

Abstract
The synaptic vesicle-associated cysteine-string protein (CSP) is important for synaptic transmission. Previous studies revealed multiple defects at neuromuscular junctions (NMJs) of csp null-mutant Drosophila, but whether these defects are independent of each other or mechanistically linked through J domain mediated-interactions with heat-shock cognate protein 70 (Hsc70) has not been established. To resolve this issue, we genetically dissected the individual functions of CSP by an in vivo structure/function analysis. Expression of mutant CSP lacking the J domain at csp null-mutant NMJs fully restored normal thermo-tolerance of evoked transmitter release but did not completely restore evoked release at room temperature and failed to reverse the abnormal intraterminal Ca2+ levels. This suggests that J domain-mediated functions are essential for the regulation of intraterminal Ca2+ levels but only partially required for regulating evoked release and not required for protecting evoked release against thermal stress. Hence, CSP can also act as an Hsc70-independent chaperone protecting evoked release from thermal stress. Expression of mutant CSP lacking the L domain restored neurotransmission and partially reversed the abnormal intraterminal Ca2+ levels, suggesting that the L domain is important, although not essential, for the role of CSP in regulating intraterminal Ca2+ levels. We detected no effects of csp mutations on individual presynaptic Ca2+ signals triggered by action potentials, suggesting that presynaptic Ca2+ entry is not primarily impaired. Both the J and L domains were also required for the role of CSP in synaptic growth. Together, these results suggest that CSP has several independent synaptic functions, affecting synaptic growth, evoked release, thermal protection of evoked release, and intraterminal Ca2+ levels at rest and during stimulation.
Keywords: neurosecretion, synapse, calcium, homeostasis, chaperone, vesicle fusion, synaptic plasticity
Introduction
The synaptic vesicle-associated cysteine-string protein (CSP) is critical for regulated neurotransmission, but its true physiological actions have been the subject of considerable controversy. Three different hypotheses have been proposed for CSP function. (1) Initially, it was suggested that CSP regulates the activity of presynaptic Ca2+ channels (Gundersen and Umbach, 1992), although it remains controversial whether CSP upregulates or downregulates Ca2+ channel activity (Gundersen and Umbach, 1992; Brown et al., 1998; Chamberlain and Burgoyne, 1998; Umbach et al., 1998; Zhang et al., 1998, 1999; Burgoyne et al., 2000; Graham and Burgoyne, 2000; Magga et al., 2000; Chen et al., 2002). (2) A direct role of CSP in exocytosis has been suggested by interactions with syntaxin and synaptotagmin (Nie et al., 1999; Wu et al., 1999; Evans and Morgan, 2002). This role is further supported by abnormal exocytosis of neuroendocrine cells overexpressing CSP and by Drosophila csp null mutants showing depressed transmitter release in the presence of abnormally increased Ca2+ levels (Brown et al., 1998; Zhang et al., 1998, 1999; Dawson-Scully et al., 2000; Graham and Burgoyne, 2000). (3) CSP could act as a use-dependent synaptic chaperone renaturing presynaptic proteins during continuous operation of the synaptic vesicle cycle (Fernandez-Chacon et al., 2004).
Like CSP knock-out mice (Fernandez-Chacon et al., 2004), adult csp null-mutant flies show a progressive neurodegeneration in the CNS, a progressive paralysis, and premature lethality (Zinsmaier et al., 1994). In addition, neuromuscular junctions (NMJs) of csp mutant Drosophila exhibit abnormal neurotransmitter release at 20°C, an additional deterioration of release at higher temperatures, and abnormal levels of intraterminal Ca2+ above 24°C (Umbach et al., 1994; Dawson-Scully et al., 2000). Because phenotypes that progressively deteriorate with increasing temperature or age are often attributable to impaired functions of chaperone proteins, the behavioral and synaptic defects of flies and mice support chaperone-like functions of CSP. However, whether all of these phenotypic defects are causally linked remains unresolved.
Previous biochemical and genetic studies suggest that the physiological functions of CSP are based on its molecular action as a cofactor of the classical chaperone heat-shock cognate protein 70 (Hsc70) (for review, see Chamberlain and Burgoyne, 2000; Zinsmaier and Bronk, 2001). In vitro, CSP binds and activates the ATPase activity of Hsc70 through its J domain, which is apparently essential and sufficient for ATPase stimulation (Braun et al., 1996; Chamberlain and Burgoyne, 1997b; Stahl et al., 1999; Zhang et al., 1999). Cooperative physiological roles of CSP and Hsc70 are further indicated by similar presynaptic defects in mutants of Drosophila csp and hsc70 (Umbach et al., 1994; Zinsmaier et al., 1994; Bronk et al., 2001). The cooperative action of CSP and Hsc70 may also require small glutamine-rich tetratricopeptide repeat protein (SGT), which interacts biochemically with Hsc70 and CSP (Tobaben et al., 2001).
Based on the well documented interaction of CSP with the ATPase Hsc70, it has been commonly assumed but not tested that CSP functions through its ability to interact with Hsc70. Accordingly, all of the functions of CSP should be mechanistically linked through a J domain mediated-interaction with Hsc70. To test the significance of J domain mediated-interactions for synaptic transmission and to gain additional insights into the causal relationship of the hypothesized synaptic functions of CSP, we conducted an in vivo structure/function analysis of CSP at nerve terminals of Drosophila NMJs. We also examined the role of the unique, highly conserved linker (L) domain of CSP, the function of which is currently unknown.
Materials and Methods
Generation of mutant constructs. All mutant constructs were made from the type II alternatively spliced csp cDNA (DCSP2) (Zinsmaier et al., 1990, 1994; Nie et al., 1999). The H45Q point mutation was constructed using a Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) with type II csp cDNA as template DNA and primers (5′-3′) GCACTCAAATACCAACCCGATAAGAACC and GGTTCTTATCGGGTTGGTATTTGAGTGC. The mutant cDNA was then EcoRI-restricted and subcloned into the cloning site of a pUAST P-element vector (Brand and Perrimon, 1993). For ΔJ-CSP, the cDNA was SalI restricted, and PCR products were made with SalI sites at the 5′ and 3′ ends and subcloned into a pKS vector. The mutant cDNA was then EcoRI/XhoI restricted and subcloned into the pUAST vector. For ΔL-CSP, the cDNA was SpeI/SalI restricted, and PCR products were amplified from the region upstream of the L domain with SpeI sites on the 5′ and 3′ ends and amplified from the region downstream of the L domain with a SpeI site on the 5′ end and a SalI site on the 3′ end. These PCR products were then subcloned into a pKS vector, EcoRI/XhoI restricted, and subcloned into a pUAST vector. For P-element-mediated germ-line transformation, the J and L deletion pUAST plasmids were injected together with the helper plasmid pUChsπΔ2-3 into 30- to 60-min-old w1118 embryos by standard methods (Spradling, 1986). The pUAST-H45Q-CSP plasmid was injected into 30- to 60-min-old yw/w1118; P[Δ2-3], Ki/+ embryos. At least two homozygous viable transgenic lines were established and crossed into a cspR1 null-mutant genetic background (Zinsmaier et al., 1994).
Crosses for analysis. Transgenic strains containing P-element insertion of UAS-CSP reporter constructs on the second chromosome were kept homozygous for the respective P element, whereas the cspR1 third chromosome was balanced with TM6TbSb. For the D42-GAL4 driver strain, a P[D42-GAL4], cspR1 double-mutant chromosome was generated by meiotic recombination. Fly strains were maintained at 25-27°C to prevent suppressors of the csp null phenotype from developing.
Expression of mutant CSP in a cspR1 null was examined in F1 progeny of crosses combining the elav- or D42-driver and reporter transgenes. For example, w1118; P[D42-GAL4], cspR1/TM6TbSb flies were crossed to w1118; P[UAS-ΔJ]; cspR1/TM6TbSb flies. csp mutant F1 larvae were identified by the absence of the dominant Tb marker and were genotypically w1118; P[UAS-ΔJ]/+; cspR1. For recording, all crosses as well as w1118 controls were kept at 20°C.
Immunohistochemistry. Antibody stainings were performed with PBS containing 0.2% Triton X-100 (PBT) after fixation with 3.5% formaldehyde essentially as described previously (Zinsmaier et al., 1994). Antibodies were diluted in PBT as follows: mouse anti-CSP at 1:100 [ab49/92 (Zinsmaier et al., 1990)], anti-mouse conjugated Cy3 at 1:500, and anti-HRP-conjugated FITC at 1:100 (both from Jackson ImmunoResearch, West Grove, PA). Fluorescence images were taken using similar parameters for control and mutant larvae with a confocal microscope system (FV300; Olympus Optical, Tokyo, Japan).
Electrophysiology. Current-clamp recordings were made from muscle 6 in the anterior ventral abdomen of climbing third-instar larvae, as described previously (Dawson-Scully et al., 2000). Dissections were made in Ca2+-free HL-3 solution (Stewart et al., 1994). The composition was as follows (in mm): 70 NaCl, 5 KCl, 20 MgCl2, 10 NaHCO3, 5 trehalose, 5 HEPES, and 115 sucrose. For recordings, HL-3 solution was supplemented with 1 mm CaCl2 and continuously superfused over the preparation. To elicit a postsynaptic response, the segmental nerve was stimulated for 0.1 ms at 2.5-3 times the stimulus amplitude required for a threshold response. Voltage signals were amplified with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA), filtered at 1 kHz, and digitized at 5 kHz directly to disk with a DigiData 1200 interface and pClamp 8.0 software (Axon Instruments). Evoked responses were analyzed with Clampfit 8.0 software (Axon Instruments), and spontaneous events were analyzed with the Mini Analysis Program (Synaptosoft, Leonia, NJ). Plots were made using Microsoft (Seattle, WA) Excel.
Calcium imaging. Measurements of Ca2+ concentrations in resting and stimulated motor neurons (type 1b boutons) on larval muscle 6 were obtained by loading the calcium indicator fura-2 conjugated to 10 kDa dextran (Molecular Probes, Eugene, OR) into the open end of the severed motor axon, as described in detail by Macleod et al. (2002). After dye loading in Schneider's insect medium, the bath solution was switched to HL-6-recording solution containing 0.5 mm Ca2+ (Macleod et al., 2002). For measurements of Ca2+ spikes derived from single action potentials, we used the indicator Oregon Green 488 BAPTA-1 conjugated to 10 kDa dextran. Acquisition of ratiometric data from fura dextran and the estimation of absolute values for Ca2+ were identical to those described recently (Macleod et al., 2004).
Two stimulation protocols were used to compare Ca2+ accumulation and clearance at room temperature (20°C) and at elevated temperature (30°C). In the first, a brief 15 s period of stimulation at 30 Hz was used. Measurements of [Ca2+]i were calculated before stimulation (resting [Ca2+]i), during stimulation, and at two time points immediately after stimulation. In the second protocol, which was used for relatively few specimens, stimulation was prolonged for 60 s at 30 Hz, and estimates of [Ca2+]i were made at 10 s intervals before, during, and immediately after stimulation.
Results
Previous analyses of csp null-mutant Drosophila revealed a variety of presynaptic defects, including reduced evoked release, reduced thermal tolerance and increased asynchronicity of evoked release, increased levels of intraterminal Ca2+ during and shortly after stimulation, and a reduced ability to maintain normal Ca2+ resting levels at high temperatures (Umbach et al., 1994; Zinsmaier et al., 1994; Heckmann et al., 1997; Umbach and Gundersen, 1997; Ranjan et al., 1998; Dawson-Scully et al., 2000). One way to examine whether all of these synaptic defects truly represent independent functions of CSP is an in vivo structure/function analysis. This approach is possible because presynaptic expression of normal CSP at csp null-mutant NMJs fully restores synaptic transmission (Nie et al., 1999; Arnold et al., 2004). The principle idea of this approach is to mutate CSP in vitro and determine whether the expression of mutant protein restores individual phenotypic defects at csp null-mutant synapses. Differential effects on some of the multiple synaptic defects in csp nulls would provide evidence for independent functions of CSP and for the in vivo significance of the mutated protein domain.
Generation and expression of mutant CSP proteins at larval NMJs
CSP contains three evolutionary conserved domains: the J domain, the L domain, and the cysteine-string domain (Fig. 1A,B). We selected for our analysis the N-terminal J domain of CSP (residues 19-82) because in vitro studies suggest a central role of the J domain for CSP function, directing the subcellular localization and activity of Hsc70 (for review, see Chamberlain and Burgoyne, 2000; Zinsmaier and Bronk, 2001). The J domain is folded like a finger, fitting into a binding pocket of heat shock protein Hsp70 or Hsc70 (for review, see Kelley, 1998, 1999). In vitro, the J domain is essential and sufficient for binding Hsc70 and stimulating ATPase activity of Hsc70 (Braun et al., 1996; Chamberlain and Burgoyne, 1997b; Zhang et al., 1999). Genetic interactions between mutations in Drosophila csp and hsc70 also provided evidence that both proteins act in a common signaling pathway (Bronk et al., 2001). The J domain also mediates interactions with Gα subunits facilitating a synprint-independent G-protein inhibition of heterologously expressed vertebrate N-type channels (Miller et al., 2003a). We deleted the entire J domain of CSP to test whether it plays a central role for CSP function (ΔJ-CSP) (Fig. 1). In addition, we generated a point mutation (H45Q) (Fig. 1B) in the highly conserved HPD tripeptide motif of the J domain because an analogous mutation in bovine CSP abolishes the activation of ATPase activity of Hsc70 (Chamberlain and Burgoyne, 1997b).
Figure 1.
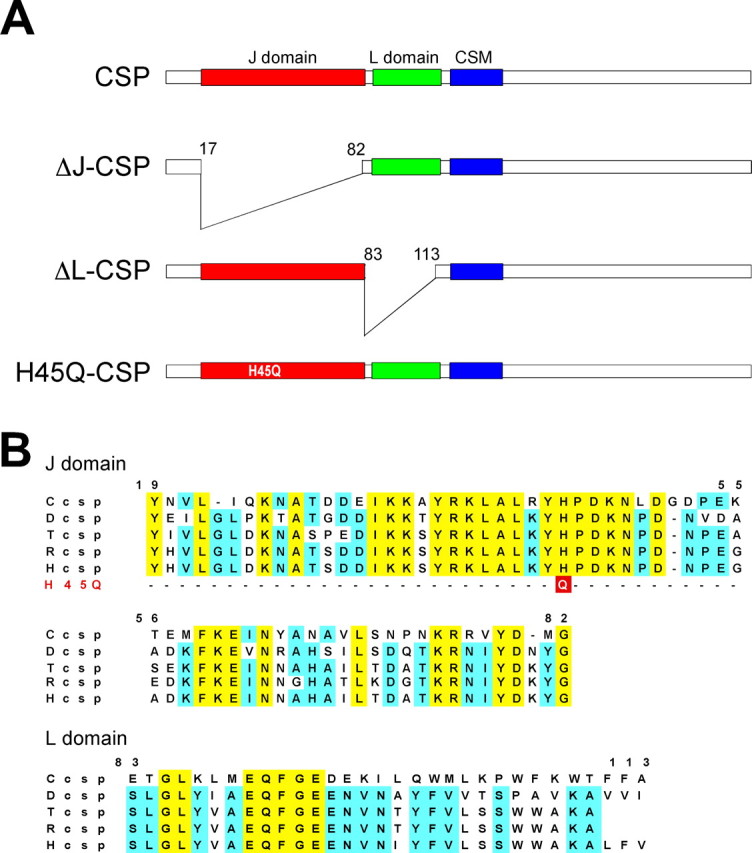
Mutations in Drosophila CSP. A, Schematic representation of CSP and its three evolutionary conserved domains: the J domain (residues 19-82), the L domain (residues 83-113), and the cysteine-string motif (CSM; residues 114-138). The J domain deletion spans residues 17-82 (ΔJ-CSP), and the L domain deletion spans residues 83-113 (ΔL-CSP). The H45Q mutation substitutes histidine45 in the conserved HPD motif with a glutamine (H45Q-CSP). B, Primary sequence of the J and L domain of CSP from Caenorhabditis elegans (Ccsp), Drosophila melanogaster (Dcsp), Torpedo californica (Tcsp), rat (Rcsp), and human (Hcsp). The position of the fly H45Q mutation in the HPD motif of the J domain is indicated.
We also selected the L domain (residues 83-113) of CSP for our analysis (Fig. 1A,B). The L domain is highly conserved between human and Drosophila CSP (21 of 31 amino acids are identical), but its synaptic significance is uncertain. Structurally, the L domain of vertebrate CSP (amino acids 83-112) binds to Gβ subunits, mediating a synprint-dependent G-protein inhibition of N-type Ca2+ channels (Miller et al., 2003a). In addition, a point mutation in the L domain reduced dominant-negative effects induced by the overexpression of bovine CSP in insulin-secreting cells (Zhang et al., 1999). To test the significance of the L domain for the synaptic functions of CSP, we engineered a CSP protein containing a deletion of the entire L domain (ΔL-CSP) (Fig. 1A).
Using standard in vitro mutagenesis approaches, we generated mutant csp cDNAs containing the respective deletions or point mutations. The mutations were verified by sequencing the complete cDNA constructs (data not shown), which also excluded unwanted second-site mutations. The mutant cDNAs were then transgenically expressed in a cspR1 null-mutant background of Drosophila (Zinsmaier et al., 1994), using the motor neuron-specific driver D42-Gal4 or the pan-neuronal driver elav-Gal4 (Lin and Goodman, 1994; Parkes et al., 1998). Expression of mutant proteins was verified by Western blot analysis and immunostainings at synaptic boutons of third-instar larval NMJs (Fig. 2A,C). When driven with the D42 driver, all examined transgenes were expressed at larval NMJs and showed a doughnut-like distribution in synaptic boutons, which is typical of normal CSP and other synaptic vesicle proteins. No expression of CSP was detectable in the absence of a driver transgene, confirming the csp null-mutant genetic background.
Figure 2.
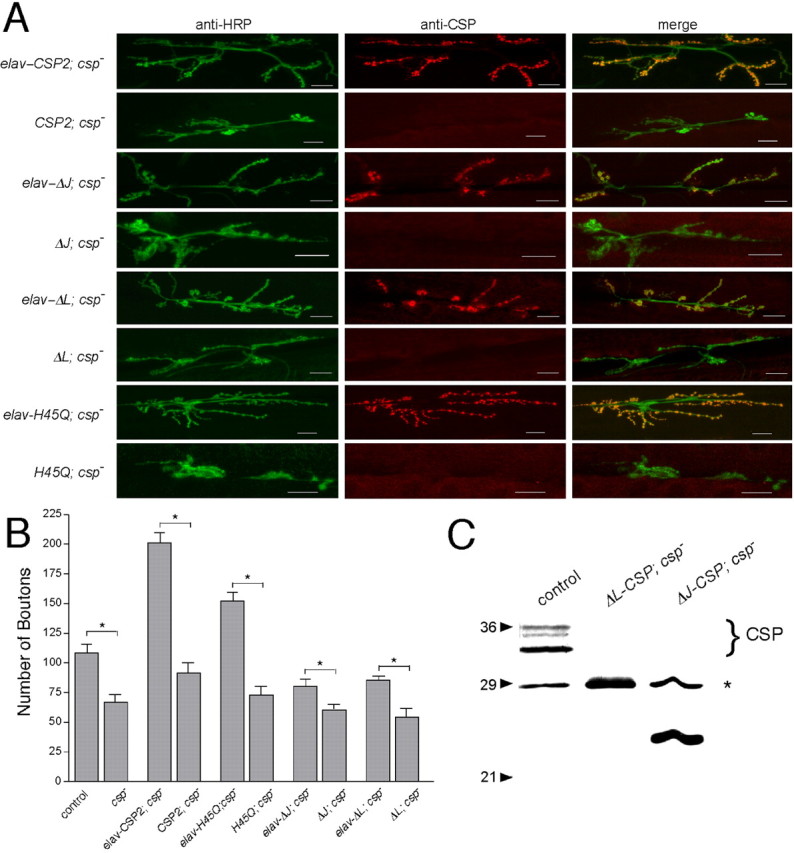
Transgenic expression of in vitro engineered mutant Drosophila CSPs. A, Mutant UAS-CSP transgenes are expressed in the presence but not in the absence of the neuron-specific elav-Gal4 driver. Double immunostainings of NMJs from csp null-mutant larvae containing the elav-Gal4 driver and a UAS-transgene encoding a normal CSP protein (elav-CSP2; csp-) or the mutant CSP proteins ΔJ-CSP (elav-ΔJ; csp-), ΔL-CSP (elav-ΔL; csp-), and H45Q-CSP (elav-H45Q; csp-). Negative controls contain only the UAS-transgene but not the Gal4 driver (CSP2; csp-, ΔJ; csp-, H45Q; csp-, and ΔL; csp-). The left column shows the fluorescence from anti-HRP antibodies marking neuronal membranes of the NMJ, the middle column shows the signal from anti-CSP antibodies, and the right column shows the merge of both signals. Note the variation in the number of synaptic boutons between the genotypes. Scale bar, 20 μm. B, Quantification of the total number of synaptic boutons (types 1b and 1s) at NMJs of csp mutants (genotypes as in A) and control flies (w1118; genetic background of csp mutations). Significant differences to control were found for csp nulls, csp nulls expressing normal CSP, and all silent transgenes (p < 0.01; NMJs, n > 6; larvae, n > 3; one-way ANOVA). In addition, comparison of individual pairs of silent and active transgenes showed significant differences for normal and all examined mutant CSP proteins: *p < 0.05 (Student's t test). Error bars represent SEM. C, Analysis of ΔJ-CSP and ΔL-CSP expression. Shown is an immunoblot of fly protein extracts from w1118 controls, csp null-mutant larvae expressing ΔL-CSP, and larvae expressing ΔJ-CSP. The endogenous CSP triplet in control flies is indicated. Asterisk indicates nonspecific protein staining. Note the smaller size of ΔL-CSP and ΔJ-CSP. ΔL-CSP is similar in size to the nonspecifically stained protein. Proteins were resolved by 11% SDS-PAGE, blotted, and immunostained for CSP with the monoclonal antibody ab49.
Elav-driven pan-neuronal expression of normal CSP partially reverted the temperature-sensitive lethality of csp null mutants at 25°C (Table 1). The partial rescue was presumably attributable to the decline of elav-driven expression levels during late pupal development. However, neither expression of ΔJ-CSP nor ΔL-CSP in csp nulls restored viability. Surprisingly, expression of both mutant CSPs actually enhanced the temperature-sensitive lethality of csp nulls. Whereas csp null mutants alone were fully viable at 18°C, only 11 or 6% of csp null mutants expressing ΔJ-CSP or ΔL-CSP survived to adulthood, respectively (Table 1). Because even repeated normalization of the genetic background did not abolish the enhanced lethality, it seems likely that this may be attributable to a dominant-negative effect of ΔJ-CSP or ΔL-CSP expression. However, this effect is presumably weak and/or easily compensated by normal CSP because overexpression of ΔJ-CSP or ΔL-CSP expression in a wild-type csp+ background had no adverse effects on viability (Table 1).
Table 1.
Lethality of csp null mutants
|
Genotype |
Viability at 18°C |
Viability at 25°C |
|---|---|---|
| cspR1 | 100% | <1% |
| elav-CSP2; cspRI | 100% | 62% |
| elav-ΔJ; cspRI | 11% | N.D. |
| elav-ΔL; cspRI | 6% | N.D. |
| elav-CSP2; +/+ | N.D. | 70% |
| elav-ΔJ; +/+ | N.D. | 105% |
|
elav-ΔL; +/+ |
N.D. |
98% |
Files containing a homozygous UAS-CSP transgene or elav-Ga14 transgene in a heterozygous cspR1 null mutant or csp + wild-type background were crossed to each other. At least 400 progeny were scored for the ratio of observed to expected flies. N.D., Not determined.
Effects of CSP mutations on synaptic growth at csp null-mutant NMJs
The loss of evoked release at cspU1 null NMJs correlates with a reduced number of synaptic boutons (Dawson-Scully, 2003). Similarly, in our study, the number of boutons was also reduced to 62 ± 6% of control at cspR1 null-mutant NMJs (p < 0.01; one-way ANOVA) (Fig. 2B). Because both csp null alleles were derived from two independent genetic screens (Eberle et al., 1998), this may suggest that the loss of synaptic boutons is caused by the loss of CSP. To further test this idea, we determined whether expression of normal CSP at csp null-mutant NMJs, which fully restores the loss of evoked release (Nie et al., 1999; Arnold et al., 2004), also reverses the loss of synaptic boutons. Surprisingly, presynaptic elav-driven expression of normal CSP at csp null-mutant NMJs not only reverted the loss of synaptic boutons but dramatically increased the number of synaptic boutons to 186 ± 8% of wild-type control (p < 0.01; one-way ANOVA) (Fig. 2B). A similar increase was observed for D42-driven expression of normal CSP (data not shown). The “overcompensation” of wild-type CSP expression may be attributable to the strong expression levels of the elav and D42 promoters.
Expression of ΔJ-CSP and ΔL-CSP at csp null-mutant NMJs reverted the loss of synaptic boutons of csp nulls, increasing the number of synaptic boutons to 74 ± 5 and 80 ± 3% of wild-type control (Fig. 2B). Compared with the effect of the silent transgenes, ΔJ-CSP and ΔL-CSP expression caused a significant increase (p < 0.05; Student's t test). However, because expression of normal CSP at csp null-mutant NMJs increased the number of boutons to ∼186% of wild type, these results suggest that the protein activity of both ΔJ-CSP and ΔL-CSP is significantly attenuated compared with the expression of normal CSP. Accordingly, both the J and L domains are important for the role of CSP in mediating synaptic growth. To test this further, we examined the effects of the point mutation H45Q in Drosophila CSP, because an analogous mutation in bovine CSP (H43Q) abolishes the J domain-mediated activation of the ATPase activity of Hsc70 in vitro (Chamberlain and Burgoyne, 1997b). Expression of H45Q-CSP at csp null-mutant NMJs increased the number of synaptic boutons to 140 ± 7% of control (p < 0.01; ANOVA) (Fig. 2B). Although the activity of H45Q-CSP was significantly higher than that of either ΔJ-CSP or ΔL-CSP, it was still reduced compared with normal CSP (p < 0.01; one-way ANOVA) (Fig. 2B). This suggests that a J domain-mediated activation of the ATPase activity of Hsc70 and the interaction with an additional unknown protein are necessary for the role of CSP in synaptic growth.
Expression of J domain-mutant CSPs incompletely restores evoked transmitter release at csp null-mutant NMJs
To determine whether expression of ΔJ-CSP restores the loss of evoked release observed at csp null-mutant NMJs, we examined the synaptic physiology of larval NMJs by whole-cell recordings of evoked excitatory junction potentials (EJPs) and spontaneous miniature excitatory junction potentials (mEJPs). EJPs were elicited by stimulating the cut nerve at 0.2 Hz in 1 mm [Ca2+]e and were the combined responses of two glutamatergic axons innervating muscle 6 (Kurdyak et al., 1994). Consistent with previous studies, evoked EJP amplitudes at csp null-mutant NMJs were 52.0 ± 6.6% of wild-type control amplitudes (genetic background, w1118) at 22°C (p < 0.01; one-way ANOVA) (Fig. 3A,B). This defect in synaptic transmission of csp nulls is fully restored by the expression of normal CSP (Nie et al., 1999; Arnold et al., 2004). Expression of normal CSP also reverses the temperature-sensitive paralysis and premature lethality of adult csp null mutants (Arnold et al., 2004).
Figure 3.
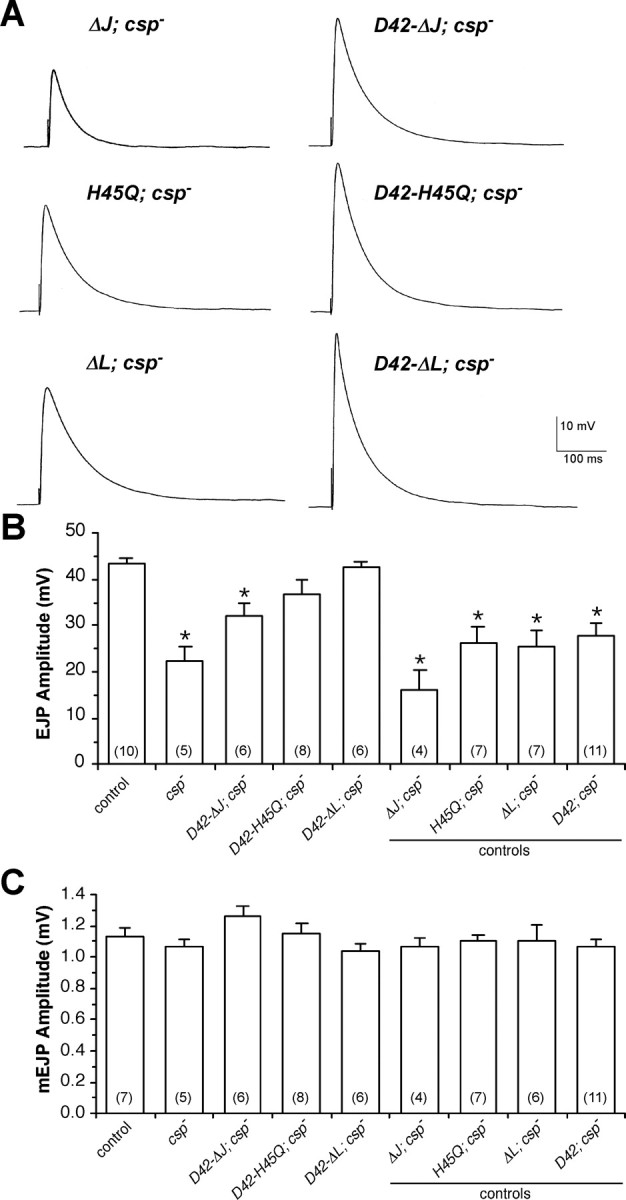
Effects of expressing ΔJ-CSP, ΔL-CSP, and H45Q-CSP on neurotransmitter release at csp null-mutant NMJs. A-C, Evoked EJPs and spontaneous mEJPs were recorded from muscle 6 in HL-3 solution at 22-23°C with 1 mm [Ca2+]e. Error bars represent SEM, and numbers in parentheses indicate the number of larvae recorded. A, Representative EJP traces are shown for csp null-mutant larvae containing silent transgenes (left column; ΔJ; csp-, H45Q; csp-, and ΔL; csp-) and active transgenes (right column) for ΔJ-CSP (D42-ΔJ; csp-), H45Q-CSP (D42-H45Q; csp-), and ΔL-CSP (D42-ΔL; csp-). Calibration for all voltage traces is indicated. B, Mean EJP amplitudes are shown for the same genotypes as in A, with the addition of csp null mutants (csp-), wild-type control (w1118), and the D42-Gal4 driver transgene (D42; csp-). *p < 0.05 indicates significant differences to wild-type control (one-way ANOVA). EJPs from csp null larvae expressing ΔJ-CSP, ΔL-CSP, or H45Q-CSP are also significantly different from EJPs from csp nulls and csp nulls containing the respective silent transgene (p < 0.05). All negative controls including csp nulls and csp nulls containing silent transgenes are statistically similar (p > 0.05). C, Mean mEJP amplitudes from larvae expressing any CSP mutant transgene were not significantly different from wild-type control (p > 0.05). Genotypes as in B.
Expression of ΔJ-CSP in motor neurons increased EJP amplitudes at csp null-mutant NMJs to 74 ± 7% of control amplitudes at 22°C (Fig. 3A,B). However, normal function was not restored because the amplitudes of EJPs resulting from ΔJ-CSP expression were significantly different from wild-type control (p < 0.05; one-way ANOVA). An independent transgene had similar effects (data not shown). Controls addressing effects of the silent ΔJ-CSP transgene and the D42 promoter transgene alone were statistically indistinguishable from csp null mutants (p > 0.05). We also examined effects of ΔJ-CSP expression on spontaneous release but found no significant change in mEJP amplitude (Fig. 3C) or mEJP frequency (data not shown).
The partial activity of ΔJ-CSP attenuating the loss of evoked release at csp null-mutant NMJs is surprising because it suggests that the deleted J domain is important but not entirely essential for the function of CSP in evoked release at 22°C. Consequently, this implies that the function of CSP in evoked release does not depend entirely on the activation of ATPase activity of Hsc70. To test this further, we also examined the effects of the point mutation H45Q in Drosophila CSP, because an analogous mutation in bovine CSP (H43Q) abolishes the J domain-mediated activation of the ATPase activity of Hsc70 in vitro (Chamberlain and Burgoyne, 1997b). Expression of H45Q-CSP in motor neurons also increased EJP amplitudes at csp null-mutant NMJs (Fig. 3A,B). Compared with wild-type controls, the mean EJP amplitude in larvae expressing H45Q-CSP was 85 ± 7% and was not significantly different (p > 0.05; one-way ANOVA). Expression of H45Q-CSP did not significantly affect mEJP amplitudes (p > 0.05) (Fig. 3C) or mEJP frequency (data not shown). The silent H45Q transgene alone or the D42 promoter transgene alone had no significant effect on EJP amplitudes (p > 0.05) (Fig. 3B).
Although H45Q-CSP expression restored EJP amplitudes to control levels, the H45Q-CSP-mediated effect was not very robust. The coefficient of variation of EJP amplitudes (mean SD/EJP) was 0.08 for control, whereas for H45Q-CSP it was 0.24. This difference suggests that H45Q-CSP expression did not fully restore synaptic transmission. Accordingly, the effect of expressing H45Q-CSP is consistent with the incomplete rescue by ΔJ-CSP expression, indicating a contributing role for the J domain and its mediated protein interactions in evoked release.
Expression of CSP containing a deletion of the L domain fully restores evoked transmitter release at csp null-mutant NMJs
Recently, Arnold et al. (2004) reported that a small deletion in the L domain only marginally impairs CSP function in Drosophila. Expression of the mutant protein rescued evoked release at larval NMJs and the premature lethality but not the temperature-sensitive paralysis in adult csp null mutants.
To test the significance of the L domain for the functions of CSP at nerve terminals more comprehensively, we examined the effects of CSP protein containing a deletion of the entire L domain (ΔL-CSP). Motor neuron-specific expression of ΔL-CSP significantly increased evoked EJP amplitudes at csp null-mutant NMJs from 52 ± 6 to 98 ± 3% of control EJPs (p < 0.01; one-way ANOVA) (Fig. 3A,B), whereas the silent ΔL-CSP transgene alone had no effect (p > 0.05). The increased EJP amplitudes were statistically indistinguishable from wild-type controls (p < 0.05). Expression of ΔL-CSP did not affect mEJP amplitude (Fig. 3C) or frequency (data not shown) in csp null mutants.
The rescue of the csp null EJP phenotype by ΔL-CSP expression was very robust without the large variation seen for the expression of H45Q mutation. The coefficient of variation for the SD of EJP amplitudes for ΔL-CSP was 0.08, similar to wild-type controls, whereas the coefficient of variation for H45Q-CSP was 0.24. This difference in variation strengthens our previous interpretation that there are likely some minor defects still associated with the expression of H45Q-CSP. We conclude that ΔL-CSP expression fully rescues evoked EJP amplitudes at csp nullmutant NMJs, whereas H45Q-CSP or ΔJ-CSP expression achieves only a partial rescue. Thus, the L domain of CSP is not required for maintaining normal evoked release, consistent with previous findings (Arnold et al., 2004).
Expression of ΔJ-CSP and ΔL-CSP restores the thermotolerance of evoked transmitter release at csp null-mutant NMJs
A hallmark of the defects at csp-deficient synapses is the thermointolerance of evoked release above 30°C, which causes an almost complete loss of evoked release compared with the ∼50% reduction of evoked release at 22°C (Fig. 4A) (Umbach et al., 1994; Dawson-Scully et al., 2000). A reasonable assumption is that this thermo-intolerance is simply a progressive deterioration of the defect at room temperature, resembling a classical chaperone defect. Because this is consistent with the presumed chaperone function of a CSP/Hsc70 complex, it would be expected that the J domain is critical for maintaining the synaptic functions of CSP at high temperatures.
Figure 4.

Effects of ΔJ-CSP and ΔL-CSP expression on the thermo-intolerance of neurotransmitter release at csp null-mutant NMJs. A-C, EJPs/mEJPs were recorded from muscle 6/7 in HL-3 solution at 22 and 32°C with 1 mm [Ca2+]e. Error bars represent SEM. Mean values are from five larvae for each group. A value of p < 0.05 was considered significant (Student's t test). A, Temperature dependence (22 and 32°C) of nerve-evoked EJP amplitudes for wild-type control, csp null-mutant larvae (csp-), csp null-mutant larvae expressing ΔJ-CSP (ΔJ-CSP; csp-), and csp null-mutant larvae expressing ΔL-CSP (ΔL-CSP; csp-). Only EJPs recorded from csp nulls show a significant difference in amplitude between 22 and 32°C (p < 0.05). Amplitudes of csp nulls and csp nulls expressing ΔJ-CSP are different from wild-type controls and csp nulls expressing ΔL-CSP at both temperatures (p < 0.05). Amplitudes from wild-type controls and csp nulls expressing ΔL-CSP are statistically similar at both temperatures (p > 0.05). B, C, Temperature dependence (22 and 32°C) of mEJP frequency (B) and mEJP amplitude (C). No significant differences were observed between all groups.
To test this prediction, we examined the effects of ΔJ-CSP or ΔL-CSP expression on evoked release at csp null-mutant NMJs at 22 and 32°C (Fig. 4A). At wild-type NMJs, increasing the temperature from 22 to 32°C reduced the average EJP amplitude by 11 ± 7%, but at csp nullmutant NMJs, EJP amplitudes were reduced by 94 ± 11%. Surprisingly, expression of ΔJ-CSP restored the thermotolerance of evoked release at csp null NMJs: compared with the average EJP amplitude of ΔJ-CSP expressing NMJs at 22°C, the EJP amplitude at 32°C was decreased by only 9 ± 15% (Fig. 4A), statistically indistinguishable from control. This shows that expression of ΔJ-CSP restored the thermo-tolerance of evoked release without fully restoring evoked release at 22°C. This result is inconsistent with the idea that the severe thermo-intolerance of evoked release is simply a progressive deterioration of a defect at room temperature, mainly because such a model requires the defect at high temperature to be more severe than the defect at low temperature. Consequently, two separate and independent mechanisms regulating evoked release must be impaired in csp nulls. In addition, these results suggest that CSP might act as an Hsc70-independent synaptic chaperone, protecting evoked release from thermal stress. This is supported by the ability of CSP to prevent thermally induced protein aggregation in vitro (Chamberlain and Burgoyne, 1997a).
Expression of ΔL-CSP at csp null-mutant NMJs also restored the thermo-tolerance of release: at 32°C, the average EJP amplitude was slightly reduced by 16 ± 9% (Fig. 4A) but was not significantly different from wild-type control (p > 0.1). In addition, no differences were observed in spontaneous quantal release between any of the constructs at any temperature (Fig. 4B,C). Neither mEJP frequency nor mEJP amplitude were significantly different between the groups at 22 or 32°C (p > 0.05). Accordingly, we conclude that neither the J domain nor the L domain of CSP is required to maintain the thermo-tolerance of evoked neurotransmitter release at larval NMJs.
Expression of ΔL-CSP but not of ΔJ-CSP counteracts the defect in asynchronous release at csp null-mutant NMJs
csp null-mutant NMJs exhibit a significant increase in the frequency of asynchronous release (Heckmann et al., 1997), which is likely a consequence of the reduction in synchronous release of csp mutants. If so, one might expect that expression of ΔJ-CSP or ΔL-CSP in csp null mutants will proportionally reverse defects in both synchronous and asynchronous release.
At wild-type Drosophila NMJs, asynchronously occurring quantal release events were rarely observed after the peak of evoked EJPs (Fig. 5A). Expression of ΔJ-CSP at csp null-mutant NMJs had little effect on the abnormally increased frequency of asynchronous release shortly after the evoked EJP (Fig. 5B,C). However, asynchronous release was much reduced at csp nullmutant NMJs expressing ΔL-CSP (Fig. 5D). This suggests that the J domain but not the L domain of CSP is important for minimizing asynchronous release.
Figure 5.
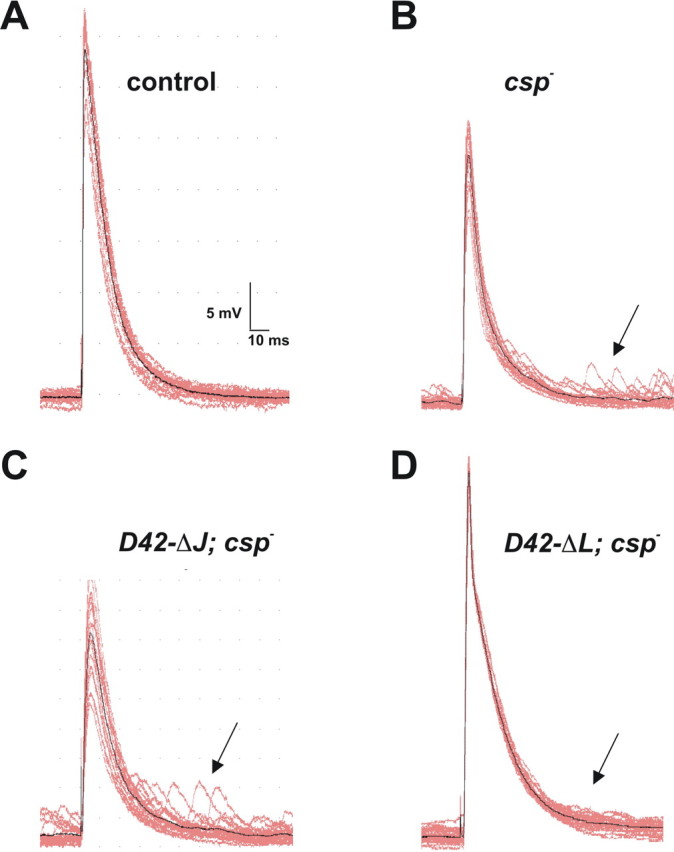
Effects of ΔJ-CSP and ΔL-CSP expression on asynchronous neurotransmitter release at csp null-mutant NMJs. A-D, Typical EJPs and subsequent mEJPs are shown from intracellular recordings of muscle 6/7 with 1 mm [Ca2+]e at 22°C, using a 1 Hz stimulation. Calibration for voltage traces is indicated. A, Wild-type control traces show little or no mEJPs shortly after the evoked EJP. B, csp- null mutants show an increased number of mEJPs (arrow) shortly after an evoked EJP, as well as a significantly reduced EJP amplitude. C, Expression of ΔJ-CSP at csp null-mutant NMJs (ΔJ-CSP; csp-) has no obvious effect on the increased occurrence of mEJPs shortly after stimulation (arrow). D, Expression of ΔL-CSP at csp null-mutant NMJs (ΔL-CSP; csp-) reduces the increased number of mEJPs occurring shortly after stimulation (arrow) and restores normal EJP amplitudes.
Expression of ΔL-CSP but not of ΔJ-CSP partially restores normal intraterminal Ca2+ levels at csp nullmutant NMJs
Previously, we showed that stimulusevoked intraterminal Ca2+ levels are abnormally increased at csp null-mutant NMJs and that csp null-mutant synaptic terminals exhibit approximately a twofold increase in intraterminal Ca2+ resting levels above 30°C but normal Ca2+ resting levels at room temperature (Dawson-Scully et al., 2000). To obtain a clearer understanding of how CSP affects Ca2+ homeostasis and whether the defects in stimulus-evoked Ca2+ levels and Ca2+ resting levels are truly linked, we examined the absolute change of intraterminal Ca2+ levels before, during, and after stimulation at 20 and 30°C at csp null-mutant NMJs expressing ΔJ-CSP or ΔL-CSP. We used the ratiometric Ca2+ indicator fura dextran excited at two separate wavelengths (340 and 380 nm) to measure intraterminal Ca2+ levels at rest and during stimulation in type 1b boutons, as described by Macleod et al. (2002).
Consistent with previous studies (Dawson-Scully et al., 2000), the average resting [Ca2+]i of wild-type control boutons was not significantly different between 20 and 30°C (p = 0.8) (Fig. 6A). In boutons of csp null mutants carrying only the D42-Gal4 driver, the average resting [Ca2+]i was significantly increased from 32.9 ± 6.5 nm at 20°C to 145.7 ± 15.1 nm at 30°C (Fig. 6A) and not different from csp nulls alone (data not shown). Motor neuron-specific expression of ΔJ-CSP had no significant effect on the elevated intraterminal [Ca2+]i of csp null-mutant NMJs. The values at 20 and 30°C were statistically indistinguishable from those of csp null mutants at 20°C (p = 0.5) and at 30°C (p = 0.4) and were similar to those of controls at 20°C (p = 0.8) but not to those of wild-type control at 30°C (p < 0.001). This suggests that the J domain of CSP is essential for the function of CSP in maintaining normal intraterminal Ca2+ resting levels at high temperatures.
Figure 6.

Effects of ΔJ-CSP and ΔL-CSP expression on intraterminal Ca2+ levels at csp null-mutant NMJs. A-D, Intraterminal Ca2+ levels were measured with the ratiometric Ca2+ indicator fura dextran excited at two separate wavelengths (340 and 380 nm) at rest and during stimulation in type 1b boutons. Error bars represent SEM, and n represents number of larvae. A, Ca2+ resting levels at 20 and 30°C are similar in larval motor nerve terminals of wild-type control (genetic background, w1118) and csp heterozygous siblings (controls) of larvae containing mutant transgenes but not in csp null mutants carrying only the D42-Gal4 driver (D42; csp-). At 30°C, expression of ΔL-CSP but not of ΔJ-CSP partially reversed the elevated resting [Ca2+]i of csp null mutants. Significant differences from wild-type control (*) and from csp null mutants (^) are indicated. Error bars represent SEM. B, Effects of ΔJ-CSP and ΔL-CSP expression at csp null-mutant NMJs on stimulus-induced Ca2+ levels at 20°C. Measurements were obtained before, during, and after stimulation at 30 Hz for 15 s (bar). No significant differences were observed. Genotypes as in A. Error bars represent SEM. C, Effects of ΔJ-CSP and ΔL-CSP expression at csp null-mutant NMJs on stimulus-induced Ca2+ levels at 30°C. Stimulation was as in B. Whereas expression of ΔJ-CSP had no significant effects on [Ca2+]i in csp null-mutant motor nerve terminals, expression of ΔL-CSP significantly reduced the abnormally increased Ca2+ levels. Genotypes as in A. Error bars represent SEM. D, Effects of prolonged stimulation on posttetanic Ca2+ resting levels in csp null-mutant (D42; csp-) and control (w1118) motor nerve terminals. Measurements were obtained before, during, and after stimulation at 30 Hz for 60 s (bar) at 30°C. After prolonged stimulation, posttetanic Ca2+ resting levels of csp null-mutant terminals did not return to pretetanic levels, which contrasts the similar pretetanic and posttetanic Ca2+ resting levels during shorter periods of stimulation (see C). Error bars represent SEM.
At 30°C, motor neuron-specific expression of ΔL-CSP partially reversed the abnormally high Ca2+ resting levels of csp nullmutant boutons to an intermediate level of 93.4 ± 8.2 nm. Statistically, Ca2+ levels at 30°C were significantly different from those seen in csp null mutants (p = 0.009) and csp null mutants expressing ΔJ-CSP (p < 0.001). Ca2+ levels at 30°C were also different from those seen in wild-type controls at 30 and 20°C (p < 0.001). At 20°C, expression of ΔL-CSP had no significant effect on the intraterminal [Ca2+]i (p = 0.5). Thus, expression of ΔL-CSP partially restored normal Ca2+ levels in csp null-mutant boutons, suggesting that the L domain of CSP has an important role in maintaining normal Ca2+ resting levels in synaptic terminals.
To examine absolute Ca2+ levels during and after stimulation, we stimulated type 1b boutons via the segmental nerve at 30 Hz for 15 s in 0.5 mm [Ca2+]e. Statistically, Ca2+ levels in csp nullmutant boutons were not significantly increased from wild-type control at 20°C during and after stimulation (p = 0.4; two-way ANOVA) (Fig. 6B). At 30°C, however, intraterminal Ca2+ levels were significantly increased during stimulation in csp nullmutant boutons compared with wild-type controls (p < 0.001; two-way ANOVA) (Fig. 6C).
Expression of ΔJ-CSP in motor neurons had no significant effects on Ca2+ levels in csp null-mutant motor terminals before, during, and after stimulation at 20 and 30°C (p > 0.05) (Fig. 6B,C). Consequently, these results indicate an essential role of the J domain for CSP function in maintaining normal stimulus-evoked Ca2+ levels at nerve terminals. In contrast, expression of ΔL-CSP at csp null-mutant NMJs partially restored normal intraterminal Ca2+ levels at 30°C (Fig. 6C). Ca2+ signals at rest, during, and after stimulation in ΔL-CSP expressing boutons reached intermediate levels, being significantly higher than those of wild-type control at 30°C (p = 0.01; two-way ANOVA) but also clearly lower than those of csp nulls (p = 0.02). Because the expression of ΔL-CSP had approximately the same effect in reducing the abnormally increased Ca2+ levels for all measured time points before, during, and after stimulation, it is likely that the defects of csp nulls in handling Ca2+ levels at rest and during stimulation are causally linked.
We also noticed some differences between our current and our previous study (Dawson-Scully et al., 2000). Ca2+ levels in csp nulls were not significantly different before and after a 15-s-long stimulation train at high temperature (Fig. 6C), whereas the previous study reported higher Ca2+ levels after stimulation than before stimulation. The difference between the two studies might be explained by the difference in the recording solutions used (HL-3 vs HL-6). The HL-6 solution was preferred in this study because it appears metabolically superior over the previously used HL-3 solution. HL-6 solution maintains the larval preparation for significantly longer periods of time, which is required for the forward-filling technique of Ca2+ indicator loading (Macleod et al., 2002). If HL-6 solution can at least to some degree counteract a use-dependent defect, then one has to expect that longer stimulation trains will eventually unmask the defect. To test this, we stimulated csp null-mutant motor nerves for longer periods (60 s) at the same frequency and found that Ca2+ levels were significantly higher after stimulation than before stimulation (p = 0.029; Student's t test) (Fig. 6D), consistent with our previous study (Dawson-Scully et al., 2000). This suggests further that mechanisms regulating Ca2+ homeostasis are progressively deteriorating in csp null-mutant boutons during prolonged stimulation and/or high intraterminal Ca2+ levels.
Ca2+ signals attributable to single action potentials are normal in csp null, ΔJ-CSP, and ΔL-CSP mutants at 22 and 32°C
The abnormal intraterminal Ca2+ levels of csp mutants could be caused by many factors, including increased Ca2+ channel activity, reduced Ca2+ clearance, defective settings of Ca2+ resting levels, or Ca2+ leakage from intracellular stores. Interestingly, vertebrate CSP facilitates a tonic G-protein-mediated inhibition of heterologously expressed N-type Ca2+ channels (Magga et al., 2000; Miller et al., 2003a). This inhibition is mediated by two distinct mechanisms, which require separate regions of CSP. N-type Ca2+ channel inhibition by CSP83-198 but not by CSP1-82 (J domain) requires the synaptic protein interaction (synprint) site of the N-type channel (Miller et al., 2003a), which is absent in invertebrate Ca2+ channels, including Drosophila (Littleton and Ganetzky, 2000; Spafford et al., 2003a,b). Hence, Drosophila CSP could mediate a synprint-independent G-protein inhibition through its J domain. Loss of this function could cause or, at least, contribute to the abnormally increased presynaptic Ca2+ levels in csp mutant motor nerve terminals.
To examine individual Ca2+ signals derived from single action potentials, we used the Ca2+ indicator Oregon Green 488 BAPTA-1 conjugated to 10 kDa dextran and confocal line scans (Fig. 7A, inset). Examining evoked Ca2+ signal amplitudes at 22 or 32°C, we found no significant differences between wild-type control, csp null mutants, and csp nulls expressing ΔJ-CSP or ΔL-CSP (p > 0.05; two-way ANOVA; data not shown). Comparing the Ca2+ signal decay time constant at 22 or 32°C, we found no significant differences between wild-type control and csp null mutants at either temperature (p > 0.1; Student's t test) (Fig. 7B). There was also no significant difference in the decay time constant of Ca2+ signals from csp null boutons between 22 and 32°C (p > 0.1; Student's t test). Similarly, no significant differences in the decay time constants were found between csp nulls expressing ΔJ-CSP or ΔL-CSP at 22 and 32°C (p > 0.1; Student's t test) or compared with all groups (p > 0.2; two-way ANOVA).
Figure 7.

Effects of csp mutations on intraterminal Ca2+ signals derived from single action potentials. A, Dextran-conjugated Oregon Green 488 BAPTA-1 fluorescence changes in response to single pulse nerve stimulation in control. After normalizing amplitudes for each animal (n = 1), a line plot for the decay phase was derived from 200-ms-long Ca2+ traces, which was fitted with a single-exponential decay equation [y = y0 + ae(-bx)] ±SEM. The time constant of decay (1/b) was compared with other groups. The inset shows the raw data trace of a confocal line scan from a synaptic bouton on muscle 6/7 and its translation into a trace. B, Average time constants of decay derived from single Ca2+ signals at 22°C (dark) and 32°C (light), as described in A. There are no significant differences in the time constants of the decay found at either 22 or 32°C between control larvae (n = 7, n = 17), csp null-mutant larvae (csp-, n = 7, n = 13), and csp null-mutant larvae expressing ΔJ-CSP (D42-ΔJ; csp-, n = 6, n = 18) or ΔL-CSP (D42-ΔL; csp-, n = 5, n = 14) in motor neurons. Error bars represent SEM.
Although these results do not support a potential role of CSP in regulating presynaptic Ca2+ channels, they also do not rule out this possibility because subtle abnormalities in Ca2+ entry of csp nulls cannot be excluded as a result of known limitations in the resolution of the current Ca2+ imaging technique (Macleod et al., 2002). However, recordings from peptidergic terminals of csp null-mutant Drosophila also showed normal presynaptic Ca2+ currents (Morales et al., 1999). In addition, normal individual Ca2+ signals at larval NMJs of Drosophila were reported after depletion of synaptic vesicles, suggesting that no significant modulation of presynaptic Ca2+ channels originates from synaptic vesicle proteins at larval Drosophila NMJs (Macleod et al., 2004). Finally, recordings from the calyx of Held terminals of very young CSP knockout mice showed normal presynaptic Ca2+ currents, although ∼30% of the Ca2+ current is derived from N-type Ca2+ channels (Fernandez-Chacon et al., 2004). Hence, a direct in vivo demonstration for a role of CSP in modulating presynaptic Ca2+ entry remains elusive.
Discussion
The independent functions of CSP at nerve terminals
Our analysis provides additional evidence that some of the synaptic functions of CSP are mechanistically independent of each other. Specifically, ΔJ-CSP expression restored a normal thermotolerance of release but only partially restored normal evoked release, whereas ΔL-CSP expression restored both defects. Together, these data support independent functions of CSP for maintaining a high efficiency of evoked release and a normal thermo-tolerance of release. An independent role in evoked release is consistent with the interaction of CSP with syntaxin and synaptotagmin (Nie et al., 1999; Wu et al., 1999; Evans and Morgan, 2002). The thermal protection of release is consistent with a role as a use-dependent general chaperone (Fernandez-Chacon et al., 2004). Thus, CSP may act as a specific chaperone regulating particular protein transitions and, as a general chaperone, renaturing proteins during continuous operation.
The loss of evoked release at csp null-mutant NMJs could be caused by a reduced number of synapses attributable to the reduced number of synaptic boutons (Dawson-Scully, 2003). However, our structure/function analysis of CSP did not show a similar correlation. Expression of ΔJ-CSP and ΔL-CSP in csp null mutants increased the number of synaptic boutons to comparable levels. However, ΔL-CSP, but not ΔJ-CSP, expression robustly rescued the defect in evoked release. In addition, expression of H45Q-CSP only partially restored normal evoked release, although it increased the number of synaptic boutons to significantly higher levels than expression of ΔL-CSP or ΔJ-CSP. The effects of mutant CSP expression on synaptic growth showed also no correlation with the effects on the thermo-tolerance of evoked release and the abnormal presynaptic Ca2+ levels. Hence, these differential effects of ΔL-CSP, ΔJ-CSP, and H45Q-CSP expression suggest that the role of CSP in synaptic growth is mechanistically independent of its other functions.
Our analysis also suggests a separation of two functions of CSP in maintaining normal presynaptic Ca2+ levels. ΔL-CSP but not ΔJ-CSP expression was able to attenuate the abnormal Ca2+ levels at csp null-mutant NMJs. Consistently, both domains mediate different biochemical interactions: the J domain binds to Hsc70 and Gα subunits, whereas the L domain binds to Gβ subunits (Braun et al., 1996; Chamberlain and Burgoyne, 1997b; Stahl et al., 1999; Zhang et al., 1999; Miller et al., 2003a). Hence, the differential effects of ΔL-CSP and ΔJ-CSP expression on presynaptic Ca2+ levels at csp mutant NMJs indicate the requirement of two mechanisms.
The J domain of CSP is not essential for all functions of CSP
Previous biochemical studies suggested a central role for the J domain, directing the subcellular localization and activity of Hsc70 for most if not all functions of CSP (for review, see Chamberlain and Burgoyne, 2000; Zinsmaier and Bronk, 2001). Our genetic analysis suggests a revision of this model because J domain mutant CSPs did not resemble null mutations for most of the synaptic functions of CSP. Most remarkably, expression of ΔJ-CSP at csp null-mutant NMJs fully restored the thermotolerance of evoked release. Because it is well documented that a deletion of the J domain abolishes any interaction of CSP with Hsc70 (Braun et al., 1996; Chamberlain and Burgoyne, 1997a,b; Stahl et al., 1999; Zhang et al., 1999), this rescue excludes any role of J domain-mediated Hsc70 interactions for this role of CSP. In addition, expression of ΔJ-CSP and H45Q-CSP at csp nullmutant NMJs attenuated the defects in evoked release and synaptic growth, indicating an important but not an essential function for J domain-mediated interactions for these synaptic roles of CSP.
Expression of ΔJ-CSP at csp null-mutant NMJs completely failed to attenuate the defects in intraterminal Ca2+ levels at rest and during stimulation. This indicates a complete loss of CSP function and an essential requirement of J domain-mediated interactions with Hsc70 for the role of CSP in regulating presynaptic Ca2+ homeostasis. Alternatively, one might argue that the large J domain deletion might affect protein folding, reducing protein stability and/or masking other functional domains of CSP. However, this alternative possibility seems unlikely because ΔJ-CSP expression fully rescued the thermo-intolerance of release, regardless of the expectation that higher temperatures will further destabilize a partially folded ΔJ-CSP protein.
Although studies using heterologous expression systems show that CSP can regulate the activity of presynaptic Ca2+ channels, it remains controversial whether CSP upregulates presynaptic Ca2+ entry by increasing the activity of Ca2+ channels (Gundersen and Umbach, 1992; Chen et al., 2002) or, alternatively, downregulates presynaptic Ca2+ entry by facilitating a G-protein-mediated inhibition of Ca2+ channels (Magga et al., 2000; Miller et al., 2003a,b). The abnormally increased intraterminal Ca2+ levels of csp loss-of-function mutants do not support a role of CSP in upregulating Ca2+ channels but are consistent with the suggested G-protein-mediated inhibition of Ca2+ channels. Because Drosophila Ca2+ channels do not contain a synprint site, the potential inhibition of fly Ca2+ channels could require the J domain of CSP, which mediates a synprint-independent G-protein inhibition of vertebrate N-type channels through its interaction with Gα subunits (Miller et al., 2003a). Consistently, the J domain of fly CSP is essential for maintaining normal presynaptic Ca2+ levels because ΔJ-CSP expression was unable to restore normal Ca2+ levels at csp null-mutant NMJs. However, we could not detect any effect of csp mutations on presynaptic Ca2+ signals triggered by single action potentials.
The significance of the L domain for the synaptic roles of CSP
The significance of the highly conserved L domain for the synaptic roles of CSP has been uncertain. Structurally, the L domain of vertebrate CSP (amino acids 83-112) binds to Gβ subunits, mediating the synprint-dependent G-protein inhibition of N-type channels (Miller et al., 2003a). We found that ΔL-CSP expression at csp mutant NMJs reversed intraterminal Ca2+ to intermediate levels, suggesting that activity of the L domain is partially required for the role of CSP in maintaining normal Ca2+ levels. Because the synprint site is absent in Drosophila Ca2+ channels (Littleton and Ganetzky, 2000), the incomplete rescue by ΔL-CSP indicates that CSP may also regulate other mechanisms of Ca2+ homeostasis that are independent of synprint site-containing Ca2+ channels.
ΔL-CSP expression also reversed the loss of synaptic boutons at csp mutant NMJs but not as effectively as expression of the normal CSP. In contrast, all of the defects in transmitter release were restored by ΔL-CSP expression, indicating that the L domain is not required for any of these functions. Hence, the L domain is only important for the role of CSP in maintaining normal presynaptic Ca2+ homeostasis and synaptic growth.
Our results are consistent with a recent study showing that expression of a smaller L domain deletion (LΔ8-CSP) restored normal amplitudes and thermo-tolerance of evoked release at csp null-mutant NMJs (Arnold et al., 2004). Interestingly, the only defect noticed after LΔ8-CSP expression was an incomplete rescue of the temperature-sensitive paralysis of adult flies (Arnold et al., 2004). This may indicate that the defect in Ca2+ homeostasis may be linked to the temperature-sensitive paralysis of csp mutant flies, which is consistent with previous observations suggesting that paralysis is not caused by a defect in evoked release (Barclay et al., 2002). However, the paralytic phenotype may also be linked to subtle defects in synaptic growth of CNS neurons.
CSP may act as an independent synaptic chaperone
The progressive deterioration of evoked release at csp null NMJs during increasing temperature resembles a classical chaperone defect and has been widely regarded as evidence for a role of the CSP-Hsc70 interaction in stabilizing proteins against thermal stress (for review, see Chamberlain and Burgoyne, 2000; Zinsmaier and Bronk, 2001). However, this is not entirely the case because ΔJ-CSP expression restored this defect. Hence, the CSP-facilitated thermo-protection of evoked release is mediated by a mechanism that is independent of the J domain and the activity of Hsc70 because the J domain is essential and sufficient for stimulating the ATPase activity of Hsc70 (Braun et al., 1996; Chamberlain and Burgoyne, 1997b; Zhang et al., 1999). A general Hsc70-independent chaperone function of CSP is supported by the ability of bovine CSP to suppress thermally induced protein aggregation in vitro, which is also not abolished by mutations of the J domain (Chamberlain and Burgoyne, 1997a). An individual synaptic chaperone function of CSP is not in conflict with the observation that evoked release is thermo-intolerant at hsc70 mutant NMJs (Bronk et al., 2001) because Hsc70 can also independently suppress protein aggregation (Langer et al., 1992; Bukau and Horwich, 1998).
Although the ΔJ-CSP-mediated rescue makes it clear that the role of CSP for the thermo-protection of evoked release does not require the J domain, it remains questionable whether this chaperone function is truly independent of the ATPase activity of Hsc70. Although all previous biochemical studies consistently suggest that the mutant ΔJ-CSP and H45Q-CSP proteins cannot directly activate Hsc70, they do not exclude the possibility that the mutant proteins are indirectly complexed with Hsc70 through a third interacting protein, such as SGT. SGT interacts individually with the C terminus of both Hsc70 and CSP and increases the ATPase activity of Hsc70 approximately threefold (Tobaben et al., 2001). Because neither the ΔJ-deletion nor the H45Q mutation are expected to interfere with CSP-SGT binding, it cannot be ruled out that a J domain mutant CSP can form a trimeric complex with SGT and Hsc70. However, to rigorously test this possibility, mutations and antibodies of Drosophila SGT protein will be required.
Footnotes
This work was supported in part by a National Research Service award (P.B.), by grants from the Canadian Institutes of Health Research (H.L.A., R.M.R.), and by National Institute of Neurological Disorders and Stroke Grant RO1NS038274 (K.E.Z.).
Correspondence should be addressed to Konrad E. Zinsmaier, Arizona Research Laboratories Division of Neurobiology, University of Arizona, Gould-Simpson Building 627, P.O. Box 210077, Tucson, AZ 85721-0077. E-mail: kez@neurobio.arizona.edu.
P. Bronk's present address: Center for Basic Neuroscience, University of Texas Southwestern Medical Center, Dallas, TX 75390.
Z. Nie's present address: Department of Neuroscience, 234d Stemmler Hall, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6974.
K. Dawson-Scully's present address: Department of Biology, University of Toronto at Mississauga, Mississauga, Ontario L5L 1C6, Canada.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252204-11$15.00/0
References
- Arnold C, Reisch N, Leibold C, Becker S, Prufert K, Sautter K, Palm D, Jatzke S, Buchner S, Buchner E (2004) Structure-function analysis of the cysteine string protein in Drosophila: cysteine string, linker and C terminus. J Exp Biol 207: 1323-1334. [DOI] [PubMed] [Google Scholar]
- Barclay JW, Atwood HL, Robertson RM (2002) Impairment of central pattern generation in Drosophila cysteine string protein mutants. J Comp Physiol 188: 71-78. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401-415. [DOI] [PubMed] [Google Scholar]
- Braun J, Wilbanks SM, Scheller RH (1996) The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem 271: 25989-25993. [DOI] [PubMed] [Google Scholar]
- Bronk P, Wenniger JJ, Dawson-Scully K, Guo X, Hong S, Atwood HL, Zinsmaier KE (2001) Drosophila Hsc70-4 is critical for neurotransmitter exocytosis in vivo. Neuron 30: 475-488. [DOI] [PubMed] [Google Scholar]
- Brown H, Larsson O, Branstrom R, Yang SN, Leibiger B, Leibiger I, Fried G, Moede T, Deeney JT, Brown GR, Jacobsson G, Rhodes CJ, Braun JE, Scheller RH, Corkey BE, Berggren PO, Meister B (1998) Cysteine string protein (CSP) is an insulin secretory granule-associated protein regulating beta-cell exocytosis. EMBO J 17: 5048-5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92: 351-366. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Graham ME, Fisher RJ (2000) Cysteine string protein controls late steps in exocytosis. Methods Find Exp Clin Pharmacol 22: 355. [Google Scholar]
- Chamberlain LH, Burgoyne RD (1997a) The molecular chaperone function of the secretory vesicle cysteine string proteins. J Biol Chem 272: 31420-31426. [DOI] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD (1997b) Activation of the ATPase activity of heat-shock proteins Hsc70/Hsp70 by cysteine-string protein. Biochem J 322: 853-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD (1998) Cysteine string protein functions directly in regulated exocytosis. Mol Biol Cell 9: 2259-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD (2000) Cysteine string protein: the chaperone at the synapse. J Neurochem 74: 1781-1789. [DOI] [PubMed] [Google Scholar]
- Chen S, Zheng X, Schulze KL, Morris T, Bellen H, Stanley EF (2002) Enhancement of presynaptic calcium current by cysteine string protein. J Physiol (Lond) 538: 383-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson-Scully K (2003) The role of cysteine string proteins at the neuromuscular junction of Drosophila. PhD thesis, University of Toronto.
- Dawson-Scully K, Bronk P, Atwood HL, Zinsmaier KE (2000) Cysteinestring protein increases the calcium sensitivity of neurotransmitter exocytosis in Drosophila. J Neurosci 20: 6039-6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle KK, Zinsmaier KE, Buchner S, Gruhn M, Jenni M, Arnold C, Leibold C, Reisch D, Walter N, Hafen E, Hofbauer A, Pflugfelder GO, Buchner E (1998) Wide distribution of cysteine string protein in Drosophila tissues revealed by targeted mutagenesis. Cell Tissue Res 294: 203-217. [DOI] [PubMed] [Google Scholar]
- Evans GJ, Morgan A (2002) Phosphorylation-dependent interaction of the synaptic vesicle proteins cysteine string protein and synaptotagmin I. Biochem J 364: 343-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Wolfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Munoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, Sudhof TC (2004) The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42: 237-251. [DOI] [PubMed] [Google Scholar]
- Graham ME, Burgoyne RD (2000) Comparison of cysteine string protein (Csp) and mutant alpha-SNAP overexpression reveals a role for Csp in late steps of membrane fusion in dense-core granule exocytosis in adrenal chromaffin cells. J Neurosci 20: 1281-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen CB, Umbach JA (1992) Suppression cloning of the cDNA for a candidate subunit of a presynaptic calcium channel. Neuron 9: 527-537. [DOI] [PubMed] [Google Scholar]
- Heckmann M, Adelsberger H, Dudel J (1997) Evoked transmitter release at neuromuscular junctions in wild type and cysteine string protein null mutant larvae of Drosophila. Neurosci Lett 228: 167-170. [DOI] [PubMed] [Google Scholar]
- Kelley WL (1998) The J-domain family and the recruitment of chaperone power. Trends Biochem Sci 23: 222-227. [DOI] [PubMed] [Google Scholar]
- Kelley WL (1999) Molecular chaperones: how J domains turn on Hsp70s. Curr Biol 9: R305-R308. [DOI] [PubMed] [Google Scholar]
- Kurdyak P, Atwood HL, Stewart BA, Wu CF (1994) Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J Comp Neurol 350: 463-472. [DOI] [PubMed] [Google Scholar]
- Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU (1992) Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 356: 683-689. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS (1994) Ectopic and increased expression of fasciclin II alters motor neuron growth cone guidance. Neuron 13: 507-523. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Ganetzky B (2000) Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron 26: 35-43. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Hegstrom-Wojtowicz M, Charlton MP, Atwood HL (2002) Fast calcium signals in Drosophila motor neuron terminals. J Neurophysiol 88: 2659-2663. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Marin L, Charlton MP, Atwood HL (2004) Synaptic vesicles: test for a role in presynaptic calcium regulation. J Neurosci 24: 2496-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JEA (2000) Cysteine string protein regulates G protein modulation of N-type calcium channels. Neuron 28: 195-204. [DOI] [PubMed] [Google Scholar]
- Miller LC, Swayne LA, Kay JG, Feng ZP, Jarvis SE, Zamponi GW, Braun JE (2003a) Molecular determinants of cysteine string protein modulation of N-type calcium channels. J Cell Sci 116: 2967-2974. [DOI] [PubMed] [Google Scholar]
- Miller LC, Swayne LA, Chen L, Feng ZP, Wacker JL, Muchowski PJ, Zamponi GW, Braun JE (2003b) Cysteine string protein (CSP) inhibition of N-type calcium channels is blocked by mutant huntingtin. J Biol Chem 278: 53072-53081. [DOI] [PubMed] [Google Scholar]
- Morales M, Ferrus A, Martinez PM (1999) Presynaptic calcium-channel currents in normal and csp mutant Drosophila peptidergic terminals. Eur J Neurosci 11: 1818-1826. [DOI] [PubMed] [Google Scholar]
- Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, Zinsmaier KE (1999) Overexpression of cysteine string protein in Drosophila reveals interactions with syntaxin. J Neurosci 19: 10270-10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL (1998) Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet 19: 171-174. [DOI] [PubMed] [Google Scholar]
- Ranjan R, Bronk P, Zinsmaier KE (1998) Cysteine string protein is required for calcium secretion coupling of evoked neurotransmission in Drosophila but not for vesicle recycling. J Neurosci 18: 956-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spafford JD, Munno DW, Van Nierop P, Feng ZP, Jarvis SE, Gallin WJ, Smit AB, Zamponi GW, Syed NI (2003a) Calcium channel structural determinants of synaptic transmission between identified invertebrate neurons. J Biol Chem 278: 4258-4267. [DOI] [PubMed] [Google Scholar]
- Spafford JD, Chen L, Feng ZP, Smit AB, Zamponi GW (2003b) Expression and modulation of an invertebrate presynaptic calcium channel alpha1 subunit homolog. J Biol Chem 278: 21178-21187. [DOI] [PubMed] [Google Scholar]
- Spradling AC (1986) P element mediated transformation. In: Drosophila: a practical approach (Roberts DB, ed), pp 175-197. Oxford: IRL.
- Stahl B, Tobaben S, Sudhof TC (1999) Two distinct domains in Hsc70 are essential for the interaction with the synaptic vesicle cysteine string protein. Eur J Cell Biol 78: 375-381. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF (1994) Improved stability of Drosophila larval neuromuscular preparations in haemolymphlike physiological solutions. J Comp Physiol 175: 179-191. [DOI] [PubMed] [Google Scholar]
- Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B (2001) A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31: 987-999. [DOI] [PubMed] [Google Scholar]
- Umbach JA, Gundersen CB (1997) Evidence that cysteine string proteins regulate an early step in the Ca2+-dependent secretion of neurotransmitter at Drosophila neuromuscular junctions. J Neurosci 17: 7203-7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JA, Zinsmaier KE, Eberle KK, Buchner E, Benzer S, Gundersen CB (1994) Presynaptic dysfunction in Drosophila csp mutants. Neuron 13: 899-907. [DOI] [PubMed] [Google Scholar]
- Umbach JA, Saitoe M, Kidokoro Y, Gundersen CB (1998) Attenuated influx of calcium ions at nerve endings of csp and shibire mutant Drosophila. J Neurosci 18: 3233-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ (1999) Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron 23: 593-605. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kelley WL, Chamberlain LH, Burgoyne RD, Wollheim CB, Lang J (1998) Cysteine-string proteins regulate exocytosis of insulin independent from transmembrane ion fluxes. FEBS Lett 437: 267-272. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kelley WL, Chamberlain LH, Burgoyne RD, Lang J (1999) Mutational analysis of cysteine-string protein function in insulin exocytosis. J Cell Sci 112: 1345-1351. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Bronk P (2001) Molecular chaperones and the regulation of neurotransmitter exocytosis. Biochem Pharmacol 62: 1-11. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Hofbauer A, Heimbeck G, Pflugfelder GO, Buchner S, Buchner E (1990) A cysteine-string protein is expressed in retina and brain of Drosophila. J Neurogenet 7: 15-29. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S (1994) Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263: 977-980. [DOI] [PubMed] [Google Scholar]