

Abstract
Ovarian cancer is the 5th most common cancer in UK women with a high relapse rate. The overall survival for ovarian cancer has remained low for decades prompting a real need for new therapies. Recurrent ovarian cancer remains confined in the peritoneal cavity in >80% of the patients, providing an opportunity for locoregional administration of novel therapeutics, including gene and viral therapy approaches. Immunotherapy is an expanding field, and includes oncolytic viruses as well as monoclonal antibodies, immune checkpoint inhibitors, and therapeutic vaccines.
Oncolytic viruses cause direct cancer cell cytolysis and immunogenic cell death and subsequent release of tumor antigens that will prime for a potent tumor-specific immunity. This effect may be further enhanced when the viruses are engineered to express, or coadministered with, immunostimulatory molecules. Currently, the most commonly used and well-characterized vectors utilized for virotherapy purposes are adenoviruses. They have been shown to work synergistically with traditional chemotherapy and radiotherapy and have met with success in clinical trials. However, pre-existing immunity and poor in vivo models limit our ability to fully investigate the potential of oncolytic adenovirus as effective immunotherapies which in turn fosters the need to develop alternative viral vectors. In this review we cover recent advances in adenovirus-based oncolytic therapies targeting ovarian cancer and recent advances in mapping immune responses to oncolytic virus therapies in ovarian cancer.
Keywords: immunnotherapy, oncolytic virus, ovarian cancer
Introduction
Ovarian cancer
Ovarian cancer (OC) is the 5th most common cancer in UK women1 and the most lethal gynaecological malignancy owing to non-specific symptoms, lack of screening tests, and advanced stage diagnosis.2 Epithelial ovarian cancer (EOC) represents approximately 90% of all OC1 and consist of the following subtypes: clear cell (10%), endometrioid (10%), mucinous invasive (3%), low grade serous (<5%), and high grade serous (70%). These all share a common anatomical location yet have differing clinicopathologic features and sites of origin.3,4 Clear cell ovarian cancer is the second most common subtype of OC. Endometriosis, a disease characterized by the ectopic growth of endometrial glands and stroma, is a risk factor for this histotype.5 The prognosis for clear cell carcinoma is worse than high grade serous ovarian cancer (HGSOC) due to poorer sensitivity to platinum-based chemotherapy6 with patient response being 15%.7 Similar to clear cell OC, endometrioid OC is also associated with endometriosis.3 Invasive mucinous cancer is often a result of metastasis to the ovary from the gastrointestinal tract, including the colon, appendix, or stomach.3 Non-epithelial malignancies of the ovary like germ cell, sex cord stromal tumors, and granulosa cell tumor (sertoli leydig cell tumor) account for ∼10% of all ovarian cancers.8
High-grade serous ovarian cancer is the most common histologic subtype of ovarian cancer. Early detection of the disease is one of the biggest challenges due to lack of early symptoms of the disease and late diagnosis with metastasis disseminated in the peritoneal cavity (peritoneal carcinomatosis).9,10 Transvaginal ultrasound and assessment of the blood cancer antigen 125 (CA125) levels are the current methods for early detection of HGSOC, but their effectiveness has been very limited.11,12 Accounting for approximately 70% of OC deaths,13,14 the 5-year survival rate has remained unchanged for over 50 years at a dismal 30%.4,14 There is compelling evidence to suggest that HGSOC may arise from secretory epithelial cells of the distal fallopian tube.13,15 A gene expression study published in 2008 reported that the expression profiles of HGSOC more closely resemble fallopian tube epithelium than ovarian surface epithelium (OSE).16 This is debated by others who have reported cortical inclusion cysts developing from the OSE as likely progenitors.4 Mullerian inclusions in the pelvic cavity and precursor cells of the ovary have also been proposed as the site of origin.15,17
Management of HGSOC involves total abdominal hysterectomy and bilateral salpingo-oophorectomy, followed by platinum-based chemotherapy, typically 3-weekly carboplatin in combination with paclitaxel for 6 cycles.2,18 Despite high (∼80%) initial response to treatment,19 >70% of patients relapse. Early peritoneal and pleural spread is a feature of HGSOC with a majority of patients presenting with Federation of Gynecology and Obstetrics stage IIC or IV disease, and, in these cases where the patients are not suitable for debulking surgery, 3 cycles of platinum-based neo-adjuvant chemotherapy followed by interval debulking surgery and adjuvant chemotherapy is the accepted approach.20 Recurrent ovarian cancer remains confined in the peritoneal cavity in >80% of the patients, providing an opportunity for locoregional administration of novel therapeutics, including gene and viral therapy approaches. Due to the current limitations of the established treatments for HGSOC there is an urgent need to develop new therapies to improve overall survival. Recently, the development of oncolytic viruses has shown great promise in treating a broad range of cancers including ovarian cancers.
Introduction to oncolytic viruses
The term oncolytic viruses (OV) is commonly employed to identify a viral strain which selectively infect and lyse tumor cells, spreading to adjacent tumor cells resulting in a self-sustaining cycle of anticancer activity. Cell lysis following viral infection, will depend on the virus used and the dose and results in cell death via various mechanisms including apoptosis, autophagy, pyroptosis, and necrosis.21 As a result, the cell death of targeted cancers is often immunogenic, releasing cancer-specific antigens leading to systemic anti-tumor immune responses.22
Both ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) viruses have been used to treat ovarian cancer. The RNA viruses selected typically display intrinsic tumor cell selectivity, while DNA viruses are engineered to have tumor selectivity. OV can be selected which either naturally target cell surface receptors aberrantly expressed on tumor cells or they can be engineered to target these receptors. Additionally, OV can also be engineered to exploit abnormal signaling pathways which are often characteristic of cancer cells. Many OV are further modified with the aim to improve their selectivity, potency, and side effect profile (Table 1).
Table 1.
Examples of oncolytic DNA and RNA viruses in cancer research
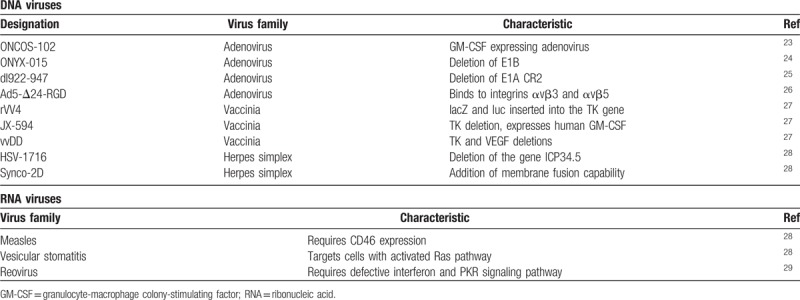
Herpes simplex virus (HSV) and adenoviruses are 2 of the most frequently studied types of OV in clinical trials. These DNA viruses are genetically engineered to have intrinsic tumor cell selectivity. For example, H101, the first government approved OV, is an adenovirus and was generated by deletion of the E1B and E3 genes. E1B inactivates p53, therefore, H101 should be unable to replicate and lyse normal cells, where p53 is active. Whereas cancer cells are typically deficient in p53, allowing H101 replication.30 Another example is T-VEC, a modified herpes simplex virus 1 (HSV-1) that has undergone a Phase III trial for the treatment of melanoma. It was developed by the deletion of nonessential genes including ICP34.5. The function of ICP34.5 includes evasion of the host antiviral response mediated by protein kinase R, frequently defective in tumor cells. This virus was shown, in stage IIIB to IV melanoma, to improve overall response rate, and overall survival.24,31 The vaccinia virus Pexa-Vec pexastimogene devacirepvec JX-594 is also an example of an oncolytic virus being tested in clinical trials against several solid tumors. It is a fourth generation targeted, armed oncolytic, and immunotherapeutic virus with disruption of the viral thymidine kinase gene,32,33 affecting the virus ability to synthesise deoxyribonucleotides and to replicate in normal cells where the reserves of deoxyribonucleotides are low compared with cancer cells.34 It is a virus engineered to express human granulocyte-macrophage colony-stimulating factor (hGM-CSF),32 which plays an important role in the maturation of specialized antigen-presenting cells.
RNA viruses have inherent tumor selectively. They may be further engineered to have more potent anti-tumor properties or to remove pathogenic activity. Reovirus, for example, begins transcription in non-cancerous cells by producing viral RNAs that lead to the activation of the PKR pathway. This pathway is important in the normal cellular anti-viral response. Reovirus therefore can target RAS-transformed cancer cells where the PKR pathway is blocked. Another RNA virus investigated as an OV is measles. Measles viruses are known to enter cells via CD46 receptor, this receptor is often over-expressed in prostate and colorectal cancerous cells. However, measles infection in humans is clinically significant and therefore modified non-pathogenic versions are required if they are to be used as OVs. An example of this is the Edmonston strain, used in vaccination, although clearly there is an issue with its use given the prevalence of immunity through vaccination.35
OVs can enter both normal and cancerous cells, although this may be less significant in normal cells if the OV in question exploits adherent expression of cell surface receptors. However once inside a cell, the ability of an OV to replicate will be reduced in a normal cell due to its anti-viral response. Normal cells are able to mount an anti-viral response, through a variety of signaling pathways, which act to detect and remove viral pathogens or induce apoptosis of the cell.21
Recent advances in oncolytic adenovirus-based therapies in ovarian cancer
Currently the most commonly used and well-characterized vectors utilized for virotherapy purposes are adenoviruses. Human adenoviruses have been extensively studied in patients with malignancies and have demonstrated no severe side effects. Due to their intrinsic biological plasticity, adenoviruses are amenable to manipulation. Serotype 5 (Ad5) in particular has proven to be an attractive vector, allowing for a variety of modifications and efficient infectivity without being associated with a serious disease.36 For optimal oncolytic activity, adenovirus are required to specifically and efficiently infect and replicate within cancer cells. The replication of human adenoviruses is initiated by E1A which is responsible for dissociating the retinoblastoma (Rb)/E2F complex. This dissociation produces free transcription factor E2F activation thereby allowing the transcription of the adenoviral E1B, E2, E3, and E4 genes.37
There are 2 broad strategies to engineer conditionally replicative adenoviruses (CRAd). Firstly, the deletions of part of the E1A and E1B genome sequence impair replication in normal cell. Typically, Ad5-based CRAd contain a 24 base pair mutation in the E1A gene disrupting the Rb binding domain resulting in an E1A protein unable to release E2F and support viral replication. In tumor cells, however, genetic defects often result in the deregulation of Rb pathway thereby complementing the deleted viral genome functions and allowing conditional replication. The second strategy relies on placing the adenoviral genes under the control of a tumor-specific or tissue-specific promoter in order to achieve selective replication in cancer cells.
Enhancing CRAd infectivity and replication
One of the main limitations of CRAd, as well as other vectors used in virotherapy, is poor or inefficient transduction into target cells due in part to low specificity, high pre-existing immunity to vectors, and poor dissemination of the vector. In the case of Ad5-based CRAd, infection is mediated by the high affinity Coxsackie and Adenovirus Receptor (CAR) and ανβ3 and ανβ5 integrins.38,39 However, recent evidence has revealed that CAR expression is highly variable across both cancer types and disease progression. CAR expression has been shown to be low in ovarian, breast, lung, and bladder cancer cells.40–42 In contrast CAR expression was reported to be upregulated in endometrium and cervix cancers as well as in breast and lung cancers.43–46 Furthermore, infection levels have been shown to correlate the expression of CAR and ανβ3 and ανβ5 in ovarian cancer cells.47–49
One approach to improve adenovirus transduction is to combine virotherapy with new drug therapies to modulate CAR expression on target cells. Ma et al50 have demonstrated that the histone deacetylase inhibitor trichostatin (TSA) can increase CAR protein levels in human oesophageal squamous cell carcinoma which ordinarily express low levels of CAR. There is also evidence that TSA upregulates CAR expression in ovarian carcinomas.51 However the variability of CAR expression across tissue and disease types makes it more attractive to select or modify adenoviral vectors in order to bypass CAR in favor of different receptors to improve cellular transduction.
An alternative approach has been to modify the fiber knobs of adenovirus vectors and create chimeric vectors. Human adenoviruses are classified into 6 distinct subgroups (A–F) with Ad5 belonging to subgroup C. One example of a chimeric adenovirus vector is the Ad5/3 fiber chimera. The fiber knob domain of serotype 3 (from subgroup B) binds primarily to CD46 and to CD80 and CD86 rather than to CAR.52,53 Kanerva et al54 have shown that this chimeric vector displays improved cytopathy in ovarian cancer cells compared with the wildtype Ad5. Additionally, the further modified Ad5/3-Delta24 chimera demonstrated enhanced cytopathic activity in murine orthotopic models of ovarian cancer.55 More recently, another Ad5-chimeric vector has been engineered by pseudotyping Ad5 with the fiber knob domain from Ad48 (from subgroup D) called Ad5/kn48. Uusi-Kerttula et al56 showed that this vector utilized CAR but not CD46 for cell entry. The vector was then further modified by the insertion of 20 to 11 peptide to redirect the vector to ανβ6 integrin. By targeting the prognostic cancer cell marker ανβ6 instead of CAR, the vector transduction was improved by ∼60 fold in epithelial ovarian cancer cells. Using the less seroprevalent Ad48 also allowed for the circumvention of pre-existing anti-Ad5 immunity. Similarly, Hulin-Curtis and colleagues modified the Ad5 vector by pseudotyping it with the Ad35 fiber thereby achieving higher transduction via CD46 into primary epithelial ovarian cancer cells despite the presence of neutralizing antibodies and factors.57
Alongside the creation of chimeric vectors, specific modifications of adenoviral constructs as proved effective in enhancing CRAd. Incorporating an RGD motif into the fiber knob domain has been shown to improve the infection of both primary ovarian cancer cells and ovarian cancer cell lines by adenovirus vectors despite low CAR expression.58 More recently Bauerschmitz et al59 demonstrated that the insertion of the RGD motif redirected viral interactions to integrins highly expressed by ovarian cancer cells, allowing CAR-independent infection enhanced oncolytic activity in orthotopic murine models of ovarian cancer. Gamble et al60 went further and engineered an Ad5 vector containing an RGD motif in both the fiber motif and the capsid protein XI. This new vector demonstrated greater oncolytic activity in vitro in ovarian cancer cell lines. Ad5-delta24-RGD vectors have shown promise in early, phase I, clinic trials.61
A different approach to improve CRAd efficacy has been to modify vectors using tissue specific promoters (TSP) in order to enhance viral replication. Rein et al62 modified an Ad5/3 chimeric vector to place E1A under the control of the TSP secretory leukoprotease inhibitor (SLPI). This Ad5/3SLPI vector displayed efficient replication and oncolytic activity in primary ovarian cancer cells, orthotopic murine models, and multiple cell lines. Additionally, this novel vector showed reduced liver toxicity increasing its potential for clinical use. Other CRAd exploiting TSP include a chimeric Ad5/3 and Ad5-RGD vector which utilize the chemokine receptor CXCR4, as well as a CRAd-S.F5/3 modified to include the surviving promoter. All of these vectors have demonstrated increased selectivity for ovarian cancer cells and improved oncolysis in ovarian cancer cell lines and murine models along with decreased hepatic toxicity.63,64
Improving adenovirus efficacy by arming vectors
In order to improve the anti-tumor efficacy of CRAd, many vectors are being modified to encapsulate therapeutic transgenes. “Arming” viral vectors in this way is designed to enhance direct cell killing, modulate tumor microenvironment (TME), and tailor immune responses. The adenoviral genome can be readily manipulated, however it can only encompass 105% of the wild-type genome, therefore, limiting the size of any inserts.65 Furthermore, any inserts must not interfere with normal viral replication.
Influencing the TME can allow viral vectors to overcome certain physical barriers that limit their effectiveness. Due to dense stromal tissue and high levels of extracellular matrix (ECM), ovarian tumors are characterized by high fluid pressures which inhibits the dissemination of CRAd limiting their efficacy. Novel CRAd armed with relaxin66 and hyaluronidase67 target the ECM in tumors and have shown promising results in oral squamous cell carcinomas and malignant gliomas. Conversely, CRAd have also been armed with tissue inhibitors of metalloproteinases (TIMP) to inhibit the degradation of the ECM to inhibit tumor invasion and proliferation. The Ad3/5-TIMP2 vector demonstrated enhanced replication and killing in primary ovarian cancer cells.68
In order to limit tumor proliferation, angiogenesis has also been targeted. An adenovirus construct was developed expressing a short hairpin RNA against VEGF (Ad-deltaB7-shVEGF) which elicited greater anti-tumor efficacy in human glioma xenografts.69 Similarly, an adenovirus vector (AdEHE2F) encoding soluble Flt-1 and Dll4 to neutralize VEGF and Notch signaling, respectively displayed enhanced anti-tumor effects and dramatic reductions in total vasculature in breast cancer.70
CRAd developed to target chemo-resistant cells or reverse resistance
A major limitation of current treatments for ovarian cancer is the development of chemotherapy resistance. Consequently, the development of novel CRAd aimed at ovarian cancers center on preferentially targeting resistant cells or reversing resistance and resensitizing cells to chemotherapy agents (Table 2). The mechanisms through which ovarian cancers acquire resistance to chemotherapy agents are still under debate.
Table 2.
Major reported strategies to enhance effectivity and the ability to overcome chemo-resistance and modulate immune response of adenovirus and vaccinia based oncolytic viruses
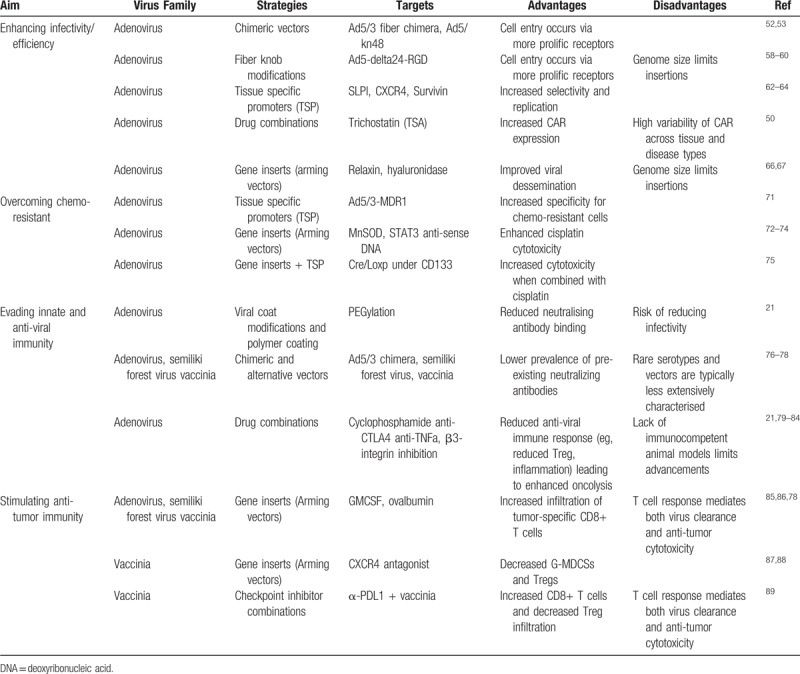
Multidrug resistance gene 1 (MDR1) overexpression is believed to be a major factor limiting anticancer drugs. Rein et al71 targeted chemotherapy resistant ovarian cancer cells by combining the TSP MDR1 (multidrug resistance gene 1) and an Ad5/3 chimeric vector. The resulting Ad5/3MDR1 constructs were shown to be a promising therapy to specifically target chemotherapy resistant cancer cells by displaying efficient oncolysis in chemo-resistant ovarian cancer cell lines, primary tumors, and orthotropic murine models.
CD133+ ovarian cancer cells have been identified as cancer stem cells which contribute to recurrence and chemo-resistance.90 Long et al75 modified an adenovirus vector to place the Cre/Loxp system under the control of a CD133 promoter to develop a new “suicide” gene therapy for ovarian cancer stem cells. They demonstrated that this new therapy initiated pro-apoptotic signaling pathways enhancing the cytotoxicity of cisplatin and promoted marked tumor suppression in xenografts. Targeting elements of the apoptotic pathway has proved to be an effective strategy to resensitise cancers to chemotherapy. Luan et al91 targeted the p53 upregulated modulator of apoptosis PUMA in ovarian cancer cell lines and increased their chemo sensitivity. Similarly, Wang et al72 increased the efficacy of cisplatin by arming an adenovirus with a manganese superoxide dismutase (MnSOD). They demonstrated that an overexpression of MnSOD resulted in a remarkable induction of apoptosis and synergised with and sensitized cells to cisplatin in ovarian cancer cells to suppress tumor growth in both in vivo and in vitro experiments.
Han et al74 were also able to enhance cisplatin cytotoxicity and reverse resistance in human ovarian cancer by targeting the signal transducer and activator of transcription-3 (STAT3). STAT3 is highly expressed in ovarian cancer and is associated with the development of resistance73,74 and by engineering an Ad5 vector to express high levels of anti-sense STAT3 DNA, they were able to selectively deplete STAT3 in ovarian cancer. This new therapy reversed cisplatin resistance in vitro and significantly enhanced cisplatin-induced apoptosis in vivo.
Clinical trials of oncolytic virus in ovarian cancer
There are numerous OV currently under investigation as potential therapeutic agents, however, to date only 1 OV has been government approved. This virus, approved in China in 2005 for nasopharyngeal cancer, is a modified adenovirus. Another herpes simplex OV, T-VEC, is expected to be approved by the Food and Drug Administration (FDA) for treatment of melanoma. They have been shown to work synergistically with traditional chemotherapy and radiotherapy.24 OV are typically administered via intratumoral or intravenous injection (Table 3).
Table 3.
Major considerations between delivery methods of oncolytic viruses in ovarian cancer
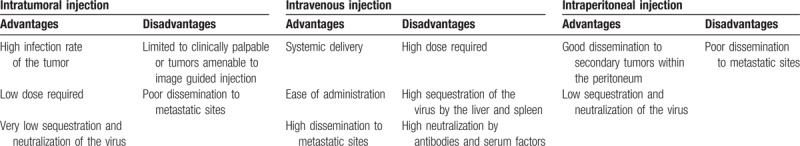
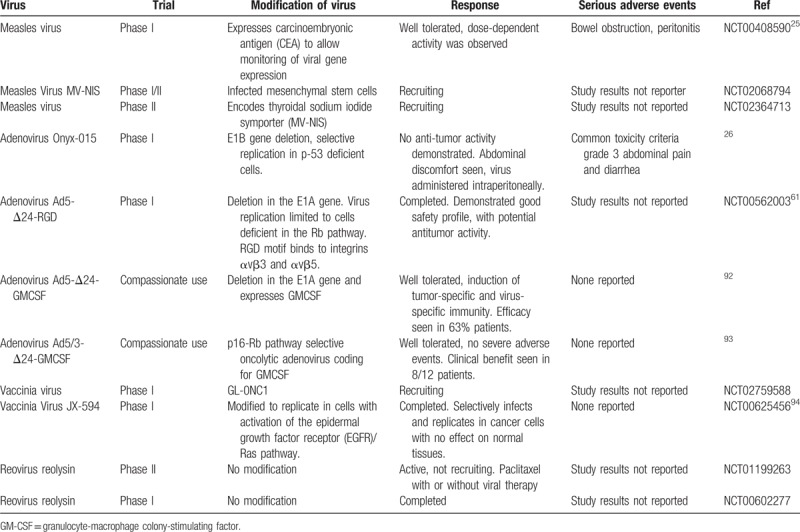
Intratumoral injection can only be given in those tumors which are either clinically palpable or amenable to imaging guided injection. Intravenous administration can be limited by sequestration of the virus by the liver and spleen.29 Although OV have yet to be approved for the treatment of ovarian cancer they have met with positive outcomes in clinical trials. Past and current clinical trials using oncolytic viruses in ovarian cancer are summarized in the table below (Table 4).
Table 4.
Oncolytic virus used in clinical trials to treat patients with ovarian cancer.
Oncolytic virus and the immune system
The effectiveness of OV depends on various factors including the TME, alterations in tumor cell signaling pathways, and expression of immunosuppressive factors as well as the innate and adaptive immune response induced by the virus.21,22 Of the innate and adaptive immune system, it is thought that the adaptive immune system plays the most important role in the anti-tumor immunity induced by OV. While tumors develop many ways to evade and dampen immune responses, OV driven cell death is immunogenic as new tumor-specific antigens are released following cell lysis. These new circulating antigens are then free to stimulate systemic anti-tumor immune response which can lead to tumor regression at sites not infected by the OV.
While viruses can help to promote an immune response against the tumor cells, on the other hand, neutralising antiviral responses may block virus replication and further infection of other tumor cells.21 Therefore, the anti-tumoral immune response is a critical part of the action of OV, but the immune activation can also lead to clearance of the virus. Strategies to prevent antibody binding include PEGylation of the viral coat and polymer coating. Additionally, low dose cyclophosphamide has been used to successfully reduce numbers of regulatory T cells (Tregs), while not impairing the cytotoxic T cell response.21,95 Tregs are also important in the prevention of tumor associated immunity. In ovarian cancer, Tregs in tumor infiltrating lymphocytes (TILs) are associated with a poor prognosis. Whereas the presence of other TILs is associated with delayed recurrence of epithelial ovarian cancer. The presence of tumor infiltrating CD3+ cells in ovarian cancer has also been found to increase patient survival and improve response to chemotherapy.29,96,97
Immune checkpoint inhibitors are able to activate lymphocytes within tumors. Immune checkpoints exist to modulate the immune response and are important in maintaining self-tolerance, however, tumors can use these pathways to aid immune resistance. The first class of immune checkpoint inhibitors approved by the FDA were cytotoxic T-lymphocyte associated antigen 4 (CTLA4) antibodies. CTLA4 is expressed by T cells and is involved in regulating the amplitude of T cell activation. Programmed cell death protein 1 (PD1) and its ligands (PDL1 and PDL2) are involved in limiting T cell activity during an inflammatory response. Immune checkpoint inhibitors could lead to a reduction of Treg numbers, and patients most likely to benefit from treatment with immune checkpoint inhibitors are those with pre-existing TILs.
Therefore, the combination of OV and immune checkpoint inhibitors is an exciting area of research. Several studies have shown that the combination of Newcastle Disease Virus (NDV) with CTLA-4 blockade in tumor bearing mice leads to regression of tumors, including those at sites distal to initial OV injection, and greater long-term survival.79–81 However, a significant limitation in OV research is the lack of immunocompetent animal models. The majority of research is carried out in immunosuppressed mice growing human tumors. This means our understanding of the immune response following OV treatment is incomplete, and it is vital to improve our understanding of this effect to further improve OV efficacy.82
Harnessing the immune component and immune limitation of adenovirus
A major advantage of oncolytic viral therapies is their unique ability to stimulate host immune responses against cancers through the release of cancer specific antigens following immunogenic cell death. These effects can be further enhanced by arming CRAd with immune-modulatory compounds and transgenes. The attempts to influence the immune response in relation to CRAd fall broadly into 2 categories, firstly in an effort to minimize and bypass the host immune response to the viral vector itself in order to enhance the efficiency of the therapy and secondly to elicit an immune response targeted towards the cancer cells to maximize cell killing and removal.
Several strategies have shown success in altering immune response to enhance the efficacy of CRAd in ovarian cancer. Combining CRAd therapies with a pharmacological inhibition of tumor necrosis factor alpha (TNF-α) has been shown to increase adenovirus activity in ovarian cancer cell lines through the suppression of apoptosis inhibitors (cIAP1 and cIAP2).83 The inflammatory response initiated by CRAd was further target by Browne et al84 by inhibiting β3-integrins in ovarian cancer xenograft models thereby showing that CRAd therapies in β3 knockout mice released significantly less pro-inflammatory cytokines and presented with lower inflammatory hepatic toxicity. In a different approach, Ashshi et al98 were able to improve virus-induced cytotoxic effects by overexpression IL24, a member of the anti-inflammatory IL10 family, by arming a CRAd with IL-24. CRAd-IL24 infection resulted in significantly higher yields of infectious particles which translated into enhanced efficacy.
One of the most successful CRAd engineered to stimulate an immune response in ovarian cancer has been ONCOS-102, an Ad5/3 chimeric adenovirus expressing granulocyte-macrophage colony stimulating factor GM-CSF. ONCOS-102 has undergone clinical trials in which the treatment resulted in a progressive infiltration and activation of tumor-specific CD8+ T cells into the tumors of selected patients while the majority of patients showed a short-term increase in systemic pro-inflammatory cytokines and a marked increase in tumor-infiltrating lymphocytes.23,85 A recent study has demonstrated that the efficacy of CRAd is mediated by T-cell responses which regulate both the clearance of viruses and enhancing anti-tumor cytotoxicity.86 These findings indicate that influencing T-cell responses is a delicate balance able to both enhance and limit CRAd effectivity.
The rapidly emerging evidence that oncolytic viruses are a valuable tool beyond their ability to selectively and directly kill cancers cells but also to induce anticancer immune responses has been guiding the field to study the immunological impact of CRAd in greater detail. However, adenoviruses present with 2 major limitations concerning immunity. Firstly, the majority of most populations will have been infected with an adenovirus at some point, often early on, in life and have developed pre-existing immunity to common adenovirus serotypes. This pre-existing immunity is manifested by the presence of neutralizing antibodies that limit future therapies. The prevalence of antibodies against the Ad5 serotype in particular is high in humans with a prevalence of 50% to 90% in the developing world.76,77 Secondly, adenoviruses are unable to replicate in mouse tissue, as a result in vivo models are limited and determining the involvement of immune components in CRAd therapies is challenging.
Other oncolytic viruses used to boost the immune system against ovarian cancer
Alternatives to adenovirus-based oncolytic viruses such as measles, herpes simplex virus, reovirus, vaccinia virus, and sindbis viruses have also been employed for virotherapy as they have shown to target and lyse the tumor cells directly.78 Unlike adenoviruses, these alternative OV lend themselves more readily to studies elucidating the role of the immune system in virotherapies.34 Advanced metastatic tumors are often immunosuppressive and therefore challenging to treat with conventional immunotherapy, which is the case with ovarian cancer of epithelial origin. The therapeutic benefit of oncolytic viruses causing direct cytolysis and cancer-specific immunity has been demonstrated in several preclinical studies and preclinical studies have further demonstrated that the combination of oncolytic viruses and antigen-specific immunotherapy leads to enhanced anti-tumor effects against ovarian tumors.
As previously mentioned, the immune response to oncolytic viruses can be a double-edged sword, on one hand, they can promote an immune response against the tumor cells but, on the other hand, neutralizing antiviral responses may block virus replication and ongoing infection of tumor cells. A study aiming to circumvent the limitation of neutralizing antibodies against oncolytic viruses demonstrated that intercalating the treatment of ovarian tumor-bearing mice with semiliki forest virus (SFV) and vaccinia virus (VV) led to mice prolonged survival compared with mice re-infected several times with the same virus. To further increase the immunological antitumor effects of the viruses, authors added the foreign antigen—ovalbumin bound (OVA) to SFV and VV. Immunocompetent C57BL/6 mice were intraperitoneally (i.p.) injected with MOSEC ovarian tumor cells and then infected with SFV-OVA or VV-OVA and vice versa, resulting in a significant enhancement of OVA-specific CD8+ T-cell immune response in the peritoneal washes as well as the spleens, suggesting accumulation of antigen-specific T-cells around the tumor site, which further increased the enhanced anti-tumor effects against ovarian cancer compared with infection with either virus.78
Using a virally-delivered CXCR4 antagonist to block the CXCL12/CXCR4 axis in combination with doxorubicin has been shown to increase survival in ovarian tumor-bearing mice by reversing the immunosuppressive phenotype of the TME while promoting antitumor immunity. In this study, the treatment of both orthotopic murine ID8-R and human CAOV2-R drug-resistant ovarian tumors in syngeneic C57/BL6 with an armed VV expressing the CXCR4 antagonist 12 hours before doxorubicin revealed reduction of metastatic spread and tumor growth in comparison to monotherapy treatment modalities. This effect was correlated with reduction of intraperitoneal recruitment of granulocyte-like myeloid derived suppressor cells granulocyte-like myeloid derived suppressor cells (G-MDSCs) (CD11+Ly6GhighLy6Clow) and Tregs (CD4+CD25+Foxp3+) and increase of CD11c+CD86+ DCs and IL-12-producing monocytes/macrophages, which was associated with higher ratios of interferon gamma (IFN-γ)-producing CD8+ T-cells to Tregs. Similarly, 50% tumor-free survival was observed in CAOV2-R-bearing SCID mice treated with CXCR4-A-Fc VV and doxorubicin compared with 10% observed in mice treated with VV-Fc and doxorubicin, indicating a higher viral replication/oncolysis in immunocompromised mice and a direct effect of CXCR4 antagonist on tumor cells.87
Cancer-initiating cells (CIC) have been demonstrated in clinical and preclinical studies to survive conventional chemotherapies and to give rise to more aggressive, recurrent ovarian tumors.99,100 Therefore, it is important to develop therapies that simultaneously target CIC and the ovarian TME that promotes their growth. In a preclinical study, researchers demonstrated that targeting the CXCL12/CXCR4-signaling axis through an oncolytic vaccinia virus (OVV) resulted in decreased CXCL12 and VEGF expression in ascitic fluid, reduced accumulation of neutrophils/G-MDSCs and increased DCs recruitment into the tumor site of the treated mice. It is known that CXCL12 induces intratumoral localization of CD24+CD25+FoxP3+ Tregs in ovarian cancer but in this study the treatment with a CXCR4 antagonist resulted in significantly lower percentages of tumor-infiltrating Tregs compared with control. The results of this study illustrate the therapeutic efficacy of an armed oncolytic vaccinia virus to target CIC as well as reducing the tumor immunosuppressive network and induction of humoral and cellular responses.88
Cancer cells are able to use immune checkpoints to avoid immune control and rejection, therefore, inhibition of these inhibitory pathways would represent a potent strategy to combat cancer. The combination of an anti α-PD-L1 to block PD1/PD-L1 immune checkpoint binding and an oncolytic vaccinia virus to activate anticancer immune response was tested by Liu et al.89 In this study, a monotherapy with VV or α-PD-L1 decreased tumor burden but a dual therapy of VV with α-PD-L1 led to a significant reduction of the tumor burden and increased mice survival compared with monotherapies in murine colon and ovarian cancer models. After 13 days of treatment, the dual therapy significantly elevated CD8+ T-cells expression by CD45+ cells, enhanced infiltration of effector T cells, and at the same time reduced Treg cells and exhausted CD8+ T-cells. The ratio of CD8+ T-cells to Treg increased significantly with the dual therapy, as well as the infiltration of IFN-γ+FoxP3−CD4+T cells over FoxP3–CD4+T cells. The levels of killer cell activity markers in the TME such as IFN-γ, granzyme B, and perforin also increased significantly. CD8+, CD4+ T cells, and IFN-γ all played essential roles in the therapeutic efficacy of the dual therapy sustaining an elicited systemic immunity. These data suggest the optimal timing is simultaneous delivery of both agents, and this supports the idea that an anti-PD-L1 approach could be incorporated into the virus without the need for delayed expression of the transgene.89
Another virus-based anticancer therapy that is being used against ovarian cancer is the reovirus which is currently undergoing phase I/II clinical trials for the treatment of ovarian cancer (Table 4). A study lead by Gujar et al101 focused on characterizing the effects of reovirus therapy on ovarian cancer and the associated immune microenvironment, as this virus had proven cell killing activity by direct oncolysis and induction of efficient antitumor immunity in lung,102 melanoma,102 and prostate cancer103 models. A study from the same authors have previously demonstrated that reovirus does not kill non-transformed, normal ovarian cells,104 making it a suitable oncolytic therapy candidate for ovarian cancer. Ovarian cancer-bearing mice had longer survival rates when treated with reovirus compared with control treated mice, accompanied by a substantial alleviation of abdominal distension and reduction of ascetic fluid volume. Additionally, tumors from reovirus-treated mice displayed intratumoral infiltration of immune cells, upregulation of IFN-γ and a favorable modulation of the frequencies of MDSC and Tregs preceding an anti-tumor immunity response. Additionally, this study demonstrated that reovirus therapy postponed peritoneal carcinomatosis when administered at the early stages of ovarian cancer, which at the moment cannot be translated fully to the clinic because most patients are diagnosed when ovarian cancer is already fully developed to its late stages.101
In a study led by Kohno et al105, the HF10 amplicon, a highly attenuated variant of the herpes simplex virus type 1 (HSV-1), containing the murine GM-CSF cytokine (mGM-CSF amplicon) had demonstrated oncosuppressive effects in a murine colorectal tumor model, therefore, they hypothesized that the same mGM-CSF amplicon would also be effective to treat disseminated ovarian cancer in a mouse model. Indeed, the treatment with mGM-CSF of orthotopic tumors of HM-1 cells (ovarian cancer cells) in B6C3F1 mice resulted in significantly prolonged mice survival than did mice treated with either vehicle or HF10 amplicon. There was also increased infiltration of CD4+ and CD8+ T immune cells into the peritumoral layer, concomitant with significant decrease of macrophages infiltration. The mGM-CSF amplicon treatment was also able to sustain an efficient memory immune response demonstrated by the capacity of splenocytes cells from mGM-CSF amplicon-treated group to induce the expression of IFN-γ and TNF-α when cocultured with untreated ovarian cancer HM1 cells, demonstrating once more the anti-tumor immune therapeutic effect of an oncolytic virus armed with an immune transgene.106
Some oncolytic viruses are currently being used to treat patients with ovarian cancer in clinical trials either at the recruiting level or in phase I/II studies (Table 4). An edmonston vaccine strain of measles virus (MV) was engineered to express the marker peptide carcinoembryonic antigen (CEA) to permit real-time monitoring of viral gene expression in tumors in a phase I clinical trial. MV attenuated strains have an excellent safety record and it was demonstrated that MV vaccine strains predominantly enter cells via the CD46 receptor, which is highly expressed in ovarian cancer cells. Patients with taxol and platinum-refractory recurrent ovarian cancer and with normal CEA levels were eligible to determine the safety and tolerability of i.p. administration of MV-CEA up to 109 TCID50. Best objective response was stable disease in 14 of 21 evaluable patients with median duration of 92.5 days and median overall survival of 12.15 months in comparison to the expected median survival of 6 months in this patient population. No evidence of induced immunosuppression was observed following measles vaccination, and all observed toxicities were grade 1 and 2, highlighting the safety of MV as an oncolytic platform and the potential of oncolytic measles therapy in recurrent ovarian cancer patients.25
Conclusion
Most ovarian cancers can be characterized by rapid proliferation, aggressive neovascularization, resistance to chemo- and radiotherapy, and marked local and systemic immunosuppression. All of these factors contribute to high tumor recurrence and current treatment inadequacies. Combining cancer immunotherapy with oncolytic viruses is one of the most promising platforms for the treatment of cancer. To date, oncolytic viruses have shown great promise in mediating direct cancer cell cytotoxicity but beyond this, OV therapies have a unique ability to elicit anti-tumor immune response. Multiple studies described in this review have demonstrated that OV can lead to increased T cell infiltration into ovarian tumors in mice and that this is associated with a favorable clinical outcome. More specifically it is known that the presence of CD8+ cytotoxic T cells delays disease progression and extends overall survival of cancer patients.
Despite significant advances in adenovirus-based therapies, there are still limitations to overcome when using adenovirus in preclinical and clinical studies. Firstly, most of the population will have been infected at some point in their lives with adenovirus, thus presenting with developed, pre-existing immunity when re-infected with this virus. The induction of antiviral immunity to adenoviruses leads to inadequate dissemination of the virus and hampers effective delivery. Secondly, the inability of adenovirus to replicate in mouse tissue presents a major hurdle to developing in vivo models for preclinical studies. The exploration of alternative viral vectors to adenovirus such as vaccinia virus and reovirus will allow for a more comprehensive understanding of how OV therapies can initiate and shape anti-tumor immune responses.
The combination of viral oncolysis and antigen-specific immunity leads to enhanced antitumor effects, culminating in antitumor immune responses that control metastatic growth. Treatment of patients with oncolytic virus will require a well-coordinated strategy that would synergistically kill tumor cells with simultaneous induction of anti-tumor immunity, reduce intratumoral recruitment of immunosuppressive elements like Tregs in favor of immunostimulatory signals (like IL-12), and enhance local tumor-specific T cell accumulation in order to sustain a durable immune response against the tumor.
Oncolytic virus based therapies hold great promise in treating a range of cancers. It will be critical for the further development and refinement of these therapies to follow new avenues beyond adenoviruses to better understand the mechanisms of action of OV-based immunotherapies, which in turn, will lead to the development of efficient combination therapies for future clinical trials.
Acknowledgments
None.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.UK CR. Types of ovarian cancer; 2017. Available at: http://www.cancerresearchuk.org/about-cancer/type/ovarian-cancer/about/types-of-ovarian-cancer. Accessed May/June 2017. [Google Scholar]
- 2.Alkema NG, Wisman GB, van der Zee AG, et al. Studying platinum sensitivity and resistance in high-grade serous ovarian cancer: different models for different questions. Drug Resist Updat 2016; 24:55–69. [DOI] [PubMed] [Google Scholar]
- 3.Vaughan S, Coward JI, Bast RC, Jr, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer 2011; 11:719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol 2013; 24 (suppl):x16–x21. [DOI] [PubMed] [Google Scholar]
- 5.Pearce CL, Templeman C, Rossing MA, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol 2012; 13:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mabuchi S, Sugiyama T, Kimura T. Clear cell carcinoma of the ovary: molecular insights and future therapeutic perspectives. J Gynecol Oncol 2016; 27:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.OVCARE. Types of Ovarian Cancer; 2017. Available at: http://www.ovcare.ca/about_ovarian_cancer/types_of_ovarian_cancer/. Accessed May/June 2017. [Google Scholar]
- 8.Colombo N, Peiretti M, Parma G, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010; 21 (suppl):v23–v30. [DOI] [PubMed] [Google Scholar]
- 9.Montfort A, Pearce O, Maniati E, et al. A strong B-cell response is part of the immune landscape in human high-grade serous ovarian metastases. Clin Cancer Res 2017; 23:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabova L, Yin C, Bupp S, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res 2012; 72:4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cesario S. Advances in the early detection of ovarian cancer: how to hear the whispers early. Nurs Womens Health 2010; 14:222–234. [DOI] [PubMed] [Google Scholar]
- 12.Neesham D. Ovarian cancer screening. Aust Fam Physician 2007; 36:126–128. [PubMed] [Google Scholar]
- 13.Bowtell DD, Bohm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015; 15:668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreuzinger C, Gamperl M, Wolf A, et al. Molecular characterization of 7 new established cell lines from high grade serous ovarian cancer. Cancer Lett 2015; 362:218–228. [DOI] [PubMed] [Google Scholar]
- 15.Perets R, Wyant GA, Muto KW, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013; 24:751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tone AA, Begley H, Sharma M, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin Cancer Res 2008; 14:4067–4078. [DOI] [PubMed] [Google Scholar]
- 17.Folkins AK, Saleemuddin A, Garrett LA, et al. Epidemiologic correlates of ovarian cortical inclusion cysts (CICs) support a dual precursor pathway to pelvic epithelial cancer. Gynecol Oncol 2009; 115:108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchetti C, Pisano C, Facchini G, et al. First-line treatment of advanced ovarian cancer: current research and perspectives. Expert Rev Anticancer Ther 2010; 10:47–60. [DOI] [PubMed] [Google Scholar]
- 19.Wiedemeyer WR, Beach JA, Karlan BY. Reversing platinum resistance in high-grade serous ovarian carcinoma: targeting BRCA and the homologous recombination system. Front Oncol 2014; 4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bohm S, Montfort A, Pearce OM, et al. Neoadjuvant chemotherapy modulates the immune microenvironment in metastases of tubo-ovarian high-grade serous carcinoma. Clin Cancer Res 2016; 22:3025–3036. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14:642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res 2014; 2:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vassilev L, Ranki T, Joensuu T, et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8+ T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015; 4:e1017702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33:2780–2788. [DOI] [PubMed] [Google Scholar]
- 25.Galanis E, Hartmann LC, Cliby WA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res 2010; 70:875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vasey PA, Shulman LN, Campos S, et al. Phase I trial of intraperitoneal injection of the E1B-55-kd-gene-deleted adenovirus ONYX-015 (dl1520) given on days 1 through 5 every 3 weeks in patients with recurrent/refractory epithelial ovarian cancer. J Clin Oncol 2002; 20:1562–1569. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Tong J, Rahman MM, et al. Oncolytic virotherapy for ovarian cancer. Oncolytic Virother 2012; 1:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartkopf AD, Fehm T, Wallwiener D, et al. Oncolytic virotherapy of gynecologic malignancies. Gynecol Oncol 2011; 120:302–310. [DOI] [PubMed] [Google Scholar]
- 29.Aghi M, Martuza RL. Oncolytic viral therapies - the clinical experience. Oncogene 2005; 24:7802–7816. [DOI] [PubMed] [Google Scholar]
- 30.Lei J, Li QH, Yang JL, et al. The antitumor effects of oncolytic adenovirus H101 against lung cancer. Int J Oncol 2015; 47:555–562. [DOI] [PubMed] [Google Scholar]
- 31.Hughes T, Coffin RS, Lilley CE, et al. Critical analysis of an oncolytic herpesvirus encoding granulocyte-macrophage colony stimulating factor for the treatment of malignant melanoma. Oncolytic Virother 2014; 3:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MK, Breitbach CJ, Moon A, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med 2013; 5:185ra63. [DOI] [PubMed] [Google Scholar]
- 33.Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013; 19:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al Yaghchi C, Zhang Z, Alusi G, et al. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy 2015; 7:1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012; 30:658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu RL, Post DE, Khuri FR, et al. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin Cancer Res 2004; 10:5299–5312. [DOI] [PubMed] [Google Scholar]
- 37.Nevins JR. Transcriptional regulation. A closer look at E2F. Nature 1992; 358:375–376. [DOI] [PubMed] [Google Scholar]
- 38.Louis N, Fender P, Barge A, et al. Cell-binding domain of adenovirus serotype 2 fiber. J Virol 1994; 68:4104–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997; 275:1320–1323. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Thorne S, Hannock J, et al. A novel assay to assess primary human cancer infectibility by replication-selective oncolytic adenoviruses. Clin Cancer Res 2005; 11:351–360. [PubMed] [Google Scholar]
- 41.Wunder T, Schmid K, Wicklein D, et al. Expression of the coxsackie adenovirus receptor in neuroendocrine lung cancers and its implications for oncolytic adenoviral infection. Cancer Gene Ther 2013; 20:25–32. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K, Shariat SF, Ayala GE, et al. Loss of coxsackie and adenovirus receptor expression is associated with features of aggressive bladder cancer. Urology 2005; 66:441–446. [DOI] [PubMed] [Google Scholar]
- 43.Dietel M, Hafner N, Jansen L, et al. Novel splice variant CAR 4/6 of the coxsackie adenovirus receptor is differentially expressed in cervical carcinogenesis. J Mol Med (Berl) 2011; 89:621–630. [DOI] [PubMed] [Google Scholar]
- 44.Giaginis CT, Zarros AC, Papaefthymiou MA, et al. Coxsackievirus and adenovirus receptor expression in human endometrial adenocarcinoma: possible clinical implications. World J Surg Oncol 2008; 6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin TA, Watkins G, Jiang WG. The Coxsackie-adenovirus receptor has elevated expression in human breast cancer. Clin Exp Med 2005; 5:122–128. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Wang S, Bao Y, et al. Coxsackievirus and adenovirus receptor expression in non-malignant lung tissues and clinical lung cancers. J Mol Histol 2006; 37:153–160. [DOI] [PubMed] [Google Scholar]
- 47.Kim JS, Lee SH, Cho YS, et al. Enhancement of the adenoviral sensitivity of human ovarian cancer cells by transient expression of coxsackievirus and adenovirus receptor (CAR). Gynecol Oncol 2002; 85:260–265. [DOI] [PubMed] [Google Scholar]
- 48.Kelly FJ, Miller CR, Buchsbaum DJ, et al. Selectivity of TAG-72-targeted adenovirus gene transfer to primary ovarian carcinoma cells versus autologous mesothelial cells in vitro. Clin Cancer Res 2000; 6:4323–4333. [PubMed] [Google Scholar]
- 49.You Z, Fischer DC, Tong X, et al. Coxsackievirus-adenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression. Cancer Gene Ther 2001; 8:168–175. [DOI] [PubMed] [Google Scholar]
- 50.Ma J, Zhao J, Lu J, et al. Coxsackievirus and adenovirus receptor promotes antitumor activity of oncolytic adenovirus H101 in esophageal cancer. Int J Mol Med 2012; 30:1403–1409. [DOI] [PubMed] [Google Scholar]
- 51.Chen G, Wang BB, Li FJ, et al. [Enhancive effect of histone deacetylase inhibitor trichostatin a on transfection efficiency of adenovirus in ovarian carcinoma cell line A2780]. Ai Zheng 2005; 24:1196–1200. [PubMed] [Google Scholar]
- 52.Short JJ, Pereboev AV, Kawakami Y, et al. Adenovirus serotype 3 utilizes CD80 (B7.1) and CD86 (B7.2) as cellular attachment receptors. Virology 2004; 322:349–359. [DOI] [PubMed] [Google Scholar]
- 53.Sirena D, Lilienfeld B, Eisenhut M, et al. The human membrane cofactor CD46 is a receptor for species B adenovirus serotype 3. J Virol 2004; 78:4454–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanerva A, Wang M, Bauerschmitz GJ, et al. Gene transfer to ovarian cancer versus normal tissues with fiber-modified adenoviruses. Mol Ther 2002; 5:695–704. [DOI] [PubMed] [Google Scholar]
- 55.Raki M, Kanerva A, Ristimaki A, et al. Combination of gemcitabine and Ad5/3-Delta24, a tropism modified conditionally replicating adenovirus, for the treatment of ovarian cancer. Gene Ther 2005; 12:1198–1205. [DOI] [PubMed] [Google Scholar]
- 56.Uusi-Kerttula H, Davies J, Coughlan L, et al. Pseudotyped alphavbeta6 integrin-targeted adenovirus vectors for ovarian cancer therapies. Oncotarget 2016; 7:27926–27937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hulin-Curtis SL, Uusi-Kerttula H, Jones R, et al. Evaluation of CD46 re-targeted adenoviral vectors for clinical ovarian cancer intraperitoneal therapy. Cancer Gene Ther 2016; 23:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanderkwaak TJ, Wang M, Gomez-Navarro J, et al. An advanced generation of adenoviral vectors selectively enhances gene transfer for ovarian cancer gene therapy approaches. Gynecol Oncol 1999; 74:227–234. [DOI] [PubMed] [Google Scholar]
- 59.Bauerschmitz GJ, Lam JT, Kanerva A, et al. Treatment of ovarian cancer with a tropism modified oncolytic adenovirus. Cancer Res 2002; 62:1266–1270. [PubMed] [Google Scholar]
- 60.Gamble LJ, Ugai H, Wang M, et al. Therapeutic efficacy of an oncolytic adenovirus containing RGD ligand in minor capsid protein IX and Fiber, Delta24DoubleRGD, in an ovarian cancer model. J Mol Biochem 2012; 1:26–39. [PMC free article] [PubMed] [Google Scholar]
- 61.Kimball KJ, Preuss MA, Barnes MN, et al. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin Cancer Res 2010; 16:5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rein DT, Breidenbach M, Kirby TO, et al. A fiber-modified, secretory leukoprotease inhibitor promoter-based conditionally replicating adenovirus for treatment of ovarian cancer. Clin Cancer Res 2005; 11:1327–1335. [PubMed] [Google Scholar]
- 63.Rocconi RP, Zhu ZB, Stoff-Khalili M, et al. Treatment of ovarian cancer with a novel dual targeted conditionally replicative adenovirus (CRAd). Gynecol Oncol 2007; 105:113–121. [DOI] [PubMed] [Google Scholar]
- 64.Zhu ZB, Lu B, Park M, et al. Development of an optimized conditionally replicative adenoviral agent for ovarian cancer. Int J Oncol 2008; 32:1179–1188. [DOI] [PubMed] [Google Scholar]
- 65.Bett AJ, Prevec L, Graham FL. Packaging capacity and stability of human adenovirus type 5 vectors. J Virol 1993; 67:5911–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee SY, Park HR, Rhee J, et al. Therapeutic effect of oncolytic adenovirus expressing relaxin in radioresistant oral squamous cell carcinoma. Oncol Res 2013; 20:419–425. [DOI] [PubMed] [Google Scholar]
- 67.Vera B, Martinez-Velez N, Xipell E, et al. Characterization of the antiglioma effect of the oncolytic adenovirus VCN-01. PLoS One 2016; 11:e0147211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang SW, Cody JJ, Rivera AA, et al. Conditionally replicating adenovirus expressing TIMP2 for ovarian cancer therapy. Clin Cancer Res 2011; 17:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoo JY, Kim JH, Kwon YG, et al. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol Ther 2007; 15:295–302. [DOI] [PubMed] [Google Scholar]
- 70.Bazan-Peregrino M, Sainson RC, Carlisle RC, et al. Combining virotherapy and angiotherapy for the treatment of breast cancer. Cancer Gene Ther 2013; 20:461–468. [DOI] [PubMed] [Google Scholar]
- 71.Rein DT, Volkmer A, Beyer IM, et al. Treatment of chemotherapy resistant ovarian cancer with a MDR1 targeted oncolytic adenovirus. Gynecol Oncol 2011; 123:138–146. [DOI] [PubMed] [Google Scholar]
- 72.Wang S, Shu J, Chen L, et al. Synergistic suppression effect on tumor growth of ovarian cancer by combining cisplatin with a manganese superoxide dismutase-armed oncolytic adenovirus. Onco Targets Ther 2016; 9:6381–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan Z, Foster R, Bell DA, et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res 2006; 12:5055–5063. [DOI] [PubMed] [Google Scholar]
- 74.Han Z, Hong Z, Gao Q, et al. A potent oncolytic adenovirus selectively blocks the STAT3 signaling pathway and potentiates cisplatin antitumor activity in ovarian cancer. Hum Gene Ther 2012; 23:32–45. [DOI] [PubMed] [Google Scholar]
- 75.Long Q, Yang R, Lu W, et al. Adenovirus-mediated truncated Bid overexpression induced by the Cre/LoxP system promotes the cell apoptosis of CD133+ ovarian cancer stem cells. Oncol Rep 2017; 37:155–162. [DOI] [PubMed] [Google Scholar]
- 76.Majhen D, Calderon H, Chandra N, et al. Adenovirus-based vaccines for fighting infectious diseases and cancer: progress in the field. Hum Gene Ther 2014; 25:301–317. [DOI] [PubMed] [Google Scholar]
- 77.Holterman L, Vogels R, van der Vlugt R, et al. Novel replication-incompetent vector derived from adenovirus type 11 (Ad11) for vaccination and gene therapy: low seroprevalence and non-cross-reactivity with Ad5. J Virol 2004; 78:13207–13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang YQ, Tsai YC, Monie A, et al. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol Ther 2010; 18:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang H, Gomez-Manzano C, Rivera-Molina Y, et al. Oncolytic adenovirus research evolution: from cell-cycle checkpoints to immune checkpoints. Curr Opin Virol 2015; 13:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 2014; 6:226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang L, Hedjran F, Larson C, et al. A novel immunocompetent murine model for replicating oncolytic adenoviral therapy. Cancer Gene Ther 2015; 22:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salako MA, Kulbe H, Ingemarsdotter CK, et al. Inhibition of the inflammatory cytokine TNF-alpha increases adenovirus activity in ovarian cancer via modulation of cIAP1/2 expression. Mol Ther 2011; 19:490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Browne A, Tookman LA, Ingemarsdotter CK, et al. Pharmacological inhibition of beta3 integrin reduces the inflammatory toxicities caused by oncolytic adenovirus without compromising anticancer activity. Cancer Res 2015; 75:2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ranki T, Pesonen S, Hemminki A, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer 2016; 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li X, Wang P, Li H, et al. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian Hamster Model. Clin Cancer Res 2017; 23:239–249. [DOI] [PubMed] [Google Scholar]
- 87.Komorowski MP, McGray AR, Kolakowska A, et al. Reprogramming antitumor immunity against chemoresistant ovarian cancer by a CXCR4 antagonist-armed viral oncotherapy. Mol Ther Oncolytics 2016; 3:16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gil M, Komorowski MP, Seshadri M, et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol 2014; 193:5327–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Z, Ravindranathan R, Kalinski P, et al. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 2017; 8:14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steg AD, Bevis KS, Katre AA, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012; 18:869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luan QC, Sun YR, Han P, et al. Ad-PUMA sensitizes ovarian cancer cells to chemotherapeutic agents. Eur Rev Med Pharmacol Sci 2015; 19:4525–4532. [PubMed] [Google Scholar]
- 92.Cerullo V, Pesonen S, Diaconu I, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res 2010; 70:4297–4309. [DOI] [PubMed] [Google Scholar]
- 93.Koski A, Kangasniemi L, Escutenaire S, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 2010; 18:1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011; 477:99–102. [DOI] [PubMed] [Google Scholar]
- 95.Cerullo V, Diaconu I, Kangasniemi L, et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther 2011; 19:1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014; 27:1–7. [DOI] [PubMed] [Google Scholar]
- 97.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003; 348:203–213. [DOI] [PubMed] [Google Scholar]
- 98.Ashshi AM, El-Shemi AG, Dmitriev IP, et al. Combinatorial strategies based on CRAd-IL24 and CRAd-ING4 virotherapy with anti-angiogenesis treatment for ovarian cancer. J Ovarian Res 2016; 9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shah MM, Landen CN. Ovarian cancer stem cells: are they real and why are they important? Gynecol Oncol 2014; 132:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Foster R, Buckanovich RJ, Rueda BR. Ovarian cancer stem cells: working towards the root of stemness. Cancer Lett 2013; 338:147–157. [DOI] [PubMed] [Google Scholar]
- 101.Gujar S, Dielschneider R, Clements D, et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol Ther 2013; 21:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gujar SA, Marcato P, Pan D, et al. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther 2010; 9:2924–2933. [DOI] [PubMed] [Google Scholar]
- 103.Gujar SA, Pan D, Marcato P, et al. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther 2011; 19:797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirasawa K, Nishikawa SG, Norman KL, et al. Oncolytic reovirus against ovarian and colon cancer. Cancer Res 2002; 62:1696–1701. [PubMed] [Google Scholar]
- 105.Kohno SI, Luo C, Nawa A, et al. Oncolytic virotherapy with an HSV amplicon vector expressing granulocyte-macrophage colony-stimulating factor using the replication-competent HSV type 1 mutant HF10 as a helper virus. Cancer Gene Ther 2007; 14:918–926. [DOI] [PubMed] [Google Scholar]
- 106.Goshima F, Esaki S, Luo C, et al. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int J Cancer 2014; 134:2865–2877. [DOI] [PubMed] [Google Scholar]