

Abstract
BACKGROUND:
FADS1 gene encodes delta 5 desaturase, a rate-limiting enzyme in the metabolism of n-3 and n-6 polyunsaturated fatty acids (PUFAs). Minor alleles of FADS1 locus polymorphisms are associated with reduced FADS1 expression and intra-hepatic fat accumulation. However, the relationship between FADS1 expression and pediatric nonalcoholic fatty liver disease (NAFLD) risk remains to be explored.
METHODS:
We analyzed FADS1 transcription levels and their association with intra-hepatic fat and histology in children, and we performed pathway enrichment analysis on transcriptomic profiles associated with FADS1 polymorphisms. We also evaluated the weight of FADS1 alleles on the response to combined docosahexaenoic acid, choline, and vitamin E (DHA-CHO-VE) treatment.
RESULTS:
FADS1 mRNA level was significantly and inversely associated with intra-hepatic fat (p = 0.004), degree of steatosis (p = 0.03), fibrosis (p = 0.05), and NASH (p = 0.008) among pediatric livers. Transcriptomics demonstrated a significant enrichment of a number of pathways strongly related to NAFLD (e.g., liver damage, fibrosis, and hepatic stellate cell activation). Compared to children who are common allele homozygotes, children with FADS1 minor alleles had a greater reduction in steatosis, fibrosis, and NAFLD activity score after DHA-CHO-VE.
CONCLUSION:
This study suggests that decreased FADS1 expression may be associated with NAFLD in children but an increased response to DHA-CHO-VE.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in children but its pathogenesis remains incompletely understood. The prevalence of pediatric NAFLD in the U.S. was estimated to be 8–17% but up to 38% among obese children.1 NAFLD is defined as a spectrum of chronic liver disorders, beginning as an accumulation of fat in the liver without significant alcohol consumption that can gradually progress to steatohepatitis (nonalcoholic steatohepatitis (NASH)) and even cirrhosis in children, leading to substantial liver injury.2,3 Also, NAFLD in children is highly associated with a series of early metabolic conditions, including obesity, insulin resistance, glucose intolerance, and cardiovascular abnormalities,4 exerting an enormous medical challenge for pediatric care.5 Understanding the etiology of pediatric NAFLD and NASH is poor especially when compared to extensive studies in adults.1,5 Given the current rapid growing population of NAFLD and its co-morbidities in children, it is an urgent need to elucidate the principle disease etiology and develop safe and effective treatment for pediatric NAFLD.
It is well recognized that n-3 polyunsaturated fatty acid (PUFA) deficiency is associated with many metabolic perturbations, including NAFLD.6–9 In humans, particularly due to the high n-6/n-3 PUFA ratio in Western diet, n-3 PUFA deficiency has been associated with NAFLD and NASH in both adults and children.10–19 Small clinical studies in children suggest that n-3 PUFA supplementation may improve NAFLD.20 However, recent large clinical trials of n-3 PUFA in adults with NAFLD have yielded mixed results.21–24 While inter-individual variability has been recognized as a key factor influencing the response to n-3 PUFA treatment,25–27 the clinical trials testing the effectiveness of n-3 PUFA in NAFLD have not considered genetic variability in n-3 PUFA metabolism among study participants.
Both n-6 and n-3 PUFA metabolism are controlled by Δ5 and Δ6 desaturases (D5D, D6D) encoded by FADS1 and FADS2 genes, respectively. D5D is the rate-limiting enzyme in converting α-linolenic acid, the major dietary n-3 PUFA precursor, into the bioactive forms eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Our recent studies indicated that FADS1 is significantly involved in hepatic fat accumulation and NAFLD.28,29 Notably, among the genes involved in this pathway, FADS1 harbors genetic alleles in strong linkage disequilibrium (LD; allele frequency ranging from 32% to 43%30 consistently identified as the most important determinants for FADS1 (i.e., D5D) activity and endogenous n-3 PUFA levels in humans.30–32 In addition, increasing evidence suggests that FADS1 low-function alleles can influence both the pharmacokinetics and pharmacodynamics of supplemented n-3 PUFA, potentially leading to inter-patient differences in response to its intake.26,27 Unfortunately, no clinical study thus far has considered these alleles in testing the effectiveness of n-3 PUFA in treating NAFLD. Further delineating this pharmacogenetic mechanism may lead to a more effective n-3 PUFA treatment for NAFLD.
In this proof-of-concept study, we evaluated the relationship between FADS1 expression and liver histology in pediatric liver samples and explored the molecular pathway associated with low-function FADS1 alleles. Further, we examined the relationship between FADS1 polymorphism and clinical response to DHA supplementation combined with choline and vitamin E (DHA-CHO-VE) in a small cohort of children with NAFLD who have completed a randomized clinical trial registered33 at ClinicalTrials.gov (ID: NCT01934777).
METHODS AND MATERIALS
Samples
Samples used in this study as well as the data analysis plan using the samples are reported in Fig. 1.
Fig. 1.
Diagram of sample sets used in this study as well as the data analysis plan
The liver tissue samples were described previously.28 Briefly, these are leftover tissue samples collected from liver transplantation donors who are deceased individuals but were willing to donate their organs for transplantation purposes. Demographic information of the donors was demonstrated in Table 1. Transcriptomic data from 55 pediatric liver tissue samples (aged 0–18years) were collected previously, among which only 22 were characterized for histologic evaluation. Genotypic data of 6 FADS1 single-nucleotide polymorphisms (SNPs;rs174576, rs1535, rs174546, rs102275, rs174537, and rs174556) for all 55 pediatric liver samples were previously collected.28
Table 1.
Biochemical and histological characteristics of the donor liver tissue samples (n = 22)
| Male sex, n (%) | 12 (54.5) |
| Age (years) | |
| Mean ± SD | 6.1 ± 4.4 |
| Median (IQR) | 4.5 (2.25–9) |
| BMI (kg/m2) | |
| Mean ± SDa | 17.7 ± 5.9 |
| Median (IQR) | 16.5 (15.5–17.6) |
| Race | |
| White | 17 (77.3) |
| Black | 5 (22.7) |
| Biochemical features | |
| Sirius red, mean ± SD (%) | 8.2 ± 4.4 |
| Histological features | |
| Total hepatic fat, mean ± SD (mg fat/mg tissue) | 0.020 ± 0.0088 |
| Steatosis (%, >5%) | 4 (18.2) |
| NASH | 4 (18.2) |
The quantification of Sirius red staining is based on the percentage of the stained area of the liver tissue section
SD standard deviation, BMI body mass index, IQR interquartile range Missing information for six participants.
Total hepatic fat content was measured using a protocol established in our previous study.34 The degree of steatosis was visually assessed based on the hematoxylin–eosin–stained tissue sections and the total collagen content was quantified by Sirius red staining. The presence of NAFLD and NASH was defined based on the previously established method by NASH-CRN.35
Study population from clinical trial
Nineteen children with NAFLD belonging to the treatment arm of DHA-CHO-VE trial were included in the study. In particular, these children presented a liver biopsy-proven diagnosis of NASH. Other causes of liver diseases were excluded before the enrollment in the trial. These included: the presence of liver disease due to any of the following: Wilson disease, hepatitis B and C, acute systemic disease, autoimmune hepatitis, hypothyroidism, cystic fibrosis, celiac disease, and suspicion of muscular dystrophy, alpha-1-antitrypsin deficiency, and metabolic inherited diseases. Patients were also excluded if body weight and carbohydrate metabolism were altered by the use of parenteral nutrition, protein malnutrition, previous gastrointestinal surgery, structural abnormalities of the gastrointestinal tract, or neurological impairment. Finally, the use of nonsteroidal anti-inflammatory drugs, antibiotics, probiotics, or antisecretory drugs capable of causing achlorhydria within the 2 months preceding enrolment were considered additional exclusion criteria. Details of the trial have been already reported.33 However, briefly, all children in the treatment arm received, every day for 6 months, pearls combining 250 mg of DHA, 39 UI of vitamin E, and 201 mg of choline (DHA-CHO-VE). Pearls were provided by DMF Dietetic Metabolic food (Italy). Concomitantly, all patients received recommendation for hypocaloric diet (25–30 kcal/kg/day) and twice weekly 1-h physical activity during the treatment and for further 6 months of follow-up. Compliance to treatments and diet was monitored through monthly visit by counting the amount of investigational product left in the bottle.
Symptoms and side effects were assessed in each visit by the investigator, who was blind to medication assignment. Complete medical histories were recorded for all participants. Collection of anthropometrical data, biochemical data, and second biopsy were performed at baseline and after 12 months from trial start. Patients and investigators were blinded before and after the assignment of the intervention. The study was registered at ClinicalTrials.gov, with the number ID: NCT01934777 on 30 August 2013. However, we underline that the current primary end point differs from the original protocol submitted to ClinicalTrials.gov as well as described in the previous study.33 The Bambino Gesù Ethics Research Committee approved the study, in accordance with the Declaration of Helsinki, and parents of the included patients gave their written informed consent to therapies and the tests performed for research purpose.
Anthropometrics
Anthropometric data were collected at baseline and after 12 months. Weight was measured by a conventional scale with a precision of 100 g and height was measured by a Harpenden stadiometer with a precision of 1 mm. Body mass index was expressed in kilograms per square meters (kg/m2).
Laboratory parameters
Blood were collected at baseline and at month 12 after an overnight fasting and immediately processed to perform the analysis of alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl-transferase, total triglycerides and cholesterol, high-density lipoprotein, low-density lipoprotein, glucose, and uric acid by standard laboratory methods. Demographic information of the donor individuals are reported in Table 3.
Table 3.
Characteristics of Italian children with NAFLD who received DHA-CHO-VE treatment
| Demographics and clinical features |
FADS genotype (rs174576) |
|||||
|---|---|---|---|---|---|---|
| CC (n = 9) |
CA+AA (n = 10) |
|||||
| Before | After | p Value | Before | After | p Value | |
| Age | ||||||
| Mean ± SD | 13.00 ± 2.45 | 14.00 ± 2.45 | 0.32 | 13.81 ± 2.13 | 14.81 ± 2.13 | 0.21 |
| Median (IQR) | 13.00 (11.00–14.00) | 14.00 (12.00– 15.00) | 13.00 (12.00–15.50) | 14.00 (13.00–16.50) | ||
| Male sex, n (%) | 5 (55.56) | 5 (55.56) | 1 | 8 (80.00) | 8 (80.00) | 1 |
| BMI (kg/m2) | ||||||
| Mean ± SD | 26.47 ± 4.09 | 26.45 ± 4.28 | 0.89 | 29.29 ± 4.07 | 29.06 ± 4.64 | 0.96 |
| Median (IQR) | 28.89 (22.84–29.88) | 27.79 (22.84–30.00) | 30.12 (26.37–30.71) | 28.44 (26.29–33.04) | ||
| BMI (percentile) | 95.7 ± 4.1 | 97.2 ± 2.4 | 0.35 | 94.1 ± 4.6 | 94.5 ± 5.9 | 0.21 |
| WC (cm) | ||||||
| Mean ± SD | 84.54 (11.62) | 84.69 (10.55) | 0.97 | 87.56 (8.89) | 92.09 (6.85) | 0.19 |
| Median (IQR) | 89.00 (78.00–90.00) | 84.00 (76.00–89.00) | 89.50 (83.00–92.30) | 94.00 (84.50–95.25) | ||
| WC (percentile) | 83.5 ± 18.9 | 82.3 ± 20.3 | 0.95 | 83 ± 12.8 | 86.8 ± 9.5 | 0.43 |
| Before | After | p Value | Before | After | p Value | |
| Biochemical features | ||||||
| Col Tot (mg/dL) | 149.78 ± 17.38 | 149 ± 16.43 | 0.75 | 132.63 ± 29.16 | 142.51 ± 27.40 | 1 |
| HDL (mg/dL) | 48.33 ± 8.75 | 47.22 ± 8.59 | 0.82 | 47.8 ± 7.48 | 43.76 ± 5.00 | 1 |
| LDL (mg/dL) | 83.78 ± 26.54 | 80.11 ± 20.99 | 0.89 | 76.7 ± 25.84 | 94.33 ± 18.59 | 0.51 |
| TG (mg/dL) | 97.78 ± 55.80 | 112.22 ± 68.01 | 0.69 | 96.36 ± 52.66 | 94.33 ± 59.83 | 0.76 |
| Uric acid (mg/dL) | 5.33 ± 0.89 | 5.13 ± 0.87 | 0.47 | 5.22 ± 1.07 | 13.57 ± 24.92 | 0.87 |
| AST (UI/mL) | 33.44 ± 8.90 | 32.89 ± 7.45 | 0.69 | 34.72 ± 14.58 | 37.33 ± 24.17 | 0.67 |
| ALT (UI/mL) | 48.33 ± 22.94 | 45.67 ± 21.23 | 0.79 | 47.9 ± 32.70 | 32.41 ± 15.54 | 0.25 |
| GGT (UI/mL) | 16.67 ± 7.00 | 16.67 ± 6.94 | 1 | 17.54 ± 9.27 | 17.33 ± 8.98 | 0.56 |
| Glycemia (mg/dL) | 86.89 ± 7.49 | 84.22 ± 8.43 | 0.93 | 85.63 ± 7.21 | 79.33 ± 22.09 | 0.9 |
| Histological features | ||||||
| Fibrosis Index | 1.78 ± 0.44 | 1.44 ± 0.52 | 0.17 | 1.81 ± 0.40 | 1.11 ± 0.33 | 0.00057 |
| Steatosis Index | 1.56 ± 0.73 | 1.33 ± 1.22 | 0.64 | 1.45 ± 0.93 | 1 ± 1.00 | 0.011 |
| Ballooning | 1.33 ± 0.50 | 0.78 ± 0.83 | 0.12 | 1.18 ± 0.87 | 0.56 ± 0.52 | 0.0054 |
| Lobular inflammation | 1.11 ± 0.60 | 0.89 ± 0.33 | 0.36 | 1.18 ± 0.40 | 1.11 ± 0.33 | 0.58 |
| Portal inflammation | 1.33 ± 0.50 | 1.22 ± 0.44 | 0.64 | 1.45 ± 0.52 | 1.22 ± 0.66 | 0.82 |
| NAS | 4 ± 1.32 | 3.38 ± 1.92 | 0.52 | 3.9 ± 1.64 | 2.66 ± 1.50 | 0.0071 |
Measurement is expressed in mean ± SD, Wilcoxon’s rank-sum test is used for the comparison of before and after treatment
In bold the significant values for p <0.05
SD standard deviation, BMI body mass index, IQR interquartile range, WC waist circumference, Col Tot total cholesterol, HDL high-density lipoprotein, LDL low-density lipoprotein, TG triglycerides, AST aspartate aminotransferase, ALT alanine transaminase, GGT gamma-glutamyl transferase, NAS NAFLD activity score
Genotyping
Genotyping of 17 SNPs (rs174545, rs174546, rs174547, rs174548, rs174549, rs174550, rs174555, rs174556, rs174560, rs174561, rs174562, rs174568, rs1535, rs174574, rs174576, rs174577, and rs174581) across the FADS locus in the NAFLD children treated with DHA-CHO-VE were performed using with Sequenom MassARRAY iPLEX Gold assays (Sequenom, San Diego, CA). However, given the high LD level among these 17 FADS SNPs (pairwise LD r2 > 0.8), we chose the FADS variant rs174576 as a representative SNP for all subsequent analyses. We chose this SNP also because it was associated with fatty acid denaturation pathway activity, whole-body fat oxidation, and DHA/EPA index in young individuals.36,37
Statistical analyses
The association between six FADS1 SNPs and transcriptome data of donor liver tissues (n = 55) was performed using R package Matrix eQTL.38 Each SNP in FADS1 region was assessed for the association with liver transcriptome using multiple linear regression model, where age, gender, and ancestry were adjusted for the association as covariates. The SNP was coded using dominant genetic model, i.e., AA: 0, AB and BB: 1. We selected genes significantly (p < 0.05) associated with all the six FADS1 SNPs for pathway analysis. Note that, given the small sample size, we used a liberal nominal p value (0.05) as a statistical significance cutoff for selecting genes for the pathway analysis so that critical pathways may not be missed. Significantly (p <0.05) enriched pathways were identified using Ingenuity Pathway Analysis (IPA®, QIAGEN, Redwood City, www.qiagen.com/ingenuity).
Demographic, biochemical, and histological features of the NAFLD children (n = 19) were compared between groups using Wilcoxon rank-sum test for continuous variables. Multiple logistic regression model was used to test the association between FADS1 genotype and the response to the treatment in terms of steatosis reduction. Genotypes were coded in dominant genetic model. Age, gender, and the genotypes of PNPLA3 (rs738409)39 and TM6SF2 (rs58542926)40 were adjusted for the association. Logistic regression was performed using the package PLINK 1.07.41 All the other analyses were performed using R statistical packages (www.r-project.org).
RESULTS
Association between FADS1 mRNA and liver histology in children We have characterized biochemical, and histologic features of donor liver tissue samples of 22 children. As summarized in Table 1, we found that 4 out of 22 individuals possess a ≥5% steatosis in their livers, and all these 4 patients were characterized as NASH or borderline NASH. The total fat content of all 22 livers is 0.02 ± 0.0088 mg fat per mg tissue, and total collagen content is 8.2 ± 4.4%.
We aimed to understand the relationship between FADS1 transcription and liver histology in children. We found that FADS1 mRNA level was significantly and inversely associated with total hepatic fat content (p = 0.004), degree of steatosis (p = 0.03), fibrosis (p = 0.05), and NASH (p = 0.008) among pediatric livers (Fig. 2).
Fig. 2.
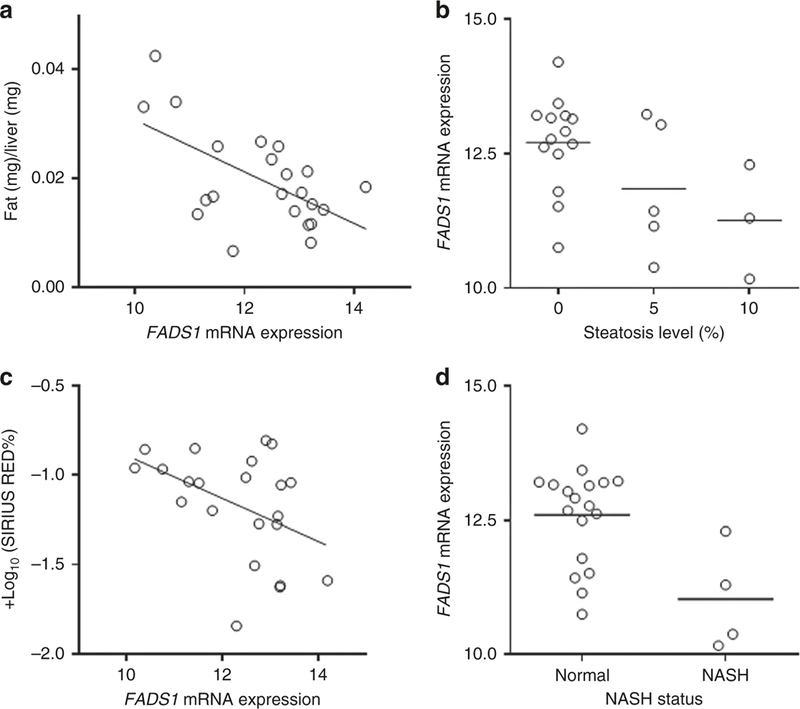
Correlation between FADS1 mRNA level and total fat content (a), steatosis level (b), fibrosis level (c), and NASH status (d)
Pathway enrichment of genes associated with FADS1 polymorphism
Our previous study has demonstrated that the minor alleles of FADS1 variants are associated with decreased hepatic FADS1 transcription.28 We hypothesized that these FADS1 low-function alleles may lead to an altered transcriptomic profile where certain pathways involved in nonalcoholic fatty liver disease may be affected. In order to test this hypothesis, we performed a genome-wide analyses on the association between FADS1 variant genotypes and mRNA expression. At a nominal significance level (p < 0.05), we identified 619 genes whose mRNA expression is significantly associated with FADS1 genotype (Supplemental Table S1 (online)). We further conducted a pathway enrichment analysis on these genes using the IPA package. Our analysis revealed a significant enrichment for a number of pathways strongly related to NAFLD: hepatic fibrosis/hepatic stellate cell Activation) (p = 0.01), protein kinase A signaling (p = 0.01), and VDR/RXR activation (p = 0.002) as well as multiple interleukin signaling pathways (p < 0.05) (Supplemental Table S2 (online)). Given the association between the minor alleles of FADS1 variants and the low FADS1 expression and function, we sought to explore whether the liver transcriptome profile associated with these minor alleles is also more likely associated with NAFLD. We then performed IPA toxicity function pathway analysis specifically for the genes whose expression is positively associated with the FADS1 minor alleles. As demonstrated in Table 2, critical toxicity function pathways, e.g., activation of liver fibrosis (p = 0.01), hepatic fibrosis (p = 0.014), and liver damage (p = 0.02), are particularly enriched. Also, hepatic fibrosis (p = 0.006) and increased liver damage (p = 0.044) are also top pathways significantly enriched among the toxicity Lists. Again, triacylglycerol biosynthesis (p = 0.046) and hepatic fibrosis/hepatic stellate cell activation (p = 0.047) are significantly enriched top canonical pathways (Table 2).
Table 2.
Top IPA enriched pathways for upregulated genes (p <0.05)
| Enriched pathways | p Value | Molecules | Functions | Disease or function annotation |
|---|---|---|---|---|
| Tox functions | ||||
| Liver enlargement, liver hepatomegaly | 0.0038 | STAT6, ESR2, MIF, GBA | Hepatomegaly | Hepatomegaly |
| Heart failure | 0.0073 | IL16 | Heart failure with preserved left ventricular ejection fraction | Heart failure with preserved left ventricular ejection fraction |
| Nephrosis | 0.0073 | TTC21B | Nephronophthisis type 12 | Nephronophthisis type 12 |
| Liver fibrosis | 0.0100 | IL1A, LEPR, MIF | Activation | Activation of hepatic stellate cells |
| Cardiac necrosis/cell death | 0.0145 | TBX5 | Apoptosis | Apoptosis of endocardial cells |
| Increased levels of CRP | 0.0145 | IL2 | Localization | Increased localization of CRP |
| Liver fibrosis | 0.0145 | IL2 | Hepatic fibrosis | Mild hepatic fibrosis |
| Liver fibrosis | 0.0145 | IL2 | Hepatic fibrosis | Moderate hepatic fibrosis |
| Congenital heart anomaly | 0.0201 | PSEN2, TBX5 | Failure of heart looping | Failure of heart looping |
| Liver damage | 0.0213 | STAT6, MIF | Damage | Damage of liver cells |
| Liver proliferation | 0.0217 | SKP2 | Growth | Arrest in growth of hepatocytes |
| Renal inflammation, Renal nephritis | 0.0217 | IL16 | IgA nephropathy | Primary IgA nephropathy |
| Kidney failure | 0.0217 | GBA | Progressive myoclonic epilepsy type 4 with renal failure | Progressive myoclonic epilepsy type 4 with renal failure |
| Renal damage | 0.0328 | IL2, ITGB6, LEPR, MIF | Damage | Damage of kidney |
| Cardiac necrosis/cell death | 0.0359 | PIN1 | Cell death | Cell death of cardiac stem cells |
| Cardiac inflammation | 0.0392 | IL2, STAT6 | Inflammation | Inflammation of heart |
| Renal atrophy | 0.0460 | AGA, OVOL1 | Atrophy of kidney | Atrophy of kidney |
| Renal proliferation | 0.0499 | SKP2 | Proliferation | Proliferation of renal tubular epithelial cells |
| Tox lists | ||||
| Hepatic fibrosis | 0.0060 | IL2, IL1A, CD40, SDC1 | ||
| Increases renal damage | 0.0214 | IL2, ITGB6, MIF | ||
| Aryl hydrocarbon receptor signaling | 0.0302 | GSTO2, DHFR, IL1A, ESR2 | ||
| Increases liver damage | 0.0437 | IL2, CD40, MIF | ||
| Canonical pathways | ||||
| Crosstalk between dendritic cells and natural killer cells | 0.0043 | IL2, ICAM3, CD40, TREM2 | ||
| dTMP de novo biosynthesis | 0.0046 | DHFR, DHFRL1 | ||
| Amyloid processing | 0.0065 | PSEN2, APH1A, CAPN2 | ||
| Granzyme A signaling | 0.0091 | HIST1H1E, H1FX | ||
| Dendritic cell maturation | 0.0095 | IL1A, COL5A3, LEPR, CD40, TREM2 | ||
| Chondroitin Sulfate degradation (metazoa) | 0.0110 | GM2A, MGEA5 | ||
| Dermatan sulfate degradation (metazoa) | 0.0110 | GM2A, MGEA5 | ||
| Regulation of the epithelial–mesenchymal transition pathway | 0.0112 | WNT8A, PSEN2, CLDN3, APH1A, TWIST1 | ||
| Estrogen-mediated S-phase entry | 0.0132 | SKP2, ESR2 | ||
| Antiproliferative role of TOB in T cell signaling | 0.0151 | IL2, SKP2 | ||
| T helper cell differentiation | 0.0158 | IL2, STAT6, CD40 | ||
| CDP-diacylglycerol biosynthesis I | 0.0166 | AGPAT3, AGPAT1 | ||
| Aryl hydrocarbon receptor signaling | 0.0224 | GSTO2, DHFR, IL1A, ESR2 | ||
| Phosphatidylglycerol biosynthesis II (non-plastidic) | 0.0240 | AGPAT3, AGPAT1 | ||
| Altered T cell and B cell signaling in rheumatoid arthritis | 0.0263 | IL2, IL1A, CD40 | ||
| Communication between innate and adaptive immune cells | 0.0282 | IL2, IL1A, CD40 | ||
| Notch signaling | 0.0309 | PSEN2, APH1A | ||
| Inhibition of matrix metalloproteases | 0.0347 | MMP26, SDC1 | ||
| Granulocyte adhesion and diapedesis | 0.0417 | CLDN3, IL1A, MMP26, SDC1 | ||
| Hematopoiesis from pluripotent stem cells | 0.0457 | IL2, IL1A | ||
| Triacylglycerol biosynthesis | 0.0457 | AGPAT3, AGPAT1 | ||
| Hepatic fibrosis/hepatic stellate cell activation | 0.0479 | IL1A, COL5A3, LEPR, CD40 | ||
Association between FADS1 alleles and clinical response to n-3 lipid supplementation in children
We postulated that, while the low-function FADS1 alleles may increase the susceptibility to NAFLD, supplementation n-3 lipids to NAFLD patients would lead to a better response in improving the liver histology. We therefore genotyped the FADS1 variants among NAFLD children who previously received DHA-CHO-VE treatment. Given the high LD level among these variants in the study cohort (r2 > 0.8), we selected rs174576 as a representative variant for the analyses. We then assessed the association between rs174576 minor allele and the improvement in liver histology after the DHA-CHO-VE treatment. We found that, as compared to the carriers of the major allele homozygotes, the minor allele carriers (heterozygotes and minor allele homozygotes) possessed a significant reduction in steatosis, fibrosis, ballooning, and NAFLD activity score (p < 0.02 for all tests;Table 3). As other NAFLD-associated genetic variants, i.e., PNPLA3 and TM6SF2, may confound this association,39,40 we further performed a multivariate analysis using a linear regression model by incorporating the genotype of these two genes. We found that the rs174576 minor allele remained to be significantly (p < 0.05) associated with more steatosis reduction after the DHA-CHO-VE treatment (Supplemental Table S3 (online)).
DISCUSSION
Our study demonstrated that reduced hepatic FADS1 transcription is associated with increased risk to NAFLD in children. The low-function alleles of FADS1 are particularly associated with alteration of molecular pathways involved in NAFLD. In addition, children with NAFLD who are carriers of FADS1 low-function alleles are the best responders to n-3 lipid supplementation. Our findings together indicate an important role of FADS1 function in the pathogenesis of NAFLD in children. Moreover, FADS1 genetic variants may be a useful pharmacogenetic marker capable of distinguishing the patients who are more likely to respond to n-3 lipid supplementation as a therapeutic strategy for NAFLD/NASH in children.
Our results provide new evidence supporting the association between the insufficiency of n-3 PUFA and NAFLD/NASH. While this association may be largely attributed to the imbalanced n-3/n-6 PUFA in the current Western diet, certain genetic perturbations of fatty acid desaturation process may exacerbate this issue. As compared to extensive dietary research, the relationship between genetic variability in fatty acid desaturation and NAFLD/NASH was less clear. This issue could be crucial in pediatric NAFLD since genetic factors may play a more significant role in disease susceptibility at the earlier stage of human development, as compared to the dietary and other environmental factors. Our previous studies have revealed that minor alleles of multiple variants across the FADS1 locus lead to reduced hepatic transcription level of FADS1, which is further associated with reduced desaturation flux of long chain fatty acids but increased total intra-hepatic fat accumulation and susceptibility to liver steatosis.28,29 Accordingly, our current study demonstrated that reduced FADS1 function in the liver is highly correlated with not only increased steatosis but also with fibrosis and NASH. This correlation may be mediated by the altered molecular pathways involved in lipid homeostasis, as well as in hepatic fibrogenesis, suggesting that insufficient FADS1 function can lead to both molecular and pathological changes in the liver even at the early developmental stages of the disease. Besides the central function of FADS1 in the liver, the gut–liver axis derangement may also be involved in the lipid homeostasis in the liver.42 Gut microbiota was found to produce PUFA43; our recent study demonstrated that FADS1 genotype may also influence the oxidized derivatives of linoleic acid that are associated with metabolic syndrome and adverse lipid protein profiles in children.44 It is thus possible that FADS1 alleles play a role in multiple systems in increasing the susceptibility to NAFLD.
Our findings also have implications in clinical management of NAFLD in children. Given the increased understanding of the key role of n-3 PUFA in NAFLD, supplementation of n-3 PUFA to NAFLD patients has long been a potential treatment and preventive option for NAFLD/NASH. Our study revealed a potential mechanism that leads to inter-patient variability in response to n-3 PUFA intake. Our findings suggest that FADS1 variants can be a pharmacogenetic marker that is useful for selecting NAFLD patients who are most likely to respond to n-3 PUFA supplementation. Our study is consistent with previous studies where FADS1 minor alleles are associated with significantly better tissue accumulation of n-3 PUFA after supplementation.26,27 This hypothesis is also supported by the recent large cohort study on using n-3 PUFA supplementation in pregnant women to prevent asthma in their children where carrying FADS1 minor alleles in the pregnant women was found to be predictive of better response in their children.45 As a particular biomarker for n-3 PUFA treatment for NAFLD has not been established, our study warranted further validation of the potential predictive value of FADS1 variants in the treatment or prevention of NAFLD in children and beyond, in particular via prospective clinical studies.
Our study was limited in several aspects. First, the small sample size leads to a low power in testing the relationship between FADS1 alleles, gene expression, and NAFLD histology or drug responses in our sample sets. A post hoc power calculation indicated that the majority of our tests is potentially under-powered (data not shown). Therefore, our findings should be limited to the observations in our cohorts and the result should be further validated in larger-scale sample sets. However, we have previously reported a strong association between FADS1 minor alleles and decreased hepatic FADS1 gene expression, whereas increased total liver fat content.28 Therefore, the findings in this study indeed validated at least in part our previous observations. Moreover, noteworthy, in our study the consistent association between FADS1 and clinical features as well as drug responses, albeit weak, collectively generated a new interesting hypothesis worth further testing in the future. Second, the age group of the donor cohort (6.1 ± 4.4 years) is different from that of the clinical study (13.00 ± 2.45 years). Therefore, the biological effect observed in the former population may not reflect that of the latter. Third, the NAFLD patients in our study were treated with a combined choline, DHA, and VE rather than n-3 PUFA as a single agent. The result thus could be confounded by the pharmacological effects of other compounds. It is thus necessary to design prospective clinical studies in a large population to specifically investigate the relationship between FADS1 alleles and clinical outcomes of n-3 PUFA supplementation. Nevertheless, our study generated interesting directions and hypotheses for understanding the pathogenesis and developing therapeutic/preventive strategies for pediatric NAFLD.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Erin G. Schuetz, St Jude Children Hospital and Dr. Mark J. Ratain, The University of Chicago for providing us the liver tissue samples for the study. This study was supported in part by R01 DK106540 to W.L. and in part by Italian Ministry of Health funds (Ricerca Corrente 2017) to V.N.
Footnotes
ADDITIONAL INFORMATION
The online version of this article (https://doi.org/10.1038/s41390–018-0132–7) contains supplementary material, which is available to authorized users.
Competing interests: The authors declare no competing interests.
Publisher's Disclaimer: Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES
- 1.Mencin AA, Loomba R & Lavine JE Caring for children with NAFLD and navigating their care into adulthood. Nat. Rev. Gastroenterol. Hepatol. 12, 617–28 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB & Angulo P The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 58, 1538–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alisi A, Manco M, Vania A & Nobili V Pediatric nonalcoholic fatty liver disease in 2009. J. Pediatr. 155, 469–74 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Bonci E, Chiesa C, Versacci P, Anania C, Silvestri L & Pacifico L Association of nonalcoholic fatty liver disease with subclinical cardiovascular changes: a systematic review and meta-analysis. Biomed. Res. Int. 2015, 213737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nobili V, Alkhouri N & Alisi A et al. Nonalcoholic fatty liver disease: a challenge for pediatricians. JAMA Pediatr. 169, 170–6 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Masterton GS, Plevris JN & Hayes PC Review article: omega-3 fatty acids - a promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 31, 679–92 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Di Minno MN, Russolillo A, Lupoli R, Ambrosino P, Di Minno A & Tarantino G Omega-3 fatty acids for the treatment of non-alcoholic fatty liver disease. World J. Gastroenterol. 18, 5839–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT & George J Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J. Hepatol. 56, 944–51 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Pacifico L, Giansanti S, Gallozzi A & Chiesa C Long chain omega-3 polyunsaturated fatty acids in pediatric metabolic syndrome. MiniRev. Med. Chem. 14, 791–804 (2014). [PubMed] [Google Scholar]
- 10.Zelber-Sagi S, Nitzan-Kaluski D & Goldsmith R et al. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): a population based study. J. Hepatol. 47, 711–7 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Shapiro H, Tehilla M, Attal-Singer J, Bruck R, Luzzatti R & Singer P The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 30, 6–19 (2011). [DOI] [PubMed] [Google Scholar]
- 12.St-Jules DE, Watters CA & Brunt EM et al. Estimation of fish and omega-3 fatty acid intake in pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 57, 627–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papandreou D, Karabouta Z, Pantoleon A & Rousso I Investigation of anthropometric, biochemical and dietary parameters of obese children with and without non-alcoholic fatty liver disease. Appetite 59, 939–44 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Puri P, Baillie RA & Wiest MM et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–90 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Elizondo A, Araya J & Rodrigo R et al. Polyunsaturated fatty acid pattern in liver and erythrocyte phospholipids from obese patients. Obesity (Silver Spring) 15, 24–31 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Zhu QQ, Lou DJ & Si XW et al. [Serum omega-3 polyunsaturated fatty acid and insulin resistance in type 2 diabetes mellitus and non-alcoholic fatty liver disease]. Zhonghua Nei Ke Za Zhi 49, 305–8 (2010). [PubMed] [Google Scholar]
- 17.Kishino T, Ohnishi H & Ohtsuka K et al. Low concentrations of serum n-3 polyunsaturated fatty acids in non-alcoholic fatty liver disease patients with liver injury. Clin. Chem. Lab. Med. 49, 159–62 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Zheng JS, Xu A, Huang T, Yu X & Li D Low docosahexaenoic acid content in plasma phospholipids is associated with increased non-alcoholic fatty liver disease in China. Lipids 47, 549–56 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Petit JM, Guiu B & Duvillard L et al. Increased erythrocytes n-3 and n-6 polyunsaturated fatty acids is significantly associated with a lower prevalence of steatosis in patients with type 2 diabetes. Clin. Nutr. 31, 520–5 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Nobili V, Carpino G & Alisi A et al. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 9, e88005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scorletti E, Bhatia L & McCormick KG et al. Effects of purified eicosapentaenoic and docosahexaenoic acids in non-alcoholic fatty liver disease: results from the *WELCOME study. Hepatology 60, 1211–21 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Sanyal AJ, Abdelmalek MF, Suzuki A, Cummings OW & Chojkier M, EPE-A Study Group. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 147, 377–84 (2014). e1. [DOI] [PubMed] [Google Scholar]
- 23.Argo CK, Patrie JT & Lackner C et al. Effects of n-3 fish oil on metabolic and histological parameters in NASH: a double-blind, randomized, placebo-controlled trial. J. Hepatol. 62, 190–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dasarathy S, Dasarathy J & Khiyami A et al. Double-blind randomized placebocontrolled clinical trial of omega 3 fatty acids for the treatment of diabetic patients with nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 49, 137–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nording ML, Yang J & Georgi K et al. Individual variation in lipidomic profiles of healthy subjects in response to omega-3 Fatty acids. PLoS ONE 8, e76575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roke K & Mutch DM The role of FADS1/2 polymorphisms on cardiometabolic markers and fatty acid profiles in young adults consuming fish oil supplements. Nutrients 6, 2290–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Hilal M, Alsaleh A & Maniou Z et al. Genetic variation at the FADS1-FADS2 gene locus influences delta-5 desaturase activity and LC-PUFA proportions after fish oil supplement. J. Lipid Res. 54, 542–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Athinarayanan S, Jiang G, Chalasani N, Zhang M & Liu W Fatty acid desaturase 1 gene polymorphisms control human hepatic lipid composition. Hepatology 61, 119–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirkov S, Myers JL, Ramirez J & Liu W SNPs affecting serum metabolomic traits may regulate gene transcription and lipid accumulation in the liver. Metabolism 61, 1523–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ameur A, Enroth S & Johansson A et al. Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 90, 809–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemaitre RN, Tanaka T & Tang W et al. Genetic loci associated with plasma phospholipid n-3 fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium. PLoS Genet. 7, e1002193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka T, Shen J & Abecasis GR et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet 5, e1000338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zohrer E et al. Efficacy of docosahexaenoic acid-choline-vitamin E in paediatric NASH: a randomized controlled clinical trial. Appl. Physiol. Nutr. Metab. 42, 948–954 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Li M, Song J, Mirkov S, Xiao SY, Hart J & Liu W Comparing morphometric, biochemical, and visual measurements of macrovesicular steatosis of liver. Hum. Pathol. 42, 356–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleiner DE, Brunt EM, Van Natta M & Behling C et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–21 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Abdelmagid SA, Clarke SE & Roke K et al. Ethnicity, sex, FADS genetic variation, and hormonal contraceptive use influence delta-5-and delta-6-desaturase indices and plasma docosahexaenoic acid concentration in young Canadian adults: a cross-sectional study. Nutr. Metab. 12, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roke K, Jannas-Vela S, Spriet LL & Mutch DM FADS2 genotype influences whole-body resting fat oxidation in young adult men. Appl. Physiol. Nutr. Metab. 41, 791–4 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Shabalin AA Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romeo S, Kozlitina J & Xing C et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozlitina J, Smagris E & Stender S et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B & Todd-Brown K et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poeta M, Pierri L & Vajro P Gut-liver axis derangement in non-alcoholic fatty liver disease. Children 4, E66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Druart C, Bindels LB & Schmaltz R et al. Ability of the gut microbiota to produce PUFA-derived bacterial metabolites: proof of concept in germ-free versus conventionalized mice. Mol. Nutr. Food Res 59, 1603–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trico D, et al. Oxidized derivatives of linoleic acid in pediatric metabolic syndrome: is their pathogenic role modulated by the genetic background and the gut microbiota? Antioxid. Redox Signal. 2017, April 7 10.1089/ars.2017.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bisgaard H, Stokholm J & Chawes BL et al. Fish oil-derived fatty acids in pregnancy and wheeze and asthma in offspring. N. Engl. J. Med. 375, 2530–9 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.