

Abstract
A set phosphonate prodrugs of a butyrophilin ligand was synthesized and evaluated for plasma stability and cellular activity. The mixed aryl acyloxy esters were prepared either via a standard sequence through the phosphonic acid chloride, or through the more recently reported, and more facile, triflate activation. In the best of cases, this class of prodrugs shows cellular potency similar to that of bis-acyloxyalkyl phosphonate prodrugs and plasma stability similar to that of aryl phosphonamidates. For example, ((((3E)-5-hydroxy-4-methylpent-3-en-1-yl) (naphthalen-2-yloxy) phosphoryl) oxy) methyl 2,2-dimethylpropanoate can activate BTN3A1 in K562 cells after just 15 minutes of exposure (at an EC50 = 31 nM) and is only partially metabolized (60% remaining) after 20 hours in human plasma. Other related novel analogs showed similar potency/stability profiles. Therefore, mixed aryl acyloxyalkyl phosphonate prodrugs are an exciting new strategy for delivery of phosphonate-containing drugs.
Keywords: butyrophilin, BTN3A1, phosphorus, phosphoantigen, prodrugs
Graphical Abstract
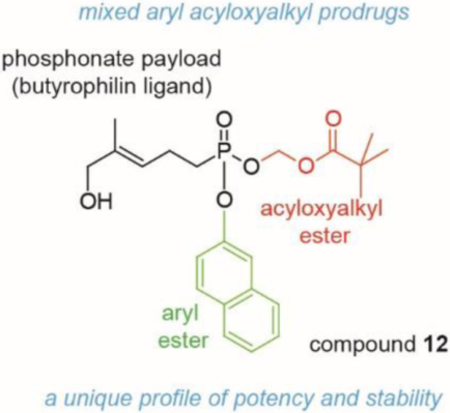
Phosphonate prodrugs.
Quick acting bis-acyloxyalkyl prodrugs are poorly stable in blood, while stable aryl-phosphonamidates are slow to act. The novel aryl acyloxyalkyl prodrug 12 displays the best of both worlds, it acts quickly (15 min EC50 = 31 nM) but is also plasma stable (t½ > 20 hr).
Introduction
Phosphates are critical to a variety of biological processes,[1] and as such the incorporation of a phosphate group or a more metabolically stable phosphonate group into a drug is often desirable to maximize the impact on biological systems.[2] At the same time, these functional groups necessarily carry a negative charge when in solution at physiological pH, which can restrict cellular entry. Therefore, development of prodrug strategies that enable facile cellular entry of phosphate- or phosphonate-containing drugs is of interest.[3]
We have investigated several prodrug analogs (Figure 1) of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP, 1),[4] an intermediate of bacterial isoprenoid biosynthesis and the most potent naturally occurring phosphoantigen.[5] The phosphoantigen prodrugs are interesting in part because they can bypass normal endocytic pathways[6] to trigger an immune response in humans through binding to the butyrophilin protein BTN3A1 and subsequently promote the functions of Vγ9Vδ2 T cells.[4a, 7] Thus butyrophilin ligands, and the Vγ9Vδ2 T cells whose proliferation they stimulate, may ultimately be of use for clinical applications relevant both to cancer and to infectious diseases.[8]
Figure 1.
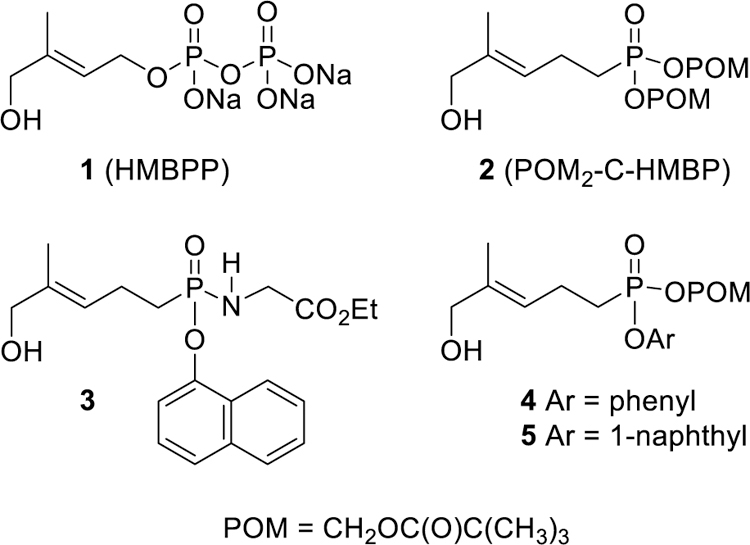
Structures of HMBPP and some phosphoantigen prodrugs evaluated herein.
The bis-pivaloyloxymethyl phosphoantigen prodrug POM2-C-HMBP (2)[9] initially was developed as a proof-of-principle compound that demonstrated the importance of cellular internalization for phosphoantigen activity.[4a] Other bis-POM compounds, while used clinically, have limitations themselves with respect to plasma stability,[10] so further investigations into other prodrug forms are important. While compound 2 remains the most efficient known phosphoantigen prodrug based on both speed and potency of triggering a T cell response,[11] it does undergo rapid plasma metabolism that could limit in vivo applications.[4e]
We also have applied the phosphonamidate strategy[12] to produce phosphoantigen prodrugs such as compound 3.[4e] Indeed, this prodrug form did improve plasma stability, resulting in potent compounds. However, the time course of activation was much slower in the phosphonamidates relative to compound 2, leaving open the question of whether further improvements in compound efficiency could be achieved while retaining plasma stability. Therefore, we chose to pursue studies into a novel mixed aryl acyloxy family of phosphonate prodrugs.[4d]
Results and Discussion
To examine the possibility that a diaryl phosphonate would function as a prodrug, and for comparison with the activity of compound 5, the di-1-naphthyl ester 9 was prepared (Scheme 1). Accordingly, treatment of compound 6[4d] with oxalyl chloride and catalytic DMF was successful in yielding the desired acid chloride 7, a process which appears to be selective for replacement of the methyl ester over the aryl ester. The acid chloride 7 then was allowed to react with 1-naphthol in the presence of triethylamine. The resulting diester 8 was subjected to allylic oxidation by treatment with selenium dioxide and tert-butyl hydroperoxide. As seen in comparable systems, the allylic oxidation of the phosphonate proceeded in poor yield but was successful in the selective synthesis of the desired E-allylic alcohol 9.
Scheme 1.
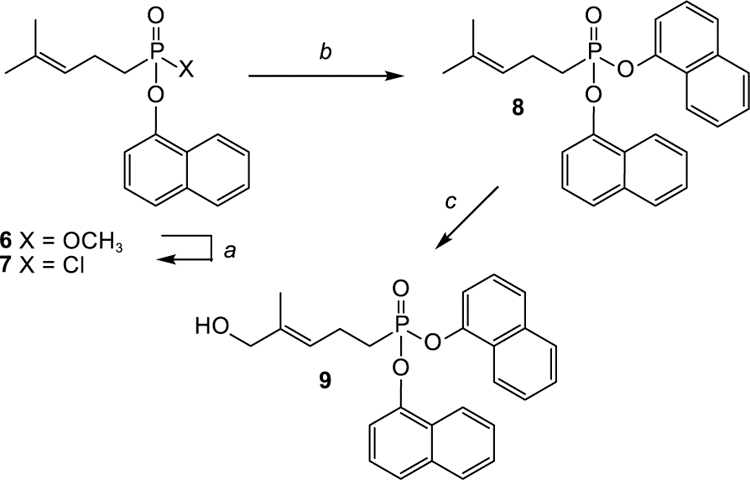
Synthesis of a diaryl phosphonate. Reagents and conditions: (a) COCl2, DMF, 40 °C; (b) 1-naphthol, Et3N, rt, 69% in two steps; (c) SeO2, tBuOOH, rt, 14%.
To investigate the metabolism of mixed aryl acyloxy prodrugs in more detail, the synthesis of several other aromatic analogs was pursued. As shown in Scheme 2, the 2-naphthyl ester 12 was prepared via a sequence parallel to that used to prepare compound 4. Thus the phosphonic acid chloride 10[4d] was allowed to react with 2-naphthol to afford the mixed diester 11. Compound 11 then was treated with POMCl and NaI to exchange the methyl ester for a POM ester. While the mechanism of this transesterification has not been explored here, given the high reactivity of POMCl one could assume that this reaction parallels the mechanism of the McKenna hydrolysis which has been studied with isotopic labels.[13] Initially the 2-naphthyl/POM mixed ester was isolated and characterized, but subsequently other mixed esters were taken directly to the oxidation after minimal purification. The resulting mixed ester was treated with selenium dioxide to obtain the E-allylic alcohol, i.e. compound 12 from the diester 11. In a parallel fashion, reaction of the acid chloride 10 with p-nitrophenol gave the mixed ester 13. Again, after introduction of the POM group (POMCl/NaI) an immediate selenium dioxide oxidation gave the desired E-allylic alcohol 14.
Scheme 2.
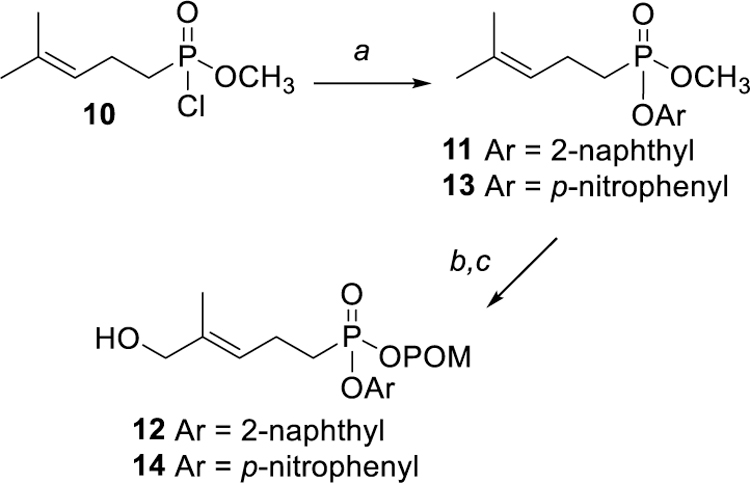
Synthesis of mixed aryl acyloxy phosphonate esters. Reagents and conditions: (a) ArOH, Et3N, 82% for 11, 35% for 13; (b) POMCl, NaI, reflux, 31% for 11, 28% for 13 ; (c) SeO2, tBuOOH, rt, 12% for 12, 9% for 14.
A recent report[14] identified an alternate method to prepare mixed phosphonate esters, and that method was employed to obtain the 2-nitronaphthyl ester 17. This synthesis utilized dimethyl homoprenylphosphonate (15) but employed triflic anhydride to initiate the ester exchange. After treatment of the dimethyl ester 15 with triflic anhydride and pyridine, addition of 2-nitro-1-naphthol rapidly gave the desired mixed aryl/methyl functionality 16 in very good yield (Scheme 3). This mixed ester then was subjected to the standard sequence of POM installation and allylic oxidation to produce the target compound 17.
Scheme 3.
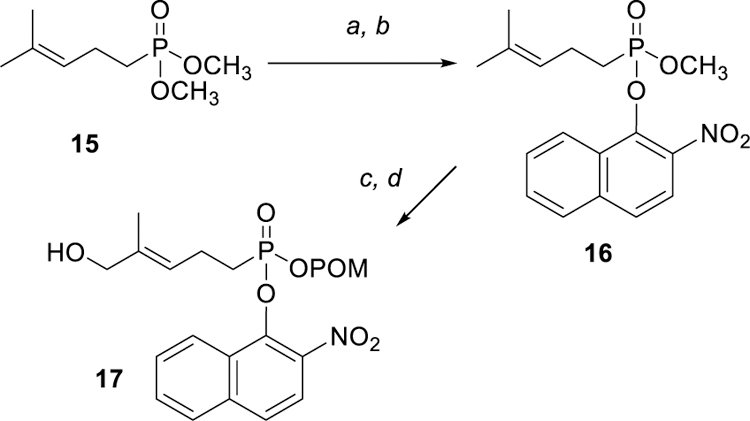
Alternate triflic anhydride mediated synthesis of mixed phosphonate esters. Reagents and conditions: (a) Tf2O, pyr, rt; (b) 2-nitro-1-naphthol, rt, 95% in two steps; (c) POMCl, NaI, reflux, 30%; (d) SeO2, tBuOOH, 15%.
The set of six aryl containing compounds first was tested for stability in 50% human plasma (Figure 2). Compounds were dissolved readily in ethanol and diluted to working stocks with water (as described in SI). With the exception of compound 9 which lacks a phosphorus stereocenter, all compounds were prepared and tested as racemic mixtures. Much like compound 2, compound 4 demonstrated low plasma stability, and was almost fully metabolized following two hours of plasma exposure. However, the naphthyl containing compounds were more stable in plasma, with 60% of 1-naphthyl analog 5 and 100% of the 2-naphthyl analog 12 remaining after two hours in plasma. In fact, compound 12 displayed 60% remaining even after 20 hours.
Figure 2.

Stability of compounds in 50% human plasma in PBS. Compounds were incubated in 50% human plasma in PBS for indicated times and quantified by LCMS. Each experiment was repeated two independent times. Bars represent mean +/− stdev.
Perhaps surprisingly, addition of a nitro group resulted in different and unpredictable effects on the plasma stability. Both compounds were more stable than the bis-acyloxyalkyl ester 2. Compound 14 also gained stability relative to compound 4, indicating that at some positions the incorporation of the nitro group can itself promote plasma stability of aryl phosphonate esters (Figure 2, Table S1). However, the nitro functionalized 1-naphthyl analog 17 was less stable compared to the non-nitro analog 5.
Notably, the bis-1-napthyl compound 9 was resistant to metabolism up to 20 hours in plasma. Without the more labile POM group present, esterases found in plasma are apparently not capable of hydrolyzing the 1-naphthyl phosphonate diester, implying that the POM group is likely to be removed first, allowing subsequent hydrolysis of the naphthyl ester. All six of the tested compounds showed higher plasma stability relative to compound 2.
With a promising plasma stability profile identified for some of these compounds, the aryl phosphonates were next tested for their ability to act as phosphoantigens and stimulate the proliferation of Vγ9Vδ2 T cells from human peripheral blood (Figure 3) following an extended 72 hour compound exposure. Here, compound 12 was active with excellent potency, though it failed to exceed that of the known 1-napthyl analog 5 (Table 1). The nitro analog 14 displayed increased potency relative to compound 4, while the nitro analog 17 displayed decreased potency relative to compound 5. The di-naphthyl analog 9 displayed only modest activity in this assay. In general, only a moderate correlation was observed between the stability at the two hour time point and potency for T cell proliferation (Pearson, r = 0.48). Together, three compounds in the set exhibit both improved stability and improved cellular potency relative to the diester 2.
Figure 3.

Expansion of Vγ9Vδ2 T cells. PBMCs were incubated with test compounds as indicated for 3 days. After 11 additional days, the % of Vγ9Vδ2 T cells was assessed by flow cytometry. Each experiment was repeated three to five independent times. Bars represent mean +/− stdev.
Table 1.
Compound activity for expansion of Vγ9Vδ2 T cells.[a]
| Compound | cLogPa | EC50 [µM] (95% CI) | vs. 2 |
|---|---|---|---|
| 2[4a] | 3.42 | 0.0054 | NA |
| 4[4d] | 3.56 | 0.014 | ND |
| 5[4d] | 4.75 | 0.00079 | 6.8 |
| 12 | 4.75 | 0.0021 (0.0000071 to 0.61) |
2.6 |
| 14 | 3.52 | 0.0040 (0.00080 to 0.020) |
1.4 |
| 17 | 4.63 | 0.021 (0.00044 to 0.98) |
0.26 |
| 9 | 6.03 | 0.27 (0.19 to 0.39) |
ND |
cLogP values were determined from www.molinspiration.com, ND = not determined, NA = not applicable. Data represent the mean and 95% confidence intervals (CI) of three to five independent experiments.
The new compounds also were evaluated for phosphoantigen activity and direct cytotoxicity against the K562 cell line. Generally, none of the six compounds displayed potent toxicity against the K562 cells. The three compounds containing unmodified naphthyl substituents (compounds 5, 12, and 9) did show some toxicity at 100 µM following a 72 hour treatment (Figure 4). This mild cytotoxicity is unlikely to be due to the free 1- or 2- naphthol, as prior studies have shown no impact of those naphthols at 100 µM.[4e]
Figure 4.

72 hour cytotoxicity of test compounds against K562 cells. K562 cells were treated with compounds as indicated, and after 72 hours the viability was assessed by cell QB assay. Each experiment was repeated three independent times. Points represent mean +/− stdev.
In order to evaluate the efficiency of prodrug activation, we turned to a co-culture model in which K562 cells are exposed to test compounds for various times, washed, and tested for their ability to stimulate interferon production by Vγ9Vδ2 T cells. The ability to washout the compound is a critical advantage of this system which enables detection of activity at very short incubation times. Compound 14 potently stimulated the production of interferon following exposure for as little as 15 minutes (Figure 5). Compound 17 also was active in this assay. However, compound 9 was not active under these conditions.
Figure 5.
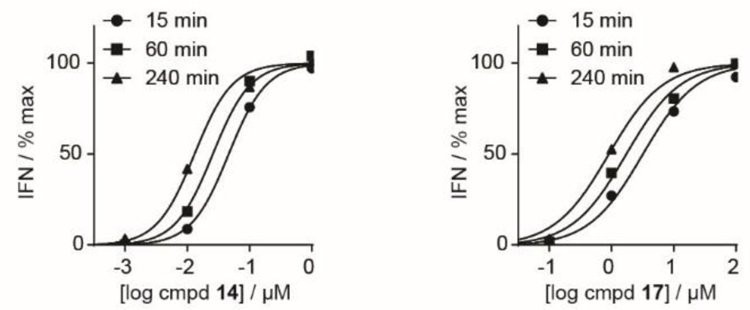
Interferon γ production by Vγ9Vδ2 T cells. K562 cells were treated with the indicated times and concentrations, then exposed to Vγ9Vδ2 T cells. After 20 hours, the interferon was quantified by ELISA. Each experiment was repeated three independent times.
The aryl acyloxy phosphonates as a group compared favorably relative to HMBPP (1) and early generation phosphoantigen prodrugs such as compounds 2 and 3 (Table 2). It is notable that the activity of HMBPP is too weak to measure in this assay due to its slow uptake and rapid metabolic decomposition.[11] Four of the five mixed aryl /POM prodrugs were more potent than compound 3 following a 15 minute exposure time (usually by greater than ten-fold). These compounds were even more potent than compound 2 at the 240 minute time point.
Table 2.
Interferon γ production by Vγ9Vδ2 T cells in response to loaded K562 cells.[a]
| Compound | 15 min EC50 [µM] (95% CI) | 60 min EC50 [µM] (95% CI) | 240 min EC50 [µM] (95% CI) |
|---|---|---|---|
| 1[11] | >100 | >100 | >100 |
| 2[11] | 0.043 | 0.030 | 0.024 |
| 3[11] | 0.54 | 0.11 | 0.020 |
| 4[11] | 0.037 | 0.023 | 0.0084 |
| 5[11] | 0.057 | 0.037 | 0.018 |
| 12[11] | 0.031 | 0.026 | 0.012 |
| 14 | 0.045 (0.035 to 0.059) |
0.025 (0.019 to 0.034) |
0.013 (0.010 to 0.016) |
| 17 | 3.1 (2.2 to 4.2) |
1.6 (1.2 to 2.2) |
0.87 (0.65 to 1.2) |
| 9 | >10 | >10 | >10 |
Data represent the mean and 95% confidence intervals (CI) of three independent experiments.
Clearly compound 2 remains the most efficient compound in this family, as it is the only compound with less than a two-fold difference between its 15 minute and 240 minute potencies (Table 2). To quantify the efficiency of prodrug activation, the data was fit to a power law function (Table 3), as previously described.[11] The power constant k1 of each of the five mixed aryl compounds more closely resembles that of compound 2 than compound 3. Thus, taken together, the mixed aryl/POM prodrug class can achieve both high plasma stability like the aryl phosphonamidates and high efficiency of activation like the bis-POM prodrug.
Table 3.
Determination of relative prodrug efficiencies.[a]
| Compound | k1 | α | R2 |
|---|---|---|---|
| 2[11] | −5.2 | 4.7 | 0.97 |
| 3[11] | 0.96 | 0.84 | 1.0 |
| 4[11] | −1.3 | 1.8 | 0.97 |
| 5[11] | −1.7 | 2.4 | 0.98 |
| 12[11] | −2.6 | 2.6 | 0.90 |
| 14 | −1.8 | 2.2 | 1.0 |
| 17 | 2.2 | 2.2 | 0.97 |
Data represent the values determined from the mean of three independent experiments.
Conclusions
In summary, we designed and synthesized a new series of aryl acyloxyalkyl phosphonate prodrugs of a butyrophilin ligand. The compounds show mid- to low-nanomolar potency after only 15 minutes exposure to K562 cells, with good stability in human plasma relative to the bis-POM prodrugs. Potency was confirmed in an orthogonal assay of Vγ9Vδ2 T cell expansion from primary human PBMCs, where 72 hour EC50 values were in the low-nanomolar range. We believe the mixed aryl acyloxyalkyl phosphonate prodrug strategy holds promise for in vivo delivery of phosphoantigens, and merits further study with other payloads, because these mixed aryl acyloxyalkyl forms exhibit both quick efficacy similar to the bis-acyloxyalkyl forms and extended plasma stability similar to the phosphonamidate forms.
Experimental Section
General Experimental Procedures.
Both THF and diethyl ether were freshly distilled from sodium and benzophenone, while acetonitrile, methylene chloride, pyridine, and triethylamine (Et3N) were distilled from calcium hydride prior to use. Toluene and DMF were dried over molecular sieves prior to use. All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions were conducted in flame or oven dried glassware under a positive pressure of argon and with magnetic stirring. The NMR spectra were obtained at 400 MHz for 1H, 75, 100, or 125 MHz for 13C, and 121 or 161 MHz for 31P. Internal standards of Si(CH3)4 (1H, 0.00 ppm) or CDCl3 (1H, 7.27 ppm; 13C, 77.2 ppm) were used. The 31P chemical shifts are reported in ppm relative to 85% H3PO4 (external standard). High resolution mass spectra were obtained at the University of Iowa Mass Spectrometry Facility. Silica gel (60 Å, 0.040–0.063 mm) was used for flash chromatography. The HPLC separations were carried out using a Beckman System Gold instrument with a model 166 variable wavelength UV detector connected to a 128 solvent module.
Naphthalen-1-yl (4-methylpent-3-en-1-yl)phosphonochloridate (7).
Oxalyl chloride (0.74 mL, 8.7 mmol) was added dropwise to a solution of methyl naphthalen-1-yl (4-methylpent-3-en-1-yl)phosphonate[15] (880 mg, 2.9 mmol) in freshly distilled dichloromethane (10 mL). Three drops of DMF were added and the reaction was warmed to 40 °C and held at this temperature for 3 days. The reaction then was allowed to cool to rt and subsequently concentrated in vacuo. The resulting material was dissolved in anhydrous dichloromethane (5 mL) and concentrated to a reddish-brown oil, which was utilized without further purification.
Bis(naphthalen-1-yl) (4-methylpent-3-en-1-yl)phosphonate (8).
1-Naphthol (833 mg, 5.8 mmol) and triethylamine (0.80 mL, 5.8 mmol) were added to anhydrous THF (25 mL) and stirred for 10 minutes to form a homogenous solution. The resulting material was added to a solution of the acid chloride 7 (2.9 mmol) in THF (50 mL) and the reaction was allowed to stir for 8 hours. The reaction then was diluted with diethyl ether (25 mL) and washed with brine (10 mL) and aqueous K2CO3 (3 × 5 mL). The organic layer was dried (Na2SO4) and filtered through celite, and the filtrate was concentrated in vacuo. The resulting red oil was purified via flash chromatography (silica, 100% hexanes–20% EtOAc in hexanes) and the product 8 was isolated as a red oil in 69% yield (831 mg): 1H NMR (400 MHz, CDCl3) δ 8.21–8.18 (m, 2H), 7.90–7.87 (m, 2H), 7.64–7.61 (m, 2H), 7.58–7.52 (m, 2H), 7.44–7.39 (m, 4H), 7.37–7.33 (m, 2H), 5.25 (br s, 1H), 2.65–2.55 (m, 2H), 2.42–2.33 (m, 2H), 1.71 (s, 3H), 1.63 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 146.5 (d, JPC = 8.8 Hz), 134.9, 133.7, 127.9, 126.7, 126.6, 126.4, 125.7, 125.0, 122.5 (d, JPC = 16.5 Hz), 121.6, 115.7, 26.7 (d, JPC = 137.2 Hz), 25.7, 21.5 (d, JPC = 4.8 Hz), 17.7; 31P NMR (121 MHz, CDCl3) δ + 25.5.
Bis(naphthalen-1-yl) ((3E)-5-hydroxy-4-methylpent-3-en-1-yl)phosphonate (9).
Selenium dioxide (166 mg, 1.5 mmol) and a 70% solution of tert-butyl hydroperoxide (0.82 mL, 6.0 mmol) were added to a solution of diester 8 (831 mg, 2.0 mmol) in dichloromethane (10 mL). The solution was stirred at rt and allowed to react for 3 days. The reaction then was diluted with dichloromethane (20 mL) and washed with saturated Na2SO3 (3 × 5 mL). The organic portions were dried (Na2SO4) and filtered, and the filtrate was concentrated. The resulting yellow oil was purified via column chromatography (silica, 30% EtOAc in hexanes to 100% EtOAc) and the product 9 was isolated as a yellow oil in 14% yield (121 mg): 1H NMR (400 MHz, CDCl3) δ 8.11–8.07 (m, 2H), 7.85–7.82 (m, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.64–7.61 (m, 2H), 7.53–7.47 (m, 4H), 7.36 (dd, J = 7.8 Hz, 2H), 5.44 (t, J = 7.2 Hz, 1H), 3.88 (s, 2H), 2.69–2.57 (m, 2H), 2.40–2.29 (m, 2H), 1.58 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 146.3 (d, JPC = 8.7 Hz), 136.9, 134.8, 127.9, 126.7, 126.5, 126.4, 125.6, 125.0, 122.5 (d, JPC = 16.7 Hz), 121.5, 115.6 (d, JPC = 3.0 Hz), 68.3, 26.4 (d, JPC = 140.4 Hz), 21.1, 13.7; 31P NMR (121 MHz, CDCl3) δ + 25.5. HRMS (ES+, m/z) calcd. for (M+Na)+ C26H25NaO4P: 455.1388; found: 455.1384.
Methyl naphthalen-2-yl (4-methylpent-3-en-1-yl)phosphonate (11).
A solution of 2-naphthol (1.91 g, 13.3 mmol) and triethylamine (1.84 mL, 13.3 mmol) in toluene (10 mL) was added dropwise to a solution of the acid chloride 10 (5.3 mmol) in toluene (10 mL) and the reaction was allowed to proceed for 15 hours. The reaction then was diluted with diethyl ether (30 mL) and quenched by addition of brine (5 mL). The organic portion was extracted four times with 1 M NaOH (5 mL), dried (MgSO4), filtered through celite, and concentrated in vacuo. The resulting oil was purified via chromatography (silica, 100% hexanes – 40% EtOAc in hexanes) and the product was concentrated to a yellow oil in 82% yield (1.32 g): 1H NMR (400 MHz, CDCl3) δ 7.79–7.78 (m, 3H), 7.68 (s, 1H), 7.46–7.41 (m, 2H), 7.35 (d, J = 8.8 Hz, 1H), 5.13 (t, J = 6.8 Hz, 1H), 3.81 (d, JPH = 11.2 Hz, 3H), 2.47–2.35 (m, 2H), 2.00–1.94 (m, 2H), 1.67 (s, 3H), 1.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 148.3 (d, JPC = 9.3 Hz), 134.0, 133.3, 130.9, 129.9, 127.7, 127.5, 126.7 125.4, 122.7 (d, JPC = 17.7 Hz), 120.5 (d, JPC = 4.4 Hz), 116.8 (d, JPC = 4.2 Hz), 52.8 (d, JPC = 6.1 Hz), 25.6, 25.6 (d, JPC = 136.9 Hz), 21.1 (d, JPC = 4.7 Hz), 17.7; 31P (161 MHz, CDCl3) δ +30.5; HRMS (ES+, m/z) calcd. for (M+H)+ C17H22O3P: 305.130; found: 305.131.
(((4-Methylpent-3-en-1-yl)(naphthalene-2-yloxy)phosphoryl)oxy)methyl 2,2-dimethylpropanoate (A).
The mixed phosphonic acid ester 11 (590 mg, 1.9 mmol) was dissolved in freshly distilled acetonitrile (1 mL), subsequently concentrated under reduced pressure 2 times, and then dissolved in acetonitrile (5 mL). Chloromethyl pivalate (0.58 mL, 3.9 mmol) and sodium iodide (580 mg, 3.9 mmol) were added and the solution was heated at reflux for 48 hours. The reaction then was allowed to cool to rt and diluted with diethyl ether (25 mL). This solution was extracted with brine (4 × 5 mL), dried (Na2SO4), filtered through celite, and concentrated in vacuo. The resulting oil was purified via flash chromatography (C18, 20% EtOAc in hexanes) and the resulting product was isolated in 31% yield (0.24 g): 1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 10.0, 9.82 Hz, 3H), 7.69 (s, 1H), 7.50 (dd, J = 6.8, 1.2 Hz, 1H), 7.45 (dd, J = 7.2, 1.2 Hz, 1H), 7.35 (ddd, J = 8.8, 2.4, 0.8 Hz, 1H), 5.74 (dd, JPH = 14.0 Hz, J = 5.2 Hz, 1H), 5.68 (dd, JPH = 12.8 Hz, J = 5.2 Hz, 1H), 5.12 (t, J = 7.2, 1.4 Hz, 1H), 2.45–2.36 (m, 2H), 2.05–1.98 (m, 2H), 1.67 (s, 3H), 1.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.7, 148.3 (d, JPC = 9.1 Hz), 136.3, 133.7, 129.8, 128.3, 127.9, 127.1, 125.8, 124.7, 122.3 (d, JPC = 17.9 Hz), 120.9, 119.3, 82.2 (d, JPC = 5.9 Hz), 38.6, 27.4 (d, JPC = 136.3 Hz), 26.7 (3C), 25.7, 20.8 (d, JPC = 4.8 Hz), 17.8; 31P (161 MHz, CDCl3) δ +29.6; HRMS (ES+, m/z) calcd. for (M+H)+ C22H30O5P: 405.1831; found: 405.1830.
((((3E)-5-hydroxy-4-methylpent-3-en-1-yl)(naphthalen-2-yloxy)phosphoryl)oxy)methyl 2,2-dimethylpropanoate (12).
Selenium dioxide (60 mg, 0.6 mmol) was added to a solution of phosphonate A (230 mg, 0.6 mmol) in freshly distilled dichloromethane (3 mL) and allowed to react for 48 hours. The reaction was diluted with dichloromethane (20 mL) and the inorganic components removed by addition of three portions of brine (5 mL). The organic portion was dried (Na2SO4), filtered through celite, and concentrated in vacuo. The resulting oil was purified by HPLC (C18, acetonitrile) to give the product in 12% yield (29 mg):1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 11.6, 8.4 Hz, 3H), 7.69 (s, 1H), 7.50 (dd, J = 6.8, 1.6 Hz, 1H), 7.45 (dd, J = 6.8, 1.2 Hz, 1H), 7.35 (ddd, J = 8.8, 2.4, 0.8 Hz, 1H), 5.74 (dd, J = 13.4, 4.8 Hz, 1H), 5.67 (dd, J = 12.4, 4.8 Hz, 1H), 5.44 (td, J = 7.2, 1.2 Hz, 1H), 4.00 (s, 2H), 2.53–2.44 (m, 2H), 2.11–2.01 (m, 2H), 1.68 (s, 3H), 1.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.3, 147.7 (d, JPC = 10.5 Hz), 136.8, 134.1, 131.2, 130.1, 127.9, 127.7, 126.9, 125.8, 123.4 (d, JPC = 17.3 Hz), 120.5 (d, JPC = 5.3 Hz), 117.2 (d, JPC = 5.2 Hz), 82.0 (d, JPC = 6.0 Hz), 68.5, 38.8, 26.9 (3C), 26.3 (d, JPC = 137.7 Hz), 20.7 (d, JPC = 5.1 Hz), 13.8; 31P (161 MHz, CDCl3) δ +29.1; HRMS (ES+, m/z) calcd. for (M+Na)+ C22H29NaO6P: 443.1599; found: 443.1602.
Methyl 4-nitrophenyl (4-methylpent-3-en-1-yl)phosphonate (13).
A solution of 4-nitrophenol (1.48 g, 10.6 mmol) and freshly distilled triethylamine (1.47 mL, 10.6 mmol) in anhydrous toluene (15 mL) was added to a solution of acid chloride 10 (5.3 mmol) in toluene (15 mL) and allowed to react overnight at rt. The reaction was diluted by addition of diethyl ether (20 mL) and quenched by addition of brine (10 mL). The organic portion was washed with a 2 M solution of NaOH (3 × 5 mL), dried (Na2SO4), and filtered through celite, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, 50% hexanes in EtOAc–100% EtOAc) and the resulting product 13 was isolated as a yellow oil in 35% yield (0.56 g): 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 5.12 (br s, 1H), 3.83 (d, JPH = 11.1 Hz, 3H), 2.44–2.32 (m, 2H), 2.04–1.93 (m, 2H), 1.69 (s, 3H), 1.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.7 (d, JPC = 8.2 Hz), 144.5, 133.6, 125.6 (2C), 122.2 (d, JPC = 16.9 Hz), 120.9 (d, JPC = 4.5 Hz, 2C), 53.0 (d, JPC = 6.9 Hz), 25.7 (d, JPC = 138.2 Hz), 25.5, 20.8 (d, JPC = 4.8 Hz), 17.6; 31P NMR (121 MHz, CDCl3) δ + 31.1.
((((3E)-5-hydroxy-4-methylpent-3-en-1-yl)(4-nitrophenoxy)phosphoryl)oxy)methyl 2,2-dimethylpropanoate (14).
The mixed ester 13 (559 mg, 1.6 mmol) was added to a solution of chloromethyl pivalate (0.59 mL, 3.9 mmol) and sodium iodide (355 mg, 2.4 mmol) in acetonitrile (5 mL). The solution was heated at reflux for 24 hours. The reaction then was allowed to cool to rt, extracted with diethyl ether (~10 mL), and the combined extracts were washed with brine (4 × 5 mL). The organic portions were combined, dried (Na2SO4), and filtered through celite, and the filtrate was concentrated in vacuo. The resulting oil was purified via flash chromatography (silica, 100% hexanes–30% EtOAc in hexanes) and the product was isolated as a yellow oil in 28% yield (174 mg) that was used directly.
Selenium dioxide (36 mg, 0.3 mmol) and a 70% solution of tert-butyl hydroperoxide (0.18 mL, 1.3 mmol) were added to a solution of diester 13 (171 mg, 0.4 mmol) in dichloromethane (5 mL). The solution was stirred at rt and allowed to react for 2 days. The reaction then was diluted with dichloromethane (20 mL) and washed with brine (3 × 5 mL). The organic portions were dried (Na2SO4) and filtered, and the filtrate was concentrated. The resulting yellow oil was purified via column chromatography (silica, 50% EtOAc in hexanes to 100% EtOAc) and the product 14 was isolated as a yellow oil in 9% yield (16 mg): 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 9.0 Hz, 2H), 7.40 (dd, J = 9.0 Hz, JPH = 1.2 Hz, 2H) 5.74 (dd, JPH = 13.5, J = 5.4 Hz, 1H), 5.67 (dd, JPH = 12.4, J = 5.4 Hz, 1H), 5.44 (td, J = 7.1, 1.3 Hz, 1H), 4.01 (s, 2H), 2.51–2.42 (m, 2H), 2.12–2.03 (m, 2H), 1.68 (s, 3H), 1.17 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.0, 155.0, 144.5, 137.0, 125.8 (2C), 122.6 (d, JPC = 16.6 Hz), 121.2 (d, JPC = 4.7 Hz, 2C), 81.9 (d, JPC = 6.2 Hz), 68.2, 38.8, 26.4 (d, JPC = 138.7 Hz), 25.7, 20.4 (d, JPC = 5.0 Hz), 13.7; 31P NMR (121 MHz, CDCl3) δ + 29.7.
Methyl 2-nitronaphthalen-1-yl (4-methylpent-3-en-1-yl)phosphonate (16).
To a solution of 15 (1.07 g, 5.5 mmol) in freshly distilled DCM (25 mL) was added triflic anhydride (1.02 mL, 6.1 mmol) and pyridine (0.89 mL, 11.1 mmol). The solution was stirred and allowed to react for 15 minutes followed by addition of 2-nitro-1-naphthol (2.10 g, 11.1 mmol). The reaction was stirred for 30 minutes and then concentrated in vacuo. The resulting oil was purified via column chromatography (silica, 40% EtOAc in hexanes) and the product 16 was isolated as a yellow oil in 95% yield (1.84 g): 1H NMR (400 MHz, CDCl3) δ 8.44–8.41 (m, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.91–7.87 (m, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.70–7.67 (m, 2H), 5.19 (tt, J = 7.0, 1.4 Hz, 1H), 3.64 (d, JPH = 11.2 Hz, 3H), 2.52–2.43 (m, 2H), 2.25–2.13 (m, 2H), 1.71 (s, 3H), 1.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 140.4 (d, JPC = 8.9 Hz), 140.1, 137.5, 136.6, 130.3, 128.8, 127.9, 127.7, 127.2 (d, JPC = 12.0 Hz), 126.0, 124.8, 121.0, 53.0 (d, JPC = 6.8 Hz), 27.1 (d, JPC = 137.9 Hz), 26.8, 21.2, 19.8; 31P NMR (121 MHz, CDCl3) δ + 32.8.
((((3E)-5-hydroxy-4-methylpent-3-en-1-yl)((2-nitronaphthalen-1-yl)oxy)phosphoryl)oxy)methyl 2,2-dimethylpropanoate (17).
According to the procedure described for preparation of compound 14, compound 16 (707 mg, 2.0 mmol) was treated with POMCl (0.33 mL, 2.2 mmol) and NaI (364 mg, 2.4 mmol) in acetonitrile at reflux. Standard work-up and purification provided the desired POM ester (273 mg, 30%). The POM ester then was allowed to react with selenium dioxide (24 mg, 0.2 mmol) and a 70% solution of tert-butyl hydroperoxide (0.12 mL, 0.9 mmol) in DCM (10 mL). Standard workup and purification provided the desired allylic alcohol 17 as a yellow oil in 15% yield (20 mg): 1H NMR (400 MHz, CDCl3) δ 8.44–8.42 (m, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.93–7.91 (m, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.74–7.71 (m, 2H), 5.64 (t, J = 7.5 Hz, 1H), 5.57 (dd, JPH = 10.8 Hz, J = 5.0 Hz, 1H), 5.41 (dd, JPH = 11.2 Hz, J = 5.0 Hz, 1H), 4.25 (s, 2H), 2.73–2.62 (m, 2H), 2.46–2.38 (m, 2H), 1.57 (s, 3H), 1.10 (s, 9H), 13C NMR (100 MHz, CDCl3) δ 177.1, 140.3 (d, JPC = 11.8 Hz), 140.1, 138.5, 136.6, 130.1, 128.6, 128.1, 127.7 (d, JPC = 2.9 Hz), 127.0 (d, JPC = 11.0 Hz), 126.2, 124.8, 121.1, 82.4 (d, JPC = 5.2 Hz), 67.0, 38.8, 27.3 (d, JPC = 139.1 Hz), 27.1, 26.9 (3C), 20.6 (d, JPC = 5.1 Hz); 31P NMR (121 MHz, CDCl3) δ + 31.0.
Supplementary Material
Acknowledgements
We appreciate the assistance of Dr. Jeremy Balsbaugh at the University of Connecticut Proteomics & Metabolomics Facility with the LCMS analysis and Dr. Wu He at the University of Connecticut Flow Cytometry Facility with the flow cytometry data. We thank the Center for Biocatalysis and Bioprocessing for a fellowship (B. J. F.) through the Predoctoral Training Program in Biotechnology (T32 GM008365). Financial support from the NIH (R01CA186935 to AJW), the Herman Frasch Foundation for Chemical Research, Bank of America, N.A., Trustee (HF17 to AJW), and the Roy J. Carver Charitable Trust through its Research Program of Excellence (01–224 to DFW), is gratefully acknowledged.
Footnotes
Supporting information for this article is given via a link at the end of the document
Notes
A.J.W. and D.F.W. own shares in Terpenoid Therapeutics, Inc. The current work did not involve the company. The other authors have no financial conflicts of interest.
References
- [1].Westheimer FH, Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- [2].a)Sevrain CM, Berchel M, Couthon H, Jaffres PA, Beilstein J Org. Chem 2017, 13, 2186–2213; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Jessen H, Synlett 2018, 29, 699–713. [Google Scholar]
- [3].Wiemer AJ, Wiemer DF, Top. Curr. Chem 2015, 360, 115–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a)Hsiao CH, Lin X, Barney RJ, Shippy RR, Li J, Vinogradova O, Wiemer DF, Wiemer AJ, Chem. Biol 2014, 21, 945–954; [DOI] [PubMed] [Google Scholar]; b)Wiemer AJ, Shippy RR, Kilcollins AM, Li J, Hsiao CH, Barney RJ, Geng ML, Wiemer DF, ChemBioChem 2016, 17, 52–55; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Shippy RR, Lin X, Agabiti SS, Li J, Zangari BM, Foust BJ, Poe MM, Hsiao CC, Vinogradova O, Wiemer DF, Wiemer AJ, J. Med. Chem 2017, 60, 2373–2382; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Foust BJ, Poe MM, Lentini NA, Hsiao CC, Wiemer AJ, Wiemer DF, ACS Med. Chem. Lett 2017, 8, 914–918; [DOI] [PMC free article] [PubMed] [Google Scholar]; e)Lentini NA, Foust BJ, Hsiao CC, Wiemer AJ, Wiemer DF, J. Med. Chem 2018, 61, 8658–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Morita CT, Jin C, Sarikonda G, Wang H, Immunol. Rev 2007, 215, 59–76. [DOI] [PubMed] [Google Scholar]
- [6].a)Kilcollins AM, Li J, Hsiao CH, Wiemer AJ, J. Immunol 2016, 197, 419–428; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Thompson K, Rogers MJ, Coxon FP, Crockett JC, Mol. Pharmacol 2006, 69, 1624–1632. [DOI] [PubMed] [Google Scholar]
- [7].a)Wang H, Morita CT, J. Immunol 2015, 195, 4583–4594; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Rhodes DA, Chen HC, Price AJ, Keeble AH, Davey MS, James LC, Eberl M, Trowsdale J, J. Immunol 2015, 194, 2390–2398; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, Breathnach R, Bonneville M, Scotet E, Adams EJ, Immunity 2014, 40, 490–500; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S, Dechanet-Merville J, Leger A, Herrmann T, Breathnach R, Olive D, Bonneville M, Scotet E, Blood 2012, 120, 2269–2279; [DOI] [PMC free article] [PubMed] [Google Scholar]; e)Boutin L, Scotet E, Front. Immunol 2018, 9, 828; [DOI] [PMC free article] [PubMed] [Google Scholar]; f)Nguyen K, Li J, Puthenveetil R, Lin X, Poe MM, Hsiao CC, Vinogradova O, Wiemer AJ, FASEB J 2017, 31, 4697–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a)Sharma A, Zumwalde NA, Gumperz JE, Methods Mol. Biol 2019, 1884, 57–72; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Zumwalde NA, Sharma A, Xu X, Ma S, Schneider CL, Romero-Masters JC, Hudson AW, Gendron-Fitzpatrick A, Kenney SC, Gumperz JE, JCI insight 2017, 2; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Castella B, Melaccio A, Foglietta M, Riganti C, Massaia M, Front. Oncol 2018, 8, 508; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Shen L, Frencher J, Huang D, Wang W, Yang E, Chen CY, Zhang Z, Wang R, Qaqish A, Larsen MH, Shen H, Porcelli SA, Jacobs WR Jr., Chen ZW, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 6371–6378; [DOI] [PMC free article] [PubMed] [Google Scholar]; e)Wiemer DF, Wiemer AJ, Biochem. Pharmacol 2014, 89, 301–312. [DOI] [PubMed] [Google Scholar]
- [9].Rasmussen M, Leonard NJ, J. Am. Chem. Soc 1967, 89, 5439–5445. [Google Scholar]
- [10].a)Naesens L, Balzarini J, Bischofberger N, De Clercq E, Antimicrob. Agents Chemother 1996, 40, 22–28; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Shaw JP, Louie MS, Krishnamurthy VV, Arimilli MN, Jones RJ, Bidgood AM, Lee WA, Cundy KC, Drug Metab. Dispos 1997, 25, 362–366; [PubMed] [Google Scholar]; c)Shaw JP, Sueoko CM, Oliyai R, Lee WA, Arimilli MN, Kim CU, Cundy KC, Pharm. Res 1997, 14, 1824–1829. [DOI] [PubMed] [Google Scholar]
- [11].Hsiao CC, Wiemer AJ, Biochem. Pharmacol 2018, 158, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McGuigan C, Pathirana RN, Balzarini J, De Clercq E, J. Med. Chem 1993, 36, 1048–1052. [DOI] [PubMed] [Google Scholar]
- [13].Blazewska KM, J. Org. Chem 2014, 79, 408–412. [DOI] [PubMed] [Google Scholar]
- [14].Huang H, Denne J, Yang CH, Wang H, Kang JY, Angew. Chem. Int. Ed. Engl 2018, 57, 6624–6628. [DOI] [PubMed] [Google Scholar]
- [15].Foust BJ, Poe MM, Lentini N, Hsiao CHC, Wiemer AJ, Wiemer DF, ACS Med. Chem. Lett 2017, 8, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.