

Similarities between a mouse model and human patient informed diagnosis and management of a novel cause of dilated cardiomyopathy.
Abstract
Neonatal heart failure is a rare, poorly-understood presentation of familial dilated cardiomyopathy (DCM). Exome sequencing in a neonate with severe DCM revealed a homozygous nonsense variant in leiomodin 2 (LMOD2, p.Trp398*). Leiomodins (Lmods) are actin-binding proteins that regulate actin filament assembly. While disease-causing mutations in smooth (LMOD1) and skeletal (LMOD3) muscle isoforms have been described, the cardiac (LMOD2) isoform has not been previously associated with human disease. Like our patient, Lmod2-null mice have severe early-onset DCM and die before weaning. The infant’s explanted heart showed extraordinarily short thin filaments with isolated cardiomyocytes displaying a large reduction in maximum calcium-activated force production. The lack of extracardiac symptoms in Lmod2-null mice, and remarkable morphological and functional similarities between the patient and mouse model informed the decision to pursue cardiac transplantation in the patient. To our knowledge, this is the first report of aberrant cardiac thin filament assembly associated with human cardiomyopathy.
INTRODUCTION
Dilated cardiomyopathy (DCM) is the most common subtype of genetic cardiomyopathy, and although estimated incidence rates vary, it likely affects 1:250 to 1:500 individuals (1). While pediatric DCM is relatively rare (2–4), it represents the most severe presentation of this disorder. Pediatric DCM has a wide variety of etiologies including infectious myocarditis, syndromic cardiomyopathies, such as neuromuscular disorders and inborn errors of metabolism, and isolated gene mutations. Historically, it has been thought that the majority of pediatric DCM cases are idiopathic (4). However, given recent advances in cardiomyopathy genetic testing, a pathogenic genetic mutation is now identified in up to 40% of all patients with DCM tested (5). The diagnostic yield of sequencing in pediatric DCM populations may be even higher. A recent study found a 54% diagnostic yield of combined exome sequencing and single-nucleotide polymorphism microarray analysis in a cohort of 95 pediatric patients with DCM (6). Given these high diagnostic rates, sequencing of known cardiomyopathy genes is now recommended for all patients with DCM (7, 8).
While mutations in thin filament proteins that regulate the interaction of actin and myosin are associated with DCM (e.g., troponins and tropomyosin), pathogenic mutations that result in dysregulation of thin filament assembly have yet to be described. The leiomodins (Lmods) are actin-binding proteins that nucleate actin filaments in vitro and regulate thin filament assembly (9–11). The Lmod family of proteins is critical for proper muscle function. Homozygous or compound heterozygous mutations in LMOD3, the isoform predominant in skeletal muscle, result in severe nemaline myopathy (NM) (12). NM is a skeletal muscle disorder characterized by muscle weakness. A biallelic mutation in LMOD1, the smooth muscle-specific Lmod, leads to megacystis microcolon intestinal hypoperistalsis syndrome (13), a rare visceral myopathy characterized by defective contractile activity of muscles that line the bladder and intestines. The predominant Lmod isoform in cardiac muscle, LMOD2, which has been reported to allow for thin filament elongation in vivo, has yet to be associated with human disease. However, Lmod2 constitutive null (knockout) mice present with short cardiac thin filaments and die at ~3 weeks due to dysfunctional, dilated hearts (11, 14). Knockout of Lmod2 in adult mice also results in abnormally short thin filaments, cardiac failure, and death (15).
Here, we report the first known case of a human cardiomyopathy caused by a biallelic mutation in LMOD2. Using genetic and molecular approaches, we dissected how a single mutation in a contractile protein results in human DCM.
RESULTS
Clinical presentation
The proband, a full-term female, presented with late decelerations and tachycardia during delivery, necessitating a cesarean section. Shortly after birth, she developed desaturations with agitation, and subsequent echocardiogram demonstrated severe biventricular dysfunction, with an ejection fraction of 14%. Her left ventricle (LV) was severely dilated with an internal diameter in systole of 21 mm and z score = 6.29 and in diastole of 28 mm and z score = 2.85 (normal z score range, −2 to 2) (Fig. 1, A to C). She required mechanical circulatory support with either extracorporeal membrane oxygenation or a Berlin Heart left ventricular assist device (LVAD) for the majority of her early life and was therefore listed for heart transplantation as an infant. While waiting, she had intermittent episodes of ventricular tachycardia and ectopy (Fig. 1D), and while on anticoagulation for her LVAD, she had a large medial cerebral artery hemorrhagic stroke. She underwent orthotopic heart transplantation at 10 months of age. Pathology of the explanted heart was consistent with the known diagnosis of DCM (Fig. 1, E and G).
Fig. 1. Representative cardiac imaging and histology from the patient.

(A to C) Echocardiograms from the second day of life, before cannulation onto extracorporeal membrane oxygenation. (A) Apical four-chamber view in systole showing a dilated left atrium, normal size right ventricle (RV), and severely dilated LV without hypertrophy. There are severely diminished RV and LV systolic function and swirling (“smoke”) within the LV cavity. With color Doppler over the mitral valve, there is mild regurgitation. The aortic valve is trileaflet but barely opening, with mild aortic insufficiency in the setting of minimal cardiac output. (B and C) Parasternal long and short axis views, demonstrating severely dilated and globular LV and severely diminished LV and RV systolic function. bpm, beats per minute. (D) Representative electrocardiogram clips of leads II and V5 demonstrating dysrhythmias experienced by the patient before transplant, including monomorphic ventricular tachycardia with a right bundle branch block morphology. (E) Areas of stretched and wavy myocardium are present (hematoxylin and eosin stain, 20×). (F) Foci of somewhat disorganized and hypertrophic myocytes with nuclear crowding are observed; nuclear enlargement and irregularity of nuclear contours as well as perinuclear clearing are quite prominent in certain regions (hematoxylin and eosin, 60×). (G) Marked fibrosis (blue) is present throughout the subendocardium of both ventricles with patchy interstitial nonreplacement-type perimyocyte fibrosis (Masson’s trichrome stain, 10×). (H) Multilayered elastic fibers (black) are also noted in the thickened subendocardium (elastic stain, 10×).
Diagnostic workup
Diagnostic evaluations completed shortly after birth included unremarkable infectious and metabolic evaluations (table S1). On clinical genetics consultation, notable findings included a family history of consanguinity. Both parents are of Mexican descent, and the proband’s great-grandfathers are half brothers. There was no family history of heart failure, cardiomyopathy, sudden cardiac death, or any other heart disease. Given her critical condition, we performed clinical rapid exome sequencing in conjunction with mitochondrial genome sequencing and deletion/duplication testing (GeneDx). Mitochondrial genome sequencing was normal.
Exome sequencing identified a biallelic homozygous nonsense variant (c.1193G > A, p.Trp398*) in a candidate gene LMOD2, which encodes Lmod2. The patient’s mother, a carrier of the LMOD2 variant, has normal cardiac function, even during pregnancy; this is consistent with Lmod2 heterozygous mice, which show no detectable phenotype, even after multiple pregnancies (11). The patient’s father, also heterozygous for the p.Trp398* variant, is clinically asymptomatic, but an echocardiogram could not be obtained. Subsequently, the proband’s parents gave birth to a second child. She does not harbor the LMOD2 variant on either allele and had a normal cardiac evaluation including an echocardiogram and electrocardiogram at 3 months of age.
As part of the transplant evaluation process, we evaluated the patient for extracardiac manifestations of disease. There were concerns that the patient, even with a transplanted heart, would suffer from skeletal myopathy since LMOD2 is expressed in skeletal muscle, albeit at much lower levels (<5%, at the protein level, in humans) (12). Muscle ultrasound at 1 month showed no overt evidence of myopathy. Creatine kinase level was also normal at 75 U/liter (normal, 60 to 305). Biopsy of the quadriceps muscle at 4.5 months of age showed nonspecific myopathic changes consistent with an infant in an intensive care unit setting. Nemaline rods were not present on biopsy.
No skeletal muscle abnormalities are observed at time of death in the Lmod2 knockout mouse model; this is likely due to the fact that in both humans and mice, Lmod3 is the predominate isoform in skeletal muscle. Given the proband’s unremarkable muscle findings and strong similarities between cardiac disease progression in the patient and Lmod2 knockout mice (11), she was listed for cardiac transplantation as described above. Additional clinical details can be found in the Supplementary Materials.
Lmod2 protein expression is severely reduced in the patient’s explanted heart
Next, we sought to understand the pathophysiologic mechanism of LMOD2-related cardiomyopathy. We had the unique opportunity to perform experiments on tissue from the patient’s explanted heart in parallel with studies in Lmod2 null mice to determine how a mutation in LMOD2 disrupts cardiac myocyte function and induces cardiomyopathy.
The LMOD2 p.Trp398* mutation encodes a premature stop codon predicted to result in a 45-kDa protein (Fig. 2A). Full-length or truncated Lmod2 was not detected in the LV of the proband’s explanted heart via Western blot analysis, whereas analysis of LV samples from three pediatric nonfailing hearts suggested Lmod2 expression increases over time (from ~1 to 4 years of age) (Fig. 2, B and C). Since Lmod2 is known to function in thin (actin) filament length regulation, we monitored the levels of other integral thin filament components. Cardiac actin, cardiac troponin I, cardiac troponin T, and tropomyosin, which assemble along the length of thin filaments, were decreased, while the thin filament capping protein tropomodulin 1 (Tmod1), which is restricted to thin filament pointed ends, was relatively unchanged in the patient’s heart (Fig. 2, B and C, and fig. S1). This result indicates an alteration in the number and/or lengths of thin filaments in the diseased heart.
Fig. 2. Lmod2 protein is not detected, mature mRNA levels are reduced, and pre-mRNA levels are largely unchanged in the patient’s LV.
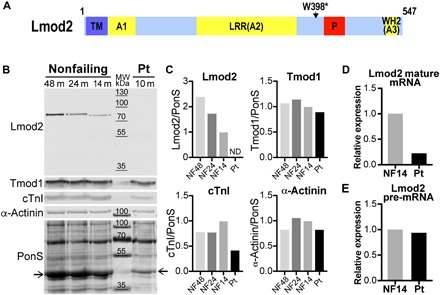
(A) Diagram of the domain layout of human Lmod2 protein with the location of the patient’s nonsense mutation (Trp398*) indicated. Lmod2 contains a tropomyosin binding domain (TM), three actin-binding domains (A1 to A3), and a proline-rich region (P). Actin-binding site 2 is composed of a leucine-rich domain (LRR), while actin-binding site 3 is a Wiskott-Aldrich syndrome protein–homology 2 (WH2) domain (34). (B) Western blots of Lmod2, Tmod1, cardiac troponin I (cTnI), and α-actinin in LV of nonfailing (NF) donor hearts at 48, 24, and 14 months of age and the patient’s (Pt) explanted heart (10 months). Total protein was stained with Ponceau S (PonS); the arrows indicate actin. MW, molecular weight. (C) Relative protein levels following normalization to total protein. Note that the 14-month-old nonfailing donor heart (closest in age to the patient) was set to 1. ND, not detected. Quantitative polymerase chain reaction (qPCR) analysis of Lmod2 mature mRNA (D) and pre-mRNA (E) expression in the LV of a 14-month-old nonfailing donor heart (black bar) or the patient’s explanted heart (gray bar).
To determine whether the lack of detectable Lmod2 protein resulted from degradation of the mutant protein or a decrease in levels of its transcript, we used reverse transcription quantitative polymerase chain reaction (RT-qPCR). Compared to an age-comparable nonfailing heart, the patient had a large decrease in mature Lmod2 mRNA in the LV (assessed using intron-spanning primers located within exons) (Fig. 2D). To determine whether the decrease in Lmod2 transcript is evident before its maturation, we performed RT-qPCR using primers located within an intron. Lmod2 pre-mRNA levels were only slightly decreased in the explanted LV (Fig. 2E).
Loss of Lmod2 results in significantly shorter thin filaments
Deconvolution immunofluorescence microscopy of LV cryosections from the patient’s heart revealed myofibrillar disarray and wide Z-discs indicated by staining for the integral Z-disc protein α-actinin (Fig. 3A); both are hallmarks of DCM. Staining for F-actin showed exceptionally short thin filaments that barely extended out from the Z-disc. Tmod1, a marker of thin filament pointed ends, localized to bands flanking the Z-disc, confirming the presence of remarkably short thin filaments. Conversely, thin filaments in a nonfailing donor heart extended from the Z-disc to center of the sarcomere where they were capped by Tmod1. We used Tmod1 localization to measure thin filament lengths. Mean thin filament length was significantly reduced in the patient’s heart (by ~70%) (Fig. 3B).
Fig. 3. The patient’s heart displays disorganized myofibrils, wide Z-discs, and extraordinarily short thin filaments.
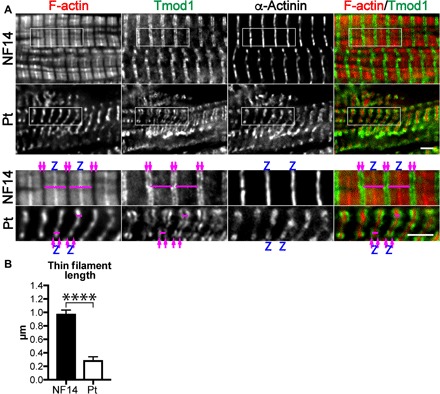
(A) Representative deconvolution immunofluorescence images of LV tissue from a 14-month-old nonfailing donor heart and explanted patient’s heart stained with fluorescently conjugated phalloidin to label F-actin and antibodies to Tmod1 (thin filament pointed end marker) and α-actinin (Z-disc marker). F-actin is in red, and Tmod1 is in green in the merged images. Magenta arrows denote thin filament pointed ends, and Z-discs are marked with a Z. Magenta bars indicate examples of thin filament arrays (one Z-disc with two bundles of thin filaments extending into opposite half-sarcomeres) that were measured. Scale bar, 2.5 μm. (B) Mean LV thin filament lengths measured via Tmod1 staining using the DDecon plugin for ImageJ. Note that the length of one thin filament is one-half of the width of the thin filament array marked in (A). Values are means ± SD; n = 35 and 39 measurements (NF14 and Pt, respectively). ****P < 0.0001, Student’s t test.
Loss of Lmod2 results in decreased maximal force production and calcium sensitivity in the patient’s cardiomyocytes
We analyzed force production and response to calcium in skinned cardiomyocytes isolated from the patient’s LV. Maximum force production in the patient’s cardiomyocytes was severely reduced compared with three nonfailing samples (patient versus NF14, 2.3 ± 0.5 mN/mm2 versus 18.6 ± 2.2 mN/mm2; Fig. 4 and table S2). Furthermore, calcium sensitivity was decreased [as seen by an increase in half maximal effective concentration (EC50)], and cooperativity (Hill coefficient) was reduced (patient versus NF14, 4.4 ± 1.0 μM versus 1.7 ± 0.1 μM and 1.7 ± 0.3 μM versus 5.1 ± 0.5 μM, respectively).
Fig. 4. Calcium-activated contractile force is reduced, and the myofilament lattice is desensitized to calcium in cardiomyocytes isolated from the patient’s heart.
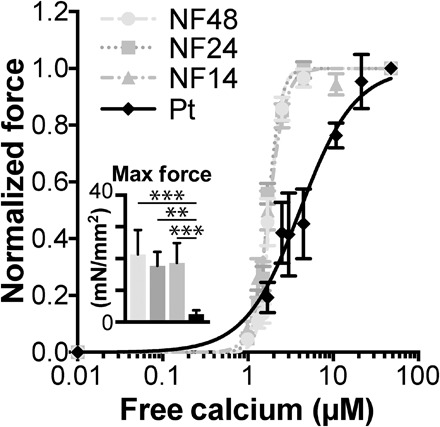
The fraction of maximum force produced at various concentrations of free calcium is plotted (sarcomere length, ~1.8 μm). Mean force at maximum calcium activation is indicated in the inset. All values are means ± SEM; n = 7, 8, 8 cells and 4 cell clusters (NF48, NF24, NF14, and Pt, respectively). **P < 0.01 and ***P < 0.001, one-way analysis of variance (ANOVA) with Tukey’s post hoc test.
When mutant Lmod2 protein is expressed in vivo, it retains partial functionality
To determine the extent to which the mutant protein is functional in vivo, we expressed the homologous mutation (mouse Lmod2 p.W405*) in Lmod2 constitutive knockout mice. Note that the expression construct was generated from complementary DNA (cDNA), so it is not susceptible to potential degradation by nonsense-mediated decay (NMD). The premature stop codon in Lmod2 is predicted to result in a protein with a C-terminal truncation that leads to loss of an actin-binding site (Wiskott-Aldrich syndrome protein–homology 2 domain) (9); thus, we predicted that it would not function effectively. Previously, we showed that introduction of green fluorescent protein (GFP)–Lmod2 via adeno-associated virus at postnatal day 4 prevents (“rescues”) cardiac dilation and juvenile lethality characteristic of Lmod2 knockout mice (11). Using this approach, we unexpectedly found that expression of GFP-Lmod2[W405*] also reduces onset of cardiac dilation and dysfunction in Lmod2 null mice, noted by a decrease in LV internal diameter in diastole and an increase in systolic performance (percent ejection fraction) compared to mice expressing GFP alone (Fig. 5 and table S3). We did not find a correlation between the amount of exogenous protein expressed and cardiac function, as mice expressing even the lowest levels of GFP-Lmod2 or GFP-Lmod2[W405*] have improved systolic performance and near normal LV geometry (fig. S2). This result demonstrates that the mutant protein (if it were to be expressed) is at least partially functional by all criteria measured.
Fig. 5. Expression of GFP-Lmod2[W405*] reduces cardiac disease progression of Lmod2 knockout mice.
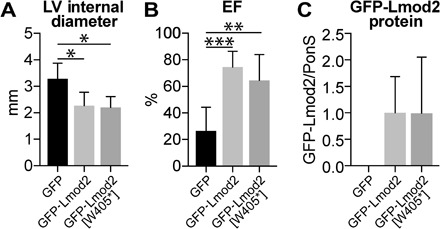
Echocardiography and Western blot analysis of Lmod2 knockout mice injected with adeno-associated virus expressing GFP (black bars), GFP-Lmod2 (light gray bars), or GFP-Lmod2[W405*] (dark gray bars). (A) LV internal diameter in diastole. (B) Systolic performance determined by left ventricular ejection fraction (EF). (C) Western blot analysis of exogenous Lmod2 protein levels in the LV of adeno-associated virus–injected mice. Relative protein levels following normalization to total protein via Ponceau S staining are shown. Values are means ± SD; n = 6, 5, and 5 (knockout + GFP, knockout + GFP-Lmod2, and knockout + GFP-Lmod2[W405*], respectively). *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA with Tukey’s post hoc test.
DISCUSSION
Proper thin filament assembly and structure are essential for efficient cardiac myocyte contraction. Mutations in integral thin filament proteins involved in actin-myosin interactions are a well-established cause of DCM [for reviews, see (5, 16–18)]. However, pathogenic mutations in proteins involved in regulating thin filament assembly in cardiac muscle have yet to be described. Here, we present the first case of DCM associated with abnormal actin filament lengths due to a biallelic loss of function mutation in LMOD2, a member of the Tmod family of actin-binding proteins (19).
Note that there are remarkable similarities between the clinical cardiac phenotype observed in the patient described in this work and mice lacking Lmod2. In both, loss of Lmod2 is compatible with normal fetal development, including appropriate birth growth parameters and no signs of fetal hydrops. However, shortly after birth, in both mice and humans, symptoms of a very early-onset, severe DCM become evident. The cardiomyopathy progresses rapidly to heart failure in the neonatal period without evidence of intervening hypertrophy. In addition, neither group demonstrates detectable evidence of skeletal muscle myopathy, likely because LMOD3, rather than LMOD2, is the major Lmod isoform in skeletal muscle. Neither mice nor human carriers, i.e., individuals with a single loss of function LMOD2 allele, have cardiac disease.
Cardiac muscles from both the Lmod2 knockout mouse model and our human patient demonstrate lack of Lmod2, short thin filaments, and disorganized myofibrils. Myocytes isolated from both species have reduced contractile force generation. Collectively, these pathologic changes provide an explanation for the severe ventricular dysfunction observed in this disorder. While the hearts of both the patient and Lmod2 knockout mice display abnormally short thin filaments, the extent of reduction is substantially more in the patient’s heart (~70% in the patient versus ~15% in the mouse). It is possible that the extensive intervention undertaken to keep the patient alive (~10 months) allowed for progressive shortening of the thin filaments. The decrease in calcium sensitivity of the patient’s isolated cardiomyocytes appears to be unique. A previous study that analyzed the contractile mechanics of isolated cardiomyocytes from nine pediatric explanted hearts with DCM and two hearts with noncompaction cardiomyopathy showed that the diseased hearts had a decrease in maximal force but an increase in calcium sensitivity compared with adult nonfailing controls (20). While the underlying cause of the difference in calcium sensitivity is unclear, the lack of Lmod2 and/or very short thin filaments could result in a different phosphorylation profile of thin filament regulatory proteins in the patient.
The lack of detectable Lmod2 protein and decrease in Lmod2 mature mRNA, but not pre-RNA, in the patient’s explanted heart suggests that the mutant mRNA is eliminated by NMD. The position of the premature stop codon in Lmod2 [>55–base pair (bp) upstream of the final exon-exon junction but not very close to the start codon] is consistent with a transcript that has the potential to be an NMD substrate (21). Furthermore, expression of a relatively small amount of truncated Lmod2 ameliorates the cardiac phenotypes displayed by Lmod2 knockout mice, supporting the conclusion that the patient does not express significant levels of the mutant protein. Together, these results also provide a potential for therapeutic intervention. Inhibition of NMD may result in enough functional Lmod2 protein to improve cardiac function in patients with similar nonsense mutations. Since NMD has been shown to be involved in normal gene regulation, global inhibition of NMD is likely deleterious (22). Thus, one possible therapeutic avenue could be to target specific components of NMD such that disease-causing transcripts containing premature stop codons are preserved with minimal effects on endogenous regulation (23, 24).
Although one limitation of this work is that only a single patient was identified with a biallelic loss-of-function mutation in LMOD2, several lines of genetic evidence support pathogenicity of this mutation (25), including the following: (i) it is a nonsense mutation; (ii) it is biallelic and inherited from two unaffected carrier parents; and (iii) it is absent (in the homozygous state) from control population databases. In addition, computational prediction algorithms and functional analysis of the variant in human myocardium strongly support pathogenicity. Furthermore, the patient underwent an extensive genetic, metabolic, and infectious workup, and no alternative etiology of her heart failure was identified.
It is likely that additional cases of LMOD2-related cardiomyopathy remain undiagnosed, given the early lethality of the phenotype and limited use of clinical exome sequencing in this patient population. Addition of LMOD2 to clinical cardiomyopathy sequencing panels could facilitate identification of additional cases. This would help define the full clinical spectrum of LMOD2-related cardiomyopathy, as it is possible that milder mutations may be associated with a more attenuated clinical course. In summary, a biallelic loss-of-function LMOD2 mutation induces profound cardiac muscle dysfunction with aberrant thin filament formation and should be considered in the differential diagnosis of severe DCM.
MATERIALS AND METHODS
Human tissue
All work was approved by the Institutional Review Board (IRB) at the University of Arizona and the IRB at the Children’s Hospital of Philadelphia. Well-characterized left ventricular tissue from three healthy donor hearts as close in age as possible to the patient was obtained from the Sydney Heart Bank (26) (University of Sydney, Australia) following approval of the Human Research Ethics Committee. Their sample codes are 5.114, 7.050, and 4.152. The tissue was transported and stored in liquid nitrogen.
Animal studies
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Arizona.
Western blot
For human tissues, a piece of LV free-wall tissue was ground on liquid N2 in a Kontes dounce tissue grinder and solubilized in urea buffer [50 mM tris-HCl (pH 6.8), 8 M urea, 2 M thiourea, 3% SDS, 75 mM dithiothreitol, 10 mM NaF, 5 mM Na3VO4, and 0.03% bromophenol blue] plus 50% glycerol and protease inhibitors at 60°C for 10 min. Mouse LV tissue was solubilized as described previously (11). Lysate was resolved on a 10% SDS gel and transferred to nitrocellulose, and Western blotting was completed as previously described (15). Primary antibodies included goat polyclonal anti–α-actinin 1 (0.5 μg/ml; AF8279, R&D Systems), mouse monoclonal anti–β-tubulin (0.7 μg/ml; 1D4A4, Proteintech), mouse monoclonal anti-GFP (0.2 μg/ml; B-2, Santa Cruz Biotechnology), mouse monoclonal anti–cardiac actin (1:1000; Ac1.20.4.2, American Research Products), rabbit polyclonal anti-Lmod2 (0.1 μg/ml; E13, Santa Cruz Biotechnology), rabbit polyclonal anti-Tmod1 (0.08 μg/ml), mouse monoclonal anti–tropomyosin-1 (1:2000; TM311, Novus Biologicals), mouse monoclonal anti–cardiac troponin I (1:1000; Mab20, Covance), and mouse monoclonal anti–troponin T (1:200; JLT-12, Sigma-Aldrich). Secondary antibodies included Alexa Fluor 680 or Alexa Fluor 790 AffiniPure donkey anti-rabbit, mouse, or goat immunoglobulin G (IgG) (1:40,000; Jackson ImmunoResearch). Blots were imaged and analyzed using the Odyssey CLx imaging system (LI-COR Biosciences).
Reverse transcription quantitative polymerase chain reaction
A piece of LV was placed in ribonuclease-free microcentrifuge tubes containing RLT lysis buffer (QIAGEN) and stainless steel beads (Green Bead Lysis Kit, Next Advance Inc.) and homogenized in a Bullet Blender (BBX24, Next Advance Inc.) at speed 10 for 4 min at 4°C. Total RNA was extracted using the RNeasy Fibrous Tissue Mini Kit, including on-column deoxyribonuclease digestion (QIAGEN). cDNA was synthesized from 30 ng of total RNA using the SuperScript IV (Thermo Fisher Scientific). An oligo deoxythymidine dT primer was used to enrich for mature mRNAs, while a random hexamer primer was used to obtain immature mRNAs. Five microliters of template cDNA (diluted 1:6) was used in a PCR with Maxima SYBR Green qPCR master mix (Thermo Fisher Scientific) on a LightCycler 480 (Roche). To determine relative gene expression, a modified ΔΔCt method was used (27). This method takes primer efficiencies (E) into account and normalizes each sample to the geometric mean of multiple reference genes. Reference genes ribosomal protein L32 (Rpl32) and tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ), shown to be stable in failing human hearts, were selected from the literature (28, 29). Primer efficiencies were determined from standard curves generated using cDNA dilutions. Primers included Lmod2 spanning intron 1 [5′-GGACATTCAGCAGAGAGGCACT-3′ (forward) and 5′-GATAAGCTCTTCTTCACTTTCCTC-3′ (reverse); 128-bp product; E = 1.834], Lmod2 located within intron 1 [5′-TGCAGATAGAGCCAGAGGGT-3′ (forward) and 5′-GGGGCCTGTCTCAAAATCCA-3′ (reverse); 95-bp product; E = 1.873], Rpl32 [5′-CACCAGTCAGACCGATATGTCAAAA-3′ (forward) and 5′-TGTTGTCAATGCCTCTGGGTTT-3′ (reverse); 64-bp product; E = 1.912], and YWHAZ [5′-ACCGTTACTTGGCTGAGGTTGC-3′ (forward) and 5′-CCCAGTCTGATAGGATGTGTTGG-3′ (reverse); 130-bp product; E = 2.064].
Immunofluorescence microscopy
LV pieces were thawed, stretched, fixed, frozen, cryosectioned, and stained as previously described (15). Primary antibodies included rabbit polyclonal anti-Tmod1 (2 μg/ml) and mouse monoclonal anti–α-actinin (1:200; EA-53, Sigma-Aldrich) antibodies. Secondary antibodies included Alexa Fluor 488–conjugated donkey anti-rabbit IgG (1:1000) and Alexa Fluor 350–conjugated goat anti-mouse IgG (1:200). Texas Red–conjugated phalloidin (1:50) was used to stain F-actin (Thermo Fisher Scientific). Images were captured using a DeltaVision RT system (GE Healthcare) with a 100× numerical aperture 1.3 objective and a charge-coupled device camera (CoolSNAP HQ, Photometrics). Images were deconvolved using SoftWoRx software and processed using Photoshop CC (Adobe). Thin filament lengths were measured by Tmod1 staining using the DDecon plugin for ImageJ (30).
Solutions
The compositions of all solutions were reported previously for single cells (31). Activating solution and relaxing solution were mixed to obtain activating solutions containing between 0.64 and 46.8 μM [Ca2] (pCa, 6.2 to 4.3).
Force-calcium relationship
Single-cell experiments were performed on an inverted microscope stage using an Aurora Scientific 803B permeabilized myocyte apparatus with some slight modifications. A 406A force transducer was used to get a wider range of minimum and maximum forces. Permeabilized single cells were acquired by mechanically separating cells from a small chunk of tissue via gentle homogenization with a tissue grinder (ULTRA-TURRAX T8; IKA), in standard relaxing solution containing 1% Triton X-100. The cells were gently pelleted with a 3-min centrifugation at 23 relative centrifugal forces. The pellet was washed three times with standard relaxing buffer to remove any remaining detergent, and the centrifugation was repeated. Sarcomere length for all samples was set to 1.8 ± 0.4 μm before activation and checked throughout the experimental procedure. Since single cells isolated from the patient did not produce measurable force, clusters of cells were used.
Force-[Ca2+] relationships were fit individually to a modified Hill equation as previously described (32) to
| (1) |
where Frel is the force as a fraction of maximum force at saturating [Ca2+] (Fmax), EC50 is [Ca2+] where the Frel is half of Fmax, and n is the Hill coefficient.
Adeno-associated virus rescue experiments
Mouse Lmod2 cDNA was cloned into pEGFP-C2 vector (Clontech). The W405* mutation was introduced into GFP-Lmod2 via site-directed mutagenesis using the following primer sequences: 5′-GTCTCCCTGATCATCTCCCAAAGTCTCCAAGAAAG-3′ (forward) and 5′-GATGATCAGGGAGACTGTCTCGGAGATGCATATGG-3′ (reverse) (33). Adeno-associated virus expressing GFP-Lmod2 or GFP-Lmod2[W405*] was generated, and 1.25 × 1012 to 2.5 × 1012 genomic copies were injected into the pericardial cavity at postnatal day 4 as previously described (11). The Lmod2 null mice were on C57BL/6J background (stock no. 000664, the Jackson laboratory). Both males and females were injected and euthanized at a time point at which the null mice begin to die (postnatal day 16 to 18) (11).
Mouse echocardiography
Isoflurane-anesthetized mice were placed in dorsal recumbence on a heated (37°C) platform. After attaining a target heart rate of 550 ± 50 beats/min, transthoracic echo images were obtained with a Vevo 770 high-resolution imaging system (VisualSonics) using a model 704 transducer array. Images were collected and stored as a digital cine loop for offline calculations. Standard imaging planes and functional calculations were obtained according to the American Society of Echocardiography guidelines. M-mode images at the level of the papillary muscles were used to determine LV wall thicknesses, chamber dimensions, and ejection fraction.
Statistics
Prism 7 was used to perform all statistical analyses (GraphPad Software). Student’s t tests were used to compare two groups (Fig. 3B). Multiple groups were compared using one-way analyses of variance (ANOVAs) with Tukey’s post hoc test (Figs. 4 and 5 and tables S2 and S3). P < 0.05 was considered significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Lines were fit to the data in fig. S2B using linear regression analysis, and correlations were considered significant if the slopes of the linear regression fits were significantly nonzero.
Supplementary Material
Acknowledgments
We would like to thank C. dos Remedios (University of Sydney) for assistance in providing us nonfailing heart samples from the Sydney Heart Bank. Funding: This work was supported by NIH R01HL108625 and R01HL123078 as well as funding from Linda and Jim Lee to C.C.G., T32GM008638 to R.C.A.-N., and T32GM07526-41 to C.M. Author contributions: R.C.A.-N., L.M., A.R., I.D.K., C.M., K.Y.L., S.W.Y., and C.K.C. completed the clinical and genetic diagnostic evaluations of the human patient. C.T.P. and C.C.G. designed experiments. C.T.P., G.P.F., R.M.M., and T.M.L. performed and analyzed experiments. R.C.A.-N., J.H.B., C.K.C., C.T.P., G.P.F., and C.C.G. drafted the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax2066/DC1
Fig. S1. The levels of thin filament proteins are reduced in the patient’s LV.
Fig. S2. Even low levels of GFP-Lmod2 or GFP-Lmod2[W405*] expression prevents onset of DCM in Lmod2 knockout mice.
Table S1. Initial diagnostic evaluations.
Table S2. Summary of mechanics data for cells isolated from nonfailing donor hearts at 48, 24, and 14 months of age and the patient’s explanted heart (10 months).
Table S3. Echocardiography data of Lmod2 knockout mice injected with adeno-associated virus.
Reference (35)
REFERENCES AND NOTES
- 1.Hershberger R. E., Hedges D. J., Morales A., Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Lipshultz S. E., Sleeper L. A., Towbin J. A., Lowe A. M., Orav E. J., Cox G. F., Lurie P. R., McCoy K. L., McDonald M. A., Messere J. E., Colan S. D., The incidence of pediatric cardiomyopathy in two regions of the United States. N. Engl. J. Med. 348, 1647–1655 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Nugent A. W., Daubeney P. E., Chondros P., Carlin J. B., Cheung M., Wilkinson L. C., Davis A. M., Kahler S. G., Chow C. W., Wilkinson J. L., Weintraub R. G.; National Australian Childhood Cardiomyopathy Study , The epidemiology of childhood cardiomyopathy in Australia. N. Engl. J. Med. 348, 1639–1646 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Wilkinson J. D., Landy D. C., Colan S. D., Towbin J. A., Sleeper L. A., Orav E. J., Cox G. F., Canter C. E., Hsu D. T., Webber S. A., Lipshultz S. E., The pediatric cardiomyopathy registry and heart failure: Key results from the first 15 years. Heart Fail. Clin. 6, 401–413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pugh T. J., Kelly M. A., Gowrisankar S., Hynes E., Seidman M. A., Baxter S. M., Bowser M., Harrison B., Aaron D., Mahanta L. M., Lakdawala N. K., McDermott G., White E. T., Rehm H. L., Lebo M., Funke B. H., The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16, 601–608 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Herkert J. C., Abbott K. M., Birnie E., Meems-Veldhuis M. T., Boven L. G., Benjamins M., du Marchie Sarvaas G. J., Barge-Schaapveld D. Q. C. M., van Tintelen J. P., van der Zwaag P. A., Vos Y. J., Sinke R. J., van den Berg M. P., van Langen I. M., Jongbloed J. D. H., Toward an effective exome-based genetic testing strategy in pediatric dilated cardiomyopathy. Genet. Med. 20, 1374–1386 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Hershberger R. E., Givertz M. M., Ho C. Y., Judge D. P., Kantor P. F., McBride K. L., Morales A., Taylor M. R. G., Vatta M., Ware S. M., Genetic evaluation of cardiomyopathy-a heart failure society of America practice guideline. J. Card. Fail. 24, 281–302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershberger R. E., Givertz M. M., Ho C. Y., Judge D. P., Kantor P. F., McBride K. L., Morales A., Taylor M. R. G., Vatta M., Ware S. M.; ACMG Professional Practice and Guidelines Committee , Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 20, 899–909 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Chereau D., Boczkowska M., Skwarek-Maruszewska A., Fujiwara I., Hayes D. B., Rebowski G., Lappalainen P., Pollard T. D., Dominguez R., Leiomodin is an actin filament nucleator in muscle cells. Science 320, 239–243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukada T., Pappas C. T., Moroz N., Antin P. B., Kostyukova A. S., Gregorio C. C., Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J. Cell Sci. 123, 3136–3145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pappas C. T., Mayfield R. M., Henderson C., Jamilpour N., Cover C., Hernandez Z., Hutchinson K. R., Chu M., Nam K. H., Valdez J. M., Wong P. K., Granzier H. L., Gregorio C. C., Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc. Natl. Acad. Sci. U.S.A. 112, 13573–13578 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuen M., Sandaradura S. A., Dowling J. J., Kostyukova A. S., Moroz N., Quinlan K. G., Lehtokari V. L., Ravenscroft G., Todd E. J., Ceyhan-Birsoy O., Gokhin D. S., Maluenda J., Lek M., Nolent F., Pappas C. T., Novak S. M., D’Amico A., Malfatti E., Thomas B. P., Gabriel S. B., Gupta N., Daly M. J., Ilkovski B., Houweling P. J., Davidson A. E., Swanson L. C., Brownstein C. A., Gupta V. A., Medne L., Shannon P., Martin N., Bick D. P., Flisberg A., Holmberg E., Van den Bergh P., Lapunzina P., Waddell L. B., Sloboda D. D., Bertini E., Chitayat D., Telfer W. R., Laquerriére A., Gregorio C. C., Ottenheijm C. A., Bönnemann C. G., Pelin K., Beggs A. H., Hayashi Y. K., Romero N. B., Laing N. G., Nishino I., Wallgren-Pettersson C., Melki J., Fowler V. M., MacArthur D. G., North K. N., Clarke N. F., Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Invest. 124, 4693–4708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halim D., Wilson M. P., Oliver D., Brosens E., Verheij J. B., Han Y., Nanda V., Lyu Q., Doukas M., Stoop H., Brouwer R. W., van IJcken I. W. F., Slivano O. J., Burns A. J., Christie C. K., de Mesy Bentley K. L., Brooks A. S., Tibboel D., Xu S., Jin Z. G., Djuwantono T., Yan W., Alves M. M., Hofstra R. M., Miano J. M., Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. U.S.A. 114, E2739–E2747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S., Mo K., Tian H., Chu C., Sun S., Tian L., Ding S., Li T. R., Wu X., Liu F., Zhang Z., Xu T., Sun L. V., Lmod2 piggyBac mutant mice exhibit dilated cardiomyopathy. Cell Biosci. 6, 38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappas C. T., Farman G. P., Mayfield R. M., Konhilas J. P., Gregorio C. C., Cardiac-specific knockout of Lmod2 results in a severe reduction in myofilament force production and rapid cardiac failure. J. Mol. Cell. Cardiol. 122, 88–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson T. M., Michels V. V., Thibodeau S. N., Tai Y. S., Keating M. T., Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280, 750–752 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Olson T. M., Kishimoto N. Y., Whitby F. G., Michels V. V., Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J. Mol. Cell. Cardiol. 33, 723–732 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Robinson P., Griffiths P. J., Watkins H., Redwood C. S., Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Conley C. A., Fritz-Six K. L., Almenar-Queralt A., Fowler V. M., Leiomodins: Larger members of the tropomodulin (Tmod) gene family. Genomics 73, 127–139 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Bollen I. A. E., van der Meulen M., de Goede K., Kuster D. W. D., Dalinghaus M., van der Velden J., Cardiomyocyte hypocontractility and reduced myofibril density in end-stage pediatric cardiomyopathy. Front. Physiol. 8, 1103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hug N., Longman D., Cáceres J. F., Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karousis E. D., Nasif S., Muhlemann O., Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA 7, 661–682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang L., Low A., Damle S. S., Keenan M. M., Kuntz S., Murray S. F., Monia B. P., Guo S., Antisense suppression of the nonsense mediated decay factor Upf3b as a potential treatment for diseases caused by nonsense mutations. Genome Biol. 19, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Usuki F., Yamashita A., Shiraishi T., Shiga A., Onodera O., Higuchi I., Ohno S., Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 110, 15037–15042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W. W., Hegde M., Lyon E., Spector E., Voelkerding K., Rehm H. L.; ACMG Laboratory Quality Assurance Committee , Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dos Remedios C. G., Lal S. P., Li A., McNamara J., Keogh A., Macdonald P. S., Cooke R., Ehler E., Knöll R., Marston S. B., Stelzer J., Granzier H., Bezzina C., van Dijk S., De Man F., Stienen G. J. M., Odeberg J., Pontén F., Linke W., van der Velden J., The Sydney Heart Bank: Improving translational research while eliminating or reducing the use of animal models of human heart disease. Biophys. Rev. 9, 431–441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J., qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina C. E., Jacquet E., Ponien P., Muñoz-Guijosa C., Baczkó I., Maier L. S., Donzeau-Gouge P., Dobrev D., Fischmeister R., Garnier A., Identification of optimal reference genes for transcriptomic analyses in normal and diseased human heart. Cardiovasc. Res. 114, 247–258 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Brattelid T., Winer L. H., Levy F. O., Liestøl K., Sejersted O. M., Andersson K. B., Reference gene alternatives to Gapdh in rodent and human heart failure gene expression studies. BMC Mol. Biol. 11, 22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gokhin D. S., Fowler V. M., Software-based measurement of thin filament lengths: An open-source GUI for distributed deconvolution analysis of fluorescence images. J. Microsc. 265, 11–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan D., Wannenburg T., de Tombe P. P., Decreased myocyte tension development and calcium responsiveness in rat right ventricular pressure overload. Circulation 95, 2312–2317 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Farman G. P., Walker J. S., de Tombe P. P., Irving T. C., Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 291, H1847–H1855 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Liu H., Naismith J. H., An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fowler V. M., Dominguez R., Tropomodulins and leiomodins: Actin pointed end caps and nucleators in muscles. Biophys. J. 112, 1742–1760 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marston S., Montgiraud C., Munster A. B., Copeland O., Choi O., Dos Remedios C., Messer A. E., Ehler E., Knöll R., OBSCN mutations associated with dilated cardiomyopathy and haploinsufficiency. PLOS ONE 10, e0138568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax2066/DC1
Fig. S1. The levels of thin filament proteins are reduced in the patient’s LV.
Fig. S2. Even low levels of GFP-Lmod2 or GFP-Lmod2[W405*] expression prevents onset of DCM in Lmod2 knockout mice.
Table S1. Initial diagnostic evaluations.
Table S2. Summary of mechanics data for cells isolated from nonfailing donor hearts at 48, 24, and 14 months of age and the patient’s explanted heart (10 months).
Table S3. Echocardiography data of Lmod2 knockout mice injected with adeno-associated virus.
Reference (35)