

SUMMARY
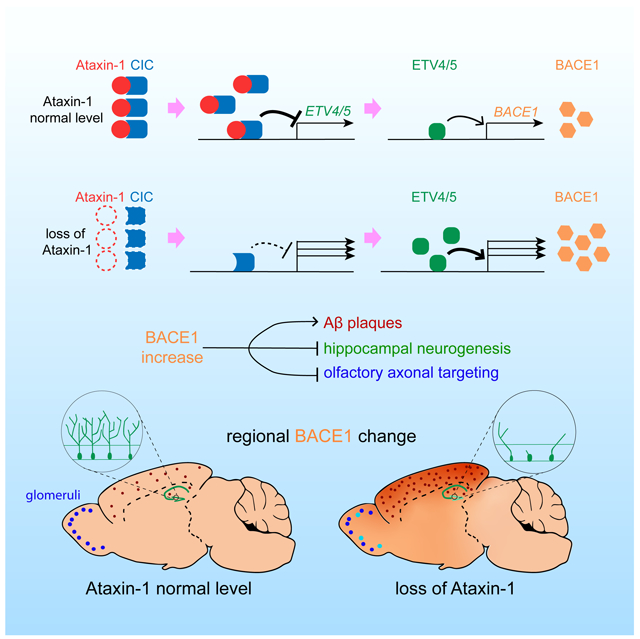
Expansion of CAG trinucleotide repeats in ATXN1 causes spinocerebellar ataxia type 1 (SCA1), a neurodegenerative disease that impairs coordination and cognition. While ATXN1 is associated with increased Alzheimer’s disease (AD) risk, CAG repeat number in AD patients is not changed. Here, we investigated the consequences of Ataxin-1 loss of function and discovered that knockout of Atxnl reduced CIC-ETV4/5-mediated inhibition of Bacel transcription, leading to increased BACE1 levels and enhanced amyloidogenic cleavage of APP, selectively in AD-vulnerable brain regions. Elevated BACE1 expression exacerbated Aβ deposition and gliosis in AD mouse models, and impaired hippocampal neurogenesis and olfactory axonal targeting. In SCA1 mice, polyglutamine-expanded mutant Ataxin-1 led to the increase of BACE1 post-transcriptionally, both in cerebrum and cerebellum, and caused axonal targeting deficit and neurodegeneration in the hippocampal CA2 region. These findings suggest loss of Ataxin-1 elevates BACE1 expression and Aβ pathology, rendering it a potential contributor to AD risk and pathogenesis.
Graphical Abstract
INTRODUCTION
After aging, family history is the strongest risk factor for Alzheimer’s disease (AD), the leading cause of dementia in the elderly (Gatz et al., 2006). Linkage analyses of multi-generational families with early-onset AD have identified nearly 300 pathogenic mutations in three genes: APP, PSEN1 and PSEN2 (Tanzi, 2012). In addition to APOE, genome-wide association studies (GWAS) have uncovered dozens of associated loci for late-onset AD (Bertram and Tanzi, 2012). More recently, efforts employing whole exome/genome sequencing have revealed AD-associated variants in several genes including TREM2 (Guerreiro et al., 2013; Jonsson et al., 2013; Steinberg et al., 2015). Bioinformatic analysis of these genes and loci are unearthing potentially important biological pathways in AD (Zhang et al., 2013). However, detailed insight into the pathogenetic mechanisms of many AD-associated GWAS gene variants remains largely unclear.
ATXN1 is one of the rare AD-associated genes (Bertram et al., 2008), and yet it has been extensively studied in another neurodegenerative disorder (Orr et al., 1993). Expanded CAG repeats, coding for a polyglutamine (polyQ) tract in the N-terminal region of Ataxin-1, cause spinocerebellar ataxia type 1 (SCA1). SCA1 is an autosomal dominant neurodegenerative disease, characterized by cerebellar atrophy with severe loss of Purkinje cells. ATXN1 alleles with more than 39 uninterrupted CAG repeats can cause SCA1, with a tight inverse correlation between the length of CAG repeats and the age of disease onset (Zoghbi and Orr, 2000). SCA1 clinically presents with the deterioration of motor coordination and balance, and progressive difficulty in swallowing and breathing in late stages. Cognitive impairments, such as frontal executive dysfunction and impaired verbal memory, also occur later in the disease course (Burk et al., 2001; Fancellu et al., 2013). While the pathogenic mechanism of the cerebellar degeneration is mainly driven by a gain of function mechanism mediated by the interaction of polyQ-expanded Ataxin-1 with its binding partner Capicua (CIC) (Rousseaux et al., 2018), the mechanism driving cognitive features remains to be investigated. Of interest is the finding that the polyQ-expanded Ataxin-1 partially loses its function in addition to the gain of function, particularly in non-cerebellar brain areas where the aggregation of the mutant protein is more evident with the disease progression (Crespo-Barreto et al., 2010; Watase et al., 2002). While the contribution of partial loss of function is minimal in the cerebellar phenotypes (Rousseaux et al., 2018), its impact remains to be determined on other SCA1 phenotypes.
In a previous family-based GWAS study (Bertram et al., 2008), we observed genome-wide significant association of ATXN1 (rs179943) with AD. A replication study by another group supported the association of the same single nucleotide polymorphism (SNP) in a well characterized group of AD patients and control individuals (Bettens et al., 2010). Moreover, two studies on the copy number variation (CNV) in ADNI (Swaminathan et al., 2011) and NIA-NCRAD (Swaminathan et al., 2012) AD cohorts revealed ATXN1 is one of several genes that harbor CNV exclusively in AD patients, but not in control individuals. These findings support the association of ATXN1 with AD; however, pathogenic variants/mutations have not been documented in AD, to date. In addition, we found the CAG repeat number in ATXN1 is not changed in DNA samples from AD families. This led us to hypothesize that loss of normal Ataxin-1 function, rather than expanded polyQ tract-mediated neurotoxicity, might underly the genetic association with AD.
To test this hypothesis, we examined Ataxin-1 knockout (KO) mice (Matilla et al., 1998) to determine whether the depletion of Ataxin-1 causes AD-related phenotypes in the brain. Ataxin-1 KO mice were previously shown to be free of any SCA1-related phenotypes; however, these mice exhibit hippocampus-dependent learning deficits, suggesting a potential role of Ataxin-1 in learning and memory (Matilla et al., 1998). In the present study, we found that loss of Ataxin-1 led to elevated BACE1 levels and enhanced Aβ pathology in the cerebrum, as well as impairments in hippocampal neurogenesis and olfactory axonal targeting. These findings reveal that decreased Ataxin-1 levels lead to an increase in amyloidogenic processing of APP, exacerbation of Aβ deposition, as well as disturbances in neuronal connectivity and adult neurogenesis. Altogether, these data suggest that reduced Ataxin-1 levels or function may contribute to an earlier clinical manifestation of AD.
RESULTS
Number of CAG trinucleotides in ATXN1 is not increased in AD patients.
Modest CAG repeat expansions in ATXN1 have been associated with schizophrenia (Culjkovic et al., 2000), while intermediate polyQ sizes in Ataxin-2 have been reported to increase risk for amyotrophic lateral sclerosis (ALS) (Elden et al., 2010). To examine whether the length of polyQ tract in Ataxin-1 is associated with the increased AD risk, we measured the CAG repeat numbers in all 438 probands of the NIMH AD family samples (Bertram et al., 2008). The mean repeat number (30.09 ± 1.42) and the distribution pattern (range of 25-38) of the 876 chromosomes of AD probands were very similar to those of normal chromosomes (29.99 ± 1.67; range of 26-36) of 525 individuals of 30 CEPH families (Jodice et al., 1994), indicating CAG repeat number in ATXN1 is not increased in AD patients (Figure 1A). These findings exclude the possibility of ATXN1 CAG repeat expansion as a risk factor for AD.
Figure 1. Increased BACE1 expression in the cerebrum of Ataxin-1 KO mice.

(A) Allele frequencies of ATXN1 with different CAG repeat lengths in the probands of NIMH AD families and healthy individuals of CEPH families. (B) Western blot analysis of APP processing in the brains of 3 month-old WT (+/+), Ataxin-1 hetero-KO (+/−) and KO (−/−) mice. RIPA buffer-soluble lysates were used to detect Ataxin-1 (ATXN1), APP, APP-CTF, ADAM10, BACE1. TBS-soluble lysates for sAPPα and sAPPβ. Actin is a loading control. Arrow head, proADAM10; *, non-specific band. (C) Densitometric quantification of western blot results. WT, relative value of 100. Values are mean ± SEM. n = 5. *p < 0.05, **p < 0.01, t-test. (D) Immunohistochemical (left) and immunofluorescence (right) staining for BACE1. Ctx, cortex; Hip, hippocampus; Cbl, cerebellum; Str, striatum; BSt, brain stem. Bar = 500 μm. (E) Ataxin-1, BACE1, and APP levels in dissected brain regions. OB, olfactory bulb. (F) Densitometry of BACE1 levels. n = 3. n.s.: not significant. ***p < 0.001. (G) RT-qPCR analysis of Atxn1 and Bace1 mRNA expression in the whole brain. n = 4. **p < 0.01, ***p < 0.001 versus WT Atxn1 mRNA; ##p < 0.01 versus WT Bace1 mRNA. (H) Bace1 mRNA levels in dissected cortex and cerebellum. n = 4. (I) Representative real-time qPCR amplification graphs for cortical cDNA. RFU, relative fluorescence unit. See also Figure S1.
Depletion of Ataxin-1 increases BACE1 levels in AD-vulnerable brain regions
To determine whether the loss of Ataxin-1 function would lead to AD-related biochemical changes, we first examined APP processing in Ataxin-1 KO mice. Genetic, biochemical and clinical data indicate that abnormal processing of APP and generation of β-amyloid (Aβ), a major component of senile plaques in AD brains, play central roles in the etiology and pathogenesis of AD (Hardy and Selkoe, 2002; Karran and De Strooper, 2016). Western blot analysis of brain lysates of 3-month-old Ataxin-1 KO mice revealed that the levels of both mature and immature APP and APP C-terminal fragments (CTFs) are not changed in the KO mice when compared to wild-type (WT) controls. In contrast, sAPPα levels were significantly decreased whereas sAPPβ levels were increased (Figures 1B, 1C, and S1A). While the expression of ADAM10, the major α-secretase in the brain (Prox et al., 2013), was largely unchanged, BACE1 (β-secretase) level was significantly elevated (~45%) in the Ataxin-1 KO brains, suggesting the increase of sAPPβ is due to increased BACE1. Given the unchanged ADAM10 level, the observed decrease of sAPPα is likely the result of reduced a-secretase cleavage of APP, owing to competitive cleavage by increased β-secretase (Luo et al., 2001; Suh et al., 2013).
Immunohistochemical staining for BACE1 revealed that, while the distribution pattern was not changed, BACE1 immunoreactivity was notably increased in the cerebrum of Ataxin-1 KO mice (Figures 1D and S1B). No changes were observed in the brain stem and cerebellum. Distinctively, while Ataxin-1 was mainly expressed in neuronal nuclei, BACE1 expression was concentrated at presynaptic terminals (e.g., mossy fibers in hippocampus) (Figures 1D and S1C). Immunoblot analysis of dissected brains confirmed the up-regulation of BACE1, selectively in the AD-vulnerable regions (Figures 1E and 1F). The increase of BACE1 level was also observed in the cortex of Ataxin-1 heterozygous KO mice (Atxn1+/−), suggesting partial loss of Ataxin-1 function could elevate BACE1 expression in the cerebrum (Figure S1D). Analysis of brain RNA by RT-qPCR showed that Bacel mRNA level is significantly increased (48%) in Ataxin-1 KO brain (Figure 1G), and that the increase is more pronounced (89%) in the cortex (Figures 1H, 1I, and S1E). No change was observed in the cerebellum.
Analysis of other organs/tissues revealed that brain is the only organ that expresses both Ataxin-1 and BACE1 at high levels. In other organs/tissues, depletion of Ataxin-1 did not change BACE1 levels (Figure S1F). Moreover, changes in BACE1 level were not observed either in embryonic brains, primary cortical neurons, or embryonic fibroblasts, all of which are derived from Ataxin-1 KO mice (Figure S1G). Together, these findings suggest that lack of Ataxin-1 plays a minimal role in BACE1 expression during brain development, and that BACE1 up-regulation by loss of Ataxin-1 occurs selectively in adult cerebrum.
Loss of Ataxin-1 increases Bace1 transcription by de-repressing Etv4/Etv5 expression in the cortex
Ataxin-1 is known to interact directly with transcription factors, such as CIC (Crespo-Barreto et al., 2010; Lam et al., 2006). Ataxin-1 can bind RNA as well via its AXH domain to potentially regulate RNA metabolism (Zoghbi and Orr, 2009). Thus, we set out to explore whether the increase of Bace1 mRNA in Ataxin-1 KO brain is caused by either loss of Ataxin-1-mediated enhanced transcription or increased mRNA stability. To test these two possibilities, we employed acute brain slice cultures that were derived from Ataxin-1 KO and WT mice. The transcriptional processes of the brain slices were modulated and measured by ex vivo application of transcription inhibitors and nucleotide analogs into artificial cerebrospinal fluid (ACSF). After 24 and 48 hr treatment with actinomycin D, we found the ratios (~ 2-fold) of Bace1 mRNA levels were not increased in the cortical slices of Ataxin-1 KO versus WT mice, as compared to vehicle-treated controls (Figures 2A and S2A), suggesting loss of Ataxin-1 does not increase the stability of Bace1 mRNA. Electrophysiological recordings from hippocampal CA1 neurons revealed that the cultured slices are biologically active 48 hr after the incubation, and that CA1 neuronal activity is barely affected by actinomycin D (Figure 2B), confirming the brain slice cultures are suitable for the ex vivo experiment. Next, we examined if depletion of Ataxin-1 would increase Bacel transcription. We measured the amount of newly synthesized mRNA that had incorporated 5-ethynyl uridine, a uridine analog, from the culture media. Similar to their steady-state levels, the amount of nascent Bace1 mRNA was higher in Ataxin-1 KO than WT cortex (Figure 2C), indicating loss of Ataxin-1 increases Bace1 transcription.
Figure 2. Increased Bace1 transcription in Ataxin-1 KO cortex.

(A) Actinomycin D or vehicle (DMSO) was treated for 48 hrs in acute brain slice cultures. Results are from two independent brain slice cultures (2 mice each for WT and Ataxin-1 KO). n (brain slices) = 3–4. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (B) Left: extracellular field potential records of multiple unit activity in CA1 areas of brain slices 48 hrs after incubation. Right: histograms representing spike amplitude distribution (bin size 10 mV) and inter-spike interval (bin size 2 ms). (C) Representative real-time qPCR amplification graphs of steady-state (total) and nascent Bace1 mRNA. Brain slices were incubated for 16 hrs in ACSF containing 5-ethynyl uridine. n (brain slices) = 4. (D) Locations of predicted CIC and PEA3 (ETV1/4/5) binding sites in Etv1/4/5 and Bace1 promoter regions, respectively. The CIC binding site in Etv1 promoter is relative distal (−1486) than those in Etv4/5. (E) CIC expression in medial cortex (left) and hippocampal CA1 (right). Bar = 50 μm. (F) mRNA levels of Etv1, Etv4, and Etv5 in the cortex and cerebellum. Gapdh was used as an internal control for RT-qPCR.All levels were presented as relative to Etv1-WT (100). Compared to cortex, the relative levels of Etv4 and Etv5 versus Etv1 mRNA are very low in cerebellum. n= 6 (cortex), 4 (cerebellum). (G) Western blot analysis for cortical levels of Ataxin-1, CIC, ETV4, ETV5, BACE1, sAPPβ, and sAPPα. (H) Cerebellar levels of ETV4 and ETV5. Vertical lines indicate that different parts within same blot were placed together for better comparison. (I) Densitometric quantification. n = 5, except ETV4 and ETV5 (n = 7). (J) Luciferase activity in HEK293T cells measured 24 hr after transfection with pGL3 plasmids harboring BACE1 promoter and vectors expressing either ETV4 or ETV5. Results from 3 independent experiments of sextuplicate transfection were normalized and combined. n = 18. See also Figure S2.
To further investigate the molecular mechanism, we hypothesized that loss of Ataxin-1 affects transcription factors that either interact with or are regulated by Ataxin-1, and changes in such transcription factors would subsequently alter Bace1 transcription. Based on this premise, we identified five transcription factors (Rora, Gfi-1, ETV1, ETV4, and ETV5) that have at least one binding motif in Bace1 promoter (Figure 2D). Rorα and Gfi-1 are known to interact with Ataxin-1; however, their expression levels were not changed in Ataxin-1 KO brains (Figure S2B). ETV1, ETV4 and ETV5 are PEA3 family transcription factors that are negatively regulated by CIC, an Ataxin-1 binding transcriptional repressor (Figure 2D) (Kawamura-Saito et al., 2006; Lee et al., 2011; Wang et al., 2017). CIC levels in Ataxin-1 KO brains were markedly lower than WT (Figures 2E and 2G), as the stability of CIC protein depends on the amount of Ataxin-1 in the nucleus (Lim et al., 2008). ETV1/ETV4/ETV5 have two binding sites (CCGGAA/T at −86 and −793 from transcription start site) in Bace1 promoter. To examine whether the increase of Bace1 transcription is mediated by this Ataxin-1-CIC-ETVs pathway, we measured the mRNA and protein expression of ETV1/ETV4/ETV5 in the cortex and cerebellum. Analysis of cortical RNA and protein levels revealed that both ETV4 (47% [RNA] and 44% [protein]) and ETV5 (61% and 67%) are significantly increased in Ataxin-1 KO mice (Figures 2F-2I, and S2C). In contrast, in the cerebellum, the protein levels of neither ETV4 nor ETV5 were increased (Figure 2H).
To confirm that ETV4 and ETV5 up-regulate BACE1 transcription through binding to the predicted motif in BACE1 promoter, we performed promoter-reporter assays. Human BACE1 promoter region, −816 to +56 from the transcription start site (Xiang et al., 2014), which contains the conserved proximal ETV binding motif, was cloned into luciferase reporter vector pGL3. Overexpression of either ETV4 or ETV5 in HEK293 cells significantly increased (~2-fold) the BACE1 promoter-dependent luciferase activity (Figures 2J and S2D). Furthermore, introduction of a mutation in the ETV binding site (from CCGGAA to CTATGA) or deletion of it significantly decreased the luciferase reporter activity (Figure S2E). These data suggest that both ETV4 and ETV5 activate BACE1 transcription via binding to the conserved motif. It is also worth noting that the core of BACE1 promoter (−126 to +56), previously reported as playing an essential role in BACE1 transcription (Xiang et al., 2014), encompasses the ETV binding site, which is conserved between human and mouse. Together, these findings suggest that loss of Ataxin-1 increases the transcription of Bace1 by decreasing nuclear CIC, which leads to an increase of ETV4/ETV5 expression and subsequent activation of Bace1 promoter. The fact that notable increases of ETV4 and ETV5 expression occurred only in the cortex, not in the cerebellum of Ataxin-1 KO mice, further supports the direct roles of ETV4 and ETV5 on Bace1 transcription.
To examine whether the increased BACE1 expression is responsible for the enhanced amyloidogenic processing of APP in Ataxin-1 KO brain, we generated a new mouse line, Atxn1−/−; Bace1+/−, that is partially depleted for BACE1 on an Ataxin-1 KO background. Compared to Ataxin-1 KO mice, BACE1 level was significantly decreased in the cortex of Atxn1−/−; Bace1+/− mice, and shifted to a level similar to that of WT mice (Figures 2G, 2I, S2F, and S2G). Concordantly, the amyloidogenic processing of APP was also significantly decreased in the cortex (sAPPβ/sAPPα ratio: WT 1.00, Atxn1−/− 1.55, Atxn1−/−; Bace1+/− 1.03). While the critical role of BACE1 in amyloidogenic processing is well established, our findings in Atxn1−/−; Bace1+/− mice show that increased BACE1 level, not other changes in Ataxin-1 KO brain, played a major role in shifting APP processing toward amyloidogenic pathway.
Shift of APP processing toward the β-secretase pathway and Aβ generation in AD mice
To assess the impact of Ataxin-1 loss of function on AD pathology, we crossed Ataxin-1 KO mice with a well-characterized AD mouse model, APPswe/PS1ΔE9 (APP-PS1) (Jankowsky et al., 2004). APP-PS1/ATXN1-KO mice, generated from APP-PS1/Atxn1+/− × Atxn1+/− crosses, did not show any notable changes in morphology or cage behavior (Figures S3A). BACE1 up-regulation was observed in 5-month-old APP-PS1/ATXN1-KO mice (Figures 3A, 3B, and S3B), similar to what was observed in the Ataxin-1 KO mice. Concordantly, the levels of endogenous mouse sAPPβ were increased (Figures 3A and S3C), but the amounts of sAPPβ-swe, which is derived from transgenic mutant APP, were barely changed (Figures 3A and 3B). One possible explanation for this unchanged sAPPβ-swe levels is that the over-produced sAPPβ-swe might be subject to further processing in the brain (Suh et al., 2013). In support of this premise, the total amount of sAPP remained at similar levels in the brain as compared to those of APP-PS1 controls. ADAM10 expression was modestly decreased in male but not changed in female APP-PS1/ATXN1-KO mice (Figure S3B). Yet, sAPPα generated from the transgenic APP was decreased by more than 60% in both male and female APP-PS1/ATXN1-KO mice, resulting in ~3-fold increase in sAPPβ/sAPPα ratio (Figure 3C).
Figure 3. Shift of APP processing toward amyloidogenic pathway in APP-PS1/ATXN1-KO mouse brains.

(A) Analysis of Ataxin-1, BACE1, ADAM10, sAPPα, sAPPβ, and sAPP levels in AD mouse brains. sAPPα and sAPPβ derived from either transgenic APPswe or endogenous APP were detected by different antibodies. (B) Densitometric quantification. Number of mice analyzed, n = 11–12. ***p < 0.001, t-test. (C) Ratio of transgenic sAPPα and sAPPβ. **p < 0.01. (D-E) Levels of APP-CTFα and - CTFβ in brains. Number in parenthesis denotes the number of mice analyzed. *p < 0.05. (F) TBS-insoluble Aβ40 and Aβ42 amounts in the brains of 5-month-old mice. The amounts of TBS-insoluble Aβ at this age were ~ 20 times higher than those of TBS-soluble Aβ. (G) Western blot analysis of detergent-soluble brain lysates with 6E10 antibody targeting N-terminal region of human Aβ. *, non-specific bands. (H) Densitometric quantification of Aβ levels. See also Figure S3.
Increase of amyloidogenic processing of APP by the depletion of Ataxin-1 was also apparent from the analysis of APP-CTFs. APP-CTFβ levels were higher in female than male, as previously reported in other AD mouse models (Sadleir et al., 2015; Schafer et al., 2007), and modestly increased in APP-PS1/ATXN1-KO mice as compared to the same sex of APP-PS1 mice (Figures 3D and 3E). APP-CTFα levels were significantly reduced in the male mice, but unchanged in the females. Nevertheless, in both sexes, the ratios of CTFβ/CTFα were significantly increased in APP-PS1/ATXN1-KO brains (283% in male, 128% in female), indicating a shift of APP processing toward amyloidogenic pathway (Figure 3E). Concordantly, ELISA measurement and immunoblot analysis showed that APP-PS1/ATXN1-KO mice have 2-3 times higher levels of Aβ40 and Aβ42 than APP-PS1 mice, and female mice have 2-3 times more Aβ than males.
Loss of Ataxin-1 increases Aβ plaque load and reactive gliosis
Immunohistochemical staining of 5-month-old mouse brain sections with anti-Aβ antibody revealed that Aβ plaque load is significantly increased (236%) in APP-PS1/ATXN1-KO versus APP-PS1 mice (Figures 4A and 4B). Increase of Aβ deposition was also evident in APP-PS1/Atxn1+/− mice, suggesting partial loss of Ataxin-1 function is sufficient to exacerbate Aβ pathology (Figure S4A). The plaque load in cortex and hippocampus increased dramatically at 9 months, and APP-PS1/ATXN1-KO mice exhibited greater plaque deposition as compared to age-matched controls (Figure 4A). Female mice harbored more plaques than males (Figures 4B), in agreement with Aβ ELISA results (Figure 3F). Immunolabeling for BACE1 revealed increased BACE1 expression in the cerebrum of APP-PS1/ATXN1-KO mice and strong BACE1 immunoreactivity around the dendritic plaques of both APP-PS1 and APP-PS1/ATXN1-KO mice (Figures 4A, 4C, and S4B).
Figure 4. Elevated Aβ plaque load and reactive gliosis in APP-PS1/ATXN1-KO mouse.

(A) Upper and middle panels: Aβ immunohistochemical staining of 5- and 9-month-old mice. Aβ plaques (arrows) are abundantly detected in cortex and hippocampus, but not in brain stem (*). Lower panel: BACE1 immunohistochemistry. BACE1 staining revealed plaque-like strong immunoreactivity (arrows). Bar = 1 mm. (B) Quantification of Aβ plaque load in the cortex and hippocampus of 5-month-old APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). In parenthesis, number of mice analyzed (n). 5–6 brain sections per mouse. *p < 0.05, ***p < 0.001, t-test. (C) Double immunofluorescence staining of Aβ plaques with Aβ and BACE1 antibodies. Arrows indicate BACE1 immunoreactivity around Aβ plaques. Bar = 100 μm. (D) Frontal cortex of 5- and 9-month-old mice immunolabeled for a microglial marker Iba1 and an astrocyte marker GFAP. Arrows, activated microglia and astrocytes. Right panel: higher magnification revealing activated microglia around Congo red-stained dendritic plaques (arrows) and hypertrophy of astrocytes around the plaques (arrows). Bar = 200 μm. (E) Quantification of GFAP+ astrocytes in the cortex of 5-month-old mice. 3–4 brain sections/mouse. See also Figure S4.
Reactive gliosis, another key pathological feature in AD brains, was also increased by the loss of Ataxin-1. In the brains of 9-month-old APP-PS1 mice, activation of microglia was evident around Congo red-stained dendritic plaques, and astrocytes were transformed into a hypertrophic morphology in the vicinity of plaques (Figure 4D). In concordance with Aβ plaque load, the amounts of activated microglia and astrocytes were higher in APP-PS1/ATXN1-KO mice than age-matched of APP-PS 1 controls (Figures 4D, 4E, S4C, and S4D), suggesting loss of Ataxin-1 potentiates gliosis though the increase of Aβ plaque deposition in the brain.
Elevated BACE1 expression inhibits the proliferation of NPCs in hippocampus
In addition to Aβ pathology, changes in APP processing or BACE1 expression could affect the generation of newborn neurons in adult brain (Chatila et al., 2018; Demars et al., 2013). In humans, impairment of neurogenesis in the hippocampus has been linked to cognitive decline in the elderly and AD (Moreno-Jimenez et al., 2019; Spalding et al., 2013). Thus, we examined whether loss of Ataxin-1, which leads to BACE1 increase, would affect hippocampal neurogenesis in adult brain. Immunolabeling of 5-month-old APP-PS1 brain sections for doublecortin (DCX), an immature neuronal marker, revealed that majority of newborn neurons in the subgranular cell layer (SGL) project their dendrites beyond granular cell layer (projecting DCX+ neurons) (Figures 5A, 5B). However, in APP-PS1/ATXN1-KO mice, the number of DCX+ neurons was significantly reduced (67%) and their dendrites were barely developed in the granular cell layer (tangential DCX+ neurons). This dramatic decrease of newborn neurons was similarly observed in Ataxin-1 KO mice that do not overexpress transgenic mutant APP and PS1 (Figure S5A). Even in APP-PS1/Atxn1+/− mice, the dendritic development of DCX+ neurons was significantly impaired, implying that partial loss of Ataxin-1 function could lead to deficits in hippocampal neurogenesis (Figure S5B). In contrast, Golgi staining of Ataxin-1 KO brains revealed no difference in the dendritic arborization of mature neurons, as compared to WT controls (Figures 5C and S5C). These findings suggest that Ataxin-1 is essential for proper dendrite development of newborn neurons, but not for mature neurons. Examination of Ki-67+ neural progenitor cells (NPCs) showed that NPC proliferation was significantly decreased (61%) in the dentate gyrus of APP-PS1/ATXN1-KO mice (Figures 5D and 5E).
Figure 5. Decrease of adult hippocampal neurogenesis by depletion of Ataxin-1.

(A) Dentate gyrus of APP-PS1 and APP-PS1/ATXN1-KO mice immunolabeled for DCX. Brackets, granular cell layer; arrows, projecting immature neurons; arrow heads, tangential immature neurons. Right panel: high resolution images. *, background DCX immunoreactivity. Bar = 100 μm. (B) Counts of DCX+ (total) and projecting DCX+ cells (projecting) in dentate gyrus per section. Number of mice analyzed, n = 10–12. ***p < 0.001, t-test. (C) Brown, Golgi-stained neurons; blue, Nissl-stained cell nuclei. (D) Ki-67 immunoreactivity (arrows) in dentate gyrus. Area between dots and dashed lines is granular cell layer. (E) Counts of Ki-67+ cells in dentate gyrus. n = 7–8. **p < 0.01. Photomicrographs (F) and quantification (G) of DCX+ neurons in the upper dentate gyrus for their cell bodies in subgranular cell layer (total), projections within granular cell layer (projecting), dendrites in molecular layer (dendrites), and projecting ratio (projecting/total). Each quantification was normalized to WT group (100). Arrows, non-specific immunoreactivity of microglia. n = 6–7. *p < 0.05, **p < 0.01, ***p < 0.001. (H) Number of Ki-67+ cells. (I) Ratio of DCX+ vs. Ki-67+ cells. n = 7–8. See also Figure S5.
To investigate whether increased BACE1 levels are responsible for the impaired hippocampal neurogenesis in Ataxin-1 KO brains, we examined Atxn1−/−; Bace1+/− mice that express reduced levels of BACE1 than Ataxin-1 KO mice. At 4 months of age, the numbers of DCX+ neurons and Ki-67+ NPCs were significantly higher (64% and 45%, respectively) in Atxn1−/−; Bace1+/− mice than in the Ataxin-1 KO mice (Figures 5F-5H and S5D), suggesting the neurogenesis is recovered in Atxn1+/−; Bace1+/− mice. Meanwhile, the ratio of dendrite-projecting DCX+ neurons in Atxn1−/−; Bace1+/− was not different from that of Ataxin-1 KO mice (Figures 5F and 5G). Moreover, the ratio of DCX+ and Ki-67+ cell numbers did not differ among WT, Ataxin-1 KO and Atxn1−/−; Bace1+/− mice (Figures 5I), suggesting loss of Ataxin-1 does not affect the differentiation of NPCs into neurons. Together, these findings suggest the elevated BACE1 expression decreases the proliferation of NPC, which leads impaired hippocampal neurogenesis. However, the defect in the dendritic development of newborn neurons was not caused by increased BACE1 expression.
Axonal path-finding is impaired in BACE1-elevated olfactory bulbs of Ataxin-1 KO mice
Many of BACE1 substrates are known to play key roles in axon guidance (Barao et al., 2015; Lee et al., 2016; Ou-Yang et al., 2018), and previous studies have shown that BACE1 KO mice exhibit disorganized glomerular structure and impaired axonal guidance in the olfactory system (Cao et al., 2012a; Hitt et al., 2012; Rajapaksha et al., 2011). Moreover, recent clinical studies have revealed that olfactory deficits correspond with cognitive decline and AD (Cao et al., 2012b; Devanand et al., 2015; Dhilla Albers et al., 2016). To further investigate the physiological impact of the increased BACE1 levels observed in Ataxin-1 KO brains, we examined if axonal path-finding was altered in the olfactory system. The olfactory system is one of the most extensively studied and well-established structures for axonal guidance in vivo (Ma et al., 2014). Each olfactory sensory neuron (OSN) in the olfactory epithelium expresses only one type of odorant receptor, and all the axons of OSNs expressing the same odorant receptor converge onto one or a few glomeruli in each olfactory bulb, where they make synapses with the dendrites of mitral and tufted cells (Fleischmann et al., 2008; Sakano, 2010). Immunostaining of tissue sections containing olfactory epithelium and bulb showed that BACE1 levels are higher in Ataxin-1 KO versus WT mice, particularly in the glomeruli (Figures 6A, 6B, and S1B).
Figure 6. Impaired axonal targeting in the olfactory bulbs of Ataxin-1 KO mice.

(A) Olfactory bulbs of WT and Ataxin-1 KO mice immunolabeled for BACE1 (green). Bar = 200 μm. (B) Upper: high resolution of WT olfactory bulb (similar region to the marked area). Lower: marked area in Ataxin-1 KO bulb. (C) Olfactory epithelium immunolabeled for MOR28. Arrows, MOR28-expressing OSNs. (D) Two serial sections of olfactory bulbs of WT (upper) and Ataxin-1 KO mice (lower) stained with MOR28 antibody. Small arrows, original locations of medial MOR28 glomeruli; large arrows, translocated MOR28 glomeruli; arrow heads, lateral MOR28 glomeruli; dashed line, midline in the olfactory system. (E) Upper: olfactory bulb of Atxn1−/−; Bace1+/− mouse labeled for MOR28. Lower: lateral MOR28 glomeruli of each mouse genotype. (F) Distance of medial MOR28 glomeruli from the midline. Number of mice/olfactory bulbs analyzed, n = 9/12 (WT), 10/14 (Atxn1−/−), 7/12 (Atxn1−/−; Bace1+/−). *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (G) Areas of medial and lateral MOR28 glomeruli.
To examine axonal targeting in the olfactory system, olfactory sections were labeled for two odorant receptors, MOR28 and M71. In all WT mice, axons of OSNs expressing MOR28 converged on two topologically fixed glomeruli (Barnea et al., 2004): one at a ventromedial position and the other at a lateral position of the olfactory bulb (Figures 6C-6E). However, in many of the Ataxin-1 KO mice, the location of ventromedial glomeruli was shifted away from the olfactory midline, indicating mis-targeting of axons to an ectopic site on the ventral side (Figures 6D and 6F). The position of lateral MOR28 glomeruli remained unchanged; however, the size of the lateral glomeruli was significantly increased (187%) (Figures 6D, 6E and 6G). In contrast to MOR28, the location and the size of M71 glomeruli on the dorsal side were unchanged in the Ataxin-1 KO mice (not shown). These results suggest that the impact by Ataxin-1 depletion on olfactory axonal targeting varies depending on the types of OSNs and the location of corresponding glomeruli (Cao et al., 2012a).
Examination of Atxn1−/−; Bace1+/− mice, in which BACE1 levels were reduced in the olfactory glomeruli, revealed that, compared to Ataxin-1 KO mice, the location of ventromedial MOR28 glomeruli was shifted back to olfactory midline, to a position similar to that of WT mice (Figures 6E-6G). In addition, the size of lateral MOR28 glomeruli became significantly smaller than that of Ataxin-1 KO mice. Together, these findings suggest the impaired axonal targeting in Ataxin-1 KO mice is at least in part due to increased BACE1 expression in the olfactory bulb.
PolyQ-expanded Ataxin-1 leads to post-transcriptional increase of BACE1 levels and degeneration of CA2 neurons
While the cerebellar pathology of SCA1 is mainly driven by a gain function of polyQ-expanded Ataxin-1 (Rousseaux et al., 2018), the polyQ expansion also leads to aggregation of mutant Ataxin-1 and causes a partial loss of Ataxin-1 function which affects some interactions (Crespo-Barreto et al., 2010; Fryer et al., 2011). Thus, we next asked if the polyQ expansion would also increase BACE1 expression in the brain. To this end, we examined Ataxin-1 154Q knockin mice (Atxn1154Q/+) that exhibit SCA1-related neurologic phenotypes, such as ataxia and premature death (Watase et al., 2002). Learning and memory deficits are also observed in this mouse, which may be relevant to the cognitive impairments in SCA1 patients. Immunoblot analysis of cortical and cerebellar lysates of 6-month-old Atxn1 154Q/+ mice revealed that the amounts of normal Ataxin-1 (2Q) in the mice are about half of WT controls (Figure 7A). PolyQ-expanded mutant Ataxin-1 is barely detected as it becomes aggregated and insoluble. As compared to WT, BACE1 levels were significantly increased both in the cortex (31%) and cerebellum (42%). Immuohistochemical staining revealed age-dependent increase of BACE1 in the cerebrum (Figure S6A) and cerebellum (Figure 7B). Interestingly, however, RT-qPCR analyses showed that Bace1 mRNA levels are not changed in the SCA1 brains at any age point tested (Figures 7C and S6B), suggesting that Ataxin-1 polyQ expansion leads to BACE1 increase via post-transcriptional mechanisms, as the disease progresses. Moreover, the previous observations of increased function of polyQ-expanded Ataxin-1 through its interaction with CIC (Rousseaux et al., 2018) and no notable decrease of CIC levels in Atxn1154Q/+ mice (Figure S6C) further support that elevated BACE1 levels in the SCA1 mice is not due to increased Bace1 transcription as observed in Ataxin-1 KO brains.
Figure 7. Elevated BACE1 levels and CA2 neurodegeneration in polyQ-expanded Ataxin-1 knockin mice.

(A) Ataxin-1 and BACE1 levels in 6-month-old Atxn1154Q/+ and WT mice. Arrow and dashed arrow indicate the positions of WT (2Q) and mutant (154Q) Ataxin-1, respectively. In bar, number of mice analyzed. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (B) BACE1 immunolabeled cerebellum of 1.5-, 4.5-, and 8-month-old Atxn1154Q/+ mice. *, molecular layer. Bar = 200 μm. (C) Bace1 mRNA levels in the cortex and cerebellum. Relative value of 100 is given to the cortex and cerebellum of WT at each age group. (D) Hippocampus of Atxn1154Q/+ and WT mice immunolabeled for BACE1. Arrows, BACE1-immunopositive mossy fiber ends. Bar = 200 μm. (E) Quantification of BACE1-immunoreactive area near CA2. In bar, number of brain section analyzed. 1–2 sections/mouse. (F) Hippocampal CA1/CA2/CA3 regions of 8-month-old mice immunolabeled for Ataxin-1 and BACE1. Arrow head, punctate staining of CA1 neurons by Ataxin-1 and DAPI. *, outward shape of CA2 region. Arrows in inset, BACE1 immunoreactivity (yellow signal) in the neuronal soma. Bar = 100 μm. (G) Immunofluorescence signals of Ataxin-1 and synaptoporin, a presynaptic marker. Arrows, synaptoporin+ presynaptic terminals. Insets, high resolution images without Ataxin-1 signal. (H) Hippocampal regions immunolabeled for PCP4 and NeuN, a neuronal marker. Bracket, CA2; *, stratum lucidum; arrow, granular neurons. (I) Counts of PCP4+ neurons and NeuN+ neurons in CA2 regions of 6-9 months-old mice. In bar, numbers of brain section analyzed. 2–4 sections/mouse. (J) Quantification of PCP4 immunoreactivity of CA2 neurons. 1–2 sections/mouse. (K) RGS14 immunoreactivity in 6 month-old mice. Arrows, RGS14+ dendrites of CA2 neurons; *, RGS14+ axons. Bar = 100 μm. (L) Dentate gyrus of 4.5-month-old mice immunolabeled for DCX. Arrows, DCX+ neurons. *, background DCX immunoreactivity. Right: counts of total and projecting DCX+ cells per dentate gyrus. n = 4. See also Figures S6 and S7.
In contrast to the modest change of BACE1 levels in other brain regions, the terminus of stratum lucidum that is immunopositive for BACE1 was markedly enlarged in the SCA1 mice, in an age-dependent manner (Figures 7D, 7E and S6D). Co-immunolabeling for BACE1 and Ataxin-1 revealed additional morphological and immunohistochemical changes in the hippocampal regions (Figure 7F). First, Ataxin-1 immunoreactivity in Atxn1 154Q/+ hippocampal neurons showed punctate nuclear signals, representing the aggregates of 154Q-mutant Ataxin-1. Second, the border of pyramidal neuronal layer between CA1 and CA2 became morphologically more distinct. The CA2 area was extended outwardly, potentially due to the widening of stratum lucidum. Third, numerous hippocampal neurons in CA2 and CA3 revealed overlapped immunoreactivity for Ataxin-1 and BACE1 in their soma. These aberrant BACE1 signals suggest that presynaptic terminals of granular neurons are formed on the cell bodies, in addition to the dendrites of pyramidal neurons (Figures 7G and S6E).
Immunolabeling for PCP4, a CA2 neuronal marker (Kohara et al., 2014), confirmed that the broadened end of stratum lucidum is adjacent to CA2 neurons (Figure 7H). Interestingly, the numbers of PCP4+ neurons and NeuN+ neurons in CA2 were significantly decreased (48% and 27%, respectively) in the SCA1 mice. Moreover, PCP4 immunoreactivity in the soma and process decreased in an age-dependent manner and almost disappeared at 8 months (Figures 7H-7J and S7A-S7G). The selective and robust vulnerability of CA2 neurons was further confirmed by Nissl staining and immunostaining for other CA2 neuronal markers, RGS14 and STEP (Figures 7K and S7H-S7K). Based on these findings, it is plausible to speculate that the selective degeneration of CA2 neurons may have caused both the aberrant axonal targeting and the expansion of the terminus of stratum lucidum, as a compensatory mechanism for the loss of synapses between CA2 neurons and granular neurons. In Ataxin-1 KO mice, neither the degeneration of CA2 neurons nor the widening of stratum lucidum was observed (Figure S7L), suggesting the CA2 phenotype in SCA1 mice is not caused by Ataxin-1 loss of function but by gain of function of polyQ-expanded Ataxin-1. Furthermore, while the SCA1 mice express similar levels of WT Ataxin-1 protein as compared to Atxn1+/− mice (Figure 7A), the proliferation of NPCs and the dendritic development of newborn neurons were severely impaired (Figures 7L and S7M). Together, these findings suggest that, while some phenotypes (e.g. increased levels of BACE1) observed in SCA1 mouse brains are comparable to those in Ataxin-1 KO brains, the underlying mechanisms for the phenotypes are different between the two mouse models. Elevated BACE1 expression and degeneration of CA2 neurons in the SCA1 mice are not caused by Ataxin-1 loss of function but mainly by gain of function of the polyQ-expanded mutant Ataxin-1, and become more pronounced with disease progression.
DISCUSSION
Spatial and temporal regulation of BACE1 expression by loss of Ataxin-1 function
A distinct and intriguing characteristic that we found from the examination of Ataxin-1 KO mouse is that the BACE1 up-regulation is limited to adult cerebrum (Figure 1). Increased BACE1 level was not observed in other brain regions, other organs/tissues, or developing brains. These spatiotemporal-specific changes in BACE1 expression provide two important insights into the role of Ataxin-1 in AD etiology and pathogenesis. First, loss of Ataxin-1 function may not interfere with BACE1-regulated functions either in developing brains or in other regions of adult brain that are spared in AD. Second, an additional factor(s) is required for Ataxin-1 to coordinate the spatiotemporal regulation of BACE1 expression in the brain.
In the developing brain, Ataxin-1 expression is very low, whereas BACE1 levels are very high as compared to those in the adult brain (Figure S1G). Therefore, it is conceivable that depletion of Ataxin-1 in the developing brain would minimally affect CIC levels and the subsequent ETV4/ETV5-mediated BACE1 transcription. While Ataxin-1 levels in the cerebellum or brain stem are comparable to those of cerebrum, BACE1 levels in these AD-resistant regions were not changed by depletion of Ataxin-1. One possible reason for this brain region-specific change in BACE1 expression is that the levels of ETV4 and ETV5 were unaltered in these brain regions, possibly due to the ability of Ataxin-1 Like (the paralog of Ataxin-1 that also interacts with CIC) to compensate for the loss of Ataxin-1 in these regions. Collectively, our findings suggest that the spatiotemporal regulation of BACE1 expression by Ataxin-1 is achieved by differential expression of both Ataxin-1 and the transcription factors that are regulated by Ataxin-1.
Importantly, CIC binding motifs in Etv4/Etv5 promoters and ETV4/ETV5 binding motif in Bace1 promoter are conserved in humans. Hence, loss of Ataxin-1 function in the human brain may likewise lead to increased BACE1 expression via a mechanism involving CIC-ETV4/ETV5-mediated transcriptional regulation. Furthermore, to our knowledge, prior to this study, there have been no previous report documenting that any known or candidate AD genes regulate BACE1 expression in the brain. In this regard, the increase of BACE1 levels by the depletion of Ataxin-1 presents a unique regulatory mechanism, by which Ataxin-1 affects AD etiology and pathogenesis in vulnerable brain regions, with minimal interference with brain development or other unrelated neurological functions.
Recent genetic studies have revealed several rare mutations that cause pathogenic loss-of-function in AD-associated genes, such as SORL1 (Pottier et al., 2012) and ABCA7 (Steinberg et al., 2015). These findings support the notion that other AD-associated genes may also harbor loss-of-function mutations, such as frame-shift, non-sense, and splicing mutations. Variants or mutations in the 5' or 3' UTR that decrease expression levels would also produce loss-of-function phenotypes. While CNVs were previously reported in ATXN1 (Swaminathan et al., 2011; Swaminathan et al., 2012), the incidence was very low and the details of the variants are unknown. In our prior CNV study (1009 subjects from 261 early/mixed-onset AD families), no case was found to harbor a CNV in ATXN1 (Hooli et al., 2014). These results suggest that a large number of AD DNA samples and a detailed genetic analysis would be required for a future study to identify pathogenic loss-of function mutations in ATXN1. Notably, effects in Atxn1+/− mice regarding increased BACE1/AP pathology and impaired neurogenesis support the notion that mutations that cause partial loss of Ataxin-1 function may increase risk for AD through persistent increase of BACE1 expression in the brain over lifetime. It is also conceivable that non-genetic factors (e.g. environment and lifestyle) that decrease the level or function of Ataxin-1 may potentiate BACE1 expression and AD pathogenesis (Ju et al., 2014).
Roles of Ataxin-1 and BACE1 in hippocampal neurogenesis and axonal targeting
Recent studies revealed that the portion of adult-born neurons in the hippocampus is substantial and that hippocampal neurogenesis decreases during aging (Boldrini et al., 2018; Knoth et al., 2010; Spalding et al., 2013), raising the possibility that attenuated neurogenesis may contribute to cognitive deficits in the elderly and AD patients. Previous studies of Ataxin-1 KO mice with behavioral and electrophysiological tests showed that Ataxin-1 is essential for hippocampal function (Matilla et al., 1998). Our findings in this study of decreased NPC proliferation and defective dendritic development of newborn neurons in the mice suggest that impaired hippocampal neurogenesis may contribute to the learning and memory deficits. In the present study, we found through the analysis of Atxn1−/−; Bace1+h mice that decreased NPC proliferation is caused by an increase of BACE1 expression in the brain. It is plausible that BACE1 affects hippocampal neurogenesis through the cleavage of its substrates (e.g. APP) (Hu et al., 2013). In Ataxin-1 KO brain, sAPPβ level was increased and sAPPα was decreased. Previous studies showed that sAPPα, but not sAPPβ, increases neurogenesis in mouse brains (Caille et al., 2004; Demars et al., 2013). This notion of differential effects of sAPPα and sAPPβ on hippocampal neurogenesis is consistent with the findings in our prior study of ADAM10 transgenic mice (Suh et al., 2013). Future investigations will be necessary to determine whether cleavages of APP and other BACE1 substrates directly affect adult hippocampal neurogenesis.
Recent studies by others also reported reduced hippocampal neurogenesis in Ataxin-1 KO (Asher et al., 2016) and Atxn1154Q/+ SCA1 mice (Cvetanovic et al., 2017). The data from Asher et al. are consistent with ours in that loss of Ataxin-1 decreases the proliferation of NPCs. In the present study, we further demonstrated that increased BACE1 is responsible for the impaired NPC proliferation. Moreover, we demonstrated that loss of Ataxin-1 produces severe defects in the dendritic development of newborn neurons, and that the dendritic defects are not mediated by increased BACE1. Additionally, our findings in the SCA1 mice suggest that the deficits in hippocampal neurogenesis of the mice are mainly due to gain of function of polyQ-expanded mutant Ataxin-1, rather than loss of function of Ataxin-1 in the brain.
In olfactory epithelium, OSNs continually regenerate and project their new axons to glomeruli throughout life, through regulated axon guidance, to form a topological map in the olfactory bulb (Sakano, 2010). This map is functional, and the disruption of the projection pattern impairs olfactory acuity (Fleischmann et al., 2008). Given the olfactory axonal deficits in Ataxin-1 KO mice are mediated by elevated BACE1 levels, and yet BACE1 is not increased in the developing brain, it is unlikely that the axonal phenotypes are developmental deficits, but rather they would occur after brain development. Notably, accumulating evidence shows that patients with mild cognitive impairment and AD dementia have difficulties with odor recognition (Devanand et al., 2015; Dhilla Albers et al., 2016), further supporting our findings in the olfactory axonal targeting are relevant to AD.
PolyQ-expanded Ataxin-1 and degeneration of CA2 neurons in SCA1 mice
Analysis of BACE1 expression in Atxn1154Q/+ SCA1 mice unexpectedly revealed interesting morphological changes in CA2 region. Importantly, the number of CA2 neurons as well as the expression of CA2-specific markers was markedly decreased in a disease progression-dependent manner. Further studies will be needed to uncover the molecular mechanism underlying the selective neurodegeneration and to assess the significance of this change in the context of SCA1 pathogenesis. Recently, studies utilizing selected genetic lesions in mice showed that CA2 neurons play key roles in social memory (Hitti and Siegelbaum, 2014; Smith et al., 2016). Given that depression is a common comorbidity in SCAs, including SCA1 (Lo et al., 2016), it is intriguing to speculate that the degeneration/dysfunction of CA2 neurons may contribute to the depressive symptoms in SCA1 patients. Detailed examination of postmortem brains is warranted to address whether comparable changes in BACE1 expression and CA2 neurodegeneration are observed in SCA1 patients.
Although the cerebellar motor syndrome of gait ataxia, dysmetria, dysarthria and oculomotor abnormalities are the most readily identifiable clinical manifestation of SCA1, deficits in cognitive function have become increasingly recognized, especially in advanced stages (Koziol et al., 2014). These cognitive deficits, including impairments in executive function and linguistic deficits (Burk et al., 2001; Moriarty et al., 2016), can partly be explained as the cerebellar cognitive affective syndrome or Schmahmann syndrome (Manto and Marien, 2015; Schmahmann and Sherman, 1998), in which cerebellar damage-induced cognitive deficits are ascribed to the functional disruption of cerebro-cerebellar connections. However, cognitive and neuropsychological deficits differ widely among various SCAs (Kawai et al., 2009), suggesting an important role by extra-cerebellar areas for the cognitive impairments. Pathology outside of the cerebellum has been reported in a small number of SCA1 cases (Rub et al., 2012), but the molecular mechanisms responsible for the cognitive impairments have remained unknown. Our findings of increased BACE1 levels and decreased neurogenesis, and the selective degeneration of CA2 neurons in the hippocampus of SCA1 mice suggest that ATXN1 CAG repeat expansion can cause cognitive impairments by affecting cerebrum directly, beyond potential changes in cerebro-cerebellar connections.
Since the discovery of expanded CAG repeats in ATXN1 of SCA1 patients (Orr et al., 1993), research efforts on Ataxin-1 have focused on identifying the pathogenic mechanism of cerebellar neurodegeneration. Our combined study employing genetic, biochemical, and anatomical analyses in mice either lacking Ataxin-1 or expressing the SCA1-causing polyQ-expanded form of the protein has provided new insight into the relationship between Ataxin-1 and vulnerability to AD. Our data suggest that loss of Ataxin-1 function increases risk for AD by increasing BACE1 expression in a region-specific manner. In contrast, we discovered that polyQ-expanded Ataxin-1 causes distinct post-transcriptional regulation of BACE1 through a gain of function mechanism leading to distinct CA2 phenotypes that might underlie the cognitive deficits in SCA1. These studies highlight the complexity by which one protein (Ataxin-1), in two different states (reduction and enhanced function), impacts the same molecule (BACE1) via contrasting mechanisms and how different regional effects in the hippocampus can lead to distinct phenotypes that could underlie cognitive impairments. A greater understanding of the detailed regulatory mechanisms by which Ataxin-1 controls BACE1 both transcriptionally and post-transcriptionally stands to provide clearer insight into AD pathogenesis, cognitive deficits in SCA1, and regional brain vulnerability in neurodegenerative diseases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jaehong Suh (suh.jaehong@mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ataxin-1 KO (Matilla et al., 1998) and 154Q knock-in mice (Atxn115AQ+) (Watase et al., 2002) in C57BL/6 genetic background were previously described. APPswe/PSEN1ΔE9 (APP-PS1) (Jankowsky et al., 2004) and BACE1 KO mice (Cai et al., 2001) in C57BL/6 background were purchased from Jackson Laboratory. APPswe/PSEN1ΔE9 mouse overexpresses two early-onset familial AD mutations: APP Swedish (K670N/M671L) mutation that shifts APP processing toward β-secretase cleavage; and mutant human presenilin-1 missing exon 9, which affects the γ-secretase cleavage site on APP and increases the ratio of Aβ42 and Aβ40 (Jankowsky et al., 2004). The amino acid sequence of the transgenic APP in this AD model is derived from mouse, with the exception of the Swedish mutation site and the Aβ region which are derived from human sequence. APP-PS1 and APP-PS1/ATXN1-KO mice were generated by crossing APP-PS1/Atxn1+/− with Atxn1+/− mice. Histological examination revealed no obvious neuroanatomical abnormalities in APP-PS1/ATXN1-KO mouse brains. In addition, the genotype ratios of live progeny at 4 weeks were close to the expected Mendelian frequency, suggesting Ataxin-1 depletion in the AD mice does not cause any major developmental defects. Atxn1−/−; Bace1+/− mice were generated by crossing Atxn1+/−2; Bace1+/− with Atxn1+/− mice and did not exhibit any notable abnormality. Genotypes of pups were determined by PCR of tail DNA. Mice were age- and sex-matched within experiments. Equal or similar numbers of male and female mice were used in all experiments. Ages of mice used for each experiment were described in the Results, Figures, and Figure Legends. All mouse generation, husbandry, and experimental procedures were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care (MGH SRAC), and conform to the NIH guidelines of the care and use of laboratory animals. Mice were housed under specific pathogen free condition in an animal facility at MGH, under the care of full-time veterinarians or veterinarian technicians.
Human DNA samples
Previously collected DNA samples from the National Institute of Mental Health (NIMH) AD Genetics Initiative Study (Blacker et al., 2003) were used. These samples are publically available. DNA samples from probands (n = 438) were analyzed for ATXN1 CAG repeat number. The full NIMH sample includes 1439 individuals (68.9% female) from 438 families with at least two affected, including 995 affected (mean age of onset 72.4 ± 7.7 years), 411 unaffected, and 34 individuals with unknown phenotypes. A clinical diagnosis of AD was confirmed at autopsy in 94% of the cases.
Primary neuronal and embryonic fibroblast cultures
Embryonic day 15 (E15) embryos with unknown genotypes, obtained by crossing male and female Atxn1+/− mice, were individually processed for the preparation of neuronal cultures. Genotypes of the embryos were later determined by PCR of tail DNA. Sexes of the embryos and cultured primary neurons/fibroblasts were not determined, as there is no reasonable background or prior study suggesting that sex of the cultured neurons/fibroblasts would affect the results of the culture experiments. For primary cortical neuronal cultures, cerebral cortices were gently triturated with a series of fire-polished Pasteur pipettes of different pore sizes. Suspended cells were plated on six-well plates (~6 × 105 cells/well) which were pre-coated with poly-D-lysine. The cultures were maintained in Neurobasal media (Invitrogen) supplemented with B27 and L-glutamine, at 37°C in a humidified 5% CO2 incubator, as described previously (Suh et al., 2011). Half of the culture medium (1 ml) was replaced every 3–4 days. Using RIPA buffer containing protease inhibitors, neuronal cultures at days in vitro (DIV) 13 were harvested for protein expression analysis. Mouse embryonic fibroblasts (MEFs) were obtained from brain-removed E15 embryos. The embryos were trypsinized, dissociated and plated in DMEM media containing 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 μg/ml). After removing the floating debris, the dividing cells were cultured in the same media for more than 2 weeks. Once the cultures became confluent, the MEF cells were sub-cultured and harvested for protein expression analysis.
HEK293T cell line
Human embryonic kidney (HEK) 293T cells were grown in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Atlanta biologicals) and 1 × penicillin-streptomycin mix (Thermo Fisher Scientific). Cell cultures were maintained at 37°C in a humidified 5% CO2 incubator. HEK293T cells were obtained from ATCC (CRL-3216). Based on the karyotype of HEK293 cells, the source fetus of the cell line was female.
METHOD DETAILS
ATXN1 CAG repeat number analysis
Forward and reverse PCR primers were custom designed and ordered for the expanded polyglutamine-encoding CAG microsatellite region of ATXN1. A gradient polymerase chain reaction (PCR) was run to identify the optimal annealing temperature. DNA samples of 438 probands of the NIMH AD families were processed in 96-well plates along with duplicates of two SCA1 positive controls. A 20μL polymerase chain reaction was run for samples and controls at 95°C for 8 min, [95°C for 30 sec, 62°C for 30 sec, 72°C for 60 sec; (repeat for a total of 40 cycles)], 72°C for 10 min. PCR solution was then loaded on to the MegaBACE Sanger sequencing machine following the protocol supplied by the manufacturer. The sample injection voltage was set to 3 kV, run voltage was 10 kV, with an injection time of 45 seconds and run-time of 75 minutes. The exact number of CAG repeats in the NIMH test samples were calculated by extrapolation, based on the two SCA1 DNA samples used as controls in every run with previously established CAG repeats of 36 and 42 CAG repeats. CAG repeat numbers of the 525 individuals of 30 CEPH families (Jodice et al., 1994) were obtained from "http://www.cephb.fr/cephdb/detail.php?s=12180" through communication with Dr. Carla Jodice at University of Rome Tor Vergata (allele 1= 26 CAG repeats).
Tissue preparation and western blot analysis
For biochemical analysis, mice were perfused with PBS and brains were stored at −80°C until use. If needed, each brain region from freshly removed brains was dissected out in ice-cold PBS and kept at −80°C. Mouse brain hemispheres were homogenized in TBS buffer containing 5 mM EDTA, 2 mM 1,10-phenanthroline and protease inhibitor mixture (Pierce). Homogenates were centrifuged at 100,000 × g for 1 hr at 4°C. The supernatants were used for analyses of secreted APP forms, while pellets extracted with RIPA buffer (1% NP40, 0.1% SDS and 0.5% sodium deoxycholate in TBS) containing protease inhibitors were used for the analysis of membrane-bound proteins. To obtain total brain lysates, including both cytosolic and membrane proteins, TBS brain lysates were homogenized in RIPA buffer containing protease inhibitors, followed by centrifugation at 13,000 χ g for 10 min. The RIPA-soluble supernatants were used as total protein lysates. Total lysates of other organs were prepared with the same procedures used for brain lysates. Protein lysates were separated on 4–12% gradient NuPAGE Novex Bis-Tris SDS Midi gel (Invitrogen), if otherwise not indicated, and transferred to PVDF membrane (iBlot, Invitrogen). Tris-acetate gel of 3–8% was used to detect CIC. Pre-stained standard (SeeBlue Plus2, Invitrogen) was used as a size marker for western blot analysis. The G12A antibody for APP C-terminal (rabbit polyclonal, targeting last 20 amino acid residues of APP) is custom-manufactured in this study (Pierce Custom Antibodies). Other antibodies used for western blotting were Ataxin-1 (76/8, NeuroMab), ADAM10 (422751, Millipore; ab124695, Abcam), mouse sAPPα (826801, Biolegend), sAPPβ (18957, IBL), sAPPβ-swe (10321, IBL), total sAPP (22C11, Millipore), BACE1 (D10E5, Cell Signaling; ab2077, Abcam), 6E10 (detects human sAPPα, 803015, Biolegend), CIC (A301-204A, Bethyl Laboratories), ETV4 (10684-1-AP, ProteinTech), ETV5 (13011-1-AP, ProteinTech), PS1 (D39D1, Cell Signaling), pan-actin (MS-1295-P0, Fisher Scientific) and GAPDH (MAB374, EMD Millipore). Immunoreactive bands on blots were detected with enhanced chemiluminescence reagents (Pierce) on X-ray film. Films were scanned and band intensities of expected sizes were quantified by densitometric analysis using ImageJ (NIH).
Immunohistochemical staining of brain sections
For histological analysis, mice were perfused with PBS and the brains were fixed with 4% paraformaldehyde (PFA) for 2–3 days, followed by incubation in 30% sucrose solution for 2–3 days until the brains sunk to the bottom. For immunohistochemical or immunofluorescence staining, sucrose-downed mouse brains were sagittally sectioned by a sliding microtome at 40 μm thick, unless otherwise indicated. For the immunostaining, at least ten brain sections encompassing a whole hemisphere of each mouse were incubated with 0.3% Triton X-100, 0.3% Donkey serum and 3% H2O2 for 20 minutes at room temperature, to increase antibody permeability and to remove endogenous peroxidase activity. Sections were washed with TBS and then incubated with 0.3% Triton X-100, 5% Donkey serum and primary antibody for overnight at 4°C. If the primary antibodies were generated from mice, brain sections were pre-incubated with MOM Mouse Ig blocking reagent (Vector Laboratories) for 1 hr at room temperature prior to the primary antibody incubation. Primary antibodies used for immunostaining are Ataxin-1 (76/8, NeuroMab), Aβ (3D6, Eli Lilly), BACE1 (D10E5, Cell Signaling), DCX (SC-8066, Santa Cruz), GFAP (Z0334, Dako), Iba1 (019-19741, Wako), Ki-67 (DRM004, Acris Antibodies), NeuN (RPCA-FOX3, EnCor Biotechnology), PCP4 (HPA005792, Atlas Antibodies), RGS14 (75-170, Neuromab), STEP (4396, Cell Signaling), synaptoporin (102 002, Synaptic Systems). For light microscopic analysis, the bound primary antibodies were detected by biotin-conjugated secondary antibodies, followed by incubation with avidin-biotin-peroxidase complex and by subsequent development of the bound peroxidase activity with 3,3’-diaminobenzidine (DAB, Vector Laboratories). Stained sections were dehydrated with increasing concentration of ethanol and mounted with Cytoseal (Thermo Fisher Scientific). Staining was visualized with a microscope (Nikon TE300), and photomicrographs were taken with an attached digital camera and associated software (Spot InSight). For confocal microscopic analysis, the primary antibody-bound sections were incubated with AlexaFluor 488 or 568-conjugated secondary antibody (Molecular Probes). DAPI (4’,6-diamidino-2-phenylindole, Sigma) was used for nuclear counter staining, and DABCO (1,4-diazabicyclo[2.2.2]octane, Sigma) was used for mounting. Fluorescence images were captured by confocal microscopes (Olympus IX70, Nikon C2; Micro Video Systems).
Real-time RT-qPCR analysis of mRNA expression
Total RNA from mouse brain tissues was isolated using RNAzol (Gibco). RNA integrity was confirmed by the detection of 28S and 18S rRNA bands in agarose gel electrophoresis. RNA was also confirmed to be free of genomic DNA contamination by PCR in the absence of reverse transcriptase. Equal amounts (1 μg) of RNA were reverse transcribed into cDNA using M-MLV reverse transcriptase (Invitrogen). The cDNA was amplified for Bace1, Atxn1, Etv1, Etv4, Etv5, Rorα and Gfi-1 with corresponding primers and internal probes (Applied Biosystems). Gapdh expression was used as an internal control in each PCR reaction to calculate deltaCT. The PCR reactions were performed at least duplicate for each sample and the average CT values were used to calculate the mRNA expression levels (CFX96 Touch Real-Time PCR Detection System, Bio-Rad). RNA isolation and RT-qPCR analysis of 1.5 month and 7–8.5 month-old SCA1 mice and age-matched controls were performed slightly differently. For those brain tissues samples, RNA was extracted using Qiagen miRNeasy Mini Kit according to the manufacturer’s instructions. Random-primed cDNA was then prepared from 1 μg total RNA using M-MLV reverse transcriptase. qPCR reactions were run for Bace1 using PowerUp SYBR Green Master Mix (Applied Biosystems). All transcripts were analyzed in triplicate and normalized to the Gapdh control. The primer sequences used for these PCR reactions can be found in the KEY Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-ADAM10 | Millipore | Cat# 422751 |
| anti-ADAM10 (clone EPR5622) | Abcam | Cat# ab124695 |
| anti-APP (clone G12A) | Pierce Custom Antibodies | this paper |
| anti-mouse sAPPα | Biolegend | Cat# 826801 |
| anti-sAPPβ | IBL | Cat# 18957 |
| anti-sAPPβ-swe | IBL | Cat# 10321 |
| anti-human sAPPα (clone 6E10) | Biolegend | Cat# 803015 |
| anti-total sAPP (clone 22C11) | Millipore | Cat# MAB348 |
| anti-Aβ (3D6) | Eli Lilly | N/A |
| anti-Aβ (6E10) | Biolegend | Cat# 803015 |
| anti-pan-actin (clone ACTN05) | Fisher Scientific | Cat# MS-1295-P0 |
| anti-Ataxin-1 (clone N76/8) | NeuroMab | Cat# 75-117 |
| anti-BACE1 (clone D10E5) | Cell Signaling Technology | Cat# 5606 |
| anti-BACE1 | Abcam | Cat# ab2077 |
| anti-CIC | Bethyl Laboratories | Cat# A301-204A |
| anti-doublecortin (DCX) | Santa Cruz Biotechnology | Cat# sc-8066 |
| anti-ETV4 | ProteinTech | Cat# 10684-1-AP |
| anti-ETV5 | ProteinTech | Cat# 13011-1-AP |
| anti-GAPDH (clone 6C5) | EMD Millipore | Cat# MAB374 |
| anti-GFAP | Dako | Cat# Z0334 |
| anti-Iba1 | Wako | Cat# 019-19741 |
| anti-Ki-67 | Acris Antibodies | Cat# DRM004 |
| anti-NeuN | EnCor Biotechnology | Cat# RPCA-FOX3 |
| anti-PCP4 | Atlas Antibodies | Cat# HPA005792 |
| anti-PS1 (clone D39D1) | Cell Signaling Technology | Cat# 5643 |
| anti-RGS14 | Neuromab | Cat# 75-170 |
| anti-STEP | Cell Signaling Technology | Cat# 4396 |
| anti-synaptoporin | Synaptic Systems | Cat# 102 002 |
| Biological Samples | ||
| Human DNA samples for Alzheimer's disease families | National Institute of Mental Health (NIMH) AD Genetics Initiative Study | Blacker et al. 2003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma | Cat# A9415 |
| 3,3’-diaminobenzidine (DAB) | Vector Laboratories | Cat# SK-4100 |
| Thioflavine S | Sigma-Aldrich | Cat# T1892 |
| Congo red | Sigma-Aldrich | Cat# C6277 |
| FD Cresyl Violet Solution | FD NeuroTechnologies | Cat# PS102-01 |
| Critical Commercial Assays | ||
| FD Rapid GolgiStain Kit, | FD NeuroTechnologies | Cat# PK401 |
| Human/Rat Abeta40 ELISA Kit | Wako | Cat# 294-62501 |
| Human/Rat Abeta42 ELISA Kit | Wako | Cat# 290-62601 |
| Click-iT Nascent RNA Capture Kit | ThermoFisher Scientific | Cat# C10365 |
| Dual Luciferase Reporter Assay System | Promega | Cat# E1910 |
| Congo Red Staining Kit | Sigma-Aldrich | Cat# HT60-1KT |
| qPCR assay for Bace1 mRNA expression | Applied Biosystems | Mm00478664_m1 |
| qPCR assay for Atxn1 mRNA expression | Applied Biosystems | Mm00485928_m1 |
| qPCR assay for Etv1 mRNA expression | Applied Biosystems | Mm00514804_m1 |
| qPCR assay for Etv4 mRNA expression | Applied Biosystems | Mm00476696_m1 |
| qPCR assay for Etv5 mRNA expression | Applied Biosystems | Mm00465816_m1 |
| qPCR assay for Rora mRNA expression | Applied Biosystems | Mm01173766_m1 |
| qPCR assay for Gfi1 mRNA expression | Applied Biosystems | Mm00515853_m1 |
| qPCR assay for Gapdh mRNA expression | Applied Biosystems | Mm99999915_g1 |
| Experimental Models: Cell Lines | ||
| Ataxin-1 WT and KO mouse embryonic fibroblasts | This paper | N/A |
| Ataxin-1 WT and KO mouse primary cortical neurons | This paper | N/A |
| HEK293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Ataxin-1 KO mice | Matilla et al., 1998 | N/A |
| Ataxin-1 154Q knock-in mice | Watase et al., 2002 | N/A |
| APPswe/PSEN1ΔE9 (APP-PS1) mice | The Jackson Laboratory | Stock # 34832-JAX |
| BACE1 KO mice | The Jackson Laboratory | Stock # 004714 |
| Oligonucleotides | ||
| Primers for Atxn1−/− mouse genotyping | primer1:CCCCTACCCACTCCTCATAT primer2:GGCCGTGATACAGTTTGCTG primer3:CCGGTGGATGTGGAATGTGT | |
| Primers for Bace1−/− mouse genotyping | WT_pr:AGGCAGCTTTGTGGAGATGGTG MT_pr:TGGATGTGGAATGTGTGCGAG Common_pr:CGGGAAATGGAAAGGCTACTCC | |
| Primers for APPswe-PSEN1ΔE9 mouse genotyping | Tg_fp:AATAGAGAACGGCAGGAGCA Tg_rp:GCCATGAGGGCACTAATCAT In_fp:CTAGGCCACAGAATTGAAAGATCT In_rp:GTAGGTGGAAATTCTAGCATCATCC | |
| Primers for Atxn1154Q/+ knockin mouse genotyping | KI_fp: GTGAGTTTGGGTCTGGCATC KI_rp: CCAAAAGTTAGGATCACAGCCC | |
| Primers for Bace1 qPCR with SYBR Green | fp:TCTTTTCCCTGCAGCTCTGT rp:ACTGCCCGTGTATAGCGAGT | |
| Primers for Gapdh qPCR with SYBR Green | fp:AGGTCGGTGTGAACGGATTTG rp:TGTAGACCATGTAGTTGAGGTCA | |
| Primers for BACE1 promoter cloning | fp:TAGCCCGGGCTCGAGAACCATACCGG CTTTTCTCCT rp:CGGAATGCCAAGCTTCCACCATAATCCAGCTCG | |
| Primers for ETV site mutagenesis | Mutation:TGGCAGGCAATCCGGCTCATAGGGCTGGAGAGGGGTCT Deletion:CCTCTCCAGCCCAGCCGGATTGCC | |
| Recombinant DNA | ||
| pGL3-Basic Vector | Promega | Cat# E1751 |
| pRL-TK | Promega | Cat# E2231 |
| pLX_TRC311 | Addgene | Cat# 113668 |
| pLX_TRC311 ETV4-S | Addgene | Cat# 74983 |
| pLX_TRC311 ETV4-L | Addgene | Cat# 74982 |
| pLX_TRC311 ETV5 | Addgene | Cat# 74984 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | |
| Axoscope | Axon Instruments | N/A |
| Mini Analysis Program | Synaptosoft | N/A |
| Origin | Microcal Software | N/A |
| TRANSFAC | http://genexplain.com/transfac/ | |
| PROMO | http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3 | |
| Other | ||
| Nikon C2 confocal microscope | Micro Video Instruments, Inc. | N/A |
| Olympus IX70 confocal microscope | Olympus | 1X70 |
| Nikon Ci Upright microscope | Nikon | Ci-L |
| Touch Real-Time PCR Detection System | Bio-Rad | CFX96 |
| In-Fusion HD EcoDry Cloning Plus system | Clontech | Cat# 638915 |
| jetPrime transfection reagent | Polyplus transfection | Cat# 114-15 |
| QuikChange Lightning Multi Site-Directed Mutagenesis Kit | Agilent | Cat# 210513 |
Acute brain slice culture and mRNA stability
Acute mouse brain slice cultures were prepared from 10-month-old WT and Ataxin-1 KO mice. Mice were anaesthetized with isoflurane and decapitated (Dzhala and Staley, 2003). After dissection, the brains were rapidly removed and placed in oxygenated (95% O2 / 5% CO2) ice-cold artificial cerebrospinal fluid (ACSF) of the following composition: NaCl 126 mM, KCl 3.5 mM, CaCl2 2 mM, MgCl2 2 mM, NaHCO2 25 mM, NaH2PO2 1.2 mM and glucose 11 mM (pH 7.4). Coronal brain slices of 400 μm thickness were cut using Leica VT-1000E vibratome (Leica). Brain slices were incubated at room temperature in the dark in oxygenated ACSF, which contained either actinomycin D (20 μM, Sigma) or DMSO as vehicle. After incubating for given period, brain slices were dissected for cortex, hippocampus and brain stem. Total RNA isolated from cortex was analyzed for Bace1 mRNA levels by RT-qPCR as described above.
Analysis of nascent mRNA synthesis
Acute brain slice cultures were treated with 5-ethynyl uridine (5EU), an analog of uridine, or vehicle (DMSO) for 3 or 16 hrs. Total RNA was isolated from cortex of the treated brain slices and used for copper-catalyzed click reaction with azide-modified biotin. The 5EU-biotin-conjucated nascent RNA transcripts were isolated with streptavidin using magnetic beads. The click reaction and the isolation of nascent RNA transcript were carried out with Click-iT Nascent RNA Capture Kit (Life Technologies) by following the manufacturer’s instructions. Briefly, ten μg of total 5EU-RNA was used for RNA biotinylation by click reaction. After the click reaction, RNA was precipitated by glycogen and ammonium acetate, and 1 μg of biotinylated RNA was used to bind to streptavidin-conjugated magnetic beads. The bead-bound RNA was then used for cDNA synthesis using Superscript VILO cDNA synthesis kit. The beads were removed and the supernatant containing the cDNA was used for qPCR as described above.
Electrophysiological recording of brain slices
For electrophysiological recordings, individual brain slice that contains hippocampus were transferred to a conventional submerged-type chamber and continuously superfused with oxygenated ACSF at 32°C and at a rate of 2–3 ml/min. Extracellular field potentials were recorded in the CA1 pyramidal cell layer using tungsten microelectrodes and multi-channel amplifier (band-pass 0.1 Hz – 10 kHz; x1000) with enhanced electromagnetic interference (EMI) noise suppression. Microelectrodes made from coated tungsten wire of 50 μm diameter (California Fine Wire Company) were used for simultaneous recordings of multiple unit activity (MUA; 500 Hz high-pass filter) and population field activity in EEG band (1–100 Hz). Root mean square (RMS) noise level with an electrode placed in the perfusion solution was typically 4–5 μV, while the amplitude of action potentials recorded from the stratum pyramidale ranged from this noise level up to 200 μV. The signals were digitized using an analogue-to-digital converter (DigiData 1322A; Axon Instruments). Sampling interval per signal was 100 μs (10 kHz). Axoscope (Axon Instruments), Mini Analysis Program (Synaptosoft) and Origin (Microcal Software) programs were used for the acquisition and data analysis. The multiple unit activity was determined from high-pass filtered (500 Hz) raw data with a spike detection algorithm (Mini Analysis Program) and verified visually. Spikes with amplitude greater than three times the root mean square noise level were accepted.
Promoter analysis for transcription factors
To search for potential binding sites of transcription factors in Bace1 and BACE1 promoters, we utilized TRANSFAC and PROMO software programs. DNA sequences from −2000 to +1000 base pair from the transcription start site were analyzed for each gene. Options in the analysis were selected for high-quality vertebrate matrices, 'minimized false-positive’ cut-offs, high cut-offs score (> 0.9) and matrix similarity (> 0.9). CIC-binding motif TGAATG(A/G)A were searched by TRANSFAC in the promoters of mouse Etv1, Etv4, Etv5 and human ETV1, ETV4, ETV5. Transcription start sites of each gene were determined based on the following UCSC reference sequences: mouse(mm9) Bace1, NM_011792.6; Etv1, NM_007960.5; Etv4, NM_001316365.1; Etv5, NM_023794.2; human(hg19) BACE1, NM_012104.4; ETV1, NM_004956.4; ETV4, NM_001079675.2; ETV5, NM_004454.2. Sequence homology between mouse and human were analyzed for PEA3 (ETV1/4/5) binding sites in BACE1 promoter and CIC binding sites in ETV1, ETV4 and ETV5 promoters. Sequences of −50 to +50 base pairs for each binding site were aligned and compared by NCBI-Needleman-Wunsch Global Align algorithm (blast.ncbi.nlm.nih.gov).
BACE1 promoter cloning and luciferase assay
BACE1 promoter region, −816 to +56 from transcription start site (same as −1273 to −400 from translation start site), was amplified from human cDNA using the primers Eco_B1U1273F 5’-TAGCCCGGGCTCGAGAACCATACCGGCTTTTCTCCT-3’ and Eco_B1U400R 5’-CGGAATGCCAAGCTTCCACCATAATCCAGCTCG-3’. The PCR product was inserted in front of the firefly luciferase reporter gene in the pGL3-Basic Vector (Promega) at XhoI/HindIII sites using In-Fusion HD EcoDry Cloning Plus system (Clontech) according to manufacturer’s instructions. The construct, pGL3-BACE1 promoter, was confirmed by DNA sequencing. The predicted ETV4/5 binding site (−96) was mutated or deleted using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent) with BACE1_ETV4/5_M 5'-TGGCAGGCAATCCGGCTCATAGGGCTGGAGAGGGGTCT-3' (for mutation) and BACE1_ETV4/5_D 5'-CCTCTCCAGCCCAGCCGGATTGCC-3' (for deletion) primers. Correct mutagenesis was confirmed by DNA sequencing. For luciferase assay, 7.5 × 104 HEK293T cells per well were seeded in 24-well plates in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Atlanta biologicals) and Penicillin-Streptomycin Mix (Thermo Fisher Scientific). One day after seeding, the cells were transfected with 100 ng pGL3-BACE1 promoter, 50 ng pRL-TK (for renilla luciferase expression, Promega), and either 200 ng of pLX_TRC311 (Addgene #113668), pLX_TRC311 ETV4-S (Addgene #74983), pLX_TRC311 ETV4-L (Addgene #74982), or pLX_TRC311 ETV5 (Addgene #74984) using jetPrime transfection reagent (Polyplus transfection) according to manufacturer’s instructions. ETV4-S is not detected in mouse and lacks a large regulatory domain as compared to ETV4-L (relevant to ETV4 of mouse). In luciferase assays, ETV4-S did not activate BACE1 promoter activity. Twenty-four hours after transfection, the growth media was removed, the cells were washed with PBS, and luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega). In all assays, relative luciferase activity of each sample was calculated as the ratio of firefly to renilla luciferase activity.
Aβ ELISA, plaque load and reactive gliosis
Mouse hemispheres were homogenized in TBS buffer containing protease inhibitors, and then centrifuged at 100,000 × g for 1 hr. The supernatants were saved for ‘soluble Aβ’ measurements. The pellets were resuspended and further homogenized in 70% formic acid (equal volume of TBS), followed by centrifugation at 100,000 × g for 1 hr. Formic acid supernatants were neutralized with 1 M Tris for the measurement of ‘insoluble Aβ’. The amounts of soluble and insoluble Aβ40 and Aβ42 were determined by sandwich ELISA using commercially available kits (#294-62501 and #290-62601, Wako), and measurements were performed minimally in duplicate. The following are antibodies used for ELISA: Aβ40 = BNT77 for capture, BA27 for detection. Aβ42 = BNT77 for capture, BC05 for detection. For Aβ plaque load analysis, floating brain sections were probed with antibody recognizing Aβ sequence (3D6, 1:400 dilution), followed by the procedure described above for DAB development and light microscopic analysis. Photomicrographs were taken for 3D6-labeled brain sections with a dissection microscope. Whole cortex and hippocampus (5–6 brain sections/mouse) were analyzed for Aβ plaque numbers and Aβ plaque-covered areas by utilizing Analyze Particles method of ImageJ. To visualize neuritic plaques in DAB-developed brain sections, sections were mounted and stained with glass wool-filtered 1% Congo red solution for 10 min. In immunofluorescence staining for Aβ plaques, neuritic plaques were visualized with 10 min of staining with 0.025% Thioflavin S. For astrocyte activation analysis, floating brain sections were probed with anti-GFAP antibody (1:200 dilution) followed by the procedure described above for DAB development. Whole cortex (3–4 brain sections/mouse) was analyzed for GFAP immunoreactivity with ImageJ program. To visualize neuritic plaques in GFAP- or Iba1-immunolabeled brain sections, sections were mounted and stained with 1% Congo red solution for 10 min.
Hippocampal neurogenesis and Golgi staining
To analyze the dendritic development of newborn neurons in the hippocampus, brain sections were labeled with anti-DCX antibody and the immunoreactive signals were developed with DAB. Photomicrographs were taken with the 10 × objective lens, and DCX+ neurons in subgranular cell layer (total DCX+ neurons) were counted for at least 3 brain sections per each mouse, containing full hippocampal formation. DCX+ neurons that have dendrites which pass through the granular neuronal layer were counted as projecting DCX+ neurons. Projecting ratio was determined by the percentile of projecting DCX+ neurons vs. total DCX+ neurons. To compare hippocampal neurogenesis among 4 month-old WT, Atxn1−/− and Atxn1−/−; Bace1+/− mice, ImageJ was used to quantify DCX+ cell bodies in the subgranular cell layer (total DCX+ cells). The amounts of projecting DCX+ neurons were determined by quantifying the areas covered by DCX-positive immunoreactivity within the granular cell layer (ImageJ). The amounts of dendrites were determined by quantifying the DCX-positive immunoreactivity within the molecular layers. The projecting ratio was calculated by dividing projecting DCX+ neurons with total DCX+ neurons. Non-specific DCX-positive immunoreactivity, which probably is representative of microglia in the molecular layer, was subtracted in the quantification of the dendrites. Average percentile of area in molecular layer covered by microglia was 1.7%. To select more comparable brain sections among different mice, sagittal sections that were between 1–2 mm away from the brain midline, containing full hippocampal formation, were selected for the analysis of DCX immunoreactivity. To analyze the NPC proliferation in the hippocampus, Ki-67+ cells in the dentate gyrus were counted for at least 3 brain sections per each mouse. In counting and quantifying DCX+ neurons and Ki-67+ cells, experimenters were blinded for the genotypes of immunostained sections. An average was drawn from at least two persons' independent cell counting.
Neuronal dendrites of mature neurons were visualized by Golgi staining. Freshly removed mouse brains were immersed in solutions for 2 weeks for impregnation, and then brains were sagittally sectioned for 100 μm thick on a cryostat (Leica) at −22°C. The tissue preparation and staining procedure were performed in accordance with the manufacturer's instructions (FD Rapid GolgiStain Kit, FD NeuroTechnologies). Nissl staining was used as counterstaining for neuronal nuclei. Golgi-stained granular neurons and their dendrite numbers were analyzed for at least 3 brain sections per each mouse, which contained full hippocampal formation.
Olfactory axonal targeting analysis
After perfusion with PBS, olfactory tissues containing the olfactory epithelium and bulbs were dissected intact and embedded in M1 (Shandon). Fourteen micrometer-thick coronal sections were cut by cryostat (Leica) and the sections were collected on SuperFrost slides (Fisher). Immunostaining was performed with antibodies targeting MOR28 and M71 odorant receptors (provided by Dr. Barnea at Brown University). Sections were post-fixed with 4% paraformaldehyde in PBS for 7 min at room temperature. All sections were washed three times in PBS for 5 min, permeabilized in 0.1% Triton X-100 in PBS (PT) for 30 min at room temperature, blocked in 5% heat-inactivated donkey serum in PT (PTS) for 1 hr at room temperature, incubated with primary antibody in PTS under a Hybrislip (Invitrogen) overnight in a humidified chamber at 4°C. Sections then washed three times with PT for 5 min, blocked with PTS for 30 min at room temperature, incubated with a fluorescent-conjugated secondary antibody in PTS with DAPI (Invitrogen, 1/5000) for 3 hrs at room temperature, washed three times in PBS for 5 min. Vectashield (Vector Laboratories) was applied to each slide and the olfactory tissue sections were coverslipped. Slides were analyzed using a Zeiss Axio Observer Z1 microscope and ImageJ program to calculate the distance from olfactory midline and the areas of glomeruli.
Analysis of CA2 neurodegeneration
Confocal photomicrographs of mouse brain sections (20 × images) double stained for NeuN and PCP4 were used to count NeuN+ neurons and PCP4+ neurons in hippocampal CA1, CA2, and CA3 regions. To count all neurons in CA2, an empty box that covers the entire CA2 area was utilized. The same size box was used for the analysis of all the photomicrographs (Figure S7A). A half-sized box was used to count NeuN+ neurons in nearby CA1 and CA3 areas. NeuN+ neurons in CA2 were counted with photomicrographs without PCP4 signal. For each mouse, 2–4 sections were analyzed for neuronal number counting. For better comparison among different mice and genotypes, sagittal brain sections that are between 1 to 2 mm away from the brain midline, which contain full hippocampal formation, were used for neuronal counting and quantification. To examine overall morphology and density of the neurons in the hippocampus, mounted brain sections were stained with Nissl (FD Cresyl Violet Solution, FD NeuroTechnologies). The width of hippocampal neuronal layers (CA1, CA2, and CA3) was measured by dividing the area of each neuronal layer by the length of that layer. To measure the width of CA2, an empty box that covers the entire CA2 area was utilized. Within the box, the area and length of CA2 were measured using ImageJ. For CA1 and CA3, boxes that cover about half of CA1 or CA3 neuronal layers (near CA2) were used to measure the areas and lengths. Width was measured by dividing the area by length of each layer.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean ± standard error of the mean (SEM). All data requiring statistical testing were analyzed using t-test (two-tailed unequal variance, Microsoft EXCEL) or one-way analysis of variance (ANOVA) followed by Tukey’s test (SPSS version 12.0). *, **, and *** denote p < 0.05, p < 0.01, and p < 0.001, respectively, in all figures. p value of < 0.05 was considered statistically significant. All of the statistical details of experiments, including the statistical tests used, exact value of n, and what n represents (e.g., number of mice), can be found in the figures and figure legends.
DATA AND CODE AVAILABILITY
This study did not generate dataset/code. All algorithms and software used in this study are publicly available and links are provided in the corresponding sections of the methods.
Supplementary Material
Figure S1. BACE1 expression in Ataxin-1 KO mice, Related to Figure 1
(A) Densitometric quantification of western blot results for Ataxin-1, ADAM10, APP, APP-CTF and PS1-CTF in WT (+/+), Ataxin-1 hetero-KO (+/−) and KO (−/−) mouse brains. Bottom right, a representative western blot image of PS1-CTF. Full length PS1 was undetectable in these samples. Values are mean ± SEM. Number of mice analyzed, n= 5. ***p < 0.001, t-test. (B) Upper panel, photomicrographs of BACE1-immunolabeled olfactory bulb and forebrain region of WT and Ataxin-1 (ATXN1) KO mice. Arrow indicates glomeruli, which showed strong immunoreactivity for BACE1. Lower, fluorescence photomicrographs of olfactory glomeruli (arrow) region immunolabeled for Ataxin-1 and BACE1. DAPI was used for nuclear staining. Brain sections from BACE1 KO were used as a negative control for the staining. Bar = 100 μm. (C) Fluorescence photomicrographs of hippocampal CA3 region of WT mice immunolabeled for Ataxin-1 (red), BACE1 (green), and DAPI (blue). Bar = 100 μm. (D) Levels of BACE1, sAPPβ and sAPPα in the cortex of WT (+/+), Ataxin-1 KO (−/−) and hetero-KO (+/−) mice. n = 5. ***p < 0.001, **p < 0.01, *p < 0.05. (E) Analysis of sAPPβ (arrow) levels in the cortex and cerebellum of WT and Ataxin-1 KO mice. Bottom, densitometric quantification of the western blot result (n = 3). *p < 0.05. n.s.: not significant. (F) Analysis of Ataxin-1 and BACE1 expression in different organs/tissues. Protein lysates from Ataxin-1 KO or BACE1 KO mice were used as controls for the band specificity. Immunoreactive bands that are in similar size with Ataxin-1 (~90 kDa) appeared in other organs/tissues beyond brain, particularly with a strong intensity in heart same. However, these bands are likely non-specific signals, as they were also detected in the lysates of Ataxin-1 KO samples for the same tissues/organs. (G) Levels of BACE1 in WT, Ataxin-1 hetero-KO and KO embryonic (embryonic day 15, left top), postnatal (P0, right top) and adult mouse brains. Vertical straight lines indicate that different parts within same blot were placed together. Ataxin-1 levels in the embryonic/postnatal brains were lower than those of adult brains. In contrast, BACE1 levels were much higher in the embryonic/postnatal brains. Bottom panel, no effect of Ataxin-1 depletion on BACE1 levels in cultured cortical neurons (DIV10) and embryonic fibroblasts that are derived from Ataxin-1 KO mouse embryos.
Figure S2. Increased Bace1 transcription in the cortex of Ataxin-1 KO mice, Related to Figure 2
(A) Actinomycin D or vehicle (DMSO) was treated for 24 or 48 hrs in acute brain slice cultures derived from WT and Ataxin-1 KO mice. Bace1 mRNA amount per each brain slice was analyzed. Gapdh was used as an internal control for RT-qPCR. Same RT-qPCR data from 48 hr-cultured slices were presented as Bace1 mRNA/total RNA in Figure 2A. n (brain slices) = 2 (24 hr, WT and ATXN1 KO), 3 (48 hr, WT), 4 (48 hr, ATXN1 KO). *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (B) RorαmRNA levels in the cortex of WT and Ataxin-1 KO mice. Levels of Gfi1 mRNA were too low to be reliably analyzed by RT-qPCR. (C) mRNA levels of Etv1, Etv4, and Etv5 in the cortex and cerebellum of WT and Ataxin-1 KO mice. Expression levels were presented as relative values to WT group (100) for each gene. Number of mice analyzed, n = 6 (cortex), 4 (cerebellum). (D) Protein expression levels of ETV4 and ETV5 in pLX_TRC311, pLX_TRC311 ETV4-L, or pLX_TRC311 ETV5-transfected HEK293T cells. (E) Luciferase reporter activity in HEK293T cells transfected with pGL3-BACE1 promoter vector. A conserved ETV4/ETV5 binding site in BACE1 promoter was mutated from CCGGAA to CTATGA or deleted. Results from 2 independent experiments of sextuplicate transfection were normalized and combined. n = 12 (2 × 6). (F) BACE1 levels in the brains of Bace1+/− (39%) as compared to WT mice. (G) Immunohisotochemical staining for BACE1 in the cortical and hippocampal regions of WT, Atxn1−/− and Atxn1−/−; Bace1+/− mice. Bar = 500 μm.
Figure S3. Amyloidogenic processing of APP in APP-PS1/ATXN1-KO mouse brains, Related to Figure 3
(A) Expected and actual genotype ratios of APP-PS1 and APP-PS1/ATXN1-KO mice at 4 weeks. (B) Densitometric quantification of western blot analysis of BACE1, ADAM10, sAPPβ (transgenic) and sAPPα (transgenic) in the brains of male and female APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). Numbers in parenthesis denote the number of mice analyzed. **p < 0.01, ***p < 0.001, t-test. (C) Endogenous sAPPβ levels in APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). Arrow, endogenous sAPPβ; *, non-specific band. n = 7. **p < 0.01. (D) TBS-soluble Aβ40 and Aβ42 levels in the brains of 5-month-old male and female APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). *p < 0.05. (E) Longer film exposure of western blot image (shown in Figure 3G) probed with 6E10 antibody revealed APP-CTFβ at ~10 kDa.
Figure S4. Effects of Ataxin-1 depletion on Aβ plaque deposition and gliosis, Related to Figure 4
(A) Quantification of area covered by Aβ plaques in the cortex and hippocampus of 5-month-old male and female APP-PS1/WT, APP-PS1/Atxn1+/− and APP-PS1/Atxn1−/− mice. Number in parenthesis denotes number of mice analyzed. Data for APP-PS1/WT and APP-PS1/Atxn1−/− mice are same as shown in Figure 4B. *p < 0.05, ***p < 0.001, t-test. (B) Hippocampal regions of APP-PS1 and APP-PS1/ATXN1-KO mice immunolabeled with 3D6 Aβ (red) antibody followed by staining with thioflavin S (green). Most of the plaques in both genotypes were thioflavin S-positive dendritic plaques. Bar = 200 μm. (C) Correlation between Aβ plaque load and GFAP+ astrocyte activation. (D) Frontal cortex of 5-month-old WT and Ataxin-1 KO mouse brains (without APPswe/PS1ΔE9 overexpression) immunolabeled for GFAP. Bar = 500 μm. Inset, high resolution image. No notable change was detected for astrocyte activation in Ataxin-1 KO mice.
Figure S5. Effects of Ataxin-1 depletion on hippocampal neurogenesis, Related to Figure 5
(A) DCX+ immature neurons in the dentate gyrus of 3-month-old WT and Ataxin-1 KO mice (without APP-PS1 overexpression). Brackets: granular cell layer. Arrows, DCX+ neurons. Right, counts of DCX+ cells in dentate gyrus. n = 3. **p < 0.01. Bar = 100 μm. (B) Impaired dendrite projection of newborn neurons in APP-PS1/Atxn1+/− mice. Left: counts of DCX+ and projecting DCX+ cells per dentate gyrus section. Right, ratio of dendrite projection of each DCX+ neuron. For a better comparison, all brain sections from three different genotypes were stained together for DCX. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (C) Number of Golgi-stained granular neurons per dentate gyrus section (left) and number of dendrites per each granular neuron (right) in the dentate gyrus of WT and Ataxin-1 KO mice. Number of mice analyzed, n = 5 (WT), 4 (ATXN1 KO). Three brain sections per mouse were used for counting Golgi-stained granular neurons in the dentate gyrus. Twenty granular neurons per section (total 60 neurons per mouse) were used for counting the number of dendrites per neuron. n.s., not significant. (D) Photomicrographs of Ki-67+ cells (arrows) in the dentate gyrus of 4-month-old WT, Atxn1−/−, Atxn1−/−; Bace1+/− mice. Bar = 100 μm.
Figure S6. Elevated BACE1 levels and aberrant axonal targeting in CA2 regions of Ataxin-1 polyQ expansion mutant mice, Related to Figure 7
(A) Immunohistochemical staining of 1.5-, 4.5-, and 8-month-old WT and Atxn1154Q/+ mouse brains for BACE1. Modest increase of BACE1 immunoreactivity in the cortex (*) and hippocampus of 4.5- and 8-month-old Atxn1154Q/+ mice as compared to age-matched WT mice. Bar = 1 mm. (B) RT-qPCR analysis of Bace1 mRNA levels in the hippocampus of 1.5- and 7–8.5 month-old mice. Number of mice analyzed, n = 4 – 9 (in bar). Gapdh was used as an internal control. Relative value of 100 is given to WT at each age group. (C) CIC levels (long and short forms) in the cortex. n.s., non-specific bands at ~ 80kDa. These bands were observed in the same blot for CIC and are shown as loading controls. Bottom: densitometric quantification of the CIC western blot result. (D) Quantification of BACE1 immunoreactivity intensity at the end of stratum lucidum. Number in bar denotes the number of brain sections analyzed. One or 2 sections/mouse. **p < 0.01, ***p < 0.001, t-test. (E) Hippocampal CA2 regions of 6-month-old Atxn1154Q/+ and WT mice immunolabeled for synaptoporin. Arrows, synaptoporin+ presynaptic terminals within hippocampal neuronal layer (dashed lines). *, synaptoporin+ presynaptic terminals of granular neurons at striatum lucidum. Bar = 100 μm. Right: counts of synaptoporin+ presynaptic terminals within CA2 neuronal layer. Number of mice analyzed, n = 3. One or 2 brain sections were analyzed for each mouse.
Figure S7. Degeneration of CA2 neurons and impaired hippocampal neurogenesis in SCA1 mice. Related to Figure 7
(A-C) Representative images for neuronal cell counting in CA1/CA2/CA3 regions of hippocampus of 6-9 month-old SCA1 (Atxn1 154Q/+) and age-matched WT mice immunolabeled for PCP4 and NeuN: yellow box for PCP4+ neurons in CA2, white box for NeuN+ neurons in CA1, and red box for NeuN+ neurons in CA3. NeuN+ neurons in CA2 were counted with photomicrographs without PCP4 signal. Arrows indicate ectopic positions of PCP4+ neurons just outside of CA2 region, suggesting overall distribution of PCP4+ neurons are not changed in the hippocampal neuronal layers of SCA1 mice. Bar = 100 μm. Analyzed numbers of mouse/brain section = 6/16. 2–4 sections/mouse. **p < 0.01. For better comparison among different mice and genotypes, sagittal brain sections that are between 1 to 2 mm away from the brain midline, which contain full hippocampal formation, were selected for the neuronal counting and quantification. (D) z-stacked images of 40 μm-thick brain sections labeled for NeuN. Arrow indicates thinner CA2 neuronal layer in SCA1 mice. Bar = 100 μm. (E) High resolution images of CA2 neurons labeled with NeuN antibody and DAPI. Arrows indicate abnormally stretched neuronal cell bodies and nuclei in the SCA1 mice. Bar = 20 μm. Photomicrographs (F) and counting (G) of PCP4+ neurons in CA2 regions of 1.5-, 4.5-, and 8-month-old Atxn1154Q/+ and WT mice. Bracket, CA2 region; *, axons of granular neurons; arrow heads, PCP4+ Purkinje cells in the cerebellum of 8-month-old mice. In contrast to CA2 neurons, majority of Purkinje cells of Atxn1154Q/+ mice expressed high levels of PCP4 at this age. Bar = 100 μm. Number in bar, number of brain sections analyzed. 1–3 sections/mouse. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (H) Nissl staining revealing less populated neurons in the CA2 of SCA1 mice (arrow in high resolution image). Bar = 100 μm. (I) Widths of neuronal layers (area divided by length) in hippocampus stained with Nissl. Number of mice/brain sections analyzed: WT (4/12), SCA1 (3/8). (J) Quantification of RGS14 immunoreactivity in hippocampal CA2 of 6-month-old Atxn1154Q/+ and WT mice. Number of mice analyzed, n = 3. (K) Hippocampal region of 6-months-old SCA1 and WT and mice immunolabeled for STEP, another CA2 specific marker. Arrows indicate complete disappearance of STEP immunoreactivity in the CA2 neurons of SCA1 mice. Bar = 100 μm. (L) Hippocampal region of 7-month-old Ataxin-1 KO and WT mice immunolabeled for PCP4. (M) Photomicrographs and counts of Ki-67+ cells (arrows) in the dentate gyrus of 6 month-old Atxn1154Q/+ and WT mice. Number of mice analyzed, n = 3.
Highlights.
Ataxin-1 loss of function increases BACE1 transcription and Aβ pathology in cerebrum.
Elevated BACE1 levels impair hippocampal neurogenesis and olfactory axonal targeting.
PolyQ-expanded mutant Ataxin-1 causes degeneration of CA2 neurons in hippocampus.
Ataxin-1 loss and polyQ-expanded Ataxin-1 lead to distinct brain regional vulnerability.
Loss of Ataxin-1 leads to increases in BACE1 levels as well as amyloidogenic cleavage of APP, leading to Aβ deposition and gliosis in mouse models of Alzheimer’s Disease.
ACKNOWLEDGEMENTS
We appreciate Dr. Carla Jodice at the University of Rome Tor Vergata for generously sharing of important information, Dr. Kevin J. Staley for equipment for brain slice culture and electrophysiological recording. We also thank Rachel Souza, Abigail Lebovitz, and Nicolas Papele and Andrea Lesinski for technical assistance. This study was supported by NIH R01 AG056775 (J.S.) and R01 MH060009 and R01 AG041856 (R.E.T.), R37 NS027699 and U54 HD083092 (H.Y.Z.), the Cure Alzheimer’s Fund (J.S. and R.E.T.), the MINDlink Foundation (J.D.S.), and the Jeffry and Barbara Picower Foundation (R.E.T. and H.Y.Z). H.Y.Z. is an investigator of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Asher M, Johnson A, Zecevic B, Pease D, and Cvetanovic M (2016). Ataxin-1 regulates proliferation of hippocampal neural precursors. Neuroscience 322, 54–65. [DOI] [PubMed] [Google Scholar]
- Barao S, Gartner A, Leyva-Diaz E, Demyanenko G, Munck S, Vanhoutvin T, Zhou L, Schachner M, Lopez-Bendito G, Maness PF, et al. (2015). Antagonistic Effects of BACE1 and APH1B-gamma-Secretase Control Axonal Guidance by Regulating Growth Cone Collapse. Cell Rep 12, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnea G, O'Donnell S, Mancia F, Sun X, Nemes A, Mendelsohn M, and Axel R (2004). Odorant receptors on axon termini in the brain. Science 304, 1468. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, et al. (2008). Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, and Tanzi RE (2012). The genetics of Alzheimer's disease. Prog Mol Biol Transl Sci 107, 79–100. [DOI] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Van Miegroet H, Gil A, Engelborghs S, De Deyn PP, Vandenberghe R, Van Broeckhoven C, and Sleegers K (2010). Follow-up study of susceptibility loci for Alzheimer's disease and onset age identified by genome-wide association. J Alzheimers Dis 19, 1169–1175. [DOI] [PubMed] [Google Scholar]
- Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, et al. (2003). Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet 12, 23–32. [DOI] [PubMed] [Google Scholar]
- Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, et al. (2018). Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk K, Bosch S, Globas C, Zuhlke C, Daum I, Klockgether T, and Dichgans J (2001). Executive dysfunction in spinocerebellar ataxia type 1. Eur Neurol 46, 43–48. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, and Wong PC (2001). BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4, 233–234. [DOI] [PubMed] [Google Scholar]
- Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, and Prochiantz A (2004). Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, and Albers MW (2012a). The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep 2, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Schrank BR, Rodriguez S, Benz EG, Moulia TW, Rickenbacher GT, Gomez AC, Levites Y, Edwards SR, Golde TE, et al. (2012b). Abeta alters the connectivity of olfactory neurons in the absence of amyloid plaques in vivo. Nat Commun 3, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatila ZK, Kim E, Berle C, Bylykbashi E, Rompala A, Oram MK, Gupta D, Kwak SS, Kim YH, Kim DY, et al. (2018). BACE1 Regulates Proliferation and Neuronal Differentiation of Newborn Cells in the Adult Hippocampus in Mice. eNeuro 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, and Zoghbi HY (2010). Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet 6, e1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic B, Stojkovic O, Savic D, Zamurovic N, Nesic M, Major T, Keckarevi D, Romac S, Zamurovi B, and Vukosavic S (2000). Comparison of the number of triplets in SCA1, MJD/SCA3, HD, SBMA, DRPLA, MD, FRAXA and FRDA genes in schizophrenic patients and a healthy population. Am J Med Genet 96, 884–887. [DOI] [PubMed] [Google Scholar]
- Cvetanovic M, Hu YS, and Opal P (2017). Mutant Ataxin-1 Inhibits Neural Progenitor Cell Proliferation in SCA1. Cerebellum 16, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demars MP, Hollands C, Zhao Kda T, and Lazarov O (2013). Soluble amyloid precursor protein-alpha rescues age-linked decline in neural progenitor cell proliferation. Neurobiol Aging 34, 2431–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanand DP, Lee S, Manly J, Andrews H, Schupf N, Doty RL, Stern Y, Zahodne LB, Louis ED, and Mayeux R (2015). Olfactory deficits predict cognitive decline and Alzheimer dementia in an urban community. Neurology 84, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhilla Albers A, Asafu-Adjei J, Delaney MK, Kelly KE, Gomez-Isla T, Blacker D, Johnson KA, Sperling RA, Hyman BT, Betensky RA, et al. (2016). Episodic memory of odors stratifies Alzheimer biomarkers in normal elderly. Ann Neurol 80, 846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, and Staley KJ (2003). Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J Neurosci 23, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancellu R, Paridi D, Tomasello C, Panzeri M, Castaldo A, Genitrini S, Soliveri P, and Girotti F (2013). Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2. J Neurol 260, 3134–3143. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Shykind BM, Sosulski DL, Franks KM, Glinka ME, Mei DF, Sun Y, Kirkland J, Mendelsohn M, Albers MW, et al. (2008). Mice with a "monoclonal nose": perturbations in an olfactory map impair odor discrimination. Neuron 60, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, et al. (2011). Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 334, 690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, and Pedersen NL (2006). Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63, 168–174. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. (2013). TREM2 variants in Alzheimer's disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hitt B, Riordan SM, Kukreja L, Eimer WA, Rajapaksha TW, and Vassar R (2012). beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem 287, 38408–38425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti FL, and Siegelbaum SA (2014). The hippocampal CA2 region is essential for social memory. Nature 508, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooli BV, Kovacs-Vajna ZM, Mullin K, Blumenthal MA, Mattheisen M, Zhang C, Lange C, Mohapatra G, Bertram L, and Tanzi RE (2014). Rare autosomal copy number variations in early-onset familial Alzheimer's disease. Mol Psychiatry 19, 676–681. [DOI] [PubMed] [Google Scholar]
- Hu X, He W, Luo X, Tsubota KE, and Yan R (2013). BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep 4, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13, 159–170. [DOI] [PubMed] [Google Scholar]
- Jodice C, Malaspina P, Persichetti F, Novelletto A, Spadaro M, Giunti P, Morocutti C, Terrenato L, Harding AE, and Frontali M (1994). Effect of trinucleotide repeat length and parental sex on phenotypic variation in spinocerebellar ataxia I. Am J Hum Genet 54, 959–965. [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjomsson S, Huttenlocher J, Levey AI, Lah JJ, et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Kokubu H, and Lim J (2014). Beyond the glutamine expansion: influence of posttranslational modifications of ataxin-1 in the pathogenesis of spinocerebellar ataxia type 1. Mol Neurobiol 50, 866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, and De Strooper B (2016). The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem 139 Suppl 2, 237–252. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Suenaga M, Watanabe H, and Sobue G (2009). Cognitive impairment in spinocerebellar degeneration. Eur Neurol 61, 257–268. [DOI] [PubMed] [Google Scholar]
- Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, et al. (2006). Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15, 2125–2137. [DOI] [PubMed] [Google Scholar]
- Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, and Kempermann G (2010). Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One 5, e8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung HY, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, et al. (2014). Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat Neurosci 17, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol LF, Budding D, Andreasen N, D'Arrigo S, Bulgheroni S, Imamizu H, Ito M, Manto M, Marvel C, Parker K, et al. (2014). Consensus paper: the cerebellum's role in movement and cognition. Cerebellum 13, 151–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, et al. (2006). ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347. [DOI] [PubMed] [Google Scholar]
- Lee Y, Fryer JD, Kang H, Crespo-Barreto J, Bowman AB, Gao Y, Kahle JJ, Hong JS, Kheradmand F, Orr HT, et al. (2011). ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell 21, 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YI, Li Y, Mikesh M, Smith I, Nave KA, Schwab MH, and Thompson WJ (2016). Neuregulin1 displayed on motor axons regulates terminal Schwann cell-mediated synapse elimination at developing neuromuscular junctions. Proc Natl Acad Sci U S A 113, E479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, and Zoghbi HY (2008). Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RY, Figueroa KP, Pulst SM, Perlman S, Wilmot G, Gomez C, Schmahmann J, Paulson H, Shakkottai VG, Ying S, et al. (2016). Depression and clinical progression in spinocerebellar ataxias. Parkinsonism Relat Disord 22, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, et al. (2001). Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4, 231–232. [DOI] [PubMed] [Google Scholar]
- Ma L, Wu Y, Qiu Q, Scheerer H, Moran A, and Yu CR (2014). A developmental switch of axon targeting in the continuously regenerating mouse olfactory system. Science 344, 194–197. [DOI] [PubMed] [Google Scholar]
- Manto M, and Marien P (2015). Schmahmann's syndrome - identification of the third cornerstone of clinical ataxiology. Cerebellum Ataxias 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla A, Roberson ED, Banff S, Morales J, Armstrong DL, Burright EN, Orr HT, Sweatt JD, Zoghbi HY, and Matzuk MM (1998). Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci 18, 5508–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, and Llorens-Martin M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- Moriarty A, Cook A, Hunt H, Adams ME, Cipolotti L, and Giunti P (2016). A longitudinal investigation into cognition and disease progression in spinocerebellar ataxia types 1, 2, 3, 6, and 7. Orphanet J Rare Dis 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr., Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, and Zoghbi HY (1993). Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4, 221–226. [DOI] [PubMed] [Google Scholar]
- Ou-Yang MH, Kurz JE, Nomura T, Popovic J, Rajapaksha TW, Dong H, Contractor A, Chetkovich DM, Tourtellotte WG, and Vassar R (2018). Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, et al. (2012). High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. [DOI] [PubMed] [Google Scholar]
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D'Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, et al. (2013). Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci 33, 12915–12928, 12928a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapaksha TW, Eimer WA, Bozza TC, and Vassar R (2011). The Alzheimer's beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener 6, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux MWC, Tschumperlin T, Lu HC, Lackey EP, Bondar VV, Wan YW, Tan Q, Adamski CJ, Friedrich J, Twaroski K, et al. (2018). ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 97, 1235–1243 e1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Burk K, Timmann D, den Dunnen W, Seidel K, Farrag K, Brunt E, Heinsen H, Egensperger R, Bornemann A, et al. (2012). Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol Appl Neurobiol 38, 665–680. [DOI] [PubMed] [Google Scholar]
- Sadleir KR, Eimer WA, Cole SL, and Vassar R (2015). Abeta reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol Neurodegener 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakano H (2010). Neural map formation in the mouse olfactory system. Neuron 67, 530–542. [DOI] [PubMed] [Google Scholar]
- Schafer S, Wirths O, Multhaup G, and Bayer TA (2007). Gender dependent APP processing in a transgenic mouse model of Alzheimer's disease. J Neural Transm (Vienna) 114, 387–394. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, and Sherman JC (1998). The cerebellar cognitive affective syndrome. Brain 121 (Pt 4), 561–579. [DOI] [PubMed] [Google Scholar]
- Smith AS, Williams Avram SK, Cymerblit-Sabba A, Song J, and Young WS (2016). Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol Psychiatry 21, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, et al. (2015). Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 47, 445–447. [DOI] [PubMed] [Google Scholar]
- Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, and Tanzi RE (2013). ADAM10 missense mutations potentiate beta-amyloid accumulation by impairing prodomain chaperone function. Neuron 80, 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J, Lyckman A, Wang L, Eckman EA, and Guenette SY (2011). FE65 proteins regulate NMDA receptor activation-induced amyloid precursor protein processing. J Neurochem 119, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Kim S, Shen L, Risacher SL, Foroud T, Pankratz N, Potkin SG, Huentelman MJ, Craig DW, Weiner MW, et al. (2011). Genomic Copy Number Analysis in Alzheimer's Disease and Mild Cognitive Impairment: An ADNI Study. Int J Alzheimers Dis 2011, 729478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM, Foroud T, Mayeux R, and Saykin AJ (2012). Analysis of copy number variation in Alzheimer's disease: the NIALOAD/NCRAD Family Study. Curr Alzheimer Res 9, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE (2012). The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, Sicinska E, Sulahian R, Goodale A, Cowley GS, et al. (2017). ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep 18, 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, Kano M, Atkinson R, Sun Y, Armstrong DL, et al. (2002). A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34, 905–919. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Meng S, Wang J, Li S, Liu J, Li H, Li T, Song W, and Zhou W (2014). Two novel DNA motifs are essential for BACE1 gene transcription. Sci Rep 4, 6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Orr HT (2000). Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23, 217–247. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, and Orr HT (2009). Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem 284, 7425–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. BACE1 expression in Ataxin-1 KO mice, Related to Figure 1
(A) Densitometric quantification of western blot results for Ataxin-1, ADAM10, APP, APP-CTF and PS1-CTF in WT (+/+), Ataxin-1 hetero-KO (+/−) and KO (−/−) mouse brains. Bottom right, a representative western blot image of PS1-CTF. Full length PS1 was undetectable in these samples. Values are mean ± SEM. Number of mice analyzed, n= 5. ***p < 0.001, t-test. (B) Upper panel, photomicrographs of BACE1-immunolabeled olfactory bulb and forebrain region of WT and Ataxin-1 (ATXN1) KO mice. Arrow indicates glomeruli, which showed strong immunoreactivity for BACE1. Lower, fluorescence photomicrographs of olfactory glomeruli (arrow) region immunolabeled for Ataxin-1 and BACE1. DAPI was used for nuclear staining. Brain sections from BACE1 KO were used as a negative control for the staining. Bar = 100 μm. (C) Fluorescence photomicrographs of hippocampal CA3 region of WT mice immunolabeled for Ataxin-1 (red), BACE1 (green), and DAPI (blue). Bar = 100 μm. (D) Levels of BACE1, sAPPβ and sAPPα in the cortex of WT (+/+), Ataxin-1 KO (−/−) and hetero-KO (+/−) mice. n = 5. ***p < 0.001, **p < 0.01, *p < 0.05. (E) Analysis of sAPPβ (arrow) levels in the cortex and cerebellum of WT and Ataxin-1 KO mice. Bottom, densitometric quantification of the western blot result (n = 3). *p < 0.05. n.s.: not significant. (F) Analysis of Ataxin-1 and BACE1 expression in different organs/tissues. Protein lysates from Ataxin-1 KO or BACE1 KO mice were used as controls for the band specificity. Immunoreactive bands that are in similar size with Ataxin-1 (~90 kDa) appeared in other organs/tissues beyond brain, particularly with a strong intensity in heart same. However, these bands are likely non-specific signals, as they were also detected in the lysates of Ataxin-1 KO samples for the same tissues/organs. (G) Levels of BACE1 in WT, Ataxin-1 hetero-KO and KO embryonic (embryonic day 15, left top), postnatal (P0, right top) and adult mouse brains. Vertical straight lines indicate that different parts within same blot were placed together. Ataxin-1 levels in the embryonic/postnatal brains were lower than those of adult brains. In contrast, BACE1 levels were much higher in the embryonic/postnatal brains. Bottom panel, no effect of Ataxin-1 depletion on BACE1 levels in cultured cortical neurons (DIV10) and embryonic fibroblasts that are derived from Ataxin-1 KO mouse embryos.
Figure S2. Increased Bace1 transcription in the cortex of Ataxin-1 KO mice, Related to Figure 2
(A) Actinomycin D or vehicle (DMSO) was treated for 24 or 48 hrs in acute brain slice cultures derived from WT and Ataxin-1 KO mice. Bace1 mRNA amount per each brain slice was analyzed. Gapdh was used as an internal control for RT-qPCR. Same RT-qPCR data from 48 hr-cultured slices were presented as Bace1 mRNA/total RNA in Figure 2A. n (brain slices) = 2 (24 hr, WT and ATXN1 KO), 3 (48 hr, WT), 4 (48 hr, ATXN1 KO). *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (B) RorαmRNA levels in the cortex of WT and Ataxin-1 KO mice. Levels of Gfi1 mRNA were too low to be reliably analyzed by RT-qPCR. (C) mRNA levels of Etv1, Etv4, and Etv5 in the cortex and cerebellum of WT and Ataxin-1 KO mice. Expression levels were presented as relative values to WT group (100) for each gene. Number of mice analyzed, n = 6 (cortex), 4 (cerebellum). (D) Protein expression levels of ETV4 and ETV5 in pLX_TRC311, pLX_TRC311 ETV4-L, or pLX_TRC311 ETV5-transfected HEK293T cells. (E) Luciferase reporter activity in HEK293T cells transfected with pGL3-BACE1 promoter vector. A conserved ETV4/ETV5 binding site in BACE1 promoter was mutated from CCGGAA to CTATGA or deleted. Results from 2 independent experiments of sextuplicate transfection were normalized and combined. n = 12 (2 × 6). (F) BACE1 levels in the brains of Bace1+/− (39%) as compared to WT mice. (G) Immunohisotochemical staining for BACE1 in the cortical and hippocampal regions of WT, Atxn1−/− and Atxn1−/−; Bace1+/− mice. Bar = 500 μm.
Figure S3. Amyloidogenic processing of APP in APP-PS1/ATXN1-KO mouse brains, Related to Figure 3
(A) Expected and actual genotype ratios of APP-PS1 and APP-PS1/ATXN1-KO mice at 4 weeks. (B) Densitometric quantification of western blot analysis of BACE1, ADAM10, sAPPβ (transgenic) and sAPPα (transgenic) in the brains of male and female APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). Numbers in parenthesis denote the number of mice analyzed. **p < 0.01, ***p < 0.001, t-test. (C) Endogenous sAPPβ levels in APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). Arrow, endogenous sAPPβ; *, non-specific band. n = 7. **p < 0.01. (D) TBS-soluble Aβ40 and Aβ42 levels in the brains of 5-month-old male and female APP-PS1 (WT) and APP-PS1/ATXN1-KO mice (KO). *p < 0.05. (E) Longer film exposure of western blot image (shown in Figure 3G) probed with 6E10 antibody revealed APP-CTFβ at ~10 kDa.
Figure S4. Effects of Ataxin-1 depletion on Aβ plaque deposition and gliosis, Related to Figure 4
(A) Quantification of area covered by Aβ plaques in the cortex and hippocampus of 5-month-old male and female APP-PS1/WT, APP-PS1/Atxn1+/− and APP-PS1/Atxn1−/− mice. Number in parenthesis denotes number of mice analyzed. Data for APP-PS1/WT and APP-PS1/Atxn1−/− mice are same as shown in Figure 4B. *p < 0.05, ***p < 0.001, t-test. (B) Hippocampal regions of APP-PS1 and APP-PS1/ATXN1-KO mice immunolabeled with 3D6 Aβ (red) antibody followed by staining with thioflavin S (green). Most of the plaques in both genotypes were thioflavin S-positive dendritic plaques. Bar = 200 μm. (C) Correlation between Aβ plaque load and GFAP+ astrocyte activation. (D) Frontal cortex of 5-month-old WT and Ataxin-1 KO mouse brains (without APPswe/PS1ΔE9 overexpression) immunolabeled for GFAP. Bar = 500 μm. Inset, high resolution image. No notable change was detected for astrocyte activation in Ataxin-1 KO mice.
Figure S5. Effects of Ataxin-1 depletion on hippocampal neurogenesis, Related to Figure 5
(A) DCX+ immature neurons in the dentate gyrus of 3-month-old WT and Ataxin-1 KO mice (without APP-PS1 overexpression). Brackets: granular cell layer. Arrows, DCX+ neurons. Right, counts of DCX+ cells in dentate gyrus. n = 3. **p < 0.01. Bar = 100 μm. (B) Impaired dendrite projection of newborn neurons in APP-PS1/Atxn1+/− mice. Left: counts of DCX+ and projecting DCX+ cells per dentate gyrus section. Right, ratio of dendrite projection of each DCX+ neuron. For a better comparison, all brain sections from three different genotypes were stained together for DCX. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (C) Number of Golgi-stained granular neurons per dentate gyrus section (left) and number of dendrites per each granular neuron (right) in the dentate gyrus of WT and Ataxin-1 KO mice. Number of mice analyzed, n = 5 (WT), 4 (ATXN1 KO). Three brain sections per mouse were used for counting Golgi-stained granular neurons in the dentate gyrus. Twenty granular neurons per section (total 60 neurons per mouse) were used for counting the number of dendrites per neuron. n.s., not significant. (D) Photomicrographs of Ki-67+ cells (arrows) in the dentate gyrus of 4-month-old WT, Atxn1−/−, Atxn1−/−; Bace1+/− mice. Bar = 100 μm.
Figure S6. Elevated BACE1 levels and aberrant axonal targeting in CA2 regions of Ataxin-1 polyQ expansion mutant mice, Related to Figure 7
(A) Immunohistochemical staining of 1.5-, 4.5-, and 8-month-old WT and Atxn1154Q/+ mouse brains for BACE1. Modest increase of BACE1 immunoreactivity in the cortex (*) and hippocampus of 4.5- and 8-month-old Atxn1154Q/+ mice as compared to age-matched WT mice. Bar = 1 mm. (B) RT-qPCR analysis of Bace1 mRNA levels in the hippocampus of 1.5- and 7–8.5 month-old mice. Number of mice analyzed, n = 4 – 9 (in bar). Gapdh was used as an internal control. Relative value of 100 is given to WT at each age group. (C) CIC levels (long and short forms) in the cortex. n.s., non-specific bands at ~ 80kDa. These bands were observed in the same blot for CIC and are shown as loading controls. Bottom: densitometric quantification of the CIC western blot result. (D) Quantification of BACE1 immunoreactivity intensity at the end of stratum lucidum. Number in bar denotes the number of brain sections analyzed. One or 2 sections/mouse. **p < 0.01, ***p < 0.001, t-test. (E) Hippocampal CA2 regions of 6-month-old Atxn1154Q/+ and WT mice immunolabeled for synaptoporin. Arrows, synaptoporin+ presynaptic terminals within hippocampal neuronal layer (dashed lines). *, synaptoporin+ presynaptic terminals of granular neurons at striatum lucidum. Bar = 100 μm. Right: counts of synaptoporin+ presynaptic terminals within CA2 neuronal layer. Number of mice analyzed, n = 3. One or 2 brain sections were analyzed for each mouse.
Figure S7. Degeneration of CA2 neurons and impaired hippocampal neurogenesis in SCA1 mice. Related to Figure 7
(A-C) Representative images for neuronal cell counting in CA1/CA2/CA3 regions of hippocampus of 6-9 month-old SCA1 (Atxn1 154Q/+) and age-matched WT mice immunolabeled for PCP4 and NeuN: yellow box for PCP4+ neurons in CA2, white box for NeuN+ neurons in CA1, and red box for NeuN+ neurons in CA3. NeuN+ neurons in CA2 were counted with photomicrographs without PCP4 signal. Arrows indicate ectopic positions of PCP4+ neurons just outside of CA2 region, suggesting overall distribution of PCP4+ neurons are not changed in the hippocampal neuronal layers of SCA1 mice. Bar = 100 μm. Analyzed numbers of mouse/brain section = 6/16. 2–4 sections/mouse. **p < 0.01. For better comparison among different mice and genotypes, sagittal brain sections that are between 1 to 2 mm away from the brain midline, which contain full hippocampal formation, were selected for the neuronal counting and quantification. (D) z-stacked images of 40 μm-thick brain sections labeled for NeuN. Arrow indicates thinner CA2 neuronal layer in SCA1 mice. Bar = 100 μm. (E) High resolution images of CA2 neurons labeled with NeuN antibody and DAPI. Arrows indicate abnormally stretched neuronal cell bodies and nuclei in the SCA1 mice. Bar = 20 μm. Photomicrographs (F) and counting (G) of PCP4+ neurons in CA2 regions of 1.5-, 4.5-, and 8-month-old Atxn1154Q/+ and WT mice. Bracket, CA2 region; *, axons of granular neurons; arrow heads, PCP4+ Purkinje cells in the cerebellum of 8-month-old mice. In contrast to CA2 neurons, majority of Purkinje cells of Atxn1154Q/+ mice expressed high levels of PCP4 at this age. Bar = 100 μm. Number in bar, number of brain sections analyzed. 1–3 sections/mouse. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. (H) Nissl staining revealing less populated neurons in the CA2 of SCA1 mice (arrow in high resolution image). Bar = 100 μm. (I) Widths of neuronal layers (area divided by length) in hippocampus stained with Nissl. Number of mice/brain sections analyzed: WT (4/12), SCA1 (3/8). (J) Quantification of RGS14 immunoreactivity in hippocampal CA2 of 6-month-old Atxn1154Q/+ and WT mice. Number of mice analyzed, n = 3. (K) Hippocampal region of 6-months-old SCA1 and WT and mice immunolabeled for STEP, another CA2 specific marker. Arrows indicate complete disappearance of STEP immunoreactivity in the CA2 neurons of SCA1 mice. Bar = 100 μm. (L) Hippocampal region of 7-month-old Ataxin-1 KO and WT mice immunolabeled for PCP4. (M) Photomicrographs and counts of Ki-67+ cells (arrows) in the dentate gyrus of 6 month-old Atxn1154Q/+ and WT mice. Number of mice analyzed, n = 3.
Data Availability Statement
This study did not generate dataset/code. All algorithms and software used in this study are publicly available and links are provided in the corresponding sections of the methods.