and permeable through GluRs, albeit in some cases weakly. Even trace Ca2+ leaking through the occasionally open channel might influence Ca2+-dependent chaper-one interactions within the microdomain of the cytosolic face of the channel pore (e.g., C-terminal domain) to accelerate ER export.
In either case, whether by structural rearrangement or ion flux through the channel, intracellular conditions are favorable to support ER quality control of GluRs at a functional level. Our results demonstrate that mutations that abolish agonist binding eliminate ER export and surface expression, whereas other mutations that merely reduce agonist binding are tolerated, at least to the extent they should still bind at low millimolar concentrations of intracellular glutamate. Because coexpression and native PAGE studies indicate the ER-retained nonfunctional subunits are oligomeric, the mechanism of retention must be downstream from the assembly process and may involve channel gating. Like-wise, the binding-deficient GluR6 mutants can contribute to the functional surface receptor population in complex with GluR6-wt subunits, indicating the restoration of function is sufficient to gain exit from the ER. Additional insights into the mechanism of ER retention will be gained from studies of other loss-of-function mutations targeting events downstream from agonist binding, including channel opening, desensitization, and ion flux through the channel pore.
Precedents and implications
The fundamental importance of ER quality control is clearly evident in cases of inborn errors of metabolism that affect secretory or plasma membrane proteins. Genetic mutations that cause even minor changes in protein structure can cause defects in protein trafficking either by ER retention of mature exportable proteins or by ER export of immature or inappropriately oligomerized intermediates (Kim and Arvan, 1998). Defects involving GluRs could lead to any number of neuropathologies from epilepsy to neurodegenerative disease. Yet, our understanding of ER quality control and ER storage diseases is incomplete. Few other cases have been described of regulation at the functional level, and these too involve cell-surface receptors that are retained in the ER and rescued by ligand binding. Examples include vasopressin V2 receptors (Morello et al., 2000), δ-opioid receptors (Petaja-Repo et al., 2002), and KATP-channel (F. Yan et al., 2004). Future studies will be informative to learn how cells monitor GluR function, or the function of other surface receptors, to understand how this process might go awry in cases leading to various neuropathologies.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant NS40347. We thank Drs. P. Seeburg and M. Mayer for the parent cDNA constructs, Dr. J. Mazurkiewicz for assistance with the confocal imaging on the LSM 510 Meta NLO laser-scanning confocal microscope, and Dr. T. Lindsley for the primary astrocyte cultures.
Correspondence should be addressed to Dr. Mark W. Fleck, Center for Neuropharmacology and Neuroscience, Albany Medical College, A-136, 47 New Scotland Avenue, Albany, NY 12208. E-mail: fleckm@mail.amc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252215-11$15.00/0
References
- Abele R, Keinanen K, Madden DR (2000) Agonist-induced isomerization in a glutamate receptor ligand-binding domain. J Biol Chem 275: 21355-21363. [DOI] [PubMed] [Google Scholar]
- Anson LC, Chen PE, Wyllie DJ, Colquhoun D, Schoepfer R (1998) Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J Neurosci 18: 581-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28: 165-181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, Gouaux E (1998) Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395: 913-917. [DOI] [PubMed] [Google Scholar]
- Ayalon G, Stern-Bach Y (2001) Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron 19: 103-113. [DOI] [PubMed] [Google Scholar]
- Berger SJ, Carter JC, Lowry OH (1977) The distribution of glycine, GABA, glutamate and aspartate in rabbit spinal cord, cerebellum and hippocampus. J Neurochem 28: 149-158. [DOI] [PubMed] [Google Scholar]
- Bettler B, Egebjerg J, Sharma G, Pecht G, Hermans-Borgmeyer I, Moll C, Stevens CF, Heinemann S (1992) Cloning of a putative glutamate receptor: a low affinity kainate-binding subunit. Neuron 8: 257-265. [DOI] [PubMed] [Google Scholar]
- Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M (2000) The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 25: 177-190. [DOI] [PubMed] [Google Scholar]
- Bowie D, Lange GD (2002) Functional stoichiometry of glutamate receptor desensitization. J Neurosci 22: 3392-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15: 453-462. [DOI] [PubMed] [Google Scholar]
- Boyd GW, Low P, Dunlop JI, Robertson LA, Vardy A, Lambert JJ, Peters JA, Connolly CN (2002) Assembly and cell surface expression of homomeric and heteromeric 5-HT3 receptors: the role of oligomerization and chaperone proteins. Mol Cell Neurosci 21: 38-50. [DOI] [PubMed] [Google Scholar]
- Calver AR, Robbins MJ, Cosio C, Rice SQ, Babbs AJ, Hirst WD, Boyfield I, Wood MD, Russell RB, Price GW, Couve A, Moss SJ, Pangalos MN (2001) The C-terminal domains of the GABAB receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J Neurosci 21: 1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Pang RT, Chow BK (2001) Functional segregation of the highly conserved basic motifs within the third endoloop of the human secretin receptor. Endocrinology 142: 3926-3934. [DOI] [PubMed] [Google Scholar]
- Deutsch C (2002) Potassium channel ontogeny. Annu Rev Physiol 64: 19-46. [DOI] [PubMed] [Google Scholar]
- Deutsch C (2003) The birth of a channel. Neuron 40: 265-276. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4: 181-191. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A (1999) Setting the standards: quality control in the secretory pathway. Science 286: 1882-1888. [DOI] [PubMed] [Google Scholar]
- Everts I, Villmann C, Hollmann M (1997) N-glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol Pharmacol 52: 861-873. [DOI] [PubMed] [Google Scholar]
- Fleck MW, Cornell E, Mah SJ (2003) Amino-acid residues involved in glutamate receptor 6 kainate receptor gating and desensitization. J Neurosci 23: 1219-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E (2003) Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J 22: 2873-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Asumssen H, Banker G (1998) Rat hippocampal neurons in low density culture. In: Culturing nerve cells (Banker G, Goslin K, eds), pp 339-390. Cambridge, MA: MIT.
- Grunewald S, Schupp BJ, Ikeda SR, Kuner R, Steigerwald F, Kornau HC, Kohr G (2002) Importance of the gamma-aminobutyric acid(B) receptor C-termini for G-protein coupling. Mol Pharmacol 61: 1070-1080. [DOI] [PubMed] [Google Scholar]
- Grunwald ME, Kaplan JM (2003) Mutations in the ligand-binding and pore domains control exit of glutamate receptors from the endoplasmic reticulum in C. elegans Neuropharmacology 45: 768-776. [DOI] [PubMed] [Google Scholar]
- Hough E, Beech DJ, Sivaprasadarao A (2000) Identification of molecular regions responsible for the membrane trafficking of Kir6.2. Pflügers Arch 440: 481-487. [DOI] [PubMed] [Google Scholar]
- Jouppila A, Pentikainen OT, Settimo L, Nyronen T, Haapalahti JP, Lampinen M, Mottershead DG, Johnson MS, Keinanen K (2002) Determinants of antagonist binding at the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit, GluR-D. Role of the conserved arginine 507 and glutamate 727 residues. Eur J Biochem 269: 6261-6270. [DOI] [PubMed] [Google Scholar]
- Kawamoto S, Uchino S, Xin KQ, Hattori S, Hamajima K, Fukushima J, Mishina M, Okuda K (1997) Arginine-481 mutation abolishes ligand-binding of the AMPA-selective glutamate receptor channel α1-subunit. Brain Res Mol Brain Res 47: 339-344. [DOI] [PubMed] [Google Scholar]
- Keinanen K, Jouppila A, Kuusinen A (1998) Characterization of the kainate-binding domain of the glutamate receptor GluR-6 subunit. Biochem J 330: 1461-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Ellisman M, Taylor P (2001) Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-subunit. J Biol Chem 276: 18384-18391. [DOI] [PubMed] [Google Scholar]
- Kim PS, Arvan P (1998) Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev 19: 173-202. [DOI] [PubMed] [Google Scholar]
- Lampinen M, Pentikainen O, Johnson MS, Keinanen K (1998) AMPA receptors and bacterial periplasmic amino-acid binding proteins share the ionic mechanism of ligand recognition. EMBO J 17: 4704-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampinen M, Settimo L, Pentikainen OT, Jouppila A, Mottershead DG, Johnson MS, Keinanen K (2002) Discrimination between agonists and antagonists by the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid-selective glutamate receptor. A mutation analysis of the ligand-binding domain of GluR-D subunit. J Biol Chem 277: 41940-41947. [DOI] [PubMed] [Google Scholar]
- Laube B, Hirai H, Sturgess M, Betz H, Kuhse J (1997) Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron 18: 493-503. [DOI] [PubMed] [Google Scholar]
- Lerma J (2003) Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci 4: 481-495. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Hennecke S, Demolliere C, Cosson P (1995) Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J Cell Biol 129: 971-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner WD, Hoch W (1999) Subtype-specific assembly of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits is mediated by their N-terminal domains. J Biol Chem 274: 16907-16916. [DOI] [PubMed] [Google Scholar]
- Ma D, Jan LY (2002) ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol 12: 287-292. [DOI] [PubMed] [Google Scholar]
- Mano I, Lamed Y, Teichberg VI (1996) A Venus flytrap mechanism for activation and desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. J Biol Chem 271: 15299-15302. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27: 97-106. [DOI] [PubMed] [Google Scholar]
- McIlhinney RA, Philipps E, Le Bourdelles B, Grimwood S, Wafford K, Sandhu S, Whiting P (2003) Assembly of N-methyl-d-aspartate (NMDA) receptors. Biochem Soc Trans 31: 865-868. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Swanson DJ, Hayward JN (1989) Light and electron microscopic localization of glutamate immunoreactivity in the supraoptic nucleus of the rat hypothalamus. Neuroscience 33: 157-167. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest 105: 887-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H (1999) Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J Neurosci 19: 7781-7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paas Y, Eisenstein M, Medevielle F, Teichberg VI, Devillers-Thiery A (1996) Identification of the amino acid subsets accounting for the ligand binding specificity of a glutamate receptor. Neuron 17: 979-990. [DOI] [PubMed] [Google Scholar]
- Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Meigel I, Lampert C, Stein T, Prezeau L, Blahos J, Pin J, Froestl W, Kuhn R, Heid J, Kaupmann K, Bettler B (2001) C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J Neurosci 21: 1189-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M (2002) Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J 21: 1628-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ (2004) NMDA receptors: subunit assembly and trafficking to the synapse. J Biol Chem 279: 9673-9676. [DOI] [PubMed] [Google Scholar]
- Reddy PS, Corley RB (1998) Assembly, sorting, and exit of oligomeric proteins from the endoplasmic reticulum. BioEssays 20: 546-554. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280: 1596-1599. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21: 3063-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH (1993) The TINS/TiPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci 16: 359-365. [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Higuchi M, Sprengel R (1998) RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev 26: 217-229. [DOI] [PubMed] [Google Scholar]
- Shank RP, Aprison MH (1988) Glutamate as a neurotransmitter. In: Glutamine and glutamate in mammals, Vol II (Kvamme E, ed), pp 3-19. Boca Raton, FL: CRC. [Google Scholar]
- Smith TC, Howe JR (2000) Concentration-dependent substrate behavior of native AMPA receptors. Nat Neurosci 3: 992-997. [DOI] [PubMed] [Google Scholar]
- Sommer B, Burnashev N, Verdoorn TA, Keinanen K, Sakmann B, Seeburg PH (1992) A glutamate receptor channel with high affinity for domoate and kainate. EMBO J 11: 1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28: 887-898. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O'Hara PJ, Heinemann SF (1994) Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron 13: 1345-1357. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Green T, Sakai R, Contractor A, Che W, Kamiya H, Heinemann SF (2002) Differential activation of individual subunits in heteromeric kainate receptors. Neuron 34: 589-598. [DOI] [PubMed] [Google Scholar]
- Teasdale RD, Jackson MR (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu Rev Cell Dev Biol 12: 27-54. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Parodi AJ (2003) Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol 19: 649-676. [DOI] [PubMed] [Google Scholar]
- Tygesen CK, Rasmussen JS, Jones SV, Hansen A, Hansen K, Andersen PH (1994) Stable expression of a functional GluR6 homomeric glutamate receptor channel in mammalian cells. Proc Natl Acad Sci USA 91: 13018-13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino S, Sakimura K, Nagahari K, Mishina M (1992) Mutations in a putative agonist binding region of the AMPA-selective glutamate receptor channel. FEBS Lett 308: 253-257. [DOI] [PubMed] [Google Scholar]
- Valluru L, Xu J, Zhu Y, Yan S, Contractor A, Swanson GT (2005) Ligand binding is a critical requirement for plasma membrane expression of heteromeric kainate receptors. J Biol Chem, 280: 6085-6093. [DOI] [PubMed] [Google Scholar]
- Wanamaker CP, Christianson JC, Green WN (2003) Regulation of nicotinic acetylcholine receptor assembly. Ann NY Acad Sci 998: 66-80. [DOI] [PubMed] [Google Scholar]
- Wo ZG, Chohan KK, Chen H, Sutcliffe MJ, Oswald RE (1999) Cysteine mutagenesis and homology modeling of the ligand-binding site of a kainate-binding protein. J Biol Chem 274: 37210-37208. [DOI] [PubMed] [Google Scholar]
- Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL (2004) Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem 279: 11096-11105. [DOI] [PubMed] [Google Scholar]
- Yan S, Sanders JM, Xu J, Zhu Y, Contractor A, Swanson GT (2004) A C-terminal determinant of GluR6 kainate receptor trafficking. J Neurosci 24: 679-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22: 537-548. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B (2001) Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci USA 98: 2431-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LM, Gu ZQ, Costa AM, Yamada KA, Mansson PE, Giordano T, Skolnick P, Jones KA (1997) (2S, 4 R)-4-methylglutamic acid (SYM 2081): a selective, high-affinity ligand for kainate receptors. J Pharmacol Exp Ther 280: 422-427. [PubMed] [Google Scholar]