

Abstract
Translation is a costly, but inevitable, cell maintenance process. To reduce unnecessary ATP consumption in cells, a fine-tuning mechanism is needed for both ribosome biogenesis and translation. Previous studies have suggested that the ribosome functions as a hub for many cellular signals such as ribotoxic stress response, mammalian target of rapamycin (mTOR), and ribosomal S6 kinase (RSK) signaling. Therefore, we investigated the relationship between ribosomes and mitogen-activated protein kinase (MAPK) activation under ribotoxic stress conditions and found that the activation of c-Jun N-terminal kinases (JNKs) was suppressed by ribosomal protein knockdown but that of p38 was not. In addition, we found that JNK activation is driven by the association of inactive JNK in the 80S monosomes rather than the polysomes. Overall, these data suggest that the activation of JNKs by ribotoxic stress is attributable to 80S monosomes. These 80S monosomes are active ribosomes that are ready to initiate protein translation, rather than polysomes that are already acting ribosomes involved in translation elongation.
Keywords: Deoxynivalenol, Emetine, JNK, Ribotoxic stress, 80S monosome
INTRODUCTION
Recently, ribosomes have received a lot of attention as hubs for cellular signals such as ribotoxins and growth factors (1–3). When the Human Genome Project was established, the number of proteins in human beings was considered to be insufficient for performing all body functions. Many biologists have suggested that multifunctional proteins and various signaling pathways emerge from an identical complex. Therefore, other than its inherent translational function, a ribosome complex acts as a scaffold protein for various kinase signaling pathways. Ribosomal protein S6 (rpS6), a component of the 40S ribosomal small subunit, accepts the signal from ribosomal S6 kinase (RSK). The phosphorylated rpS6 transmits the signal to the ribosome in response to different mitogens such as growth factors and insulin (2, 4). S6 kinase is also a downstream effector of the mammalian target of rapamycin (mTor), which is a key regulator of mammalian cell growth (5). Ribosomal protein S3 (rpS3) is located on a beak portion of the ribosomal small subunit and plays a role in translation initiation (6). Recently, rpS3 was demonstrated to have several extra-ribosomal functions in inflammation (7), metastasis (8), NF-κB signaling (9), and DNA repair (10, 11). Furthermore, phosphorylation of rpS3 is controlled by various signaling proteins such as extracellular signal-regulated kinase (Erk), protein kinase C delta (PKCδ), protein kinase B (PKB/Akt), and protein phosphatase 2A (PP2A) (12–15). The receptor for activated protein C kinase-1 (RACK1), a component of the 40S ribosomal subunit, is a scaffold protein in many signaling pathways such as protein kinase C (PKC) and focal adhesion kinase (FAK) (14–17). Thus, RACK1 is considered a receptor for extracellular signaling in the ribosomal complex.
Ribotoxins cause sequence-specific damage to the α-sarcin/ricin loop of 28S rRNA (16, 17). The damage blocks peptidyl transferase activity and inhibits the translation activity of the ribosome (17, 18). Previous studies have demonstrated that the ribosome acts as a scaffold for mitogen-activated protein kinases (MAPKs) such as p38 mitogen-activated protein kinase (p38) and c-Jun N-terminal kinases (JNK) under ribotoxic stress (18, 19). Bae et al. reported that the translocation of p38 to the ribosome increases when the macrophages are treated with deoxynivalenol (DON) (20).
The activation of JNK and p38 induced by ribotoxins is essential for active ribosomes. Many researchers have reported that translation inhibition suppresses the activation of JNK and p38 under ribotoxic stress (21). Here, we have demonstrated that the 80S monosome is important for JNK activation under ribotoxic stress.
RESULTS
Knockdown of ribosomal proteins decreased JNK activation induced by ribotoxic stress
A previous study has shown that JNK activation induced by a low dose of ultraviolet C (UVC; 45 J/m2) is attenuated by RACK1 knockdown (22). To apply this phenomenon to ribotoxic stress, RACK1 knockdown HT1080 cells were treated with various ribotoxins such as a high dose of UV (Fig. 1A), DON (Fig. 1B), and anisomycin (Fig. 1C). Then, activation of the stress-activated protein kinases JNK and p38 was examined in a time-dependent manner. In all the cases, JNK activation was significantly reduced in the RACK1 knockdown cells when compared with the scramble (SC) knockdown cells (Fig. 1). In contrast, activation of p38 did not change significantly in all the cases. Next, we investigated whether attenuated JNK activation occurred under the decreased state of other 40S ribosomal proteins due to rpS3 and rpS6 knockdown. The activation of JNK was attenuated by a decrease in ribosomal proteins, as in the case of RACK1 knockdown (Fig. 1D). Therefore, we suggest that knockdown of most ribosomal proteins confer cells with similar properties that inhibit JNK activation under ribotoxic stress.
Fig. 1.

Ribotoxic stress-induced JNK activation is decreased by RACK1 depletion. HT1080 cells transfected with scramble (SC) or RACK1 siRNA were treated with 150 J/m2 UV (A), 2 μg/ml DON (B), or 2 μg/ml anisomycin (C) and incubated for the indicated times. The densitometric ratio of pJNK/JNK is shown below the blots (n = 3). Error bars, standard deviation; *P < 0.05; **P < 0.01; NS, not significant. (D) HT1080 cells transfected with the indicated siRNAs were irradiated with 150 J/m2 UV, followed by harvesting at the indicated times. The cell lysates were analyzed with immunoblotting.
Knockdown of ribosomal proteins caused a serious decrease of 80S monosomes in the ribosomal profile
To characterize the effects of ribotoxic stress on the ribosomes, we performed translational profiling of the ribosomes under various stresses. We performed ultracentrifugation analyses with a 5% to 45% (w/v) linear sucrose gradient (Fig. 2A). Cells with various stresses exhibited different abnormal ribosome profiles when compared with the unstressed cells (Fig. 2B). DON and UV showed a distinct ribosome profile with a significant increase in the 60S ribosomal subunit and 80S monosome peak, whereas anisomycin showed a slight decrease in 80S monosome and increase in the polysome. According to previous studies (23, 24), anisomycin and DON can slightly increase polysome fractions by inducing ribosomal stalling in the mRNA templates. In contrast, a translation inhibitor, emetine, induced a significant decrease in 80S monosomes and increase in the polysomes (Fig. 2C). Interestingly, emetine inhibits ribosomal peptidyl transferase activity but does not induce the activation of JNK. Emetine has previously been reported to disrupt JNK activation induced by ribotoxins, such as anisomycin (16, 25) and UV (26). These results suggest that JNK activation induced by ribotoxic stress requires a normal number of 80S monosomes but not polysomes, and JNK activation subsequently is blocked by a decrease in 80S monosomes. This indicates that the 80S monosome is “an active ribosome,” which is active to translate associated mRNAs. However, once 80S monosomes are initiated for elongation, they become “acting ribosomes” and JNK cannot be activated. To confirm this suggestion, we next examined the ribosome profiles of rpS3, ribosomal protein L13 (rpL13), and RACK1 siRNA transfected cells. The rpS3 and rpS6 knockdown cells exhibited an abnormal ribosome profile and significant decrease in 80S monosomes and mild increase in 60S ribosomal subunits (Fig. 2C), which is consistent with the results of our previous study (27). Additionally, the rpL13 siRNA transfected cells showed a striking decrease in 80S monosomes (Fig. 3A). The 80S monosomes were reduced in the RACK1 knockdown cells, but the rest of the ribosome profiles did not change significantly when compared with the control cells. (Fig. 2C). Furthermore, we confirmed that the UV-induced JNK activation was inhibited by knockdown of two 60S ribosomal proteins, rpL13 and ribosomal protein L30 (rpL30) (Fig. 3B and C). Thus, we concluded that a normal number of active 80S monosomes are required for ribotoxic stress-induced JNK activation, probably because active 80S monosomes remain ready for the next action.
Fig. 2.

Comparison of ribosomal profiles under various stress conditions and after the knockdown of ribosomal proteins. (A) Scheme of a ribosome profiling method using a linear sucrose gradient. (B) HT1080 cells were treated with 2 μg/ml DON, 2 μg/ml anisomycin, or 150 J/m2 UV. The untreated HT1080 cells were used as the control, and the profile was designated “Mock.” (C) Scramble, each ribosomal protein siRNA, and emetine-treated HT1080 cells were fractionated in a linear sucrose gradient, as described in the Materials and Methods. The polysome, monosome (80S), and ribosomal subunit (40S, 60S) regions are marked in the ribosome profile.
Fig. 3.
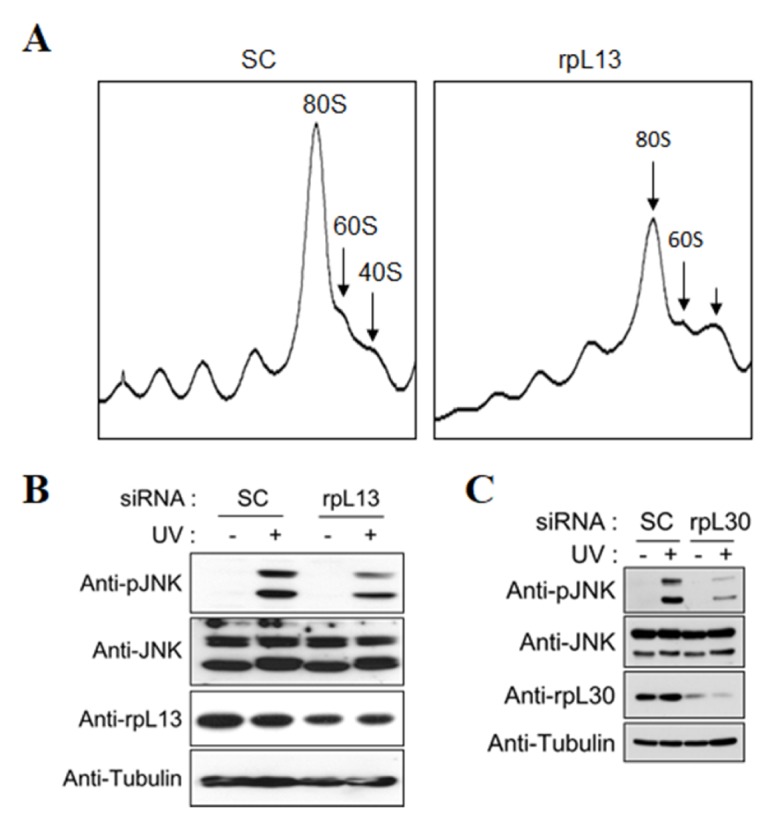
Knockdown of 60S ribosomal protein decreases the activation of JNK by UV irradiation. (A) Cell lysates from HT1080 cells transfected with scramble or rpL13 siRNA were fractionated in a linear sucrose gradient, as described in the Materials and Methods. The regions of the polysome, monosome (80S), and ribosome subunits (40S, 60S) are indicated in the ribosome profile. (B, C) HT1080 cells transfected with rpL13 (B) or rpL30 (C) siRNA were treated with 150 J/m2 UV and incubated for 1 h. These cell extracts were resolved using SDS-PAGE and analyzed with immunoblotting.
JNK activation by ribotoxic stress occurred in the monosomes but not the polysomes
Our findings suggested that the ribotoxic stress-induced JNK activation is initiated in the 80S monosomes. To support this hypothesis, we first examined the presence of JNK in each separate ribosomal fraction by using a sucrose cushion. The mRNA-associated ribosomes, such as polysomes and 80S monosomes, can be easily pelleted by sedimentation of lysates through 20% sucrose cushion. The mRNA-free ribosomes, such as the 40S and 60S ribosomal subunits, were predominantly located in the middle fractions of the 20% sucrose cushion. JNK was detected in the middle fraction, but not in the ribosome pellet, and mostly disappeared because of RACK1 knockdown (Fig. 4A). However, the location pattern of p38 in the middle fraction was similar in both scramble (SC) and RACK1 knockdown cells. In addition, we confirmed a reduction in UV-induced JNK activation under RACK1 knockdown conditions by performing in vitro kinase analysis with the middle fraction of the sucrose cushion (Fig. 4B). Next, we investigated the existence of JNK in the ribosomal fractions separated by linear sucrose gradient centrifugation. For comparison of ribosome distribution in the normal and UV-irradiated cells, we quantified the polysomes, 80S monosomes, and 60S and 40S ribosomal subunits. UV irradiation significantly increased the number of monosomes and decreased the number of polysomes (Fig. 4C), which is consistent with our previous results. Under UV irradiation, unphosphorylated JNK disappeared in the 80S monosome fractions, and phosphorylated JNK began to appear in the non-ribosomal fractions (Fig. 4D and E). Therefore, we concluded that the activated JNK may have been released from the active ribosome, which is ready to participate in the process of translational elongation.
Fig. 4.

UV-induced JNK activation by the 80S monosome is attenuated by translation initiation inhibitors. (A) HT1080 cells were transfected with scramble or RACK1 siRNA and treated with different ribotoxins (2 μg/ml DON, 2 μg/ml anisomycin, or 150 J/m2 UV) for the indicated times. The cell extracts were subjected to ultracentrifugation by using a 20% sucrose cushion. The ribosome-containing pellet, middle fraction, and non-ribosomal supernatant were collected separately. For immunoblot analysis, the ribosome pellets were resuspended in SDS-PAGE sample buffer, and the middle fractions were precipitated with TCA/acetone and mixed with the SDS-PAGE sample buffer. (B) HT1080 cells transfected with scramble or RACK1 siRNA were irradiated with 150 J/m2 UV. After 1 h, non-ribosomal and middle fractions were isolated by ultracentrifugation. Kinase assays were performed by mixing immunoprecipitated JNK of each fraction with GST-cJun in the presence of γ-32P. (C) Normal or UV-irradiated HT1080 cells were fractionated in a linear sucrose gradient, as described in the Materials and Methods. Distribution (%) of ribosome content (right) in the ribosomal fractions was calculated by measuring the area in each fraction on the basis of the ribosome profile (left). Error bars, standard deviation; ***P < 0.001; NS, not significant (n = 3). (D, E) Each fraction was resolved using 10% SDS-PAGE and subjected to immunoblot analysis with the indicated antibodies (D). The relative amount of JNK in the 80S monosome was obtained by measuring the signal intensities of fractions 5 and 6. Error bars, standard deviation; *P < 0.05 (n = 3) (E). (F) HT1080 cells were pre-treated with 25 μg/ml cycloheximide (CHX), 20 μM emetine (Eme), and 5 μM NSC119889 (NSC) for 30 min and then irradiated with 150 J/m2 of UV. After 1 h, the cell lysates were subjected to immunoblot analysis by using the indicated antibodies.
Next, although emetine, an inhibitor of translation, decreased ribotoxic stress-induced JNK activation, it is unclear whether the inhibition of all translation steps had the same effect as emetine. Therefore, we investigated UV-induced JNK activation by using various protein synthesis inhibitors. NSC119889 inhibits eIF2 ternary complex (eIF2-GTPMet-tRNAi Met) formation in the translation initiation step. Emetine inhibits protein synthesis by binding to the 40S ribosomal subunit, but the exact mechanism has not yet been elucidated. Cycloheximide inhibits eEF2-mediated tRNA translocation by binding to the 60S ribosomal subunit (28). As shown in Fig. 4F, NSC119889, and not cycloheximide, had the same negative effect on UV-induced JNK activation as emetine. Therefore, we propose that blocking translation initiation results in the inhibition of ribotoxic stress-induced JNK activation.
DISCUSSION
Recently, the ribosome, a translation machinery for protein biosynthesis, was reported to act as a scaffold for various kinase signaling pathways. Eukaryotic cells respond to ribotoxic stimuli in two ways: inhibition of protein translation or activation of MAPK signaling (16). Translation inhibition impairs the peptidyl transferase activity of the ribosomes by cleavage of the 3′-end of 28S rRNA, the binding region of aminoacyl tRNA. Then, activation of JNK and p38 occurs in active ribosomes. However, it has not been determined whether ribotoxin-sensitive active ribosomes are polysomes or 80S monosomes. The former undergoes mRNA translation, and the latter is present on the mRNA ready for translation. We propose the polysome is an acting ribosome, and the 80S monosome is an active ribosome.
Here, we have confirmed that knockdown of ribosomal proteins inhibits ribotoxic stress-induced JNK activation, as after treatment with emetine (Fig. 1D). Then, we confirmed that the knockdown of ribosomal proteins, rpS3, rpS6, and rpL13, resulted in a common decrease in the 80S monosome and polysome fractions by using ribosome profile analysis. In accordance with our previous data (27), 80S monosomes decreased because of ribosomal protein knockdown, but no significant changes were observed in the polysome fractions (Fig. 2). Emetine causes a serious decrease in 80S monosomes. Thus, we suggest that the decrease in 80S monosomes is responsible for the inhibition of ribotoxic stress-induced JNK activation.
We were interested in the correlation between the ribosomal peptidyl transferase activity and UV-induced JNK activation because various translational inhibitors have different effects on the inhibition of UV-induced JNK activation (Fig. 2B). We considered the ribonucleolytic activity of ribotoxin in the process of peptide bond formation in ribosomes. Most ribotoxins, such as anisomycin, DON, and UV, induce a malfunction in the peptidyl transferase activity of the ribosome. Thus, ribotoxin can inhibit the formation of peptide bonds between P-position loaded amino acids and A-position loaded amino acids. Indeed, anisomycin has been reported to bind to the A-site and inhibit the formation of peptide bonds. The translation inhibition mechanism of cycloheximde inhibits eEF2-mediated translocation from A-site to P-site through the binding of E-sites in the 60S ribosomal subunit (28). Emetine can bind to 40S and 60S ribosomal subunits and 80S monosomes and can inhibit peptide bond formation at high concentrations. Therefore, it seems that the 80S monosome immediately before the initiation of polypeptide chain elongation is important for ribotoxin-induced JNK activation (Fig. 4D).
Many kinases bind to ribosomes and regulate ribosomal functions through phosphorylation of the ribosomal proteins or ribosomal components (29–32). Although the upstream kinases involved in JNK and p38 activation induced by ribotoxic stress are not fully understood, several studies provide clues. The mixed lineage kinase (ZAK) plays a pivotal role in ribotoxic stress responses (33). Under ribotoxic stress conditions induced by doxorubicin, activation of JNK and p38 was suppressed by ZAK inhibition. In addition, double-stranded RNA-dependent protein (PKR) (34) and hematopoietic cell kinase (Hck) (35) have been suggested as upstream kinases for MAPK activation under ribotoxic stress. These kinases have been reported to activate MAPK signaling pathways involved in JNK and p38 activation in response to a variety of ribotoxins (16, 36). Finally, PKC, the RACK1 binding protein, can stimulate the MAPK signaling pathway in response to diverse stimuli and is recruited into ribosomes through RACK1 interaction (22). However, further studies are required to understand the mechanism underlying ribotoxic stress-induced MAPK activation.
Here, we tried to understand the relationship between ribotoxic stress-induced MAPK activation and active ribosomes. Both JNK and p38 were activated under ribotoxic stress, which is consistent with the findings of previous studies; however, interestingly, JNK activation was significantly affected by impaired ribosome biogenesis. In addition, we found that 80S monosomes are essential for JNK activation under ribotoxic stress, indicating 80S monosomes with associated mRNAs are active ribosomes.
MATERIALS AND METHODS
Detailed materials and methods are available in Supplementary Data.
Ribosomal fractionation and pelleting
For ribosomal fractionation and profiling, HT1080 cells were treated with 100 μg/ml cycloheximide for 30 min and lysed in 1 ml of hypotonic lysis buffer (1.5 mM KCl, 2.5 mM MgCl2, 20 mM Tris-HCl (pH 7.4), 0.5% sodium deoxycholate, 1% Triton X-100, 1.25 mM DTT, and 40 Units/ml RNasin). After centrifugation at 12,000 ×g for 15 min to remove the mitochondria and cell debris, the supernatants were layered on top of a 5% to 45% (w/v) sucrose gradient set in dilution buffer (80 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl (pH 7.4), and 1 mM DTT) and centrifuged with a Beckman SW41Ti rotor at 149,000 × g for 2.5 h at 4°C. The absorbance of the sucrose gradients was measured, and each fraction was collected, as described previously (37). For ribosomal pelleting, the HT1080 cells were lysed in 2 ml of hypotonic lysis buffer. After centrifugation, the supernatant was layered over a sucrose (20% (w/v)) cushion and centrifuged at 149,000 × g for 2.5 h. The ribosome-containing pellet and middle fraction and non-ribosomal supernatant were collected separately.
Supplementary Information
ACKNOWLEDGEMENTS
This work was supported in part by NRF-2017R1E1A 1A01074101 and NRF-2017R1D1A1B03029708 and a Korea University Grant. Consulting service from the MCRB (Seoul, Korea) was kindly appreciated.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Klein S, Morimoto T, Rifkin DB. Characterization of fibroblast growth factor-2 binding to ribosomes. Growth Factors. 1996;13:219–228. doi: 10.3109/08977199609003223. [DOI] [PubMed] [Google Scholar]
- 2.Stolovich M, Tang H, Hornstein E, et al. Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the phosphatidylinositol 3-kinase-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol. 2002;22:8101–8113. doi: 10.1128/MCB.22.23.8101-8113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jetzt AE, Cheng JS, Li XP, Tumer NE, Cohick WS. A relatively low level of ribosome depurination by mutant forms of ricin toxin A chain can trigger protein synthesis inhibition, cell signaling and apoptosis in mammalian cells. Int J Biochem Cell Biol. 2012;44:2204–2211. doi: 10.1016/j.biocel.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chauvin C, Koka V, Nouschi A, et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014;33:474–483. doi: 10.1038/onc.2012.606. [DOI] [PubMed] [Google Scholar]
- 5.Mahoney SJ, Dempsey JM, Blenis J. Cell signaling in protein synthesis ribosome biogenesis and translation initiation and elongation. Prog Mol Biol Transl Sci. 2009;90:53–107. doi: 10.1016/S1877-1173(09)90002-3. [DOI] [PubMed] [Google Scholar]
- 6.Passmore LA, Schmeing TM, Maag D, et al. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol Cell. 2007;26:41–50. doi: 10.1016/j.molcel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 7.Ahn EH, Kim DW, Kang HW, et al. Transduced PEP-1-ribosomal protein S3 (rpS3) ameliorates 12-O-tetradecanoylphorbol-13-acetate-induced inflammation in mice. Toxicology. 2010;276:192–197. doi: 10.1016/j.tox.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Kim SH, Kim J. Reduction of invasion in human fibrosarcoma cells by ribosomal protein S3 in conjunction with Nm23-H1 and ERK. Biochim Biophys Acta. 2006;1763:823–832. doi: 10.1016/j.bbamcr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Wan F, Anderson DE, Barnitz RA, et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Chubatsu LS, Admon A, Stahl J, Fellous R, Linn S. Implication of mammalian ribosomal protein S3 in the processing of DNA damage. J Biol Chem. 1995;270:13620–13629. doi: 10.1074/jbc.270.23.13620. [DOI] [PubMed] [Google Scholar]
- 11.Jung SO, Lee JY, Kim J. Yeast ribosomal protein S3 has an endonuclease activity on AP DNA. Mol Cells. 2001;12:84–90. [PubMed] [Google Scholar]
- 12.Kim HD, Lee JY, Kim J. Erk phosphorylates threonine 42 residue of ribosomal protein S3. Biochem Biophys Res Commun. 2005;333:110–115. doi: 10.1016/j.bbrc.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 13.Kim TS, Kim HD, Kim J. PKCdelta-dependent functional switch of rpS3 between translation and DNA repair. Biochim Biophys Acta. 2009;1793:395–405. doi: 10.1016/j.bbamcr.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Lee SB, Kwon IS, Park J, et al. Ribosomal protein S3, a new substrate of Akt, serves as a signal mediator between neuronal apoptosis and DNA repair. J Biol Chem. 2010;285:29457–29468. doi: 10.1074/jbc.M110.131367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim TS, Kim HD, Shin HS, Kim J. Phosphorylation status of nuclear ribosomal protein S3 is reciprocally regulated by protein kinase C{delta} and protein phosphatase 2A. J Biol Chem. 2009;284:21201–21208. doi: 10.1074/jbc.M109.018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iordanov MS, Pribnow D, Magun JL, et al. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/MCB.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olombrada M, Rodriguez-Mateos M, Prieto D, et al. The acidic ribosomal stalk proteins are not required for the highly specific inactivation exerted by alpha-sarcin of the eukaryotic ribosome. Biochemistry. 2014;53:1545–1547. doi: 10.1021/bi401470u. [DOI] [PubMed] [Google Scholar]
- 18.Ouyang DY, Wang YY, Zheng YT. Activation of c-Jun N-terminal kinases by ribotoxic stresses. Cell Mol Immunol. 2005;2:419–425. [PubMed] [Google Scholar]
- 19.Liu P, Wang YY, Qi X, Gu Q, Geng M, Li J. Undecylprodigiosin induced apoptosis in P388 cancer cells is associated with its binding to ribosome. PLoS One. 2013;8:e65381. doi: 10.1371/journal.pone.0065381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae HK, Pestka JJ. Deoxynivalenol induces p38 interaction with the ribosome in monocytes and macrophages. Toxicol Sci. 2008;105:59–66. doi: 10.1093/toxsci/kfn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iordanov MS, Choi RJ, Ryabinina OP, Dinh TH, Bright RK, Magun BE. The UV (Ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol Cell Biol. 2002;22:5380–5394. doi: 10.1128/MCB.22.15.5380-5394.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. RACK1 mediates activation of JNK by protein kinase C [corrected] Mol Cell. 2005;19:309–320. doi: 10.1016/j.molcel.2005.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grollman AP. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J Biol Chem. 1967;242:3226–3233. [PubMed] [Google Scholar]
- 24.Zhou HR, He K, Landgraf J, Pan X, Pestka JJ. Direct activation of ribosome-associated double-stranded RNA-dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: a new model for ribotoxic stress response induction. Toxins (Basel) 2014;6:3406–3425. doi: 10.3390/toxins6123406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampieri CL, Nuttall RK, Young DA, Goldspink D, Clark IM, Edwards DR. Activation of p38 and JNK MAPK pathways abrogates requirement for new protein synthesis for phorbol ester mediated induction of select MMP and TIMP genes. Matrix Biol. 2008;27:128–138. doi: 10.1016/j.matbio.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Magun BE. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J Biol Chem. 1998;273:15794–15803. doi: 10.1074/jbc.273.25.15794. [DOI] [PubMed] [Google Scholar]
- 27.Kim HD, Kim TS, Kim J. Aberrant ribosome biogenesis activates c-Myc and ASK1 pathways resulting in p53-dependent G1 arrest. Oncogene. 2011;30:3317–3327. doi: 10.1038/onc.2011.47. [DOI] [PubMed] [Google Scholar]
- 28.Schneider-Poetsch T, Ju J, Eyler DE, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu S, Kumar KU, Kaufman RJ. Identification and requirement of three ribosome binding domains in dsRNA-dependent protein kinase (PKR) Biochemistry. 1998;37:13816–13826. doi: 10.1021/bi981472h. [DOI] [PubMed] [Google Scholar]
- 30.Simsek D, Tiu GC, Flynn RA, et al. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell. 2017;169:1051–1065 e18. doi: 10.1016/j.cell.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grosso S, Volta V, Vietri M, Gorrini C, Marchisio PC, Biffo S. Eukaryotic ribosomes host PKC activity. Biochem Biophys Res Commun. 2008;376:65–69. doi: 10.1016/j.bbrc.2008.08.118. [DOI] [PubMed] [Google Scholar]
- 32.Bae H, Gray JS, Li M, Vines L, Kim J, Pestka JJ. Hematopoietic cell kinase associates with the 40S ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol Sci. 2010;115:444–452. doi: 10.1093/toxsci/kfq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauter KA, Magun EA, Iordanov MS, Magun BE. ZAK is required for doxorubicin, a novel ribotoxic stressor, to induce SAPK activation and apoptosis in HaCaT cells. Cancer Biol Ther. 2010;10:258–266. doi: 10.4161/cbt.10.3.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou HR, Lau AS, Pestka JJ. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol Sci. 2003;74:335–344. doi: 10.1093/toxsci/kfg148. [DOI] [PubMed] [Google Scholar]
- 35.Zhou HR, Jia Q, Pestka JJ. Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the SRC family kinase Hck. Toxicol Sci. 2005;85:916–926. doi: 10.1093/toxsci/kfi146. [DOI] [PubMed] [Google Scholar]
- 36.Laskin JD, Heck DE, Laskin DL. The ribotoxic stress response as a potential mechanism for MAP kinase activation in xenobiotic toxicity. Toxicol Sci. 2002;69:289–291. doi: 10.1093/toxsci/69.2.289. [DOI] [PubMed] [Google Scholar]
- 37.Kim HD, Kim TS, Joo YJ, et al. RpS3 translation is repressed by interaction with its own mRNA. J Cell Biochem. 2010;110:294–303. doi: 10.1002/jcb.22537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.