

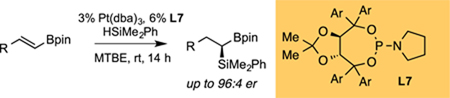
Abstract
A Pt-catalyzed enantioselective hydrosilylation of alkenylboronates is described. This reaction occurs with high regio- and enantioselectivity, providing a convenient route to chiral non-racemic geminal silylboronates. These compounds are useful reagents in stereoselective synthesis.
Keywords: If you are submitting your paper to a journal that requires keywords, provide significant keywords to aid the reader in literature retrieval.
GRAPHICAL ABSTRACT:
Geminal bimetallic reagents are endowed with distinctive reactivity patterns that allow them to engage in an array of complexity-generating processes. While bimetallic reagents based on Zn, Cr, and Cu species have received steady attention over the past 20–30 years1, recent studies have focused intensely on the synthesis and utility of geminal bis(boronates) in both catalytic and non-catalytic processes.2 In this regard, the utility of symmetric geminal bimetallic reagents (A, Scheme 1) in stereoselective catalysis requires the agency of a catalyst that can not only effect chemical transformation, but can also discriminate the enantiotopic metal groups of the substrate.3 Alternatively, nonracemic, non-symmetrical geminal dimetallic reagents (B, Scheme 1) might engage in a broader array of both catalytic and non-catalytic bond constructions wherein the unique reactivity of each C-M group can be employed to allow differential functionalization of the stereogenic carbon atom. In spite of the significant synthesis utility of non-symmetric dimetallic reagents, there are few methods available for their catalytic enantioselective preparation. While Hall and Yun established catalytic4 methods that provide access to non-symmetric geminal bis(boronates) (i.e. Scheme 1, B, M1=B(pin); M2=B(dan)), we considered that nonracemic geminal bis(metallates) bearing different metal centers might provide the most diverse reactivity.5 In this connection, pioneering studies by Hoveyda and coworkers6 (Scheme 1, eq. 1) established an efficient catalytic synthesis of nonracemic geminal silylboronates utilizing copper-boron addition to substituted styrenyl silanes. To provide enantiomerically-enriched non-functionalized geminal silylboronates Lan, Liu and co-workers7 introduced a copper-catalyzed aldehyde diboration followed by deoxygenative gem-silylation (Scheme 1, eq. 2). While this latter route is effective, yields are often modest and it requires the use of both a B-B and a B-Si bonded reagent. Herein, we describe a catalytic enantioselective hydrosilylation of readily accessible alkenyl boronates. Importantly, this process applies to a broad range of substrates and can be telescoped directly from alkyne precursors in a tandem hydroboration/hydrosilylation format. For this reason, and because it employs simple silanes and boranes as reagents, the process described herein represents a powerful, scalable route to important synthetic intermediates.
Scheme 1.
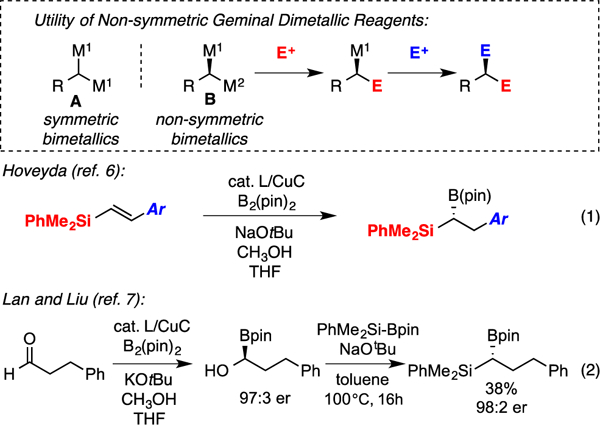
Utility and Previous Catalytic Syntheses of Nonracemic Geminal Dimetallic Reagents
On the basis of our studies on platinum-catalyzed diboration of alkenyl boronates8 we were prompted to consider the prospects for an aligned Pt-catalyzed hydrosilylation of this substrate class.9 This approach to silylboronate synthesis would have the attractive feature of accommodating a broad array of alkenyl substituents while avoiding the use of diboron and silylboron reagents. Of note, successful development of this process could rely on well-precedented “hydride-first” insertion10 where production of a stable α-boryl-organoplatinum intermediate might dictate reaction regiocontrol.
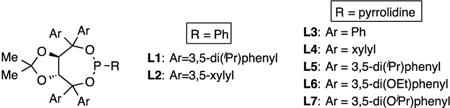
To initiate studies, reaction conditions and catalysts similar to those employed for the diboration of alkenyl boronates were employed, but the diboron reagent was replaced with a silane. Specifically, Pt(dba)3, phosphonite ligand L1, and dimethylphenylsilane were employed in the reaction and found to furnish the hydrosilylation product with complete conversion and high regioselectivity (> 20:1 rr), but only modest enantiocontrol (Table 1, entry 1). Use of less sterically encumbered xylyl-substituted phosphonite L2 (Table 1, entry 2) showed a notable increase in enantioselectivity. Further increases in enanioselectivity were obtained by employing pyrrolidine-derived phosphoramidite ligands L3-L4 (Table 1, entries 3–4). With this class of ligands, selectivity is still optimal with meta-substituted arenes on the TADDOL backbone and, of those substituents, 3,5-dialkoxysubstituted ligands L6-L7 (Table 1, entries 6–7) were consistently most effective. (see Supporting_Information for full ligand optimization). After further analysis of reaction conditions (Table 1, entries 8–12), it was concluded that MTBE as the solvent and L7 as the ligand were optimal conditions to explore the generality of the reaction.
Table 1. Catalyst Studies in Alkenyl Boronate Hydrosilylation.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R= | ligand | solvent | temp (°C) | yield (%)a | erb |
| 1 | -CH2CH2Ph | L1 | THF | 70 | 95 | 62:38 |
| 2 | -CH2CH2Ph | L2 | THF | 70 | 96 | 85:15 |
| 3 | -CH2CH2Ph | L3 | THF | 70 | 96 | 86:14 |
| 4 | -CH2CH2Ph | L4 | THF | 70 | 99 | 88:12 |
| 5 | -CH2CH2Ph | L5 | THF | 70 | 99 | 72:38 |
| 6 | -CH2CH2Ph | L6 | THF | 70 | 87 | 8:92c |
| 7 | -CH2CH2Ph | L7 | THF | 70 | 80 | 90:10 |
| 8 | -CH2CH2Ph | L6 | MTBE | r.t. | 89 | 6:94c |
| 9 | -CH2CH2Ph | L6 | toluene | r.t. | 94 | 7:93c |
| 10 | -CH2CH2Ph | L6 | cyclohexane | r.t. | 99 | 6:94c |
| 11 | -n-butyl | L7 | cyclohexane | r.t. | 73 | 95:5 |
| 12 | -n-butyl | L7 | MTBE | r.t. | 99 | 95:5 |
Yields determined by NMR analysis with comparison to an internal standard.
Enantiomer ratios (er) determined by SFC analysis of the corresponding silyl alcohol
Reaction carried out using ent-L6.
To survey the scope of the alkenyl boronate hydrosilylation, Pt(dba)3 and ligand L7 were pre-complexed for 15 minutes at 80°C, cooled to room temperature, and then used as catalyst for a room temperature reaction between the alkenyl boronate and silane. As noted in Table 2, good yields and enantioselectivities were obtained for many substrates. Various alkyl substituents can be tolerated (1, 2, 3), however β-branched groups result in diminished reactivity and enantioselectivity (4, 5, 6). Relocating the steric bulk further from the alkene reestablishes high reactivity and enantioselectivity (7). The platinum-catalyzed hydrosilylation can tolerate various functional groups such as alkyl halides (8, 9), protected alcohols (10), and heterocycles (15, 16). Notably, the hydrosilylation is selective towards alkenyl boronates over terminal alkenes (11) and ketones (14), with only trace alkene hydrosilylation observed. Ester functional groups in various positions (12, 13, 14) are tolerated, but it is worth noting that for 12, the regioisomeric hydrosilylation product was formed in equal amount. In addition to dimethylphenylsilane, benzyldimethylsilane11 (17) and diphenylmethylsilane (18) also show high reactivity and moderate to good enantioselectivity. Product 18 highlights the ability of the method to be applied to pharmaceutically-relevant problems: after oxidation of the phenyl groups this compound would furnish silane-diol motifs that are often utilized as tetrahedral intermediate bioisosteres.12
Table 2.
Scope of Pt-Catalyzed Enantioselective Hydrosilylation[a]
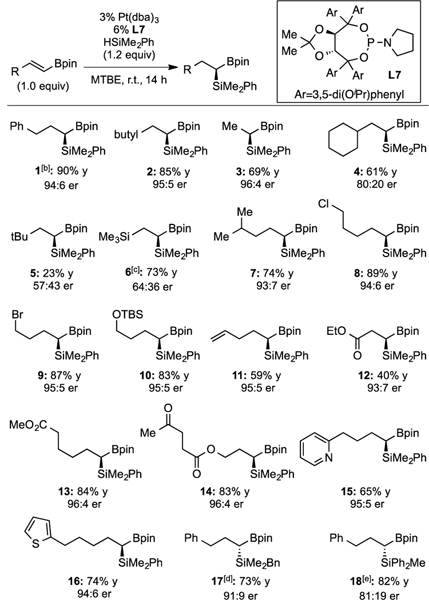 |
See text and Supporting_Information for experimental details. Both the yield and the enantiomer ratio (er) represent the average value for two experiments.
Reaction run using L6.
Reaction conducted at 70°C.
Reaction run using HSiMe2Bn and ent-L6.
Reaction run using HSiPh2Me and ent-L6.
As mentioned above, the synthesis of geminal bis(metallates) bearing different metal groups can allow for diverse functionalization by taking advantage of the increased reactivity of boron relative to silicon in 1,2-metallate rearrangements.13 As depicted in Figure 1 (eq. A), direct oxidation and amination14 of the organoboronate provided the silyl alcohol (19) and silyl amine (20) products with retention of configuration. These types of products have been shown to be useful as chiral auxiliaries15 and as amino acid bioisoteres16, respectively. A modified procedure for the Matteson homologation17 afforded the vicinal silyl boronate 21 with good yield, a compound that itself might be used in further coupling reactions. To highlight this feature and demonstrate the reactivity differences of the metal groups, vicinal silylboronate 21 was subjected to amination to afford vicinal silyl amine 22 (eq. B), and a modified Tamao-Fleming oxidation18 resulted in the formation of oxazolidinone 23 after cyclization on the –Boc group. The two-step sequence towards 23 occurred in good yield and 98% enantiospecificity.
Figure 1.
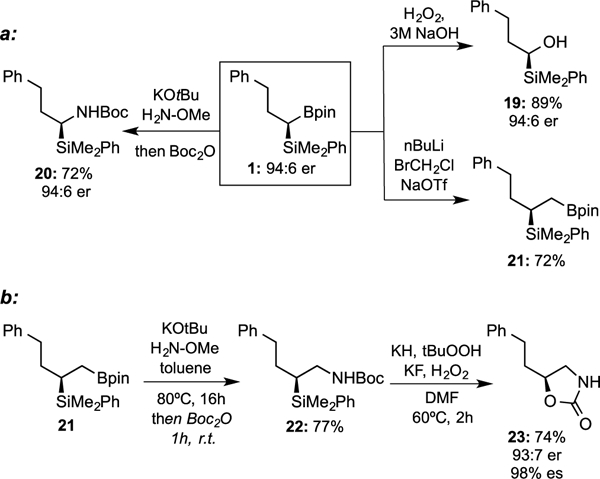
a: Direct transformation of silylboronate to chiral silyl alcohol, silyl amine, and vicinal silylboronate. b: Telescope sequence from vicinal silylboronate to oxazolidinone by amination and oxidation.
To probe the utility and robustness of the hydrosilylation, a one-pot procedure starting from 3-phenyl-1-propyne was conducted as shown in Figure 2. In this experiment, dicyclohexylborane–catalyzed hydroboration of 3-phenyl-1-propyne,19 followed by platinum-catalyzed hydrosilylation in toluene and, lastly, in situ amination afforded the silylamine 20 in good yield and enantioselectivity. This procedure highlights the utility of the enantioselective platinum-catalyzed hydrosilylation for the practical synthesis of compounds of significance for the pharmaceutical industry, and the insensitivity of the reaction towards different solvents.
Figure 2.
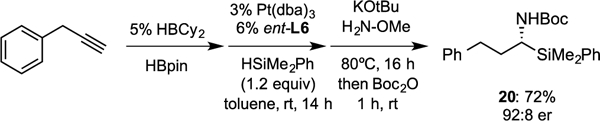
Single-flask sequential catalytic alkyne hydroboration/hydrosilylation/amination for the synthesis of silylamine 20 from 3-phenyl-1-propyne.
To further examine the practical utility of this method for large-scale dry-box free synthesis, the reaction in Figure 3 was conducted. As depicted, the metal and ligand were weighed in the open atmosphere, then the flask purged with nitrogen, heated to 80°C for 15 minutes, cooled, and the alkenyl boronate and silane introduced for reaction at room temperature. Notably, the catalyst loading could be reduced to one-percent and the solvent volume reduced while maintaining high reactivity and enantioselectivity. This experiment gave comparable yield and selectivity as smaller scale reactions conducted in the glove box.
Figure 3.
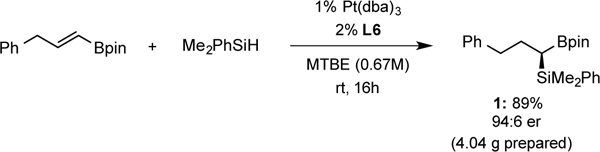
Large-scale, dry-box-free enantioselective platinum-catalyzed hydrosilylation.
Although the mechanism of platinum-catalyzed hydrosilylation has been extensively studied,10 alkenyl boronates are unique substrates for this reaction and probing the mechanism was of interest. To probe this insertion mode, deuterodimethylphenylsilane20 was synthesized and employed in the reaction. As depicted in Figure 4, the enantioselective platinum-catalyzed deuterosilylation resulted in a single product diastereomer in high enantioselectivity. Analysis of the reaction product by NMR (see Supporting Information for details) showed no deuterium scrambling, which supports the hypothesis that olefin insertion into a platinum hydride forms a stabilized α-boryl-organoplatinum intermediate, and this feature governs the regiocontrol of the hydrosilylation.
Figure 4.
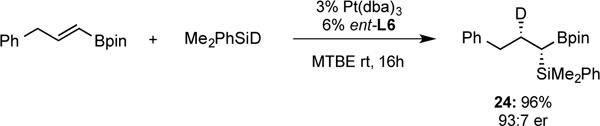
Regioselectivity of the deuteriosilylation of a vinyl boronate under Pt-phosphoramidite catalysis.
In conclusion, we have demonstrated the ability of a platinum complex to catalyze the hydrosilylation of alkenyl boronates with high regio- and enantioselectivity. The geminal silylboronate products are shown to be versatile synthetic building blocks by divergent functionalization of the Si and B groups. Further studies on the hydrosilylation of other π-systems are ongoing.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (GM-59417).
Footnotes
ASSOCIATED CONTENT
Supporting Information
(Word Style “TE_Supporting_Information”). (Word Style “TE_Supporting_Information“). A listing of the contents of each file supplied as Supporting_Information should be included. For instructions on what should be included in the Supporting_Information as well as how to prepare this material for publication, refer to the journal’s Instructions for Authors.
The Supporting_Information is available free of charge on the ACS Publications website.
brief description (file type, i.e., PDF)
brief description (file type, i.e., PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Marek I; Normant J-F Chem. Rev. 1996, 96, 3241–3268. [DOI] [PubMed] [Google Scholar]
- (2).a) Xu L; Zhang S; Li P Chem. Soc. Rev. 2015, 44, 8848–8858. [DOI] [PubMed] [Google Scholar]; b) Rygus JPG; Crudden CM J. Am. Chem. Soc. 2017, 139, 18124–18137 [DOI] [PubMed] [Google Scholar]
- (3).a) Sun C; Potter B; Morken JP J. Am. Chem. Soc. 2014, 136, 6534–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Potter B; Szymaniak AA; Edelstein EK; Morken JP J. Am. Chem. Soc. 2014, 136, 17918–17921. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Potter B; Edelstein EK; Morken JP Org. Lett. 2016, 18, 3286–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Joannou MV; Moyer BS; Meek SJ J. Am. Chem. Soc. 2015, 137, 6176–6179. [DOI] [PubMed] [Google Scholar]; e) Murray SA; Green JC; Tailor SB; Meek SJ Angew. Chem. Int. Ed. 2016, 55, 9065–9069. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shi Y; Hoveyda AH Angew. Chem. Int. Ed. 2016, 55, 3455–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).a) Lee CH; McDonald R; Hall DG Nat. Chem. 2011, 3, 894–899. [DOI] [PubMed] [Google Scholar]; b) Feng X; Jeon H; Yun J Angew. Chem. Int. Ed. 2013, 52, 3989–3992. [DOI] [PubMed] [Google Scholar]
- (5).For a recent non-catalytic asymmetric route to geminal silylboronates, see: a) Bootwicha T; Feilner JM; Meyers EL; Aggarwal VK Nat. Chem. 2017, 9, 896–902. [DOI] [PubMed] [Google Scholar]; b) Aggarwal VK; Binanzer M; de Ceglie MC; Gallanti M; Glasspoole BW; Kendrick SJF; Sonawane RP; Vazquez-Romero A; Webster MP Org. Lett. 2011, 13, 1490–1493. [DOI] [PubMed] [Google Scholar]; c) Barsamian AL; Wu Z; Blakemore PR Org. Biomol. Chem. 2015, 13, 3781–3786. [DOI] [PubMed] [Google Scholar]
- (6).Meng F; Jang H; Hoveyda AH Chem. Eur. J. 2013, 19, 3204–3214. [DOI] [PubMed] [Google Scholar]
- (7).Wang L; Zhang T; Sun W; He Z; Xia C; Lan Y; Liu CJ Am. Chem. Soc. 2017, 139, 5257–5264. [DOI] [PubMed] [Google Scholar]
- (8).Coombs JR; Zhang L; Morken JP J. Am. Chem. Soc. 2014, 136, 16140–16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For other Pt-catalyzed asymmetric hydrosilation of alkenes, see: a) Yamamoto K; Hayashi T; Kumada M J. Am. Chem. Soc 1971, 93, 5301–5302. [Google Scholar]; b) Yamamoto K; Hayashi T; Zembayashi M; Kumada M J. Organomet. Chem. 1976, 118, 161–181. [Google Scholar]; For asymmetric hydrosilation of ketones, see: a) Yamamoto K; Hayashi T; Kumada M; J. Organomet. Chem. 1972, 46, C65–C67. [Google Scholar]; b) Hayashi T; Yamamoto K; Kumada M J. Organomet. Chem. 1976, 112, 253–262. [Google Scholar]
- (10).a) Chalk AJ; Harrod JF J. Am. Chem. Soc. 1965, 87, 16–21. [Google Scholar]; b) Chalk AJ; Harrod JF J. Am. Chem. Soc. 1965, 87, 1133–1133. [Google Scholar]; c) Taylor RB; Roy AK J. Am. Chem. Soc. 2002, 124, 9510–9524. [DOI] [PubMed] [Google Scholar]; d) Taige MA; Ahrens S; Strassner T J. Organomet. Chem. 2011, 696, 2918–2927. [Google Scholar]; e) Meister TK; Riener K; Gigler P; Stohrer J; Herrmann WA; Kuhn FE ACS Catal. 2016, 6, 1274–1284. [Google Scholar]
- (11).For the utility of BnMe2Si- group in cross-coupling, see: a) Machacek MR; Ball ZT; Trost BM Org. Lett. 2003, 5, 1895–1898. [DOI] [PubMed] [Google Scholar]; b) Tymonko SA; Denmark SE J. Am. Chem. Soc. 2005, 127, 8004–8005. [DOI] [PubMed] [Google Scholar]; c) Nakao Y; Hiyama T Chem. Soc. Rev. 2011, 40, 4893–4901. [DOI] [PubMed] [Google Scholar]
- (12).a) Nielsen L; Skrydstrup T J. Am. Chem. Soc. 2008, 130, 13145–13151. [DOI] [PubMed] [Google Scholar]; b) Min GK; Hernandez D; Skrydstrup T Acc. Chem. Res. 2013, 46, 457–470. [DOI] [PubMed] [Google Scholar]
- (13).Millan A; Grigol Martinez PD; Aggarwal VK Chem. Eur. J. 2017, 24, 730–735. [DOI] [PubMed] [Google Scholar]
- (14).This is a modification of the following reference and its details will be reported separately: Mlynarski SN; Karns AS; Morken JP J. Am. Chem. Soc. 2012, 134, 16449–16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cossrow J; Rychnovsky SD Org. Lett. 2002, 4, 147–150. [DOI] [PubMed] [Google Scholar]
- (16).a) Reḿond E; Martin C; Martinez J; Cavelier F Chem. Rev. 2016, 116, 11654–11684. [DOI] [PubMed] [Google Scholar]; b) Ramesh R; Reddy DS J. Med. Chem. 2017, ASAP, 10.1021/acs.jmedchem.7b00718. [DOI] [Google Scholar]
- (17).(a) Mah RWH; Matteson DS J. Am. Chem. Soc. 1963, 85, 2599–2603. [Google Scholar]; (b) For effect of sodium triflate: Lovinger GJ; Aparece MD; Morken JP J. Am. Chem. Soc. 2017, 139, 3153–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).a) Smitrovich JH; Woerpel KA J. Org. Chem. 1996, 61, 6044–6046. [Google Scholar]; b) Walleser P; Bruckner R Eur. J. Org. Chem. 2014, 15, 3210–3224. [Google Scholar]
- (19).Shirakawa K; Arase A; Hoshi M Synthesis, 2004, 11, 1814–1820. [Google Scholar]
- (20).Takale BS; Tao SM; Yu XQ; Feng XJ; Jin T; Bao M; Yamamoto Y Org. Lett. 2014, 16, 2558–2561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.