

Abstract
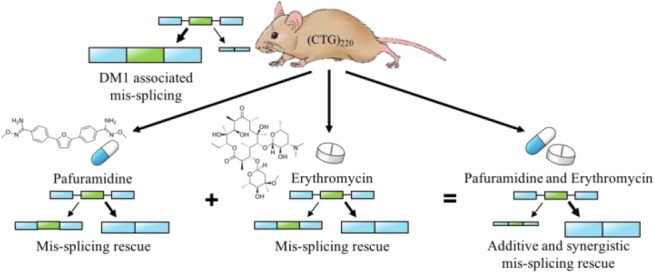
Myotonic dystrophy type 1 (DM1) is a multisystemic disease that presents with clinical symptoms including myotonia, cardiac dysfunction, and cognitive impairment. DM1 is caused by a CTG expansion in the 3′ UTR of the DMPK gene. The transcribed expanded CUG-repeat RNA sequester the muscleblind-like (MBNL) and up-regulate the CUG-BP Elav-like (CELF) families of RNA-binding proteins leading to global mis-regulation of RNA processing and altered gene expression. Currently, there are no disease-targeting treatments for DM1. Given the multistep pathogenic mechanism, combination therapies targeting multiple aspects of the disease mechanism may be a viable therapeutic approach. Here, as proof-of-concept, we studied a combination of two previously characterized small molecules, erythromycin and furamidine, in two DM1 models. In DM1 patient-derived myotubes, the rescue of mis-splicing was observed with little to no cell toxicity. In a DM1 mouse model, a combination of erythromycin and the prodrug of furamidine (pafuramidine), administered orally, displayed both additive and synergistic mis-splicing rescue. Gene expression was only modestly affected, and over 40% of the genes showing significant expression changes were rescued back toward WT expression levels. Further, the combination treatment partially rescued the myotonia phenotype in the DM1 mouse. This combination treatment showed a high degree of mis-splicing rescue coupled with low off-target gene expression changes. These results indicate that combination therapies are a promising therapeutic approach for DM1.
Keywords: myotonic dystrophy, toxic RNA, combination therapeutic, furamidine, erythromycin
Introduction
Myotonic dystrophy type 1 (DM1) is the most prevalent form of adult onset muscular dystrophy. It is a multisystemic disease that presents with clinical symptoms including myotonia, muscle weakness and wasting, cardiac dysfunction, digestive issues, cataracts, insulin resistance, and cognitive impairment.1 DM1 is caused by an expansion of a CTG repeat tract in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene, which gives rise to CUG-repeat RNA with a toxic gain-of-function.2−4 These repeat RNAs are retained in the nucleus and form ribonuclear foci that disrupt the normal functions of RNA-binding proteins belonging to the muscleblind-like (MBNL) and the CUG-BP Elav-like (CELF) families.5−8 The sequestration of MBNL proteins and the upregulation of CELF proteins cause changes in alternative splicing, translation, polyadenylation, microRNA processing, and mRNA localization.9−12 Subsequently, expression of CUG-repeat RNA leads to a developmental remodeling of the transcriptome, primarily through abnormal alternative splicing and altered gene expression.13 Abnormal regulation of alternative splicing is a molecular hallmark of DM1 and can be linked directly to multiple disease symptoms, such as mis-splicing of muscle-specific chloride channel (CLCN1), cardiac troponin T (TNNT2) and insulin receptor (INSR) mRNAs resulting in myotonia, cardiac defects, and insulin resistance, respectively.6,14,15 Further, the DM1 repeats have been shown to undergo bidirectional transcription and repeat-associated non-ATG (RAN) translation, which may have roles in disease pathogenesis.16,17
There are currently no disease-targeting treatments available for DM1; however, several therapeutic strategies have been developed to ameliorate the effects of toxic RNA in DM1 models. These strategies include genome editing using CRISPR/Cas9 to eliminate the expanded CTG repeats or to insert polyadenylation signals upstream of the CTG repeats resulting in reduced expression of CUG repeat containing transcripts,18−20 inhibiting transcription from the CTG repeats using deactivated Cas9/CRISPR,21 degrading the CUG-repeat RNA or disrupting the MBNL-CUG RNA interaction using antisense oligonucleotides (ASOs), CRISPR/Cas9, siRNAs, miRNAs, ribozymes, and peptides,22−28 and increasing MBNL levels via exogenous expression.29 Therapeutic approaches based upon small molecules, which have recently been developed, offer several advantages over other therapeutic strategies. Small molecules can be administered orally and generally have better tissue delivery with shorter half-lives, making rapid reversal of treatment easy in the case of toxicity. Small molecules typically have longer shelf lives than biologics, lower costs associated with manufacturing, opportunities for repurposing, and most notably, are amenable to high-throughput screening and optimization using medicinal chemistry-based approaches. Excitingly, there are a number of small molecules being used to target different aspects of the DM1 disease mechanism. Small molecules are being used to modulate the production or stability of the CUG repeats.30−34 Several small molecules have been found to interrupt the MBNL-CUG RNA interaction or up-regulate MBNL protein levels.35−37 There are also small molecule inhibitors that target specific kinases, such as GSK3B and H-Ras, that have been shown to rescue molecular markers of DM1.38,39
Considering that DM1 has a complex pathogenic mechanism, we hypothesized that small molecules administered in combination could be used to target different or multiple aspects of the disease mechanism with improved activity compared to the compounds administered individually. Combination therapies are commonly used to treat infectious diseases such as tuberculosis, leprosy, malaria, and most notably, HIV/AIDS, as they reduce the risk of development of drug resistance.40−43 By the same token, combination therapies have been used to treat various types of cancer, as tumors are less likely to have resistance to multiple drugs simultaneously.44 Further, combination treatments have been suggested as a potential therapeutic strategy to treat Alzheimer’s.45 The use of combination therapies in other diseases motivated us to test this approach for DM1.
Two promising small molecules that have been studied as potential therapeutics for DM1 are erythromycin, an FDA-approved antibiotic, and furamidine, a trypanocide agent.46−49 The prodrug of furamidine, pafuramidine, went through phase III clinical trials to treat African sleeping sickness.49,50 Previously, erythromycin was shown to bind fluorescein-labeled (CUG)100 RNA in a fluorescence titration assay and interrupt the MBNL-CUG RNA interaction via an electrophoretic mobility shift assay (EMSA).46 It reduced ribonuclear foci and reversed mis-splicing events in cell models of DM1. Furthermore, erythromycin administrated orally or through intraperitoneal injection rescued mis-splicing and improved myotonia in a DM1 mouse model. It was proposed that erythromycin rescued molecular phenotypes of DM1 by disrupting the MBNL-CUG RNA interaction via binding the CUG-repeat RNA, thereby releasing sequestered MBNL proteins. Furamidine rescued DM1-associated mis-splicing in patient-derived myotubes and a mouse model of DM1.47 Furamidine was shown via EMSA to disrupt the MBNL-CUG RNA complex, suggesting that it reduced CUG ribonuclear foci through this mechanism. Consistent with this model, furamidine was shown, using isothermal calorimetry (ITC), to bind CUG RNA. Furamidine was also recently shown to bind expanded CAG repeat RNA with similar affinity to CUG RNA.51 Through a currently unknown mechanism, furamidine was shown to up-regulate MBNL1 and MBNL2 protein levels in DM1 myotubes. RT-qPCR data showed that furamidine reduced CUG-containing transgene transcript levels in a DM1 mouse model when administered via intraperitoneal injection. Furamidine is purposed to work through multiple mechanisms to alleviate the molecular phenotypes of DM1: inhibition of the MBNL-CUG RNA interaction, upregulation of MBNL1 and 2 proteins, and potentially affect the transcription and/or stability of CUG RNA. Here, as a proof-of-concept, we used a combination of erythromycin and furamidine in two different DM1 models to determine if greater mis-splicing rescue could be achieved with the combination versus either drug alone.
In DM1 patient-derived myotubes, we observed additive mis-splicing rescue and no cell toxicity using combination treatments in the nanomolar (nM) concentration range for furamidine and in the micromolar (μM) concentration range for erythromycin. These combination treatments also reduced CUG ribonuclear foci and up-regulated MBNL1 and MBNL2 transcript and protein levels. Global analysis of splicing using RNA-seq in the DM1 patient-derived myotubes showed that the combination treatment rescued nearly three times the number of mis-splicing events compared to furamidine or erythromycin treatment alone. In the HSALR DM1 mouse model, a transgenic mouse expressing the human skeletal actin (HSA) gene with approximately 220 CUG repeats,52 we orally administered a combination of erythromycin and the methoxyamidine prodrug of furamidine known as pafuramidine. RT-PCR and RNA-seq were used to assess the combination’s activity on splicing and gene expression compared to that of HSALR mice treated with either drug alone. The combination treatment rescued more than twice the number of total mis-splicing events compared to either drug alone with an average percent rescue of ∼73% and displayed both additive and synergistic mis-splicing rescue. Minimal effects on gene expression were observed with the combination treatment. Notably, over 40% of the differentially expressed genes with the combination treatment in the HSALR mice were rescued, to varying degrees, back toward wild-type (WT) expression levels. Further, the combination showed a higher degree of mis-splicing rescue and lower differential gene expression changes than a higher dose of pafuramidine alone in the HSALR mouse. The combination treatment also reduced ribonuclear foci abundance, reduced HSA transgene transcript levels, and increased MBNL protein levels. Increased expression of the chloride channel, Clcn1, and improved myotonia were also observed in the HSALR mice. These results indicate that combination therapies are a promising therapeutic approach for DM1 and support targeting multiple aspects of the DM1 disease pathway to alleviate the molecular phenotypes of DM1.
Results
Combination of Erythromycin and Furamidine Displayed Additive Mis-splicing Rescue in DM1 Patient-Derived Myotubes with No Cell Toxicity
We had previously used myoblast lines derived from a non-DM individual (DM-04) and a DM1 patient (DM-05) containing approximately 2900 CTG-repeats to determine the mechanism by which furamidine rescued mis-splicing.47,53 These same cell lines were used to determine if a combination of erythromycin and furamidine would be more effective in rescuing mis-splicing than either drug alone. Using concentration ranges based upon previously published work with furamidine and erythromycin,46,47 we tested 0.25–1 μM furamidine in combination with 25–100 μM erythromycin. After the myoblasts were differentiated to myotubes for 7 days, drug treatments were carried out for 4 days. To assess the effect of combination treatments on endogenous splicing events, RT-PCR analysis was performed for the exon-skipping (ES) events MBNL1 exon5, MBNL2 exon5, NUMA1 exon2, and SYNE1 exon137. These events were previously shown to have consistent differential splicing between the non-DM control and DM1 myotubes.47 The difference in the percent spliced in (ΔPSI) for each treatment was calculated by taking the difference in inclusion of the exon of interest between the non-DM myotubes and DM1 myotubes with and without treatment. Figure 1 displays the absolute value of the ΔPSI (|ΔPSI|) for each ES event with treatments using 0.25, 0.5, 0.75, and 1 μM furamidine in combination with 25 and 50 μM erythromycin. The closer the value is to zero, the greater is the mis-splicing rescue, meaning the PSI with treatment is returning to non-DM inclusion levels. The splicing analysis for the full concentration range tested, including 75 and 100 μM erythromycin, can be found in the Supporting Information, Figure S1. Higher concentrations of erythromycin did not lead to increased mis-splicing rescue.
Figure 1.

Combination of furamidine and erythromycin displays an additive rescue of mis-splicing in DM1 patient-derived myotubes. Absolute value of the percent spliced-in difference between non-DM control and DM1 myotubes (|ΔPSI|) with and without treatment determined via RT-PCR. Cells were treated with either furamidine (FM) alone or erythromycin (EM) or a combination of both. (A) MBNL1 exon5, (B) MBNL2 exon5, (C) NUMA1 exon2, and (D) SYNE1 exon137 events all displayed additive mis-splicing rescue after 4 days of treatment with the combination. Treatment concentrations of 0.5 μM FM alone, 25 μM EM alone, and the combination at those concentrations with the mean % rescue ± standard deviation above are denoted with arrows for each event.
Notably, the combination treatments displayed additive mis-splicing rescue. For example, the MBNL1 exon5 event showed a 24 ± 4% rescue with 0.5 μM furamidine (Figure 1A), where percent rescue is the difference in exon inclusion levels between the untreated and treated DM1 myotubes divided by the difference between the non-DM myotubes and untreated DM1 myotubes multiplied by 100 (eq 1 in Materials and Methods). With 25 μM erythromycin, the MBNL1 exon5 event showed a 26 ± 2% rescue (Figure 1A). Consistent with additive mis-splicing rescue, the combination of 0.5 μM furamidine and 25 μM erythromycin showed 54 ± 4% rescue of MBNL1 exon5 (Figure 1A). A similar additive effect was observed for the MBNL2 exon5 event, which displayed a 34 ± 4, 45 ± 1, and 74 ± 4% rescue with 0.5 μM furamidine, 25 μM erythromycin, and the combination, respectively (Figure 1B). At the same treatment concentrations, the NUMA1 exon2 event displayed a 14 ± 3, 18 ± 4, and 36 ± 3% rescue, respectively (Figure 1C), and SYNE1 exon137 event displayed a 32 ± 2, 25 ± 1, and 63 ± 4% rescue, respectively (Figure 1D). Additive rescue was observed for all combination treatments shown in Figure 1; however, 50 μM erythromycin in combination with 0.25–1 μM furamidine did not display a dose dependent increase in mis-splicing rescue. To assess cell toxicity, we performed cell viability studies using an absorbance-based assay. Importantly, all combinations tested displayed little to no cell toxicity in DM1 myotubes (Figure S2). Interestingly, the addition of furamidine at higher erythromycin concentrations increased cell viability and overcame the slight toxicity caused by erythromycin alone. At 50 μM erythromycin, cell viability was reduced to 0.69 ± 9 relative to untreated DM1 myotubes. The addition of 0.25–1 μM furamidine in a combination with 50 μM erythromycin increased cell viability to the same level as untreated DM1 myotubes (Figure S2).
To assess mis-splicing rescue globally, we used RNA-seq to measure the PSI of alternatively spliced cassette exons in non-DM and untreated DM1 myotubes, along with DM1 myotubes treated with 25 μM erythromycin, 0.5 μM furamidine, or the combination treatment. This combination treatment was chosen because these were the lowest concentrations for which a higher degree of mis-splicing rescue and lower cell toxicity relative to either drug alone was observed. Additionally, these treatment concentrations were within the range where dose-dependent mis-splicing rescue was observed. Consistent with our RT-PCR data, ES events MBNL1 exon5, MBNL2 exon5, NUMA1 exon2, and SYNE1 exon137 all showed additive mis-splicing rescue with the combination treatment (Figure S3). Other events that displayed additive mis-splicing rescue included ES events ADD3 exon13 and FN1 exon26 (Figure 2A,B). The ADD3 exon13 event displayed a 40 ± 13, 45 ± 8, and 76 ± 9% rescue with 25 μM erythromycin, 0.5 μM furamidine, and the combination, respectively (Figure 2A). At the same treatment concentrations, the FN1 exon26 event showed a 22 ± 1, 20 ± 0, and 39 ± 1% rescue, respectively (Figure 2B). Further, mis-splicing events related to muscle wasting (BIN1) and insulin resistance (INSR) in DM1 patients were rescued by the combination treatment,6,54 as well as many other MBNL-dependent splicing events, such as MBNL2 exon7 and CLASP1 exon19 (Figure S4).55
Figure 2.

Combination treatment rescues more mis-splicing events in DM1 patient-derived myotubes than either drug alone. Splicing analysis of ES events determined via RNA-seq of non-DM control and DM1 myotubes treated with either 25 μM erythromycin (EM, green) or 0.5 μM furamidine (FM, blue) alone or a combination of both (EM+FM, magenta). The PSI values are shown for (A) ADD3 exon13 and (B) FN1 exon26 events. Mean % rescue ± standard deviation values are displayed below each graph. Both events display additive mis-splicing rescue after 4 days of treatment with the combination. (C) Global analysis of ES events that showed a greater than 10% change in PSI between non-DM control and DM1 myotubes were evaluated for mis-splicing rescue (p < 0.01, FDR < 0.1). The number of ES events that showed mis-splicing rescue (black bar) of > 10%, over-rescue (golden rod bar) of > 110% or mis-rescue (gray bar) of < −10% for each treatment are displayed. The average percent rescue for all “rescued” ES events for a given treatment is displayed above the black bar.
When ES events were compared between non-DM and untreated DM1 myotubes, a total of 1075 ES events were mis-regulated with a greater than 10% change in PSI (p < 0.01, FDR < 0.1); however, not all of these events were validated specifically as DM1-associated mis-splicing events. Of these events, 36 showed at least a 10% rescue with 25 μM erythromycin treatment and 41 events with 0.5 μM furamidine (p < 0.01, FDR < 0.1, Figure 2C). Interestingly, the combination treatment showed a greater than additive number of ES events rescued at 121 events (p < 0.01, FDR < 0.1, Figure 2C). The average percent rescue for all ES events using 25 μM erythromycin, 0.5 μM furamidine, and the combination were ∼61, ∼ 64, and ∼68%, respectively (Figure 2C). In contrast, all treatments caused some of the ES events to be shifted further from control inclusion levels. We classified events that showed a percent rescue of greater than 110% as “over-rescue” and those that had a percent rescue of less than −10% as “mis-rescue” events. Erythromycin caused the over-rescue of 18 events and the mis-rescue of 2 events and furamidine caused 18 events to be over-rescued and 1 event to be mis-rescued (p < 0.01, FDR < 0.1, Figure 2C). The combination treatment caused 36 events to be over-rescued and 21 events to be mis-rescued (p < 0.01, FDR < 0.1, Figure 2C).
The greater than 2-fold number of ES events rescued by the combination versus either drug alone suggested that there could be synergistic effects on mis-splicing rescue with the combination treatment and that it may not be as simple as additive rescue (Figure 2C). To investigate potential synergistic effects, we compared the theoretical percent rescue assuming purely additive effects to the actual percent rescue for the 121 ES events rescued by the combination treatment in the DM1 myotubes. We calculated the theoretical percent rescue by adding the percent rescues of erythromycin and pafuramidine alone. We then compared that to the actual percent rescue observed with the combination treatment. Figure S5 shows a scatter plot of the theoretical percent rescue of erythromycin and furamidine (gray circles) and the actual percent rescue of the combination (magenta squares) ordered by increasing theoretical percent rescue. If purely additive rescue was observed for all 121 ES events, adding the percent rescues of each drug alone would equal the percent rescue with the combination treatment. Many events that were predicted to be over-rescued or mis-rescued by the theoretical percent rescue were actually within the normal range of mis-splicing rescue based upon the actual percent rescue with the combination treatment. These events are represented by the gray circles outside of the dotted lines in Figure S5. Further, there was also a group of events that displayed worse actual rescue than the theoretical percent rescue. These data support the idea that the drug interactions of the combination are more complicated than a simple additive model.
We next defined synergistic rescue as those events having a greater than 30% difference in the percent rescue in the theoretical versus the actual percent rescue of the combination treatment. The actual percent rescue was required to fall outside of the standard deviation. A 30% difference in the percent rescue was selected because this was the first difference for which a deviation from additive rescue was observed. When we assessed events individually, the high error made it difficult to determine if the combination treatment had broad synergistic effects; however, multiple events did fall within our definition of synergy. For example, the DCAF6 event displayed a −4 ± 18, 1 ± 5, and 42 ± 20% rescue with 25 μM erythromycin, 0.5 μM furamidine, and the combination, respectively (Figure S6A), and the AGRN event showed a 74 ± 26, 79 ± 12, and 92 ± 15% rescue, respectively (Figure S6B). Other events such as CDK10 and HOOK3 showed similar trends in rescue (Figure S6C,D). These events suggest that the combination had synergistic effects; however, many events had high error and more precise measurements are required. For instance, the MIS12 event, the combination treatment reduced the slight over-rescue with erythromycin at 108 ± 44% and the over-rescue with furamidine treatment at 137 ± 21% rescue to 80 ± 80% rescue with combination treatment; however, the combination rescue was not statistically significant due to high error (Figure S6E). Further, the SORBS2 event showed a −19 ± 62, −12 ± 76, and 106 ± 15% rescue with erythromycin, furamidine, and the combination, respectively (Figure S6F). This is a greater than 30% difference in the percent rescue of the predicted additive rescue and the actual rescue of the combination treatment with the SORBS2 event, but it did not fall outside of the standard deviation.
Combination Treatment Rescued Gene Expression Changes in DM1 Patient-Derived Myotubes
We had previously shown that furamidine treatment in a DM1 mouse model partially rescued the gene expression changes associated with the expression of CUG repeats,47 so we assessed the degree of gene expression rescue for all drug treatments in the DM1 patient-derived myotubes. Erythromycin and furamidine treatments alone rescued 910 (21% of genes mis-regulated with erythromycin treatment) genes and 853 genes (18% of genes mis-regulated with erythromycin treatment), respectively, by more than 10% back to the non-DM1 expression levels (Table S1). The combination treatment rescued 1275 genes, equating to 14% of genes mis-regulated with the combination treatment. A high proportion of genes were over-rescued and mis-rescued with all treatment at 42, 44, and 44% of genes mis-regulated with erythromycin, furamidine, and the combination treatment, respectively (Table S1). The gene expression changes are much higher in the DM1 myoblasts than previously observed in the DM1 mouse model when treated with furamidine,47 which may be due to the inability of the cell to compensate for slight changes in the cellular environment versus a whole organism.
We also assessed the expression of DMPK via RT-qPCR in the treated DM1 myotubes. We found that furamidine was able to modestly, but significantly, reduce the levels of DMPK transcripts to 0.88 ± 0.02 and 0.91 ± 0.03-fold relative to untreated at 0.5 and 0.75 μM, respectively (Figure S7). Surprisingly, erythromycin treatment reduced the levels of DMPK by approximately 0.5-fold relative to untreated at all concentrations of treatment, including in combination with furamidine (Figure S7). It should be noted that this assay does not differentiate between the normal and CUG-repeat containing DMPK transcripts.
Combination Treatment Did Not Display Additive Effect on Foci Reduction in DM1 Patient-Derived Myotubes
As both furamidine and erythromycin have been shown to compete with MBNL binding to CUG-repeat RNA,46,47 we used RNA fluorescent in situ hybridization (FISH) to assess the impact of the combination treatment on ribonuclear foci formation. The foci number in at least 100 nuclei were counted per treatment, per experiment (blinded). Representative FISH images for treatments in the DM1 myotubes are shown in Figure S8. Furamidine treatment displayed a significant reduction in foci at every concentration tested (Figure S9), with a maximum reduction of approximately 30% from 2.97 ± 0.10 foci per nuclei in untreated DM1 myotubes to 2.06 ± 0.08 observed at 1 μM. Erythromycin treatments also showed significant reductions in foci formation at both concentrations tested. Maximum reduction occurred with 50 μM erythromycin to 2.19 ± 0.02 foci per nuclei (Figure S9). It is important to note that the combination treatments did not result in a significant reduction in foci abundance beyond that of 25 or 50 μM erythromycin treatments alone (Figure S9).
Combination Treatment Increases MBNL1 and MBNL2 Protein Levels in DM1 Patient-Derived Myotubes
Furamidine has been shown to increase MBNL1 and MBNL2 transcript levels with subsequent increases in MBNL1 and MBNL2 proteins within the concentration range used for the combination treatments.47 To confirm that furamidine increased MBNL transcripts and that the addition of erythromycin did not alter this increase, we performed RT-qPCR to measure expression levels of MBNL1 and MBNL2 for all treatments. As expected, furamidine increased MBNL1 and MBNL2 transcripts to similar levels as previously described,47 with levels reaching 1.76 ± 0.21-fold and 1.52 ± 0.12-fold relative to that of untreated, respectively, at 1 μM furamidine (Figure S10A,B). The addition of erythromycin in the combination treatments did not significantly impact the levels of MBNL1 and MBNL2 transcripts. The combination of 1 μM furamidine and 50 μM erythromycin showed levels of MBNL1 and MBNL2 transcripts at 1.80 ± 0.14-fold and 1.70 ± 0.13-fold, respectively, relative to that of untreated (Figure S10A,B). We also confirmed that MBNL1 and MBNL2 protein levels increased in DM1 myotubes with the combination treatments (Figure S11). Furamidine treatments of 0.25, 0.5, and 0.75 μM alone increased levels of MBNL1 and MBNL2 proteins to similar levels as previously described.47 With 0.5 μM furamidine treatment, MBNL1 and MBNL2 levels peaked at 114 ± 2% and 125 ± 8%, respectively, relative to that of untreated DM1 myotubes (Figure S11). Erythromycin treatment alone did not affect MBNL1 protein levels, but did decrease MBNL2 protein levels at 50 μM to 81 ± 4% (Figure S11). All combination treatments showed the same trends in MBNL1 and 2 protein levels as the corresponding furamidine treatment (Figure S11).
Orally Administered Combination Treatment Displayed Additive Mis-Splicing Rescue and Reduced HSA Transgene Levels in HSALR DM1 Mice
Next, we tested the activity of the combination treatment on CUG RNA toxicity in the HSALR DM1 mouse model. These mice express approximately 220 CUG repeats in skeletal muscle in the context of the HSA gene.52 As oral administration of both compounds would be easier for patients and a more viable therapeutic method for DM1, we synthesized the methoxyamidine prodrug version of furamidine known as pafuramidine.56 A description and scheme of the synthesis of pafuramidine can be found in the Supporting Information. After optimization of the oral doses for the combination treatment, we treated HSALR mice with 600 mg kg–1 erythromycin and/or 10 mg kg–1 pafuramidine in corn oil via oral administration for 14 days and used quadriceps muscle for subsequent analyses. Control HSALR mice were treated with corn oil via oral administration and WT FVB mice were used as nondisease controls. RT-PCR splicing analysis for Atp2a1 exon22 yielded percent rescues of 44 ± 15, 57 ± 16, and 84 ± 6% with 600 mg kg–1 erythromycin, 10 mg kg–1 pafuramidine, and the combination, respectively (Figure 3A). Clcn1 exon7a showed percent rescues of 56 ± 9, 73 ± 9, and 98 ± 11%, respectively (Figure 3B). We tested two other well-known MBNL-dependent mis-splicing events, Clasp1 exon13 and Nfix exon7, via RT-PCR. Clasp1 displayed percent rescues of 25 ± 10, 35 ± 17, and 73 ± 24%, respectively (Figure 3C). Nfix showed percent rescues of 40 ± 8, 49 ± 12, and 83 ± 10%, respectively (Figure 3D).
Figure 3.

Combination of erythromycin and pafuramidine rescues multiple mis-splicing events and reduces HSA transgene levels in HSALR DM1 mice. PSI between WT and HSALR mice with and without treatment determined via RT-PCR. Mice were treated with either 600 mg kg–1 erythromycin (EM, green), 10 mg kg–1 pafuramidine (PaF, blue) alone, or a combination of both (EM+PaF, magenta) per oral administration daily for 14 days. (A) Atp2a1 exon22, (B) Clcn1 exon7a, (C) Clasp1 exon13, and (D) Nfix exon7 mis-splicing events all displayed additive rescue with combination treatment. Mean % rescue ± standard deviation values are displayed below each graph. Expression levels in HSALR mice with and without treatment determined via RT-qPCR. PaF (blue) treatment alone and the combination (magenta) reduced (E) HSA transgene levels and did not affect (F) endogenous Dmpk levels, while EM (green) treatment alone did not significantly reduce either. Mean relative expression ± standard deviation values are displayed below each graph.
Previously, furamidine was shown to reduce HSA transcript levels in HSALR mice, possibly through transcription inhibition via binding of CTG-repeat DNA.47 We performed RT-qPCR analysis to assess HSA transgene levels to determine if pafuramidine treatment also reduced CTG-containing transgene transcripts. In line with our previous furamidine findings, pafuramidine significantly reduced HSA transgene levels to 0.68 ± 0.07-fold relative to HSALR control (Figure 3E) and did not affect Dmpk transcript levels (Figure 3F). Interestingly, the combination treatment significantly reduced HSA transgene levels beyond that of pafuramidine alone to 0.48 ± 0.09-fold of HSALR control levels (Figure 3E). Erythromycin treatment did not affect HSA transgene (Figure 3E) or endogenous Dmpk levels (Figure 3F) in the HSALR mice.
RNA-seq analysis was used to assess global mis-splicing rescue achieved by the combination of erythromycin and pafuramidine in the HSALR mouse. Libraries were prepared from the quadriceps muscles of the same WT, control HSALR, erythromycin-treated, pafuramidine-treated, and combination-treated HSALR mice used for the RT-PCR splicing and RT-qPCR expression analyses. Consistent with the RT-PCR analysis, the RNA-seq data showed additive rescue of the mis-splicing of Atp2a1 exon22, Clcn1 exon7a, Clasp1 exon13, and Nfix exon7 events with oral treatment in the HSALR mouse (Figure S12). Many additional events showing mis-splicing rescue were also identified. These included, Dcaf6 exon12 that had mis-splicing rescues of 37 ± 2, 24 ± 21, and 79 ± 17% with 600 mg kg–1 erythromycin, 10 mg kg–1 pafuramidine, and the combination, respectively (Figure 4A). Ldb3 exon11 showed percent rescues of 27 ± 7, 26 ± 16, and 61 ± 17%, respectively (Figure 4B). Mpdz exon27 showed percent rescues of 12 ± 15, 24 ± 24, and 98 ± 22%, respectively (Figure 4C), and Wdr7 exon17 displayed percent rescues of 25 ± 10, 35 ± 17, and 73 ± 24%, respectively (Figure 4D). Further, mis-splicing events related to muscle wasting (Bin1 and Cacna1s)54,57 and two events associated with early splicing changes in DM patients (Camk2b and Ryr1)58 were rescued by oral administration of the combination (Figure S13).
Figure 4.

Combination treatment displays additive mis-splicing rescue in HSALR DM1 mouse model. Splicing analysis of ES events determined via RNA-seq on HSALR mice treated with 600 mg kg–1 erythromycin (EM, green) or 10 mg kg–1 pafuramidine (PaF, blue) or a combination of both (EM+PaF, magenta) per oral administration daily for 14 days. (A) Dcaf6 exon12, (B) Ldb3 exon11, (C) Mpdz exon27, and (D) Wrd7 exon17 mis-slicing events displayed greater rescue with combination treatment. Mean % rescue ± standard deviation values are displayed below each graph.
Combination Treatment Displayed a High Degree of Mis-splicing Rescue and Rescued Gene Expression Changes in HSALR DM1 Mice
Comparison of global ES events between WT mice and HSALR control mice identified a total of 692 events with mis-regulation of a 10% or greater change in PSI (p < 0.01, FDR < 0.1). Not all of these events have been validated as DM1-associated mis-splicing events. Of these events, 64 showed at least a 10% rescue with 600 mg kg–1 erythromycin treatment and displayed an average percent rescue of ∼53% (p < 0.01, FDR < 0.1, Figure 5A). With 10 mg kg–1 pafuramidine, 58 events showed at least a 10% rescue with an average percent rescue of ∼64% (p < 0.01, FDR < 0.1, Figure 5B). As in the DM1 patient-derived myotubes, the combination treatment in the HSALR mice rescued a greater than additive number of ES events at 142 with an average percent rescue of ∼73% (p < 0.01, FDR < 0.1, Figure 5C). We tested a higher dose of pafuramidine alone in the HSALR mice to determine how this higher dose compared to the combination treatment. At a dose of 15 mg kg–1 pafuramidine, 84 events showed at least a 10% rescue with an average percent rescue of ∼70% (p < 0.01, FDR < 0.1, Figure 5D). Erythromycin caused 7 of the 692 ES events to be over-rescued and mis-rescued 1 event with a greater than 10% change in PSI (p < 0.01, FDR < 0.1, Figure 5A). Pafuramidine at 10 mg kg–1 caused over-rescue of 10 events and mis-rescue of 7 events (p < 0.01, FDR < 0.1, Figure 5B). The combination treatment caused 24 events to be over-rescued and 2 events to be mis-rescued (p < 0.01, FDR < 0.1, Figure 5C). The higher dose of pafuramidine caused 13 events to be over-rescued and 2 events to be mis-rescued (p < 0.01, FDR < 0.1, Figure 5D). On the basis of these data, the combination treatment rescued more than an additive number of mis-splicing events and had a higher overall percent rescue than either drug alone, including the higher dose of pafuramidine. However, there was a modest increase in over-rescued mis-splicing events.
Figure 5.

Combination treatment rescues more mis-splicing events than either drug alone and does not globally disrupt gene expression in the HSALR DM1 mouse model. Global analysis of ES events that showed a greater than 10% change in PSI between WT and HSALR mice were evaluated for mis-splicing rescue (p < 0.01, FDR < 0.1) with (A) 600 mg kg–1 erythromycin, (B) 10 mg kg–1 pafuramidine (PaF), (C) the combination (EM+PaF), and (D) 15 mg kg–1 pafuramidine (PaF) per oral administration daily for 14 days. The number of ES events that showed mis-splicing rescue (black bar) of > 10%, over-rescue (golden rod bar) of > 110% or mis-rescue (gray bar) of < −10% for each treatment are displayed. The average percent rescue for all “rescued” ES events for a given treatment is displayed above the black bar. Global differential gene expression analysis determined via RNA-seq for (E) 600 mg kg–1 erythromycin, (F) 10 mg kg–1 pafuramidine (PaF), (G) the combination (EM+PaF), and (H) 15 mg kg–1 pafuramidine (PaF) versus control HSALR mice. The number of genes that showed expression rescue (black bar) of > 10%, over-rescue (golden rod bar) of > 110% or mis-rescue (gray bar) of < −10% for each treatment are displayed. Off-target (light gray bar) are genes that showed a greater than 10% change in expression and are not typically differentially expressed between WT and HSALR mice (p < 0.1). MAplots of differential gene expression analysis determined via RNA-seq for (I) 600 mg kg–1 erythromycin, (J) 10 mg kg–1 pafuramidine (PaF), (K) the combination (EM+PaF), and (L) 15 mg kg–1 pafuramidine (PaF) versus control HSALR mice. Red dots represent gene with significantly altered expression (p < 0.1). Gray dots represent genes that were not significantly differentially expressed.
Next, the RNA-seq data were analyzed to determine the effect of the combination treatment on global gene expression changes versus erythromycin and pafuramidine alone in the HSALR mouse. Erythromycin and 10 mg kg–1 pafuramidine treatments alone only modestly affected gene expression with 0.39% (104 genes) and 0.25% (68 genes) of genes showing significant changes in expression, respectively, versus the HSALR control mice (Figure S14A,B). The combination treatment had a greater impact on gene expression with 2.1% (563 genes) of genes with a significant change in expression levels (Figure S14C). Although there was a more than additive effect on gene expression with the combination treatment, the higher dose of pafuramidine at 15 mg kg–1 had dramatically more gene expression changes than the combination. Pafuramidine at 15 mg kg–1 caused 5.8% (1525 genes) of genes to change significantly in expression versus the HSALR control mice (Figure S14D).
Previously, we had shown that furamidine treatment in the HSALR mouse enabled rescue gene expression changes associated with the expression of CUG repeat RNA,47 so we assessed the degree of gene expression rescue for all drug treatments in this study. Erythromycin and 10 mg kg–1 pafuramidine treatments rescued 43 and 30 genes, respectively, by more than 10% back to the WT expression levels (Figure 5E,F). Notably, 232 of the 563 genes that changed with the combination treatment were rescued by more than 10% back to the expression levels found in the WT mice (Figure 5G). Interestingly, the higher dose of pafuramidine only rescued 304 of the 1525 genes showing significant changes in expression (Figure 5H). Genes that were over-rescued and mis-rescued by more than 10% were also assessed. Erythromycin treatment resulted in the same number of genes being over-rescued and mis-rescued at 10 of the 104 genes differentially expressed (Figure 5E). There were 8 and 2 of the 67 genes differentially expressed with 10 mg kg–1 pafuramidine treatment that were over-rescued and mis-rescued, respectively (Figure 5F). Over-rescued and mis-rescued genes were also identified with the combination treatment, corresponding to 96 and 14, respectively, of the 563 differentially expressed genes (Figure 5G). The higher dose of pafuramidine over-rescued and mis-rescued 140 and 158 genes, respectively (Figure 5H). The off-target genes are those that showed a greater than 10% change in expression and are not typically differentially expressed between WT and HSALR mice (p < 0.1, Figure 5E–H). Erythromycin, 10 mg kg–1 pafuramidine, and the combination had 41, 27, and 221 off-target genes displaying differential expression (Figure 5E–G). The high dose of pafuramidine had over 4 times the number of off-target differentially expressed genes versus the combination at 923 genes (Figure 5H). When corrected for the rescued expression, changes in differential gene expression were reduced to 0.23% (61 genes), 0.14% (37 genes), and 1.24% (331 genes), with erythromycin, 10 mg kg–1 pafuramidine, and combination treatment, respectively (Figure 5I–K). The gene expression changes for 15 mg kg–1 pafuramidine treatment were reduced to 4.6% (1221 genes) after correction for rescue (Figure 5L). Notably, the majority of off-target gene expression changes were modest with less than 2-fold changes in expression. These data show that a higher degree of mis-splicing rescue coupled with fewer differential gene expression changes can be achieved with the combination treatment over higher doses of pafuramidine alone.
Combination Treatment Displayed Synergistic Mis-splicing Rescue in HSALR DM1 Mice
The greater than 2-fold number of ES events rescued by the combination versus either drug alone suggested the possibility of synergistic mis-splicing rescue (Figure 5C). We performed the same analysis as in the DM1 myotubes. We compared the predicted additive percent rescue to the actual percent rescue for the 142 ES events rescued by the combination in the HSALR mice. We calculated the predicted additive percent rescue and compared that to the actual percent rescue of the combination. Figure 6A shows a scatter plot of the predicted additive percent rescue of erythromycin plus furamidine (gray circles) and the actual percent rescue of the combination (magenta squares) ordered by increasing predicted additive percent rescue. There were many events rescued by the combination that corresponded to over-rescue or mis-rescue when the predicted additive percent rescue was calculated (gray circles outside of the gray dotted lines in Figure 6A). These events included Tnnt3 and Tpd52l2 (Figure 6A). When these events were individually assessed, the Tnnt3 event displayed a 35 ± 16, −160 ± 1, and 46 ± 17% rescue with 600 mg kg–1 erythromycin, 10 mg kg–1 pafuramidine, and the combination, respectively (Figure 6B). At the same treatment concentrations, the Tpd52l2 event showed a 56 ± 10, 307 ± 0, and 102 ± 14% rescue, respectively (Figure 6C). The same definition of synergistic rescue as described above was used for this analysis. The combination clearly had a synergistic effect on the Tnnt3 and Tpd52l2 events. Further, many more events showed synergistic mis-splicing rescue, especially where the addition of erythromycin was able to overcome over-rescue or mis-rescue caused by pafuramidine treatment, as with Lrch3, Ppp2r5c, and Syne1 events (Figure S15A–C). There were also a few events that had modest over-rescue or mis-rescue with erythromycin treatment and were rescued with the combination treatment, such as the Pyroxd2 event which displayed a 118 ± 0, 66 ± 14, and 109 ± 16% rescue with 600 mg kg–1 erythromycin, 10 mg kg–1 pafuramidine, and the combination, respectively (Figure S15D). In total, 36 of the 142 events rescued with the combination treatment displayed synergism.
Figure 6.

Combination treatment displays synergistic mis-splicing rescue in HSALR DM1 mice. Splicing analysis of ES events determined via RNA-seq on HSALR mice treated with 600 mg kg–1 erythromycin (EM, green) or 10 mg kg–1 pafuramidine (PaF, blue) or a combination of both (EM+PaF, magenta) per oral administration daily for 14 days. (A) Scatter plot displaying the theoretical percent rescue based upon a purely additive effect by adding the percent rescues of erythromycin and pafuramidine alone (gray circles) and the actual percent rescue with the combination treatment (magenta squares) for the 142 ES events rescued by the combination treatment ordered by increasing theoretical percent rescue. Gray dotted lines mark 10–110% rescue. The arrows denote the (B) Tnnt3 and (C ) Tpd52l2 mis-splicing events which displayed synergistic rescue with combination treatment. Mean % rescue ± standard deviation values are displayed below each graph.
Combination Treatment Decreased Ribonuclear Foci and Increased Mbnl1 and 2 Protein Levels in HSALR DM1 Mouse Model
The expression of CUG repeats in HSALR mice causes them to exhibit several DM1-like characteristics, including ribonuclear foci formation, myotonia, and abnormal muscle histology.52 Both erythromycin and pafuramidine reduced the abundance of ribonuclear foci-positive nuclei in the quadriceps muscle of HSALR mice (Figure 7A). Representative FISH images for treatments in the HSALR mice are shown in Figure 7B. In the 300 nuclei counted for each treatment, erythromycin reduced the percentage of nuclei with foci from 42 ± 2% in untreated HSALR muscle to 35 ± 3%, and pafuramidine reduced foci-positive nuclei abundance to 26 ± 3% (Figure 7A). Similar to that observed in the DM1 myotubes, the combination treatment did not display additive reduction of foci-positive nuclei below that of either drug alone at 27 ± 4% (Figure 7A).
Figure 7.

Combination treatment reduces ribonuclear foci-positive nuclei in the HSALR DM1 mouse model. (A) Quantification of the percent of nuclei with ribonuclear foci in quadriceps muscle of HSALR mice determined via in situ hybridization of CUG-repeat RNA. A reduction in the percent of nuclei with ribonuclear foci was observed with treatments of 600 mg kg–1 erythromycin (EM, green) or 10 mg kg–1 pafuramidine (PaF, blue) or a combination of both (EM+PaF, magenta) per oral administration daily for 14 days. Mean percent of nuclei with ribonuclear foci ± standard deviation values are displayed below each graph. (B) Fluorescent in situ hybridization microscopy in quadriceps muscle of untreated (HSALR), erythromycin (HSALR + EM), pafuramidine (HSALR + PaF), and combination-treated (HSALR + EM + PaF) HSALR mice against CUG RNA using a Cy3-(CAG)8 probe (red). DAPI shown in blue. Larger box inset in lower right hand corner is magnification of smaller boxed region to clearly show foci.
As Mbnl protein levels can affect foci formation,59,60 we also evaluated the levels of Mbnl1 and Mbnl2 transcripts with each treatment relative to that of untreated HSALR controls using the RNA-seq data (Figure S16). The transcript levels of Mbnl1 were not significantly changed with erythromycin, pafuramidine, or combination treatment (Figure S16A). Mbnl2 levels were increased to 1.34 ± 0.05, 1.46 ± 0.17, and 1.68 ± 0.23-fold with erythromycin, pafuramidine, and combination treatment, respectively (Figure S16B). When protein levels were measured via Western blot, Mbnl1 protein levels were increased to 109 ± 9, 125 ± 11, and 144 ± 15% relative to that of HSALR control mice with erythromycin, pafuramidine, and combination treatment, respectively (Figure S17A). Mbnl2 protein levels were not affected by erythromycin treatment; however, Mbnl2 levels increased to 120 ± 11 and 113 ± 5% relative to that of HSALR control mice with pafuramidine and the combination treatment, respectively (Figure S17B).
Combination Treatment Partially Rescued Myotonia Phenotype in HSALR DM1 Mouse Model
Mis-splicing of the voltage-dependent chloride channel, Clcn1, in skeletal muscle has been shown to cause the myotonia in DM1.14 Because we achieved significant rescue of Clcn1 exon7a with the combination treatment (Figure 3B), we graded the severity of myotonia in HSALR mice using electromyography. In the control HSALR mice, we observed grade 3 myotonia in the quadriceps muscle, indicating frequent repetitive discharges with nearly all electrode insertions (Figure 8A). When treated with erythromycin or pafuramidine alone, the myotonia decreased slightly, displaying a mix of grade 2 and 3, for which grade 2 is myotonic discharge in > 50% but not in 100% of the insertions (Figure 8A). The combination reduced all mice tested to grade 2 (Figure 8A). Further, we performed immunofluorescent (IF) staining against the chloride channel to determine if the combination treatment displayed increased expression of Clcn1. Indeed, versus untreated control HSALR mice, increased staining for Clcn1 at the membrane was observed in the quadriceps muscle of HSALR mice treated with either erythromycin or pafuramidine (Figure 8B). The combination treatment showed slightly increased staining for Clcn1 over that of erythromycin and pafuramidine alone (Figure 8B), which corresponds with the increased mis-splicing rescue of Clcn1 exon7a and myotonia rescue with the combination treatment.
Figure 8.

Combination treatment partially rescues myotonia in the HSALR DM1 mouse model. (A) Myotonia in the quadriceps muscle of HSALR mice treated with erythromycin (EM, green), pafuramidine (PaF, blue), or the combination (EM+PaF, magenta) determined via electromyography. (B) Immunofluorescence in quadriceps muscle of untreated (HSALR), erythromycin (HSALR + EM), pafuramidine (HSALR + PaF), and combination-treated (HSALR + EM + PaF) HSALR mice against chloride channel, Clcn1 (green). DAPI shown in blue.
Discussion and Conclusions
Considering the multistep nature of the DM1 pathogenic mechanism, the idea of combination therapies to target multiple aspects of the disease mechanism based upon small molecules, or other lead therapeutic strategies, is a viable therapeutic approach. Here, as proof-of-concept, we studied a combination of two previously characterized small molecules, erythromycin and furamidine, in two different DM1 models to determine the potential of the combination approach for DM1 and if this approach was better or worse compared to an individual compound.
Demonstrating additive mis-splicing rescue, and suggesting synergistic effects, the combination treatment rescued a greater than 2-fold number of events compared to either drug alone in both DM1 patient-derived myotubes and DM1 HSALR mouse model. Among the mis-splicing events rescued by the combination in DM1 myotubes are events correlated to muscle wasting (BIN1) and insulin resistance (INSR) in DM1 patients, as well as many other MBNL-dependent splicing events, such as MBNL1 exon7 and CLASP1 exon19.6,7,55 The RT-PCR and RNA-seq splicing analysis was tested in the one non-DM control versus one DM1 myoblast line containing ∼2900 CTG repeats to which we had access; however, it would be interesting to assess the effect of the combination treatment in multiple patient derived cell lines with varying repeat lengths. In the HSALR mouse model, events related to muscle wasting (Bin1 and Cacna1s) and two common events associated with early changes in DM patients (Camk2b and Ryr1) were partially rescued with oral administration of the combination of erythromycin and pafuramidine.54,57,58 Further, rescue of the Clcn1 exon7a mis-splicing event with the combination treatment increased expression of the chloride channel in the quadriceps muscle of HSALR mice, resulting in partial rescue of the myotonia phenotype.
We hypothesized that using small molecules targeting multiple aspects of the DM1 disease mechanism would result in greater mis-splicing rescue than either drug alone. Erythromycin was previously shown to rescue DM1-associated mis-splicing by competing with MBNL binding to the CUG-repeat RNA.46 Furamidine was proposed to work through a three-pronged mechanism: reduction of CUG repeats via transcription inhibition and/or stability, inhibition of the MBNL1-CUG complex and up-regulation of MBNL protein levels.47 As both compounds were shown to interrupt MBNL1-CUG complex formation, it was interesting that an additive reduction in ribonuclear foci with the combination treatment was not observed in either DM1 model, suggesting that the combination appears to work through multiple mechanisms to rescue mis-splicing. Driven by furamidine, the combination treatment increased MBNL1 and MBNL2 protein levels in both models, which likely contributed to the observed mis-splicing rescue. Further, the combination treatment significantly reduced HSA transgene levels, and therefore the load of CUG repeats, beyond that of pafuramidine alone. These data are consistent with the model that these compounds work through multiple mechanisms; 1) reduction of CUG repeats, 2) inhibition of the MBNL1-CUG complex, and 3) up-regulation of MBNL protein levels. This combination rescued more mis-splicing in these two DM1 models than either compound alone.
The rescue of mis-splicing events defined as synergistic (36 of the 142) in the HSALR mouse are interesting because many of these events were over-rescued or mis-rescued in the presence of one molecule (primarily pafuramidine), and the addition of erythromycin rescued these events. Our current working model for this effect is that the greater than 50-fold addition of erythromycin displaced pafuramidine from its off-target binding sites. These binding sites could include sites on the pre-mRNAs that were over- and mis-rescued, and the binding of erythromycin does not have the same impact as that of pafuramidine on splicing. Testing this model is beyond the scope of this work, but will be of interest in future studies.
Identifying concentration windows in which target engagement is maximized and off-target effects are minimized is a primary concern in the development of small molecule therapeutics. Here, we showed that using a higher dose of pafuramidine alone, at 15 mg kg–1, had a 4-fold increase in differential gene expression changes and a much lower proportion of gene expression rescue versus the combination of 600 mg kg–1 erythromycin and 10 mg kg–1 pafuramidine via oral administration in the HSALR mouse model. Further, 15 mg kg–1 pafuramidine rescued only 84 ES skipping events versus the 142 events rescued by the combination treatment. This combination treatment represents the highest degree of mis-splicing rescue coupled with the lowest off-target gene expression changes compared to all other small molecules that we have tested globally in the HSALR mouse model.30,31,47 These data support the idea that a higher degree of mis-splicing rescue with fewer off-target gene expression changes can be achieved with the combination treatment compared to individual treatments using the same small molecules.
Currently, erythromycin is an FDA-approved antibiotic, and the prodrug of furamidine, pafuramidine, previously advanced to phase III clinical trial for African sleeping sickness. Erythromycin is often prescribed at doses at or above 50 mg kg–1 per day for mild to moderate infections.61 In the Phase III clinical trials, pafuramidine was used at ∼4.5 mg kg–1 per day.50 Here, we administered doses of 600 mg kg–1 per day erythromycin in combination with 10 mg kg–1 per day pafuramidine in the HSALR mice. These concentrations equate to a human equivalent dose (HED) of 48 mg kg–1 erythromycin and 0.8 mg kg–1 pafuramidine.62 The dose of erythromycin used is in line with current doses being prescribed, and the dose of pafuramidine is greater than 5-fold lower than that used in the Phase III clinical trials. Taken together with the high degree of mis-splicing rescue and the low off-target gene expression changes observed, a combination of erythromycin and pafuramidine may be a promising approach for a clinical trial as an orally administered therapy for DM1.
In the future, it will be exciting to determine if combination therapies can be used as therapeutic approaches for other microsatellite expansion diseases with similar mechanisms such as myotonic dystrophy type 2, c9orf72 ALS-FTD, various spinocerebellar ataxias, and Fuchs’ Corneal Dystrophy.63 The potential for combination therapies based upon small molecules is exciting due to their ease of oral administration, shorter half-lives and longer shelf lives than biologics, generally better tissue delivery, and lower costs associated with manufacturing. Of particular interest will be small molecules that cross the blood brain barrier to help combat the cognitive symptoms associated with DM1 and other diseases. Coupling small molecules with other promising therapeutics such as ASOs could be another exciting next step in combination therapeutics for DM1.
Materials and Methods
Culturing of DM1 Myotubes
Primary patient and control myoblast cell lines were derived from muscle biopsies under a University of Florida-approved IRB protocol with informed consent from all subjects. Approximately 1 × 105 myoblasts were plated in 12-well plates in SkGM-2 BulletKit growth medium (Lonza). Cells were allowed to reach > 90% confluency and were then differentiated for 7 days in DMEM/F-12 50/50 medium (Corning) supplemented with 2% v/v donor equine serum (Hyclone). Treatments were carried out by switching to fresh SkGM-2 BulletKit growth medium and adding the indicated concentrations of drug. Myotubes were harvested after 4 days of drug treatment.
RT-PCR Splicing Analysis
RNA was isolated from cells using an Aurum Total RNA mini kit (Bio-Rad) according to the package insert with on-column DNase1 treatment. For RT-PCR splicing analysis in mouse model, RNA was TRIZol extracted from quadriceps muscle of HSALR mice treated orally with either corn oil, 10 mg kg–1 pafuramidine, and/or 600 mg kg–1 erythromycin. For all samples, RNA concentrations were determined using a NanoDrop (Thermo) and reverse transcribed with SuperScript VI with random hexamer primers (IDT). The cDNA was then subjected to a polymerase chain reaction for 32 cycles using the primer sets listed in Table 1. Resulting PCR products were run via capillary electrophoresis on a fragment analyzer using the 1–500 bp DNF-905 kit (Advanced Analytical). Quantification was done using the integration values of the electropherogram peaks corresponding to inclusion and exclusion products from the Prosize 2.0 software (Advanced Analytical). To determine the percent rescue of a given ES event, eq 1 was used, where DM1_PSI denotes the PSI of untreated DM1 myotubes or HSALR mice, WT_PSI denotes the PSI of control myotubes or wild type mice, and drug_PSI denotes the PSI of DM1 myotubes or HSALR mice treated with indicated drug.
| 1 |
Table 1. Primers Used for RT-PCR Splicing Analysis.
| target | forward primer | reverse primer |
|---|---|---|
| Human MBNL1 exon5 | 5′- AGGGAGATGCTCTCGGGAAAAGTG | 5′- GTTGGCTAGAGCCTGTTGGTATTGG |
| Human MBNL2 exon5 | 5′- ACAAGTGACAACACCGTAACCG | 5′- TTTGGTAAAGGATGAAGAGCACC |
| Human NUMA1 exon2 | 5′- AAGTATGAGGGTGCCAAGGT | 5′- CTTCAGCTTCTGCTGCTGCA |
| Human SYNE1 exon137 | 5′- GACAAAGATTTCTACCTCCGGGG | 5′- CCCAGTTGTCGGATCTGTGACTC |
| Mouse Atp2a1 exon22 | 5′- GCTCATGGTCCTCAAGATCTCAC | 5′- GGGTCAGTGCCTCAGCTTTG |
| Mouse Clcn1 exon7a | 5′- TGAAGGAATACCTCACACTCAAGG | 5′- CACGGAACACAAAGGCACTG |
| Mouse Clasp1 exon13 | 5′- CAAATCTGTGTCGACGACAGGA | 5′- GCTGAGACTGTGAAACCACTTTGG |
| Mouse Nfix exon7 | 5′- CCATCGACGACAGTGAGATGG | 5′- CTGGATGATGGACGTGGAAGG |
RT-qPCR for Expression Analysis
Quantitative real-time PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) according to the package insert. Samples were run on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) and analyzed using the Quantitative-Comparative (CT) method. The levels of MBNL1, MBNL2, and DMPK mRNA in DM1 myotubes were normalized to GAPDH mRNA and displayed graphically as relative mRNA levels setting the untreated mRNA levels to 1. The levels of human skeletal actin (HSA) mRNA and Dmpk mRNA in HSALR mice were normalized to Gtf2b mRNA and displayed graphically as relative mRNA levels setting the untreated mRNA levels to 1. Two different primer sets were used to assess HSA mRNA levels in HSALR mice and the values were averaged. All primer sets used are shown in Table 2.
Table 2. Primers Used for RT-qPCR Expression Analysis.
| target | forward primer | reverse primer |
|---|---|---|
| Human ACTA1 set 1 | 5′- GAGGCTCAGAGCAAGAGAG | 5′- TCGTTGTAGAAGGTGTGGTG |
| Human ACTA1 set 2 | 5′- GGAGCGCAAATACTCGGTG | 5′- CATTTGCGGTGGACGATGG |
| Mouse Gtf2b | 5′- CTTCATGTCCAGGTTCTGCTCC | 5′- GGAACCAAGTCCAGCTCCAC |
| Human DMPK | 5′- CACGTTTTGGATGCACTGAGAC | 5′- GATGGAGGGCCTTTTATTCGCG |
| Human MBNL1 | 5′- CGCAGTTGGAGATAAATGGACG | 5′- CACCAGGCATCATGGCATTG |
| Human MBNL2 | 5′- CCTGGTGCTCTTCATCCTTTAC | 5′- GTGAGAGCCTGCTGGTAGTG |
| Human GAPDH | 5′- AATCCCATCACCATCTTCCA | 5′- TGGACTCCACGACGTACTCA |
Toxicity Analysis in Cell Culture
Approximately 1 × 104 myoblasts were plated in 96-well plates and treated as described above to culture DM1 myotubes. After 7 days of differentiation and 4 days drug treatment, media was replaced and PrestoBlue cell viability reagent (Thermo) was added to the cells according to the package insert. The treated cells were incubated at 37 °C and 5% CO2 and protected from light for 3 h. Absorbance at 570 and 600 nm was read on a BioTek Cytation 3 plate reader. The 570 nm/600 nm absorbance ratios were calculated for all samples with a background subtraction of the average 570 nm/600 nm values of no-cell plus furamidine control wells. All samples were normalized by setting the background subtracted, nontreated cell samples to 1.
Fluorescent in Situ Hybridization Microscopy
Myotubes or slices of vastus lateralis (quadriceps) muscle from HSALR mice were fixed with 4% (w/v) PFA, permeabilized using 70% (v/v) ethanol, and prehybridized for 30 min at 37 °C. Myotubes were probed for 4 h at 50 °C with a Cy3-(CAG)8 probe (IDT). Slides were washed with 42 °C prewarmed 40% (v/v) formamide in 2X SSC and mounted using ProLong Diamond Antifade mountant with DAPI (Life Technologies). Myotubes were imaged on a Zeiss LSM 840 confocal scanning microscope with a 40× water objective. The number of nuclear foci for each cell was quantified using Fiji. In DM1 myotubes, the foci were counted blind in at least 100 nuclei per replicate (at least 300 nuclei over three replicates) for all treatments. For the muscle from HSALR mice, the number of total nuclei was divided by the number of nuclei containing foci to calculate the percent number of nuclei with foci and at least 100 nuclei per replicate (at least 300 nuclei over three replicates) were counted blind for all treatments.
Western Blot Analysis
Protein from myotubes or mouse vastus lateralis (quadriceps) muscle was harvested in RIPA buffer supplemented with 1 mM PMSF and 1X SigmaFast protease inhibitor (Sigma-Aldrich). After centrifugation at 12 000 rpm for 15 min at 4 °C, the supernatant was used to determine protein concentration with the Pierce BCA Protein Assay kit (Thermo). A total of 10 μg of protein was denatured for 5 min at 98 °C and run on a precast 10% SDS-PAGE mini gel (Bio-Rad) at 200 V for 40 min in 1× running buffer (25 mM Tris base pH 8.3, 192 mM glycine, 0.1% (w/v) SDS). Gel was transferred onto a low fluorescence PVDF membrane (Bio-Rad) for 1 h at 25 V in 1× transfer buffer (25 mM Tris base pH 8.3, 192 mM glycine, 20% (v/v) methanol). The membrane was blocked for 1 h using SeaBlock (Thermo) and then incubated overnight with primary antibodies [1:2000 MBNL1 (MB1a, Wolfson Centre for Inherited Neuromuscular Disease), 1:500 MBNL2 (3B4, Santa Cruz), 1:1000 GAPDH (14C10, Cell Signaling)]. Blots were incubated at room temperature (RT) for 1 h with secondary antibodies [1:7500 Goat anti-Rabbit IRDye @680 (Li-Cor), 1:7500 Goat anti-Mouse IRDye @800 (Li-Cor)], washed in TBS-T, and imaged on an Odyssey CLx imager (Li-Cor). Blots were analyzed using ImageStudio Lite (Li-Cor). The relative levels of MBNL were calculated by first normalizing lanes within the same blot using the GAPDH signal and then by normalizing levels of MBNL in the untreated cells to 1.
Chemical Synthesis of Pafuramidine
See Supporting methods section in the Supporting Information.
Combination Treatment of Mice
Mouse handling and experimental procedures were performed in accordance with the Osaka University guidelines for the welfare of animals, and were approved by the institutional review board. Homozygous HSALR transgenic mice of line 20b (FVB inbred background) have been described previously.52 Age (< 4 months old), gender- and sibling-matched mice were treated with 10 mg kg–1 pafuramidine and/or 600 mg kg–1 erythromycin diluted in corn oil daily for 14 days via oral administration. Control mice were treated with corn oil alone. After the treatments, mice were sacrificed, and the vastus lateralis (quadriceps) muscle was reserved. Total RNA extraction from mouse muscles, cDNA synthesis, and polymerase chain reaction (PCR) amplification were performed as described previously.46 Electromyography was performed under general anesthesia as described previously. Briefly, at least 10 needle insertions were performed in the vastus muscle, and myotonic discharges were graded on a four-point scale: 0, no myotonia; 1, occasional myotonic discharge in ≤ 50% of needle insertions; 2, myotonic discharge in > 50% of insertions; and 3, myotonic discharge with nearly all insertions.
Immunofluorescent Microscopy
Frozen vastus lateralis (quadriceps) muscle from HSALR mice was sectioned into 10 μm slices onto slides and fixed with 10% (v/v) buffered formalin, permeabilized using 1:1 methanol/acetone and blocked using Background Sniper (Biocare Medical). Slides were incubated with 1:100 rabbit anti Clcn1 (Alpha Diagnostic International) overnight at 4 °C. Samples were incubated with 1:1000 goat antirabbit Alexa Fluor 488 (Thermo Fisher) for 1 h at RT and mounted using ProLong Diamond Antifade mountant with DAPI (Life Technologies). Samples were imaged on a Zeiss LSM 840 confocal scanning microscope with a 40× water objective.
RNA-seq Library Preparation
RNA quality was checked via capillary electrophoresis on a fragment analyzer using the RNA Analysis DNF-471 kit (Advanced Analytical). The NEBNext Ultra II Directional RNA Library Prep Kit for Illumina with NEBNext rRNA Depletion Kit was used to prepare RNA-seq libraries, with a total of 500 ng input RNA from each sample. The manufacturer’s protocols were followed, with the following exceptions: 40× adaptor dilutions were used, all bead incubations were done at room temp, a 4× lower concentration of index primers was used, and 10 cycles of library amplification were performed. The resulting libraries were pooled in equimolar amounts, quantified using the KAPA Library Quant Kit for Illumina, quality checked via capillary electrophoresis on the fragment analyzer using the NGS Analysis DNF-474 kit (Advanced Analytical), and were sequenced using paired-end, 75 base pair sequencing on the Illumina NextSeq 500 massively parallel sequencer at the University of Florida Center for NeuroGenetics.
Splicing Analysis from RNA-seq Data
Raw reads were checked for quality and aligned to GRCm38.p5 mouse or human genome using STAR (version 2.5.1b). After reads were aligned, rMATS (version 3.2.5)64 was used to analyze isoform abundances and compared to three-wild type samples.65 ES events were considered significant with an FDR < 0.1 and p < 0.01. Events were considered mis-spliced in the WT vs DM1 model (DM1 myotubes or HSALR mice) data sets if the PSI change was ≥ 10% for a given ES event. Of those events, a percent rescue of ≥ 10% were considered “rescue” with drug treatment, “over-rescue” ≥ 110%, “mis-rescue” ≤ −10%, and “off-target” ES events were those not in the WT vs DM1 model events that had a change in PSI ≥ 10%. To determine the percent rescue of a given ES event, eq 1 was used as described in RT-PCR Splicing Analysis.
Transcriptome Analysis
Raw reads were checked for quality and aligned to GRCm38.p5 mouse or human genome using STAR (version 2.5.1b)66 and a .gtf file generated from Version M16 Genecode or human gene models, respectively. Uniquely aligning paired sequences were input to Stringtie (version 1.3.4d) and the prepDE.py script (offered with Stringtie package) was used to generate genecounts. Differential expression analysis was performed with DESeq2 (version 1.16.1).67 Differential expression was considered significant with p < 0.1. Of those events, a percent rescue of ≥ 10% were considered “rescue” with treatment, “over-rescue” ≥ 110%, “mis-rescue” ≤ −10%, and “off-target” gene expression events were those not in the WT vs HSALR events. To determine the rescue of a given differentially expressed gene, eq 2 was used, for which WT_EXP denotes difference in expression of WT mice versus untreated HSALR mice, drug_EXP denotes difference in expression of untreated HSALR mice versus HSALR mice treated with indicated drug.
| 2 |
Statistical Analyses
Data are expressed as mean ± standard deviation. All data shown are the summary of three or more biological replicates and statistical analyses were completed in Prism 7. For data sets for which three or more groups were analyzed simultaneously, two-tailed student’s t test was used to determine statistical significance and associated p-value. Statistical values used: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Acknowledgments
Special thanks to other members of the Berglund lab, specifically Kaalak Reddy, for helpful discussions, experimental advice, and comments on the manuscript. Thank you to Fanjo Ivankovic of the Swanson lab for providing WT FVB mouse tissue for RNA-seq and to John Cleary for cryo-sectioning of mouse tissue and helpful troubleshooting of IF assay. Thank you to Tammy Reid for assisting with drug treatment in DM1 myotubes for western blot analysis. Also, thank you to the UF Center for NeuroGenetics, especially the Ranum, Swanson, and Wang laboratories, for general support and guidance.
Glossary
Abbreviations
- ASO
antisense oligonucleotide
- CELF
CUG-BP Elav-like family of RNA binding proteins
- CLCN
muscle-specific chloride channel gene
- DM1
myotonic dystrophy type 1
- MBNL
muscleblind-like family of RNA binding proteins
- DMPK
dystrophia myotonica protein kinase gene
- ES
exon-skipping
- HSA
human skeletal actin gene
- INSR
insulin receptor gene
- μM
micromolar
- PSI
Percent Spliced In
- RAN
repeat-associated non-ATG translation
- TNNT2
cardiac troponin T gene
- WT
wild-type
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00020.
Methods for chemical synthesis of pafuramidine; supporting tables; supporting figures and associated legends (PDF)
Author Contributions
J.R.J. and J.A.B. conceived the project, analyzed results, and wrote the manuscript. J.R.J. characterized combination treatment and established treatment ranges in DM1 myotubes and performed RT-PCR splicing, RT-qPCR expression, cell viability, Western blot analyses, FISH assays, IF assays, prepared the RNA-seq libraries, and performed RNA-seq bioinformatics analyses. H.Y. and R.W.H. synthesized pafuramidine. M.N. characterized combination treatments, performed RT-PCR splicing, and collected samples for IF, FISH, and RNA-seq analysis in DM1 mouse model.
These studies were supported by funding from the MDA (516314) and NIH (AR059833) to J.A.B., JSPS KAKENHI (15K15339 and 16H05321) to M.N., and NSF predoctoral fellowship (DGE-1315138 and DGE-1842473) to J.R.J.
The authors declare the following competing financial interest(s): J. Andrew Berglund and the University of Oregon have patented diamidines for treating myotonic dystrophy (U.S. Patents 8463049 and 20130281462).
Notes
Computational Resources. The data sets generated and/or analyzed during the current study are available on the NCBI Sequence Read Archive. Human myotube data sets: non-DM (DM-04) myotubes (SRR7726419, SRR7726421, SRR7726422), DM1 (DM-05) myotubes (SRR7726417, SRR7726418, SRR7726420), DM1 (DM-05) myotubes treated with 25 μM EM (SRR9711282, SRR9711284, SRR9711285) or 0.5 μM FM (SRR9711280, SRR9711281, SRR9711283) and combination (SRR9711277, SRR9711278, SRR9711279). Mouse data sets prepared from quadriceps muscle: FVB WT (SRR7707863, SRR7707864, SRR7707865), untreated control HSALR (SRR9720668, SRR9720669, SRR9720670), HSALR treated with 600 mg kg–1 erythromycin (SRR9720665, SRR9720666, SRR9720667), 10 mg kg–1 pafuramidine (SRR9720663, SRR9720664, SRR9720672), 15 mg kg–1 pafuramidine (SRR9722308, SRR9722309, SRR9722310) or a combination of 10 mg kg–1 pafuramidine and 600 mg kg–1 erythromycin (SRR9720661, SRR9720662, SRR9720671).
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Harper P. S. (2001) Myotonic Dystrophy, W.B. Saunders Company, London. [Google Scholar]
- Mahadevan M.; Tsilfidis C.; Sabourin L.; Shutler G.; Amemiya C.; Jansen G.; Neville C.; Narang M.; Barcelo J.; O’Hoy K.; et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255, 1253–1255. 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Fu Y. H.; Pizzuti A.; Fenwick R. G. Jr.; King J.; Rajnarayan S.; Dunne P. W.; Dubel J.; Nasser G. A.; Ashizawa T.; de Jong P.; et al. (1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Brook J. D.; McCurrach M. E.; Harley H. G.; Buckler A. J.; Church D.; Aburatani H.; Hunter K.; Stanton V. P.; Thirion J. P.; Hudson T.; et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 69, 385. 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Miller J. W.; Urbinati C. R.; Teng-Umnuay P.; Stenberg M. G.; Byrne B. J.; Thornton C. A.; Swanson M. S. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448. 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur R. S.; Philips A. V.; Cooper T. A. (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29, 40–47. 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- Philips A. V.; Timchenko L. T.; Cooper T. A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741. 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- Timchenko N. A.; Patel R.; Iakova P.; Cai Z. J.; Quan L.; Timchenko L. T. (2004) Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J. Biol. Chem. 279, 13129–13139. 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- Du H.; Cline M. S.; Osborne R. J.; Tuttle D. L.; Clark T. A.; Donohue J. P.; Hall M. P.; Shiue L.; Swanson M. S.; Thornton C. A.; Ares M. Jr. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 17, 187–193. 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A.; Andersen H. S.; Doktor T. K.; Okamoto T.; Ito M.; Andresen B. S.; Ohno K. (2012) CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2, 209. 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E. T.; Cody N. A.; Jog S.; Biancolella M.; Wang T. T.; Treacy D. J.; Luo S.; Schroth G. P.; Housman D. E.; Reddy S.; Lecuyer E.; Burge C. B. (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150, 710–724. 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko N. A.; Cai Z. J.; Welm A. L.; Reddy S.; Ashizawa T.; Timchenko L. T. (2001) RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 276, 7820–7826. 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- Osborne R. J.; Lin X.; Welle S.; Sobczak K.; O’Rourke J. R.; Swanson M. S.; Thornton C. A. (2009) Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 18, 1471–1481. 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A.; Takahashi M. P.; Jiang H.; Beck C. L.; Bowers W. J.; Moxley R. T.; Cannon S. C.; Thornton C. A. (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 10, 35–44. 10.1016/S1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- Dixon D. M.; Choi J.; El-Ghazali A.; Park S. Y.; Roos K. P.; Jordan M. C.; Fishbein M. C.; Comai L.; Reddy S. (2015) Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 9042. 10.1038/srep09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T.; Gibbens B.; Doty N. S.; Gomes-Pereira M.; Huguet A.; Stone M. D.; Margolis J.; Peterson M.; Markowski T. W.; Ingram M. A.; Nan Z.; Forster C.; Low W. C.; Schoser B.; Somia N. V.; Clark H. B.; Schmechel S.; Bitterman P. B.; Gourdon G.; Swanson M. S.; Moseley M.; Ranum L. P. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U. S. A. 108, 260–265. 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D. H.; Thienes C. P.; Mahoney S. E.; Analau E.; Filippova G. N.; Tapscott S. J. (2005) Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489. 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Hao L.; Wang H.; Santostefano K.; Thapa A.; Cleary J.; Li H.; Guo X.; Terada N.; Ashizawa T.; Xia G. (2018) Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 26, 2617–2630. 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G.; Gao Y.; Jin S.; Subramony S. H.; Terada N.; Ranum L. P.; Swanson M. S.; Ashizawa T. (2015) Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33, 1829–1838. 10.1002/stem.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Agtmaal E. L.; Andre L. M.; Willemse M.; Cumming S. A.; van Kessel I. D. G.; van den Broek W.; Gourdon G.; Furling D.; Mouly V.; Monckton D. G.; Wansink D. G.; Wieringa B. (2017) CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 25, 24–43. 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto B. S.; Saxena T.; Oliveira R.; Mendez-Gomez H. R.; Cleary J. D.; Denes L. T.; McConnell O.; Arboleda J.; Xia G.; Swanson M. S.; Wang E. T. (2017) Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell 68, 479–490. e475. 10.1016/j.molcel.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Bennett C. F.; Cooper T. A. (2012) RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U. S. A. 109, 4221–4226. 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M.; Leger A. J.; Pandey S. K.; MacLeod A. R.; Nakamori M.; Cheng S. H.; Wentworth B. M.; Bennett C. F.; Thornton C. A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115. 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscianska E.; Witkos T. M.; Kozlowska E.; Wojciechowska M.; Krzyzosiak W. J. (2015) Cooperation meets competition in microRNA-mediated DMPK transcript regulation. Nucleic Acids Res. 43, 9500–9518. 10.1093/nar/gkv849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M.; Sobczak K.; Lueck J. D.; Osborne R. J.; Lin X.; Dirksen R. T.; Thornton C. A. (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339. 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak K.; Wheeler T. M.; Wang W.; Thornton C. A. (2013) RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 21, 380–387. 10.1038/mt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois M. A.; Lee N. S.; Rossi J. J.; Puymirat J. (2003) Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol. Ther. 7, 670–680. 10.1016/S1525-0016(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Batra R.; Nelles D. A.; Pirie E.; Blue S. M.; Marina R. J.; Wang H.; Chaim I. A.; Thomas J. D.; Zhang N.; Nguyen V.; Aigner S.; Markmiller S.; Xia G.; Corbett K. D.; Swanson M. S.; Yeo G. W. (2017) Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170, 899–912. e810. 10.1016/j.cell.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia R. N.; Shin J.; Yuan Y.; Beattie S. G.; Wheeler T. M.; Thornton C. A.; Swanson M. S. (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 103, 11748–11753. 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonrod L. A.; Nakamori M.; Wang W.; Carrell S.; Hilton C. L.; Bodner M. J.; Siboni R. B.; Docter A. G.; Haley M. M.; Thornton C. A.; Berglund J. A. (2013) Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 8, 2528–2537. 10.1021/cb400431f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siboni R. B.; Nakamori M.; Wagner S. D.; Struck A. J.; Coonrod L. A.; Harriott S. A.; Cass D. M.; Tanner M. K.; Berglund J. A. (2015) Actinomycin D Specifically Reduces Expanded CUG Repeat RNA in Myotonic Dystrophy Models. Cell Rep. 13, 2386–2394. 10.1016/j.celrep.2015.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Disney J. L.; Parkesh R.; Nakamori M.; Thornton C. A.; Disney M. D. (2012) Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem. Biol. 7, 1984–1993. 10.1021/cb3001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushechnikov A.; Lee M. M.; Childs-Disney J. L.; Sobczak K.; French J. M.; Thornton C. A.; Disney M. D. (2009) Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc. 131, 9767–9779. 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.; Luu L. M.; Peng S.; Serrano J. F.; Chan H. Y.; Zimmerman S. C. (2015) Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J. Am. Chem. Soc. 137, 14180–14189. 10.1021/jacs.5b09266. [DOI] [PubMed] [Google Scholar]
- Rzuczek S. G.; Colgan L. A.; Nakai Y.; Cameron M. D.; Furling D.; Yasuda R.; Disney M. D. (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 13, 188–193. 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Bodycombe N. E.; Haskell K. M.; Sun Y. L.; Wang E. T.; Morris C. A.; Jones L. H.; Wood L. D.; Pletcher M. T. (2017) A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 26, 3056–3068. 10.1093/hmg/ddx190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. H.; Nguyen L.; Peh J.; Luu L. M.; Sanchez J. S.; Richardson S. L.; Tuccinardi T.; Tsoi H.; Chan W. Y.; Chan H. Y.; Baranger A. M.; Hergenrother P. J.; Zimmerman S. C. (2014) Targeting toxic RNAs that cause myotonic dystrophy type 1 (DM1) with a bisamidinium inhibitor. J. Am. Chem. Soc. 136, 6355–6361. 10.1021/ja5012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laustriat D.; Gide J.; Barrault L.; Chautard E.; Benoit C.; Auboeuf D.; Boland A.; Battail C.; Artiguenave F.; Deleuze J. F.; Benit P.; Rustin P.; Franc S.; Charpentier G.; Furling D.; Bassez G.; Nissan X.; Martinat C.; Peschanski M.; Baghdoyan S. (2015) In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther.--Nucleic Acids 4, e262 10.1038/mtna.2015.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oana K.; Oma Y.; Suo S.; Takahashi M. P.; Nishino I.; Takeda S.; Ishiura S. (2013) Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci. Rep. 3, 2142. 10.1038/srep02142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanna A. S.; Smith B. M.; Cowan D.; Menzies D. (2013) Fixed-dose combination antituberculosis therapy: a systematic review and meta-analysis. Eur. Respir. J. 42, 721–732. 10.1183/09031936.00180612. [DOI] [PubMed] [Google Scholar]
- Smith C. S.; Aerts A.; Saunderson P.; Kawuma J.; Kita E.; Virmond M. (2017) Multidrug therapy for leprosy: a game changer on the path to elimination. Lancet Infect. Dis. 17, e293 10.1016/S1473-3099(17)30418-8. [DOI] [PubMed] [Google Scholar]
- Cui L.; Su X. Z. (2009) Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-Infect. Ther. 7, 999–1013. 10.1586/eri.09.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone V.; Thakkar N.; Jacobson J. M.; Wigdahl B.; Krebs F. C. (2011) Combinatorial approaches to the prevention and treatment of HIV-1 infection. Antimicrob. Agents Chemother. 55, 1831–1842. 10.1128/AAC.00976-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayat Mokhtari R.; Homayouni T. S.; Baluch N.; Morgatskaya E.; Kumar S.; Das B.; Yeger H. (2017) Combination therapy in combating cancer. Oncotarget 8, 38022–38043. 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt B.; Bernhardt T.; Moeller H. J.; Heuser I.; Frolich L. (2004) Combination therapy in Alzheimer’s disease: a review of current evidence. CNS Drugs 18, 827–844. 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]
- Nakamori M.; Taylor K.; Mochizuki H.; Sobczak K.; Takahashi M. P. (2016) Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 3, 42–54. 10.1002/acn3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenquin J. R.; Coonrod L. A.; Silverglate Q. A.; Pellitier N. A.; Hale M. A.; Xia G.; Nakamori M.; Berglund J. A. (2018) Furamidine Rescues Myotonic Dystrophy Type I Associated Mis-Splicing through Multiple Mechanisms. ACS Chem. Biol. 13, 2708–2718. 10.1021/acschembio.8b00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siboni R. B.; Bodner M. J.; Khalifa M. M.; Docter A. G.; Choi J. Y.; Nakamori M.; Haley M. M.; Berglund J. A. (2015) Biological Efficacy and Toxicity of Diamidines in Myotonic Dystrophy Type 1 Models. J. Med. Chem. 58, 5770–5780. 10.1021/acs.jmedchem.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B. P.; Boykin D. W. (1977) Synthesis and antiprotozoal activity of 2,5-bis(4-guanylphenyl)furans. J. Med. Chem. 20, 531–536. 10.1021/jm00214a014. [DOI] [PubMed] [Google Scholar]
- Pohlig G.; Bernhard S. C.; Blum J.; Burri C.; Mpanya A.; Lubaki J. P.; Mpoto A. M.; Munungu B. F.; N’Tombe P. M.; Deo G. K.; Mutantu P. N.; Kuikumbi F. M.; Mintwo A. F.; Munungi A. K.; Dala A.; Macharia S.; Bilenge C. M.; Mesu V. K.; Franco J. R.; Dituvanga N. D.; Tidwell R. R.; Olson C. A. (2016) Efficacy and Safety of Pafuramidine versus Pentamidine Maleate for Treatment of First Stage Sleeping Sickness in a Randomized, Comparator-Controlled, International Phase 3 Clinical Trial. PLoS Neglected Trop. Dis. 10, e0004363 10.1371/journal.pntd.0004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes F.; Massari S.; Bochicchio A.; Schorpp K.; Schilling J.; Weber S.; Offermann N.; Desantis J.; Wanker E.; Carloni P.; Hadian K.; Tabarrini O.; Rossetti G.; Krauss S. (2018) Reducing Mutant Huntingtin Protein Expression in Living Cells by a Newly Identified RNA CAG Binder. ACS Chem. Neurosci. 9, 1399–1408. 10.1021/acschemneuro.8b00027. [DOI] [PubMed] [Google Scholar]
- Mankodi A.; Logigian E.; Callahan L.; McClain C.; White R.; Henderson D.; Krym M.; Thornton C. A. (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773. 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- Xia G.; Santostefano K. E.; Goodwin M.; Liu J.; Subramony S. H.; Swanson M. S.; Terada N.; Ashizawa T. (2013) Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell. Reprogramming 15, 166–177. 10.1089/cell.2012.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier C.; Klein A. F.; Hammer C.; Vassilopoulos S.; Ivarsson Y.; Toussaint A.; Tosch V.; Vignaud A.; Ferry A.; Messaddeq N.; Kokunai Y.; Tsuburaya R.; de la Grange P.; Dembele D.; Francois V.; Precigout G.; Boulade-Ladame C.; Hummel M. C.; Lopez de Munain A.; Sergeant N.; Laquerriere A.; Thibault C.; Deryckere F.; Auboeuf D.; Garcia L.; Zimmermann P.; Udd B.; Schoser B.; Takahashi M. P.; Nishino I.; Bassez G.; Laporte J.; Furling D.; Charlet-Berguerand N. (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 17, 720–725. 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- Wagner S. D.; Struck A. J.; Gupta R.; Farnsworth D. R.; Mahady A. E.; Eichinger K.; Thornton C. A.; Wang E. T.; Berglund J. A. (2016) Dose-Dependent Regulation of Alternative Splicing by MBNL Proteins Reveals Biomarkers for Myotonic Dystrophy. PLoS Genet. 12, e1006316 10.1371/journal.pgen.1006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakunov S. A.; Bakunova S. M.; Wenzler T.; Ghebru M.; Werbovetz K. A.; Brun R.; Tidwell R. R. (2010) Synthesis and antiprotozoal activity of cationic 1,4-diphenyl-1H-1,2,3-triazoles. J. Med. Chem. 53, 254–272. 10.1021/jm901178d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z. Z.; Yarotskyy V.; Wei L.; Sobczak K.; Nakamori M.; Eichinger K.; Moxley R. T.; Dirksen R. T.; Thornton C. A. (2012) Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum. Mol. Genet. 21, 1312–1324. 10.1093/hmg/ddr568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M.; Sobczak K.; Puwanant A.; Welle S.; Eichinger K.; Pandya S.; Dekdebrun J.; Heatwole C. R.; McDermott M. P.; Chen T.; Cline M.; Tawil R.; Osborne R. J.; Wheeler T. M.; Swanson M. S.; Moxley R. T. 3rd; Thornton C. A. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 74, 862–872. 10.1002/ana.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E.; Gallardo F.; Beaudoin M.; Menard C.; Chartrand P. (2011) Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell Sci. 124, 1703–1714. 10.1242/jcs.073270. [DOI] [PubMed] [Google Scholar]
- Dansithong W.; Paul S.; Comai L.; Reddy S. (2005) MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 280, 5773–5780. 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- CLSI . (2018) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 11th ed., Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- Nair A. B.; Jacob S. (2016) A simple practice guide for dose conversion between animals and human. J. Basic Clin Pharm. 7, 27–31. 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Ashizawa T. (2017) RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 44, 17–29. 10.1016/j.gde.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S.; Park J. W.; Lu Z. X.; Lin L.; Henry M. D.; Wu Y. N.; Zhou Q.; Xing Y. (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 111, E5593–5601. 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt D. A.; Singhal N.; Kim M. L.; Cramer M. L.; Crowe K. E.; Xu R.; Jia Y.; Adair J.; Martinez-Pena Y. V. I.; Akaaboune M.; White P.; Janssen P. M.; Martin P. T. (2017) Deletion of Pofut1 in Mouse Skeletal Myofibers Induces Muscle Aging-Related Phenotypes in cis and in trans. Mol. Cell. Biol. 37, e00426-16. 10.1128/MCB.00426-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. D.; Nacu S. (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I.; Huber W.; Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.