

Abstract
The microsporidium, Anncaliia algerae (Brachiola algerae), is a eukaryotic obligate intracellular parasite first isolated from mosquitoes and is an important opportunistic human pathogen that can cause morbidity and mortality among immune-compromised individuals including patients with AIDS and those undergoing chemotherapy. There is little known about the Microsporidia-host cell interface in living host cells, due to current approaches being limited by the lack of fluorescent reporters for detecting the parasite lifecycle. Here, we have developed and applied novel vital fluorescent parasite labeling methodologies in conjunction with fluorescent protein-tagged reporters to track simultaneously the dynamics of both parasite and host cell specific components, including the secretory and endocytic trafficking pathways, during the entire infection time period. We have found dramatic changes in the dynamics of host secretory trafficking organelles during the course of infection. The Golgi compartment is gradually disassembled and regenerated into mini-Golgi structures in parallel with cellular microtubule depolymerization. Importantly, we find that Microsporidia progeny are associated with these de novo formed mini-Golgi structures. These host structures appear to create a membrane bound niche environment for parasite development. Our studies presented here provide novel imaging tools and methodologies that will facilitate in understanding the biology of microsporidial parasites in the living host.
Keywords: Anncaliia algerae, Arf-1-GFP, DRAQ5, Golgi, HeLa cells, live imaging, microtubules, organelle hijacking
MICROSPORIDIA are a phylum of obligate intracellular spore-forming parasites containing over 200 genera and approximately 1,400 species (Becnel et al. 2014). They have a very broad host range (invertebrates and vertebrates) and are opportunistic human pathogens that can cause morbidity and mortality among immuno compromised individuals. As intracellular parasites, they are dependent upon their host for access to nutritional products and have evolved several ways to obtain the required metabolites, which in turn have reduced their need to produce many of the biochemicals necessary for their development. As a result of this reduced need to produce their own metabolites, there has been a reduction in their physiological machinery (Corradi et al. 2010; Katinka et al. 2001), biochemical pathways, and their genomes are small (Biderre et al. 1994; Katinka et al. 2001). Gene sequencing data has indicated that much of the microsporidian cell machinery has also been reduced as a response to their reduced biochemical manufacturing needs, this has been described as functional minimization (Williams 2009; Williams et al. 2014).
Due to the great diversity of microsporidia and the broad range of hosts, their host-parasite interface is quite variable depending on the genus. These interfacial relationships can be divided into four categories; direct contact with the host cytoplasmic organelles, indirect contact due to parasite induced isolation, indirect contact by host-produced isolation, indirect contact by host- and parasite-produced isolation (Cali and Takvorian 2014). Regardless of the type of interface, the parasite is dependent upon the host for its nutritional needs and those organisms which develop in direct contact within the host cytoplasm often are observed in close contact or abutted to host organelles such as mitochondria and ER (Cali and Takvorian 2014). Although the relationship between the host organelles and parasite are currently poorly understood, it is clear that as intracellular obligate parasites, nutrition, energy, and possibly cellular components are being provided by the host. Since the use of electron microscopy to study the microsporidia in the late 1960s many reports commented or described the clustering of host mitochondria near developing parasites (Scanlon et al. 2004). Recently, Hacker et al. (2014) demonstrated mitochondrial binding to the parasitophorous vacuole of Encephalitozoon cuniculi to supply ATP to developing proliferative parasites and Troemel et al. (2008) demonstrated interaction with host cell intermediate filaments disrupting their assembly and producing gaps which enable Nematocida parisii spores to exit without host cell lysis.
While fluorescence-imaging studies have been used for microsporidial spore identification (Cali and Takvorian 2014), some aspects of spore germination (Cali and Takvorian 2014) and identification of polar filament/tube components (Bouzahzah et al. 2010) little has been reported on their interaction within the host cell because live micro-sporidial dynamics have largely not been possible. Because microsporidia have not been able to be genetically altered/modified/manipulated or transfected for live cell studies, their interaction with the host cell has remained somewhat enigmatic. In addition, few of the microsporidia can be grown in host cell cultures, and in cases where they can be cultured, many of the cell types are neither easily transfectable with fluorescent protein- tagged expression constructs nor are antibodies available with which to monitor the spatio-temporal distribution of host cell components. The objective of this project was to develop a simple fluorescent technique that will allow us to culture and identify a living microsporidium and investigate the mechanisms of its development within a host cells for which molecular tools are available, such as HeLa cells. We selected Anncaliia algerae, because it is a microsporidium which develops in direct contact with the host cell cytoplasm, grows in a multitude of host cell culture types, is diplokaryotic and has been identified as an opportunistic human pathogen that can cause morbidity and mortality among immuno compromised individuals (Cali et al. 2004, 2005).
Here, we demonstrate that A. algerae can be successfully labeled with the nuclear vital stain DRAQ-5 (Smith et al. 1999) and this dye can be used to monitor the spatio-temporal dynamics of A. algerae during infection and growth in human HeLa cells. By monitoring the live-cell dynamics of Arf1-GFP, a small Ras family GTPase that is critical for Golgi apparatus biogenesis and maintenance (Altan-Bonnet et al. 2004; Hsu et al. 2010; Niu et al. 2005) we discovered a close spatio-temporal interaction between the host secretory pathway and the pathogen.
MATERIALS AND METHODS
Parasite spore source, collection and purification
Anncaliia algerae spores were provided by Dr Albert H. Undeen from his original mosquito isolate and have been propagated in RK13 (rabbit kidney) cell cultures maintained at 32 °C in our laboratory for approximately 20 yr (Cali et al. 2002, 2004). The parasite spores are collected from culture media, concentrated by centrifugation, and stored in sterile distilled water at 4 °C. Spores used in these experiments were counted with a hemocytometer (three times/sample and averaged).
Cell culture and DNA transfection
Cell lines
HeLa cells were maintained in DMEM supplemented with 10% HIFBS, 2 mM L-glutamine, 1% P/S at 37 °C/5%CO2. During the experiment, cells were maintained in imaging media (DMEM without Phenol Red supplemented with 10% HIFBS and 25–50 mM HEPES pH 7.3; termed complete imaging culture media [CICM]).
Plasmids
Arf1-GFP, and Arf1-RFP (Jennifer Lippincott-Schwartz, National Institutes of Health [NIH]). Plasmids were transfected into Hela cells using FuGENE6 Transfection Reagent (Roche, IN) and incubated for 16–24 h at 37 °C/ 5% Carbon Dioxide (CO2) (Hsu et al. 2010).
Anncaliia algerae in HeLa cell culture
HeLa cells were incubated at 32 °C for 24 h to allow the cells to acclimate to the lower temperature. Anncaliia algerae spores (~2 × 105 per dish) were introduced to the cultures and maintained for 1 −8 d. These cultures were fixed on postinfection days 1, 3, 5, and 8 and stained with DRAQ-5 for imaging. Uninfected labeled cultures were fixed for controls.
Labeling spores with DRAQ5
DRAQ-5 is a fluorescent DNA dye compatible for use in live or fixed samples, its excitation at 633 nm emits fluorescence at ~700 nm (far red light). Live imaging: A. algerae spores were suspended in 1 × PBS from the stock at 1 × 107 spores per ml. DRAQ-5™ (Bio Status Limited, Leicestershire, UK) was diluted in the spore solution at 1:1,000 ratio; and incubated for 1 h at room temperature (RT) or overnight at 4 °C in an orbital shaker. Labeled spores were rinsed two times with 1 × PBS; labeled spores were then resuspended in 1 × PBS and stored at 4 °C until ready to use. Fixed samples: DRAQ-5 was diluted to 5 μM in 1 × PBS and added to samples. Samples were incubated for 20 min at 37 °C; then rinsed once with PBS and mounted.
Live-cell imaging of Arf1-GFP cells/DRAQ-5 labeled microsporidia
HeLa cells were grown on coverslip-bottomed Lab-Tek chambers (Thermo Fisher, Buffalo, NY) at 1 × 104 cells per ml and incubated overnight. Cells were transfected with 1 μg of Arf1-GFP plasmid with Fugene 6 transfection reagent and then were incubated for 16–24 h. Subsequently, cells were inoculated with 2 × 105 DRAQ5 labeled spores per dish and incubated overnight then rinsed twice to remove free spores. Samples were imaged every day for 8 d.
Microtubule interactions with A. algerae
Cultures of HeLa cells grown on coverslips were exposed to unlabeled A. algerae spores when the cells were approximately 70% confluent. Cultures were fixed and immuno labeled for β-tubulin on postinoculation (PI) days 3 and 5. Controls were uninfected cultures processed the same way. Nocodazole, an inhibitor of microtubule polymerization, was applied to uninfected HeLa cells. Samples were fixed after 30 min exposure to 5 μg/ml Nocodazole solution to provide a control for β-tubulin disruption and later processed for staining or immunofluorescence labeling.
Microtubule and Golgi interaction
Uninfected HeLa cells transfected with Arf1-RFP were incubated over ice for 10 min with 5 μg/ml nocodazole solution (nocodazole was diluted in CICM from a 5 mg/ml stock solution) and further incubated for 1 h at 37 °C/5% CO2 to allow cells to reach a steady state. Samples were fixed in 4% paraformaldehyde followed by immune fluorescence labeling of β-tubulin.
Immunofluorescence labeling
Cells plated on coverslips, were fixed with 3.7% FA or PFA solution in 1 × PBS for 10 min at RT, and then washed three times with 1×PBS. Primary antibody was diluted (1:200) in incubation buffer (0.2% saponin; 1% BSA; in 1 × PBS) and incubated for 1 h at RT; then rinsed three times 5 min each with 1 × PBS. Fluorophore tagged secondary antibodies were diluted (1:500–1,000), incubated for 1 h at RT followed by rinsing three times 5 min each with 1 × PBS.
Primary antibodies
Chicken anti-GFP antibody (Millipore, Bedford, MA), rabbit anti-YFP (Rmanujan Hedgeat the MRC Laboratory of Molecular Biology, Cambridge, UK) and mouse anti-βTubulin (Santa Cruz Biotechnology, Dallas, TX).
Secondary antibodies
Fluorescently labeled goat anti-chicken, goat anti-mouse, and goat anti-rabbit antibodies were commercially obtained (Invitrogen Corp., Carlsbad, CA; Jackson Immuno research Labs Inc., West Grove, PA).
Fixation
Methanol
Remove media and allow samples to air dry, add 100% Methanol and incubate for 10 min at RT. Remove the methanol and allow the samples to air dry. Add 1 × PBS and proceed with staining or immunofluorescence labeling.
Formaldehyde and paraformaldehyde
Remove media and add 4% solution in 1 × PBS. Incubate for 10 min at RT rinse four times with 1 × PBS and proceed with staining or immunofluorescence labeling.
Imaging and analysis
All imaging was performed on a Zeiss LSM 510 META confocal laser scanning confocal microscope (Carl Zeiss, Thornwood, NY). The LSM510 system was equipped with a 488–561Argon laser and a 633 HeNe laser, a heating stage, and an incubator with temperature, humidity and CO2 control for live-cell imaging. Images were obtained using a 63× oil immersion DIC Plan Apochromat objective lens with a numerical aperture of 1.4; using emission filters BP500–550IR for 488 and 561 nm light, BP650–710IR for the 633 nm light, and pinhole size of 248 μm,. Live cells were observed and imaged on the microscope stage in a temperature, CO2, and humidity controlled environmental chamber (Hsu et al. 2010). Images were analyzed with LSM or ImageJ software Imagej.NIH.gov.
RESULTS
The microsporidium, A. algerae was selected for this study for multiple reasons; one is its(paired abutted) nuclei which allows it to be easily distinguishable with DRAQ5 inside the host cytoplasm, additionally it can be cultured in many cell lines. In our laboratory A. algerae has been successfully cultured and propagated in RK-13 cells but the use of this cell line is limited due to the difficulties in transfection, the availability of fluorescent protein-tagged rabbit proteins as well as antibodies to native rabbit proteins. HeLa cells alternatively, are the most widely used human cell line for which there is a vast number of fluorescent protein-tagged expression constructs and antibodies available. Thus, we selected this cell line to test if it can be used for A. algerae infections.
HeLa cells grown at 37 °C/5%CO2 were plated on 12 well plates containing glass cover slips at 1 × 104 cells/ ml/well, and incubated overnight at 32 °C/5%CO2. HeLa cells were then infected with 1 × 105 spores/ml, incubated for 24 h at 32 °C, and rinsed to remove any free spores left from the initial inoculum. These cultures were methanol fixed, labeled with DRAQ5, a nuclear stain, with far-red fluorescence emission. The DRAQ-5 labeled parasite and host nuclei were visible with fluorescence microscopy (Fig. 1A, B). Inoculated cultures were fixed, stained, and observed every day for eight consecutive days. Microscopic analysis showed that the parasite was able to infect and replicate in HeLa cells. On the first day postexposure to the parasite, both intact and germinating spores were easily identified in the culture medium and inside the cells. After spore germination, the sporoplasm which contains the paired abutted nuclei could also be observed within the host cell cytoplasm (Fig. 1B). By day 3, we were able to identify proliferative forms inside the cells, characterized by multiple visible elongated diplokaryotic nuclei (Fig. 1C). At day 5, HeLa cells are filled with newly formed parasites (Fig. 1D).
Figure 1.
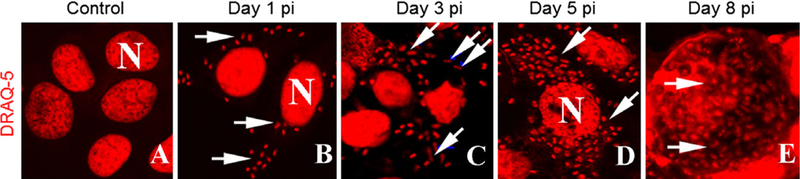
HeLa cells infected with Anncaliia atgerae were fixed at different time points during infection and fluorescently stained with DRAQ5. A. ControlHeLa cells (no infection) with nuclei stained (N). B. Inoculated HeLa cells at day 1, nuclei (diplokarya) of organisms appear elongated (arrows). They are located both at the surface and inside of the host cells. C. At day 3 postinoculation (PI), parasites are inside the cells and actively dividing (arrows). D. Day 5 PI, host cells are filled with developing stages and newly formed spores. E. Day 8, host cells are filled with red stained nuclei of developing stages and spores. Arrows = DRAQ5 stained parasite nuclei; N = host nuclei.
We next tested whether DRAQ5 could be used as a live spore fluorescent probe during infection. We incubated A. algerae spore suspensions with DRAQ5 prior to inoculation and determined if the labeled live spores were able to germinate and infect HeLa cells. Indeed prelabeled live microsporidia were identified inside HeLa cells following inoculation, indicating that DRAQ5 did not interfere in the germination of the spore and infection of the host cell (Fig. 2).
Figure 2.
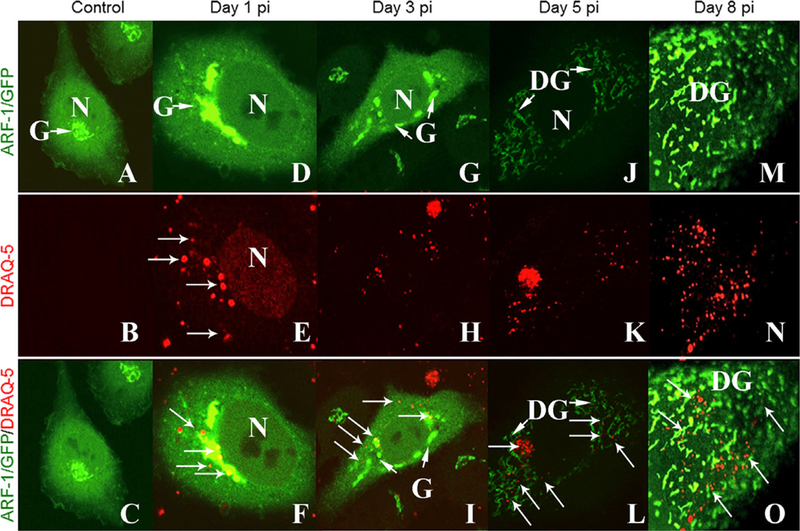
Live imaging of microsporidia and Golgi Reorganization. Plated HeLa cells were transfected with Arf1-GFP DNA plasmid before inoculating the cultures with DRAQ5 labeled Anncaliia algerae, and imaged at different time points during infection. A-C. ControlHeLa cells (No infection) showing the distribution of Arf-1, in the Golgi compartment. Note its perinuclear location. D-F. Day 1 postinfection, Arf-1 distribution is similar to the control cells and there are organisms (arrows) in the cells. G-I. On the third day PI, the Golgi fragments and shows a perinuclear distribution. Parasites are visible near the host nucleus and Golgi. J-L. Day 5 PI, the Golgi is disorganized and fragmented into mini-Golgi like structures that are redistributed throughout the cytosol coinciding with the parasite distribution. M-O. By the eighth day, the new mini-Golgi structures are clearly visible allover the host cell cytosol and are associated with parasite progeny (arrows). Arrows = DRAQ5 stained parasite nuclei; DG = mini-Golgi; G = Golgi; N = host nuclei.
Given this, we next determined if we could follow the infection of DRAQ5 labeled microsporidia in conjunction with HeLa cells expressing GFP-tagged expression constructs of host cell proteins. We focused on the little known dynamics of the host secretory pathway, in particular the Golgi apparatus, with respect to the microsporidial development, during infection. HeLa cells were transfected with Arf1-GFP plasmid to label the Golgi (Fig. 2A) and were then infected with DRAQ5 labeled live spores. After infection, the cells were washed to remove any ungerminated spores from the culture. At postinfection days 1, 3, 5, and 8, the cultures were imaged.
At day 1 postmicrosporidial inoculation (PI), the Arf-1 labeled Golgi is in a perinuclear position similar to that of control HeLa cells (Fig. 2D). A few A. algerae DRAQ5 labeled spores are visible at the periphery of the cell and some sporoplasms appear to be abutted to the host nucleus and localized near the Golgi (Fig. 2D, E). Some residual DRAQ5 from the spore inoculum carried over into the host cultures, lightly staining the host nucleus (Fig. 2E). By day 3, some segments of Golgi were still in perinuclear locations but they appeared as independent narrow entities and some were located distal to the host nucleus. Several labeled developing parasites appear to be co-localized with both the perinuclear and distal Golgi structures (Fig. 2G–F). Major morphological and positional changes to the host Golgi appeared 5 d PI. The Golgi now appeared fragmented and diffused throughout the host cytoplasm. Thin wisps of Arf-1 label were still located in the perinuclear region but the majority of label was now distributed distal to the nucleus (Fig. 2J). Intermixed with these fragmented mini-Golgi structures were numerous DRAQ5 labeled microsporidia (Fig. 2K, L). This indicated that the original DRAQ5 labeled spore inoculum had successfully undergone mitosis to produce large numbers of progeny and hence the DRAQ5 label had not interfered with cell cycle dynamics and despite repeated parasite cell division, adequate label remained to visualize parasites (Fig. 2K). By 8 d PI, the mini-Golgi like structures were clearly visible throughout the host cell cytosol (Fig. 2M) and associated with the various stages of DRAQ-5 labeled parasite progeny (Fig. 2N, O). The parasites have greatly increased in number and appear to be closely intermingled with the Arf-1 labeled mini-Golgi like structures (Fig. 2N).
Microtubule experiments
The formation of the mini-Golgi like structures suggests alteration of the cytoskeleton. Microtubules are well known to be required for the formation and maintenance of the perinuclear Golgi stacks and disrupting these results in the formation of mini-Golgi stacks adjacent to ER exit sites as well as overall cellular architecture. Therefore, we subsequently investigated the effects of microsporidial infection on microtubules.
For these experiments, HeLa cells were grown at 32 °C as above, however, the controls (Fig. 3A–C) and A. algerae exposed cultures were fixed, labeled with an anti-body against β-tubulin, and stained with DRAQ5 before mounting. At day 3 of infection, the distribution of the microtubules is similar to the control cells and a number of parasites are in the cytoplasm (Fig. 3D–F). However, by day 5, the microtubule organization was completely disrupted and parasites were present throughout the cytoplasm (Fig. 3G–I). Application of nocodazole, a microtubule disrupter, was added to uninfected HeLa cells (control); they display a complete disruption of the microtubule organization similar to the disruption observed in the A. algerae infected cells. In addition, the perinuclear Golgi apparatus was dismantled and replaced by numerous mini-Golgi stacks throughout the cytoplasm (Fig. 3J, L).
Figure 3.
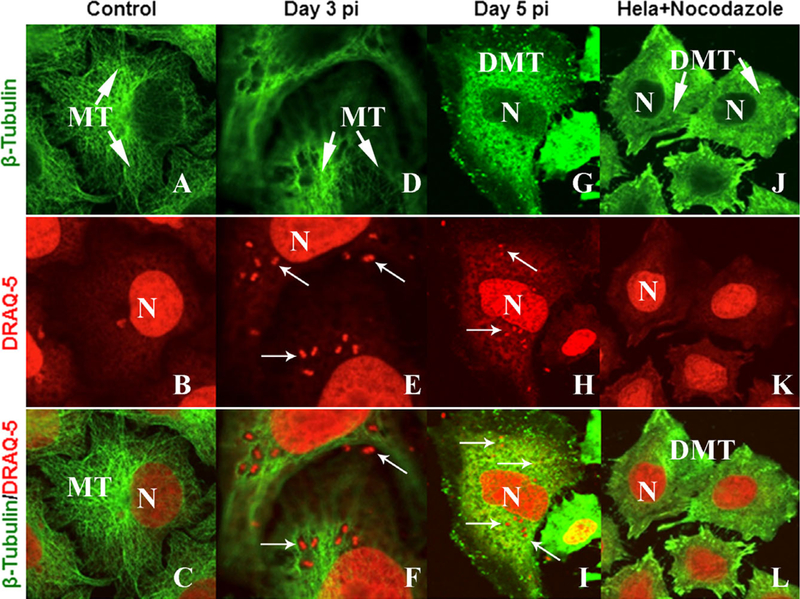
Microtubule reorganization. Plated HeLa cells were stained for β tubulin to show microtubule distribution in control cells and compared with Anncaliia algerae infected or Nocodazole treated cells. DRAQ5 was added to the samples to stain host and parasite nuclei. A-C. Control HeLa cells with microtubules (MT) in their normal organization and distribution. D-F. Three days post A. algerae infection. The organization of microtubules has changed in association with the parasites (arrows). G-I. Day 5 PI the microtubules no longer present in elongated patterns instead they appear disorganized while parasites are distributed throughout the cytoplasm (arrows). J-L. Microtubules of uninfected HeLa cells treated with nocodazole for 30 min appear similar to the disorganization induced by the microsporidian. N = host nuclei; arrows = DRAQ5 stained parasite nuclei; MT = microtubules
To study the effect of disrupted microtubules on Golgi, uninfected Arf1-RFP transfected HeLa cells were fixed and labeled for β-tubulin. These cells contained well organized microtubules and perinuclear Golgi (Fig. 4A–C). A second set of these cultures were treated with nocodazole. The p-tubulin labeled microtubules were depolymerized throughout the cell (Fig. 4D). The Arf1-RFP labeled Golgi demonstrated the formation of mini-Golgi scattered in the cytoplasm (Fig. 4E). These nocodazole experiments demonstrate that the disruption of microtubules correlates with the formation of mini-Golgi stacks (Fig. 4F), similar to the effect of microsporidial infection.
Figure 4.
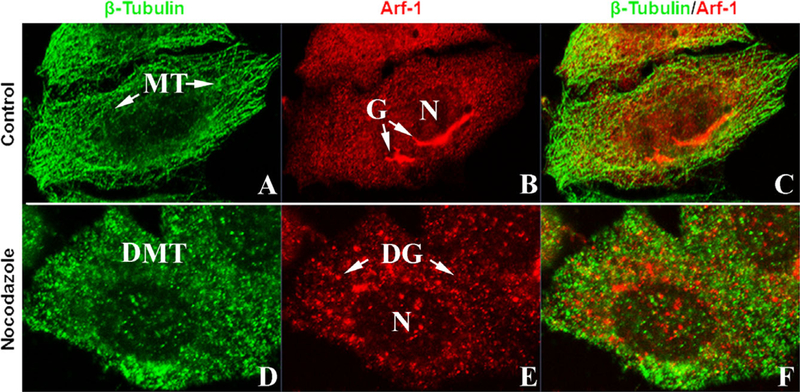
Nocodazole disruption of microtubules and Golgi in HeLa cells. Plated HeLa cells were transfected with Arf-1/RFP DNA plasmid and stained for β tubulin to show microtubule distribution. A. ControlHeLa cells fluorescent stained for β tubulin. Note the presence of organized networks of microtubules. B. ControlHeLa cells with RFP labeled Golgi. Note the perinuclear location of the Golgi. C. Co-localization of β tubulin and Golgi in controlcells. Images D-F. Nocodazole treated HeLa cells. D. HeLa cellcytoplasm is filled withscattered clumps of β-tubulin labeled mate- rialfrom the disrupted/depolymerized microtubules. E. Golgi disruption forming mini-Golgi. F. Co-localization of b-tubulin labeled disrupted microtubules and Arf-1 labeled mini-Golgi. DG = mini-Golgi; DMT = depolymerized microtubules; G = Golgi; MT = microtubules; N = nucleus.
DISCUSSION
As obligate intracellular parasites, Microsporidia have become almost totally dependent upon their host cell’s metabolism and organelles for all stages of their life cycle from initial infection through spore formation, the only stage that can survive outside the cell (Weiss and Becnel 2014). During the last 100 yr of morphological observations with various types of light and electron microscopes, most studies have been limited to observing aspects of the life cycles and host cell interactions of fixed material. The utilization of DRAQ-5 labeled spores and transfected cells has enabled us to follow the infective cycle and parasite-host organelle interaction in live cells.
The study of the microsporidian life cycle and the parasite’s interaction with the host cellular components is important for our understanding of how these minimalist organisms have evolved and adapted strategies to not only survive in a hostile environment, but also how to utilize their hosts machinery for their reproduction. In this study, we investigated new fluorescent methodologies to label microsporidia, live and fixed, in conjunction with fluorescent protein-tagged host proteins to track simultaneously the dynamics of both parasite and host cell specific components during the entire infection period in fixed and live HeLa cells.
After successfully cultivating A. algerae in HeLa cells, we utilized DRAQ5 to stain both the microsporidian and host cell nuclei in fixed cultures (Fig. 1). DRAQ5 is a deep red fluorescing agent, a synthetic anthraquinone with a high affinity for DNA and a capacity to rapidly enter living cells or stain fixed cells. DRAQ5 is optimally excited by red-light emitting sources and yields a deep red emission spectrum at 633 nm (Edward 2009; Smith et al. 1999; Wiltshire et al. 2000). We demonstrate here that DRAQ5 could penetrate the spore wall to label the diplokarya while not adversely affecting the spores ability to germinate. With this novel tool, we were able to inoculate HeLa cell cultures and visualize the dynamics of live microsporidial development from sporoplasms through spore formation enabling us to follow the multiple intracellular cell divisions up to 8 d in culture (Fig. 2). Notably, this developmental cycle coincides with previous studies of unlabeled A. algerae development in RK-13 cells (Cali et al. 2004; Santiana et al. 2015; Takvorian et al. 2005) and thus further supports DRAQ5 as a tool for monitoring live microsporidial dynamics.
DRAQ5 labeled A. algerae also enable us to identify a novel spatio-temporal interaction between the developing parasites and the host secretory pathway. The normally perinuclear host Golgi apparatus was disassembled into “mini-Golgi like” structures, redistributed throughout the cytosol (Altan-Bonnet et al. 2004; Cole et al. 1996) which coincided with the parasite distribution, as indicated by the presence of the DRAQ5 labeled organisms (Fig. 2). We observed by day 5 PI, that the host microtubule organization was also severely disrupted similar to uninfected HeLa cells treated with nocodazole, a microtubule polymerization inhibitor (Fig. 3). Furthermore, the Golgi apparatus appeared disassembled with the Arf1-GFP labeling now as punctate organelles distributed across the cytoplasm (Fig. 2). Previously, it has been reported that cellular microtubule depolymerization results in a block in anterograde transport of secretory membranes out of the ER to the microtubule organizing center while retrograde trans-port from the Golgi back to the ER continues largely unchanged leading to the disruption of the Golgi apparatus and redistribution of Golgi enzymes to ER exit sites where they emerge to form “mini-Golgi” stacks (Cole et al. 1996; English and Voeltz 2013; Niu et al. 2005).
The appearance of the Golgi in infected cells looks very much like the appearance of the Golgi in uninfected cells when the microtubules are intentionally depolymerized (Fig. 2 E, 4F), suggesting that the presence of the microsporidium, has a microtubule disrupting effect leading to the reorganization of the Golgi into many mini-Golgi-like structures.
Notably, we found that the redistribution of the Golgi apparatus into mini-Golgi like structures throughout the cytosol was spatio-temporally associated with A. algerae organisms, with each Golgi puncta now abutting DRAQ5 labeled parasites (Fig. 2). While further investigations will be needed, this close juxtaposition between the mini-Golgi stack and the developing microsporidium suggests possible trafficking of secretory components from the host to the parasite.
Results from this study demonstrate a novel labeling methodology for the study of living A. algerae to successfully observe the progression of parasite development inside live and fixed animal cells. Our studies represent an important step in understanding the biology/pathology of microsporidial parasites in living host cells.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH grant no. RO1-AI31788 and NIH grant no. R01-A1091985.
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
LITERATURE CITED
- Altan-Bonnet N, Sougrat R & Lippincott-Schwartz J 2004. Molecular basis for Golgi maintenance and biogenesis. Curr. Opin. Cell Biol., 16(4):364–372. doi: 10.1016/j.ceb.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Becnel JJ, Takvorian PM & Cali A 2014. Checklist of available generic names for microsporidia with type species and type hosts In: Weiss LM & Becnel JJ (ed.), Microsporidia Pathogens of Opportunity. Wiley Blackwell Press, Hoboken, NJ: Appendix A. p. 671–686. [Google Scholar]
- Biderre C, Pages M, Metenier G, David D, Bata J, Prensier G & Vivares CP 1994. On small genomes in eukaryotic organisms: molecular karyotypes of two microsporidian species (Protozoa) parasites of vertebrates. C. R. Biol., 317(5):399–404. [PubMed] [Google Scholar]
- Bouzahzah B, Nagajyothi F, Ghosh K, Takvorian PM, Cali A, Tanowitz HB & Weiss LM 2010. Interactions of Encephalitozoon cuniculi polar tube proteins. Infect. Immun., 78:2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali A & Takvorian PM 2014. Developmental morphology and life cycles of the microsporidia In:Weiss LM & Becnel JJ (ed.), Microsporidia-Pathogens of Opportunity, Vol. 2 Wiley Blackwell Press, Hoboken, NJ: p. 71–133. [Google Scholar]
- Cali A, Weiss LM & Takvorian PM 2002. Brachiola algerae spore membrane systems, their activity during extrusion, and a new structural entity, the multilayered interlaced network, associated with the polar tube and the sporoplasm. J. Eukaryot. Microbiol., 49(2):164–174. [DOI] [PubMed] [Google Scholar]
- Cali A, Weiss LM & Takvorian PM 2004. An analysis of the Microsporidian genus Brachiola, with comparisons of human and insect isolates of B. algerae. J. Eukaryot. Microbiol., 51 (6):678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali A, Weiss LM & Takvorian PM 2005. A review of the development of two types of human skeletal muscle infections from microsporidia associated with pathology in invertebrates and cold-blooded vertebrates. Folia Parasitol., 52(½):51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J & Lippincott- Schwartz J 1996. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell, 7(4):631–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi N, Pombert JF, Farinelli L, Didier ES & Keeling PJ 2010. The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat. Commun., 1(6):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edward R 2009. Use of DNA-specific anthraquinone dyes to directly reveal cytoplasmic and nuclear boundaries in live and fixed cells. Cells, 27:391–396. [DOI] [PubMed] [Google Scholar]
- English AR & Voeltz GK 2013. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol, 5(4):a013227. doi: 10.1101/cshper-spect.a013227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker C, Howell M, Bhella D & Lucocq J 2014. Strategies for maximizing ATP supply in the microsporidian Encephalito-zoon cuniculi: direct binding of mitochondria to the para-sitophorous vacuole and clustering of the mitochondrial porin VDAC. Cell. Microbiol. doi: 10.1111/cmi.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu N-Y, Ilnytska O, Belov G, Santiana M, Chen Y-H, Takvorian PM, Pau C, dervan Schhaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, Frank JM, vankup-peveld FJ & Altan-Bonnet N 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell, 141(5):799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, Delbac F, El Alaoui H, Peyret P, Saurin W, Gouy M, Weissenbach J & Vivares CP 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414(6862):450–453. [DOI] [PubMed] [Google Scholar]
- Niu TK, Pfeifer AC, Lippincott-Schwartz J & Jackson CL 2005. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell, 16(3):1213–1222. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiana M, Pau C, Takvorian PM & Cali A 2015. Analysis of the beta-tubulin gene and morphological changes of the microsporidium Anncaliiaalgerae both suggest Albendazole sensitivity. J. Eukaryot. Microbiol., 62(1):60–68. doi: 10.1111/jeu.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon M, Leitch G, Visvesvara G & Shaw A 2004. Relationship between the host cell mitochondria and the para-sitophorous vacuole in cells infected with Encephalitozoon microsporidia. J. Eukaryot. Microbiol., 51(1):81–87. [DOI] [PubMed] [Google Scholar]
- Smith PJ, Wiltshire M, Davies S, Patterson LH & Hoy T 1999. A novel cell permeant and far red-fluorescing DNA probe, DRAQ5, for blood cell discrimination by flow cytometry. J. Immunol. Methods, 229(1–2):131–139. [DOI] [PubMed] [Google Scholar]
- Takvorian PM, Weiss LM & Cali A 2005. The early events of Brachiola algerae (Microsporidia) infection: spore germination, sporoplasm structure, and development within host cells. Folia Parasitol., 52:118–129. [DOI] [PubMed] [Google Scholar]
- Troemel ER, Felix M-A, Whiteman NK, Barriere A & Ausubel FM 2008. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol, 6:2736–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LM & Becnel JJ, ed. 2014. Microsporidia-Pathogens of Opportunity, 2nd edn, Vol. 1 Wiley Blackwell Press, Hoboken, NJ: p. 709. [Google Scholar]
- Williams BAP 2009. Unique physiology of host-parasite interactions in microsporidia infections. Cell. Microbiol, 11:1551–1560. [DOI] [PubMed] [Google Scholar]
- Williams BAP, Dolgikh VV & Sokolova YY 2014. Microsporidian biochemistry and physiology. In: Weiss LM & Becnel JJ (ed.), Microsporidia-Pathogens of Opportunity, Vol. 9 Wiley Blackwell Press, Hoboken, NJ: p. 245–260. [Google Scholar]
- Wiltshire M, Patterson LH & Smith PJ 2000. A novel deep red/low infrared fluorescent flow cytometric probe, DRA5NO, for the discrimination of intact nucleated cells in apoptotic cell populations. Cytometry, 39:217–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.