

Summary
Betaglycan (BG) and endoglin (ENG), homologous co-receptors of the TGF-β family, potentiate the signaling activity of TGF-β2 and inhibin A, and BMP-9 and −10, respectively. BG exists as monomer and forms 1:1 growth factor (GF) complexes, while ENG exists as a dimer and forms 2:1 GF complexes. Herein, the structure of the BG orphan domain (BGO) reveals an insertion that blocks the region that the endoglin orphan domain (ENGO) uses to bind BMP-9, preventing it from binding in the same manner. Through binding studies with domain-deleted forms of TGF-β and BGO, as well as small angle X-ray scattering data, BGO is shown to bind its cognate growth factor in an entirely different manner compared to ENGO. The alternative interfaces likely engender BG and ENG with the ability to selectively bind and target their cognate GFs in a unique temporal-spatial manner, without interfering with one another or other TGF-β family GFs.
Keywords: transforming growth factor beta (TGF-β), betaglycan, endoglin, cell surface receptor, co-receptor, cell signaling, cardiac development, vascular development, X-ray crystallography, SAXS, SPR
Graphical Abstract
ETOC Blurb
Kim et al. determine the structure of the betaglycan orphan domain (BGO) and show that the edge β-strand that endoglin uses to bind its cognate growth factor is blocked by an insertion. Binding studies and SAXS show that BGO binds its cognate growth factor differently compared to endoglin.
Introduction
Temporal and spatial control of cell signaling is critical for normal development of embryos and for tissue maintenance in adults. The signaling molecules that regulate temporal-spatial control include those of the Wnt and Notch families, but also those of the transforming growth factor-beta (TGF-β, family, which have essential roles in patterning (Bier and De Robertis, 2015; Zinski et al., 2018) and organogenesis (Mullen and Wrana, 2017) in embryos, and maintenance of the heart (Goumans and Ten Dijke, 2018), vasculature (Goumans et al., 2018; Roman and Hinck, 2017), and musculoskeletal system (Walker et al., 2016) in adults. TGF-β family signaling proteins have greatly diversified, with more than 30 family members in humans. The proteins of the family can be classified as belonging either to the ancestral bone morphogenetic protein (BMP)/growth and differentiation factor (GDF) branch of the family, or the more recently evolved TGF-β/activin branch (Hinck et al., 2016). The proteins of both branches bind and bring together two type I and two type II receptors to initiate a transphosphorylation cascade and intracellular Smad signaling (Hata and Chen, 2016; Wrana et al., 1994). TGF-β family signaling proteins are well-suited to enable temporal-spatial control as multiple mechanisms exist for targeting them with high spatial and temporal resolution. These range from cell- and tissue-specific expression of receptors, co-receptors, or soluble antagonists specific for a subset of TGF-β family growth factors (GFs), to proteolytic release of the mature signaling GF dimers from their inhibitory pro-domains (Brazil et al., 2015; Hinck et al., 2016; Nolan and Thompson, 2014; Robertson and Rifkin, 2013).
The membrane-anchored co-receptors of the TGF-β family, betaglycan (BG) and endoglin (ENG), do not directly participate in the transphosphorylation cascade that leads intracellular Smad signaling, but have essential roles in promoting receptor binding and signaling by select family members (Brown et al., 2005; Lewis et al., 2000; Lopez-Casillas et al., 1993; Nickel et al., 2018; Scharpfenecker et al., 2007). The co-receptor BG has been shown to promote binding of low-affinity TGF-β2 to the TGF-β type II receptor, TβRII (KD roughly 20 μM for binding to the monomeric TβRII ectodomain (Baardsnes et al., 2009)), and to restore cellular sensitivity to concentrations comparable to that of high-affinity TGF-β1 and −β3 (KD roughly 50 – 150 nM for binding to the monomeric TβRII ectodomain (Cheifetz et al., 1990; Lopez-Casillas et al., 1993; Radaev et al., 2010)). TGF-β2 is essential for transformation of endothelial progenitor cells required for normal development and maintenance of the heart and liver and knockout of either TGF-β2 or BG in mice leads to similar defects in heart and liver development (Sanford et al., 1997; Stenvers et al., 2003). TGF-β2 and BG therefore function together to enable signaling essential for the proper development and maintenance of these organs. BG is also expressed by gonadotropic cells in the anterior pituitary and binds the TGF-β family heterodimer inhibin A (inhA) to promote activin type II/IIB receptor (ActRII or ActRIIB) binding, which is essential for inhibiting activin-stimulated follicle stimulating hormone-β (FSH²) secretion by gonadotropic cells in the anterior pituitary (Lewis et al., 2000; Li et al., 2018; Wiater et al., 2006). The co-receptor ENG is expressed on the surface of vascular endothelial cells and selectively binds BMP-9 and BMP-10 to promote binding of the type I receptor, activin-like kinase 1 (Alk1), and signaling (Alt et al., 2012; Castonguay et al., 2011; Scharpfenecker et al., 2007). This signaling stimulates migration of the endothelial cells and is essential for normal development and maintenance of the vasculature, as shown by vascular abnormalities known as arteriovenous malformations, which are the result of germline mutations in either Alk1 or ENG (Goumans et al., 2018; Roman and Hinck, 2017).
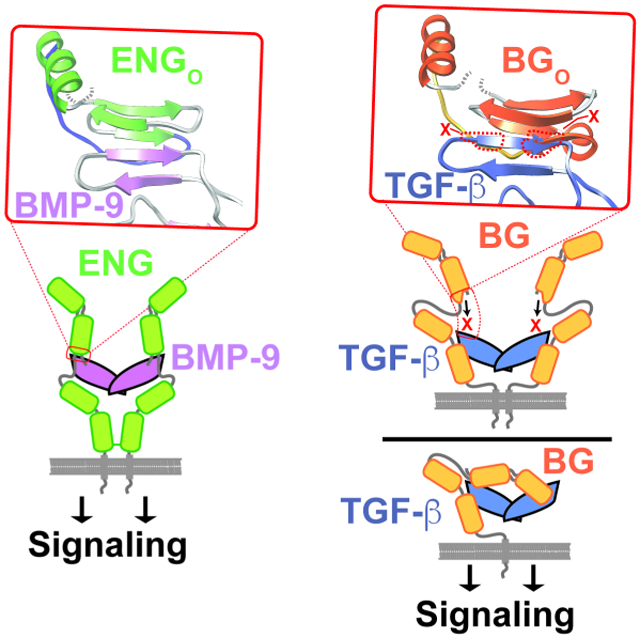
The extracellular domains of BG and ENG are comprised of two subdomains, the N-terminal membrane-distal orphan domain and the membrane-proximal zona pellucida, or ZP domain (Fig. 1A, D) (Alt et al., 2012; Esparza-Lopez et al., 2001; Mendoza et al., 2009). The structure of the BG orphan domain, BGO, is not known, but is likely tandem β-sandwichesdesignated O-D1 and O-D2, similar to the ENG orphan domain, ENGO, whose structure was recently reported (Saito et al., 2017). The BG zona pellucida domain, BGZP, is likely comprised of tandem immunoglobulin-like domains, designated BGZP-N and BGZP-C, based on available structures of BGZP-C from rat (Lin et al., 2011) and mouse (Diestel et al., 2013) and homology to the ENG zona pellucida domain, ENGZP, whose structure was also recently reported (Saito et al., 2017). Through biochemical studies, rat BG, which has 5 cysteines within its orphan domain and 10 in its ZP domain, is found on cells as a monomer (Lopez-Casillas et al., 1993) and binds TGF-β homodimers asymmetrically with an overall 1:1 stoichiometry (Villarreal et al., 2016) using both its orphan domain and the C-terminal portion of its zona pellucida domain, BGZP-C (Henen et al., 2018; Mendoza et al., 2009; Villarreal et al., 2016) (Fig. 1A). Human ENG, on the other hand, which has 5 cysteines in its orphan domain and 11 in its ZP domain, is found on cells as a disulfide-linked dimer and binds BMP-9 and -10 using only its orphan domain (Alt et al., 2012). Recent structural studies revealed that ENGO binds BMP-9 homodimers with 2:1 stoichiometry by forming a super β-sheet with finger 4 of the GF through an exposed β-strand, β6, in domain 1 (O-D1) (Fig. 1C) (Saito et al., 2017). ENG’s ZP domain is known to form a disulfide-linked dimer (Alt et al., 2012) and thus ENG binds BMP-9/-10 dimers in an antibody-like manner, in which the disulfide-linked ZP domain forms the Fc-domain and the orphan domain the “antigen-binding” Fab domain (Saito et al., 2017) (Fig. 1D).
Figure 1. Schematic of BG and ENG binding to their cognate GFs.

A. Proposed model for BG binding of TGF-β2 homodimer with 1:1 stoichiometry (Villarreal et al., 2016). Orphan domain binds around the center of the dimer without interfering with either TβRII site, while the ZP-C domain binds in one of the TβRII sites, leaving one TβRII site accessible for binding. B. Complexes and sub-complexes shown by SEC, SEC-MALS, and SPR that led to the model shown in panel A. C. Structure of the 2:1 hENGO:BMP-9 complex determined by crystallography (Saito et al., 2017). Super β-sheet formed by finger 4 of BMP-9 and β6 of O-D1 of hENGO is shown in the inset. D. Schematic of 2:1 ENG:BMP-9 complex, in which the orphan domain binds symmetrically, and only through orphan domain (O-D1), to finger 4 of the growth factor (GF). ZP-domain, which is covalently dimerized by formation of a disulfide bond in its ZP-C domain, does not directly contact the GF and is connected to the orphan domain by a flexible linker.
The objective of this study was to identify the structural basis for the different modes of GF binding by BG and ENG. Towards this end, the high-resolution structure of zebrafish BGO was determined. This showed that BGO has the same two-domain architecture as ENGO, but differs in that the loop connecting β-strand 7 to α-helix 1 in O-D1 includes an additional α-helix and β-strand. The additional strand, β8, pairs with putative O-D1 GF-interacting β6, sequestering β6 and likely preventing it from pairing with finger 4 of the GF, as does ENGO. To investigate if this modification engendered BGO with an alternative manner of binding, binding studies were carried out with BGO O-D1 and O-D2 and an engineered TGF-β monomer. These studies, together with accompanying small angle X-ray scattering measurements, supported an alternative 1:1 manner of binding in which BGO recognizes more than just the finger region and does so by utilizing both O-D1 and O-D2 to bind the GF. The alternative interfaces between BG and TGF-β and ENG and BMP-9 likely engender these otherwise structurally similar co-receptors with the ability to selectively bind their cognate GFs, enabling them to selectively target the heart, liver, and anterior pituitary and vasculature, respectively, without interference from one another or TGF-β family GFs.
Results
Zebrafish and rat BG bind human TGF-β2 in the same overall manner
Previous surface plasmon resonance (SPR), size exclusion chromatography (SEC), and isothermal titration calorimetry (ITC) binding studies showed that the full-length rat BG (rBG) ectodomain (rBGO-ZP) bound TGF-β2 dimers with 1:1 stoichiometry, with the orphan and ZP-C domain (rBGO and rBGZP-C, respectively) both directly binding the GF (Mendoza et al., 2009; Villarreal et al., 2016) (Fig. 1A). SPR, ITC, and SEC showed that this asymmetric manner of binding, which blocked one of the TβRII binding sites, but left the other accessible, was driven by the orphan domain, which alone bound TGF-β dimers with 1:1 stoichiometry and did not interfere with TβRII binding (Villarreal et al., 2016) (Fig. 1B). SPR, SEC, and structural studies, showed in contrast, that the human endoglin (hENG) ectodomain (hENGO-ZP) bound BMP-9 dimers symmetrically with 2:1 stoichiometry, with only the orphan domain (hENGO) bound to the GF (Alt et al., 2012; Saito et al., 2017) (Fig. 1C-D). To decipher the origins of these disparate modes of binding, crystallization of rBGO was pursued. Though unsuccessful, parallel efforts with the zebrafish betaglycan (zfBG) orphan domain (zfBGO), which has 54% identity with rBGO (Fig. S1A), succeeded.
Previously, zfBG had been shown to bind TGF-β, but when knocked down in zebrafish embryos, it had effects on blood vessel development (Kamaid et al., 2015), a phenotype not seen in BG null mice (Stenvers et al., 2003), raising questions as to whether zfBG was a true ortholog of mammalian BG. SPR experiments with the full-length zebrafish BG ectodomain, zfBGO-ZP, and its component domains, zfBGO and zfBGZP-C, and immobilized human TGF-β2 (hTGF-β2) were therefore performed to determine whether zfBG possessed the same binding properties as rBG (Fig. 2A-C). Similar experiments with rBGO-ZP, rBGO, and rBGZP-C were also performed to enable a direct comparison (Fig. S2A-C). In accord with previous results (Kamaid et al., 2015; Mendoza et al., 2009; Villarreal et al., 2016), the rat and zebrafish full-length BG ectodomain bound hTGF-β2, but in contrast to either the orphan or ZP-C domain alone, both of which also bound hTGF-β2, the full-length ectodomains dissociated more slowly, presumably due to bivalent binding. Kinetic analysis of the sensorgrams showed that the KDs were 5 nM for zfBGO-ZP, 27 nM for zfBGO, and 480-1100 nM for zfBGZP-C, 3 – 10 fold weaker compared to rBGO-ZP, rBGO, and rBGZP-C, which bound with KDs of 0.5 nM, 10 nM, and 90 – 610 nM (Table 1). Human and rat TGF-β2 differ at 3 positions, while human and zebrafish TGF-β2 differ at 10 positions (Fig. S1B), which may account for the lower affinities of the zfBG constructs compared to rat.
Figure 2. Characterization of zfBG binding.

A-C. SPR sensorgrams of zfBGO-ZP (A), zfBGO (B), and zfBGZP-C (C) binding to immobilized TGF-β2. Kinetic fits are shown in red over duplicate experimental curves in black. D-E. Complexes formed between zfBGO (D) and zfBGO-ZP (E) with TGF-β2DM and TβRII in solution as assessed using SEC. Shown in the insets are non-reducing SDS-PAGE gels of the major peaks that eluted. F-G. SEC-MALS analysis of the TGF-β2DM:TβRII:zfBGO (F) and TGF-β2DM:TβRII:zfBGO-ZP (G) complexes, with the blue trace corresponding to the UV absorbance and the red data points the molecular mass. Observed/anticipated masses for the TGF-β2DM:TβRII:zfBGO and TGF-β2DM:TβRII:zfBGO-ZP complexes are 89.6 kDa/88.2 kDa and 127 kDa/121 kDa, respectively. See also Figure S2.
Table 1.
SPR binding constants for zfBG and rBG, and their subdomains, to TGF-β2
| Surface | Analyte |
ka1 (M−1 s−1) |
kd1 (s−1) |
KD1 (nM) |
ka2 (M−1 s−1) |
kd2 (s−1) |
KD2 (nM) | Rmax1 (RU) | Rmax2 (RU) |
km (m/s)1 |
|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β2 | zfBGO-ZP | 3.50±0.05 × 105 | 0.00173±0.00002 | 4.95±0.13 | n.d.2 | n.d. | n.d. | 69.1±0.3 | n.d. | 5.0±0.9 × 109 |
| TGF-β2 | zfBGO | 1.066±0.007 × 106 | 0.0288±0.0002 | 27.0±0.4 | n.d. | n.d. | n.d. | 144.8±0.5 | n.d. | 1.5±0.1 × 109 |
| TGF-β2 | zfBGZP-C | 4.8±0.7 × 105 | 0.51±0.01 | 1100±200 | 2.0±0.3 × 104 | 9.3±0.4 × 103 | 480±80 | 93±9 | 105±2 | |
| TGF-β2 | rBGO-ZP | 1.478±0.006 × 106 | 0.000755±0.000001 | 0.511±0.002 | n.d. | n.d. | n.d. | 83.5±0.2 | n.d. | 1.0±0.3 × 1010 |
| TGF-β2 | rBGO | 6.03±0.02 × 105 | 0.00596±0.00002 | 9.88±0.04 | n.d. | n.d. | n.d. | 176.0±0.4 | n.d. | 1.0±0.4 × 1010 |
| TGF-β2 | rBGZP-C | 3.25±0.20 × 104 | 0.0029±0.0001 | 90.0±0.8 | 2.60±0.04 × 105 | 1.57±0.20 × 10−1 | 610±70 | 150±9 | 60±9 |
km, mass transfer rate constant
n.d., not determined
Binding stoichiometry and effects on TβRII binding were assessed by combining zfBGO-ZP or zfBGO with TGF-β:TβRII binary complex and analyzing the complexes that formed by SEC and SEC-multiangle light scattering (SEC-MALS). To enable a direct comparison, the same experiments were also performed with rBGO-ZP and rBGO; to minimize disassociation of TβRII, TGF-β2 K25R K94R or TGF-β2DM was used, which binds TβRII with high affinity (Baardsnes et al., 2009; De Crescenzo et al., 2006). In accord with previous findings with rBG (Villarreal et al., 2016), the orphan and full length ectodomains of both zebrafish and rat BG formed ternary complexes, as shown by a peak in the SEC profiles (labeled a) that eluted earlier than either the corresponding BG domain (labeled b) or TGF-β2DM:TβRII binary complex (labeled c) (Fig. 2D-E, Fig. S2D-E). In accord with previous findings with rBG (Villarreal et al., 2016), the apparent molecular weights of ternary complexes were consistent with the 1:1:1 TGF-β2DM:TβRII:BGO-ZP complex anticipated for the full-length ectodomain (127 ± 1 kDa and 129 ± 2 kDa observed vs. 121 kDa and 123 kDa anticipated for zfBG and rBG, respectively) and the 1:2:1 TGF-β2DM:TαRII:BGO complex anticipated for the orphan domain (89.6 ± 1.8 kDa and 94.9 ± 2.0 kDa observed vs. 88.2 kDa and 91.1 kDa anticipated for zfBG and rBG, respectively) (Fig. 2F-G, Fig. S2F-G). In summary, the binding data shows that zfBG binds TGF-β2 in the same overall manner as rBG (Villarreal et al., 2016), indicating that it is a true ortholog of mammalian BG and justifying it as a model system for deciphering the function of mammalian BG.
Structure of zfBGO
The crystal structure of zfBGO was determined to 2.10 Å and 2.38 Å from orthorhombic (P212121) and tetragonal (P41) crystal forms, respectively (Table 2). The initial efforts to determine the structure focused on the tetragonal form, but these were unfruitful as molecular replacement with hENGO, which has 20% sequence identity with zfBGO (Fig. S3A) proved unsuccessful, as did phasing with heavy atoms, primarily due to difficulty reproducing the crystals. To overcome these obstacles, we reasoned that zfBGO, which has six cysteines in its orphan domain (Fig. S1A), has two free cysteines which might be forming disulfide-linked aggregates. This hypothesis is based on the observation that four of the six cysteines of zfBGO are conserved with the four cysteines in hENGO that form two disulfide bonds, while the other two cysteines are at positions expected to be far apart in the three-dimensional structure (Fig. S3A). Therefore, zfBGO was pre-treated with iodoacetamide prior to SEC and final purification. This led to more consistent crystal formation, in an orthorhombic space group, and enabled preparation of two derivatives, one platinum and one iodide. Through multiple isomorphous replacement with anomalous scattering (MIRAS) phasing (Table 2), this led to an interpretable map, a portion of which is shown in Fig. S4. This map enabled building of a nearly complete model, which in turn enabled solution of the tetragonal form by molecular replacement. The asymmetric unit of the tetragonal and orthorhombic forms contained 1 and 2 molecules, respectively. The tetragonal form and one of the chains in the orthorhombic form, Chain A, were well ordered, with weak electron density in only a few loop regions (Table S3). The other chain in the orthorhombic form, Chain B, had overall weaker density and higher B-factors (Table 2), especially in domain 2 and was difficult to model, with additional regions of weak density compared to Chain A (Table S3). The weak density in this region appears to be due the nature of the lattice packing, which as shown in Figure S5, places domain 2 of chain B in a solvent void without lattice contacts to either domain of either chain (Fig. S5).
Table 2.
X-ray data collection and refinement
| Data collection | ||||
|---|---|---|---|---|
| Crystal Form | Tetragonal (native) |
Orthorhomic (native) |
Orthorhombic (Pt) |
Orthorhombic (I) |
| X-ray Source | Adv. Photon Source SER-CAT 22-ID | Adv. Photon Source SER-CAT 22-ID | Adv. Photon Source SER-CAT 22-ID | Rigaku FR-E generator and RAXIS HTC image plate detector |
| Space group | P41 | P212121 | P212121 | P212121 |
| Cell dimensions | 62.9, 62.9, 114.3 | 63.5, 107.3, 113.2 | 64.0, 106.5, | 63.1, 107.5, 113.4 |
| a, b, c (Å) | 90, 90, 90 | 90, 90, 90 | 113.7 | 90, 90, 90 |
| α, β, γ (°) | 90, 90, 90 | |||
| Wavelength (Å) | 1.0000 | 0.9692 | 0.9795 | 1.5418 |
| Resolution (Å) | 35.11-2.38 (2.47-2.38)1 | 40.98 - 2.10 (2.16 - 2.10)1 | 54.87-3.00 (3.18 - 3.00)1 | 22.18-3.20 (3.46-3.20)1 |
| Rmerge | 0.080 (1.784) | 0.126 (2.49) | 0.118 (0.586) | 0.159 (0.510) |
| Rpim | 0.046 (1.100) | 0.053 (1.069) | 0.069 (0.341) | 0.059 (0.193) |
| CC1/2 | 0.998 (0.520) | 0.998 (0.565) | 0.992 (0.932) | 0.998 (0.967) |
| I/σI | 10.6 (1.1) | 10.8 (1.3) | 8.5 (2.4) | 16.0 (5.4) |
| Completeness (%) | 99.4 (99.8) | 99.9 (99.6) | 100.0 (100.0) | 99.7 (99.9) |
| Redundancy | 7.5 (6.8) | 11.6 (11.2) | 7.3 (7.3) | 14.5 (14.7) |
| Phasing | ||||
| Number Sites | 7 | 20 | ||
| Resolution | 33.2-2.80 | 33.2-2.80 | ||
| Phasing Power2 (acentric/centric) | ||||
| Isomorphous | 0.558/0.614 | 0.721/0.712 | ||
| Anomalous | 0.566/- | 0.349/- | ||
| Cullis R-factor3 (acentric/centric) | ||||
| Isomorphous | 0.777/0.788 | 0.936/0.875 | ||
| Anomalous | 0.698/- | 0.940/- | ||
| Overall Figure of Merit (acentric/centric) | 0.52/0.49 | |||
| Refinement | ||||
| Molecules/ASU | 1 | 2 | ||
| No. reflections | 17730 | 45850 | ||
| Rwork/ Rfree | 0.198/0.229 | 0.233/0.258 | ||
| No. atoms | ||||
| Protein | 2,442 | 4803 | ||
| Water | 8 | 79 | ||
| B-factor (Å2) | ||||
| Chain A | 101.3/105.4 | 58.5/62.7 | ||
| Backbone/Overall | ||||
| Chain B | 79.9/83.9 | |||
| Backbone/Overall | ||||
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.010 | 0.010 | ||
| Bond angles (°) | 1.27 | 1.17 | ||
| Ramachandran statistics - favored, allowed, outliers (%) | 98.0, 2.0, 0.0 | 98.0, 1.4, 0.6 | ||
| PDB accession code | 6MZN | 6MZP | ||
Highest resolution shell is shown in parentheses.
Phasing Power = < [∣Fh(calc)∣/E]> where Fh(calc) is the calculated heavy atom structure factor amplitude and E is the phase-integrated lack of closure error.
Cullis R-factor = <E>/<∣ Fph -Fp ∣> where E is the phase-integrated lack of closure error, Fph is structure factor amplitude of the derivative and Fp structure factor amplitude of the native.
The structure of zfBGO is comprised of two β-sandwich domains arranged in an arch-like manner (Fig. 3A). The two β-sandwich domains are each comprised of 10 β-strands and share the same fold (Fig. 3B, D-E). They are decorated with two α-helices, one in the loop following β7 (α1), and the other packed against the convex surface of the arch-like structure formed by the two β-sandwiches (α2) (Fig. 3A, D-E). One of the unusual features is that the N-terminus of the protein is comprised of two extended segments, each roughly 10 amino acids in length, connected by a bend: the N-terminal half is tethered to the N-terminal β-strand (β1) of orphan domain 1 (O-D1) by a disulfide bond (C31-C225), while the C-terminal half is paired with the C-terminal β-strand of O-D1 (β10) and extends into orphan domain 2 (O-D2) (Fig. 3A, C-D). A similarly structured extended “exit” segment is present in O-D2, where after exiting the C-terminal β-strand of O-D2 (β10), it is tethered to the N-terminal β-strand (β1) by a disulfide bond (C55-C201), paired with the C-terminal β-strand (β10), and extends into O-D1 (Fig. 3A, C, E). One further point is that a sequence alignment of the repeating element present in zfBGO, that is the O-D1 exit sequence followed by the O-D2 β-sandwich and the O-D2 exit sequence followed by the O-D1 β-sandwich, shows that these share significant (20%) sequence identity (Fig. S1C), and thus are likely the result of gene duplication. One important consequence is that similar to hENGO (Saito et al., 2017), zfBGO is pseudosymmetric, not only in terms of its architecture, but also its sequence.
Figure 3. Structure of zfBGO.

A. Structure of zfBGO with “exit” sequences for O-D1 and O-D2 in green and lavender, respectively. Positionally conserved disulfide in the O-D1 and O-D2 subdomains, C31-C225 and C55-C201, are labeled SS1 and SS2, respectively. Single free cysteines in O-D1 and O-D2, C277 and C150, respectively, are also shown. Residue numbering can be deduced by reference to the secondary structure/residue numbering shown for zfBGO in Fig. S3. B. Overlay of zfBGO O-D1 and O-D2 with the β-strands of O-D1 depicted in orange, the β-strands of O-D2 depicted in tan, the α-helices of O-D1 and O-D2 in blue, and the exit sequences of O-D1 and O-D2 in green and lavender, respectively. C. Schematic showing the overall architecture of zfBGO. D-E. Similar orientations of the zfBGO O-D1 (D) and O-D2 (E) subdomains, illustrating the conservation of their folds and secondary structure. See also Figures S1, S4, S5, S6, S7, and Table S3.
The paired antiparallel β-strands that connect O-D1 and O-D2, seen previously in hENGO (Saito et al., 2017), represent another unique structural feature and might be important in restricting the orientation of O-D1 and O-D2. To gauge the extent of this restriction, the two zfBGO orthorhombic structures and one zfBGO tetragonal structure were aligned by minimizing coordinate differences for one domain, but not the other. This showed that the structures of individual domains were on the one hand very consistently determined, with RMSD deviations well under 1.0 Å for both O-D1 and O-D2 when all three structures were compared (Fig. S6A). The relative domain orientations in the three structures, on the other hand, were more variable, with displacements ranging from 1.3 Å at the centroid of unaligned domain when the orthorhombic Chain A and tetragonal structures were compared, to 3.7 Å when orthorhombic Chain A and Chain B were compared (Fig. S6B-D). This suggests some degree of hinge bending, though evidently this must be limited, as solution-based small angle X-ray scattering (SAXS) measurements with zfBGO showed that the experimental scattering curves, as well as P(r) function and calculated electron density, correlated well with the three zfBGO structures or parameters calculated from them (Fig. S7A-C, G-H). The rat BG orphan domain also appeared to have an overall structure similar to the zebrafish orphan domain as the experimental SAXS data for rBGO also correlated very well with the three zfBGO structures or parameters calculated from them (Fig. S7D-F, I-J).
Structural comparison of zfBGO and hEngO
The overall architecture and domain structures of zfBGO and hENGO are similar (Fig. 4A-C), despite only 20% sequence identity (Fig. S3A). There are nonetheless differences, the most prominent of which is a shift of O-D2 downward and toward the concave surface of O-D1 in zfBGO (Fig. 4A). This shift, which corresponds to 17.6 Å displacement in the centroid of the zfBG O-D2 relative to hENG O-D2 when the residues of O-D1 are aligned, results an archlike structure in zfBGO, but not hENGO (Fig. 4A). The primary reason for the more arch-like structure in zfBGO compared to hENGO is the O-D1 exit sequence. In zfBG O-D1, after exiting the domain, the β-strand of the exit sequence bends upward before extending into the N-terminal β-strand of O-D2 (Fig. S8). In contrast, in hENG O-D1, the exit sequence is shorter and has a sharp bend downward before it leads into N-terminal β-strand of O-D2 (Fig. S8). Importantly the lengthening of the exit sequence and opposite direction of the bend as it exits the domain leads to very different positioning of the N-terminal β-strand of O-D2 in hENGO and zfBGO, and thus relative positioning of O-D1 and O-D2 (Fig. S8). In order to accommodate the difference in domain orientation, the β-strand of the O-D2 exit sequence of zfBGO pairs mostly with O-D1 exit sequence, while that of hENGO pairs with the C-terminal β-strand of O-D2 and the O-D1 exit sequence (Fig. S8).
Figure 4. Structural comparison of zfBGO and hENGO.

A. Comparison of the overall structures of zfBGO and hENGO, whose strands and helices are shaded orange and blue and green and blue, respectively. Changes in the relative domain orientation of zfBGO and human ENGO (green β-strands, cyan helices) is shown in the right most subpanel in which only the backbone atoms of O-D1 have been superposed. B-C. Superposition of zfBGO and hENGO O-D1 (B) or O-D2 (C). Distinct orientation of the O-D1 “meander” loop connecting β8-α2 in zfBGO and β7-α1 in hEngO is highlighted. Structures are shaded as in panel A, with additional shading of the β8-α2 meander in zfBGO-D1 and β7-α1 meander in hENGO-D1 in gold and dark red, respectively. D. Region of super β-sheet formation in the hENGO:BMP-9 complex, but only with the β-strands corresponding to fingers 3 and 4 (F3 and F4) of BMP-9 shown and the segment of hENGO extending from β5 to α1 shown. E. Depicts the same as that shown in panel D, but with zfBGO in place of hENGO; structure shown was generated by superimposing zfBGO onto hENGO in the structure of the hENGO:BMP-9 complex and then by undisplaying hENGO. Superposition highlights clashes (red circles) between zfBGO β8 and finger 4 of BMP-9 for this mode of binding. F. Overlay of the two P212121 zfBGO models and one P41 zfBGO model in the region including an additional α-helix (α1) and β-strand (β8) compared to hENGO. G. 2mFo-DFc electron density for P212121 zfBGO (chain A) extending from β6 to α2; map shown was calculated with phenix.maps with a bulk solvent correction and anisotropic scaling and was contoured at 1σ. See also Figures S1, S3, and S8.
Though more subtle, another difference pertains to the loop region following β7. In hENG O-D1, immediately after β7 there is a sharp bend followed by an extended meander that leads into the 3-turn α-helix (α1) packed against the convex surface of the sandwich (Fig. 4B). In zfBG O-D1, after β7 there is an insertion of a 1-turn helix (α1) which traverses the gap between the concave and convex surfaces of the sandwich (Fig. 4B). Immediately following α1, there is an insertion of a β-strand (β8), which pairs with β6 on the convex surface, followed by an extended meander that connects into 3-turn α2 packed against convex surface (Fig. 4B). Importantly, in hENGO, the absence of β8 and altered positioning of the meander leaves O-D1 edge β-strand 6 accessible on the convex surface, while in zfBGO, the O-D1 α1-β8 insertion sequesters β6, making it inaccessible.
The structure of the 2:1 hENGO:BMP-9 complex shows that BMP-9 and hENGO bind through a super β-sheet formed between the exposed finger region of BMP-9, specifically finger 4 β7 and the exposed edge β-strand, β6, of hENG O-D1 (Fig. 1C, 4D). Through positioning of the zfBGO structure onto the structure of hENGO structure at the hENGO:BMP-9 interface, zfBGO O-D1 β8 is shown to sterically clash with BMP-9 finger 4 β7 (Fig. 4E). The region corresponding to β6-β7-α1-β8 in zfBG O-D1 is consistently determined by the three zfBGO structures (Fig. 4F) and is well-defined by the electron density in zfBGO (Fig. 4G), and therefore it is unlikely that β8 would be displaced by GF binding. Thus, zfBGO is unlikely to use β6 to bind its cognate growth factor, TGF-β2, in an hENGO-like manner.
Binding of zfBGO to the TGF-β mini-monomer
Through prior functional and structural studies, mmTGF-β2-7M, an engineered form of TGF-β that includes the finger region and cystine knot, but not the heel helix, was shown to maintain the same high affinity for TβRII as TGF-β1 and TGF-β3 and to have a structure, both alone and as bound to TβRII, essentially indistinguishable from that of TGF-β1 and TGF-β3 (Fig. S9A) (Kim et al., 2017). The intact finger region in mmTGF-β2-7M can therefore be used to determine if zfBGO recognizes and binds the finger region of TGF-β2, similar to hENGO, or whether it recognizes and binds features beyond those in mmTGF-β2-7M. To test this, zfBGO was combined with equimolar amounts of mmTGF-β2-7M:TβRII binary complex and analyzed by SEC. The mmTGF-β2-7M:TβRII binary complex was used for these experiments, rather than mmTGF-β2-7M alone, because it has improved solubility compared to mmTGF-β2-7M alone and because models showed that TβRII should not interfere with binding of zfBGO to mmTGF-β2-7M (Fig. S9B). The SEC chromatogram of the ternary mixture corresponded to that of zfBGO alone and the mmTGF-β2-7M:TβRII binary complex alone (Fig. 5A), which indicated that zfBGO binds mmTGF-β2-7M:TβRII weakly. To further assess binding, the SPR response was measured as increasing concentrations of zfBGO was injected over immobilized mmTGF-β2-7M (Fig. 5B). This led to a barely detectable response, consistent with the weak binding detected by SEC. There were, however, robust responses in control experiments in which TβRII was injected over mmTGF-β2-7M (Fig. 5E) or zfBGO was injected over immobilized TGF-β2 (Fig. 5F). The fitted disassociation constants for TβRII:mmTGF-β2-7M and zfBGO:TGF-β2 interactions were 63 and 15 nM nM, respectively (Table 3), comparable to the previously reported values (Table 1 and Kim, et. al., 2017 (Kim et al., 2017)). These results show that zfBGO binds mmTGF-β2-7M weakly and must therefore attain its high affinity for TGF-β2 by recognizing more than just finger 4 of the GF.
Figure 5. Binding of monomeric TGF-β by BGO.

A,C. SEC chromatograms to assess binding of zfBGO (A) or rBGO (C) to the mmTGF-β2-7M:TβRII binary complex. Chromatograms of the mmTGF-β2-7M:TβRII:BGO ternary complex, the mmTGF-β2-7M:TβRII binary complex, TβRII, and BGO are shown in blue, magenta, green, and orange, respectively. Shown in the inset in panel C is a non-reducing SDS-PAGE gel of the major peaks that eluted. B, D. SPR sensorgrams for binding of zfBGO (B) or rBGO to immobilized mmTGF-β2-7M. E. SPR sensorgrams for binding of TβRII to mmTGF-β2-7M. Kinetic fit is shown in red over the experimental data shown in black. F-G. SPR sensorgrams for binding of zfBGO (F) or rBGO (G) to immobilized TGF-β2. Kinetic fit is shown in red over the experimental data shown in black. SPR experiments shown in panels B, D, and E were performed using the same neutravidin-coupled CM5 sensor chip with captured biotinylated avi-mmTGF-β2-7M. See also Figure S9.
Table 3.
SPR binding constants for receptor binding to TGF-β2 or mmTGF-β2-7M
| Surface | Analyte | ka1 (M−1 s−1) | kd1 (s−1) | KD1 (μM) |
ka2 (M−1 s −1) |
kd2 (s−1) | KD2 (μM) | Rmax1 (RU) | Rmax2 (RU) | km (m/s)1 |
|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β2 | zfBGO | 5.28±0.04 × 105 | 7.72 ± 0.08 × 10−3 | 0.015 ± 0.002 | n.d.2 | n.d. | n.d. | 181±9 | n.d. | |
| TGF-β2 | rBGO | 3.74±0.07 × 103 | 3.45±0.05 × 10−3 | 0.009 ± 0.002 | n.d. | n.d. | n.d. | 212±6 | n.d. | |
| mmTGF-β2-7M | TβRII | 2.03± 0.03 × 106 | 1.28±0.03 × 10−1 | 0.063± 0.001 | n.d. | n.d. | n.d. | 71±5 | n.d. | |
| TGF-β2 | zfBGO-D1 | 9.3±0.3 × 103 | 0.114±0.004 | 12.3±0.4 | n.d. | n.d. | n.d. | 20.8±0.4 | n.d. | 1.2±0.3 × 106 |
| TGF-β2 | zfBGO-D2 | 5.2±0.3× 103 | 0.0064±0.0017 | 1.2±0.3 | 2.8±0.7 × 105 | 2.7±3.9 × 103 | 0.0083±0.0100 | 18±0.3 | 1.8±0.3 |
Km, mass transfer rate constant
n.d., not determined
To determine if this property was also shared with rat BG orphan domain (rBGO), the same experiments were performed, but using rBGO rather than zfBGO. The SEC experiments showed that unlike zfBGO, rBGO was able to form a detectable ternary complex with mmTGF-β2-7M and TβRII as these three proteins were each present in a peak (labeled “a”) that eluted before either rBGO or the mmTGFβ2-7M:TβRII binary complex alone (Fig. 5C). The formation of this complex is, however, evidently a result of weak binding as SPR experiments showed that when rBGO was injected over immobilized mmTGFβ2-7M, only a very weak response was obtained relative to that of TβRII, which itself is only one-third the mass of rBGO (Fig. 5D, E). The weak response when rBGO was injected over immobilized mmTGFβ2-7M, which could not be fitted to derive a reliable binding constant, stood in contrast the robust response when rBGO was injected over TGF-β2 (Fig. 5G), Which could be readily fitted to derive a binding constant (Table 3). The fact that rBGO did form a detectable complex with mmTGF-β2-7M and TβRII by SEC, while zfBGO did not, may be because of the higher overall affinity of rBGO for TGF-β2 relative to zfBGO (Fig. 2, Fig. S2, Table 1).
Binding of zfBGO-D1 and zfBGO-D2 to TGF-β2
The structural differences relative to hENGO revealed by the zfBGO crystal structure, together with impaired binding of zfBGO to the TGF-β mini monomer, suggest that zfBGO binds TGF-β2 in a manner different than hENGO binds BMP-9. To determine whether zfBGO uses both of its subdomains to contact TGF-β2, SEC and SPR binding experiments were performed using the zfBGO-D1 and zfBGO-D2 subdomains. zfBGO-D2 was made as a secreted protein in HEK-293 cells, similar to the full-length zfBGO, by expressing a construct that began a few residues before it entered O-D2 and ended a few residues after it exited (Table S2). zfBGO-D1 was made similarly by expressing a construct that included most of the N-terminal exit sequence, an artificial 2-residue Thr-Asp dipeptide, the C-terminal part of the O-D2 exit sequence, and the O-D1 β-sandwich fused C-terminally to human serum albumin and an engineered thrombin cleavage site (Table S2). Prior to the final purification step for zfBGO-D1, the fusion protein was treated with thrombin, which enabled separation of zfBGO-D1 from albumin.
To assess binding, TGF-β2DM:TβRII binary complex was combined with zfBGO, zfBGO-D1, or zfBGO-D2 and analyzed by SEC. The results showed that in contrast to full-length zfBGO, zfBGO-D1 and zfBGO-D2 both failed to form a peak that eluted earlier than the TGF-β2DM:TβRII binary complex or zfBGO-D1/zfBGO-D2 alone (Fig. 6A-B). To further investigate, SPR experiments were performed in which zfBGO-D1 and zfBGO-D2 were injected over the same immobilized TGF-β2 used for the experiments shown in Figs. 2 and S2. Though the concentrations of zfBGO-D1, and zfBGO-D2 injected were the same as those used for zfBGO, the maximal responses obtained were nonetheless only 10 – 20% of that compared to zfBGO (Fig. 6C-D, Fig. 2B). Through kinetic analysis, the data for O-D1 could be fitted to a 1:1 model, with a KD of 12 μM, while the data for O-D2 could only be fit to a heterogenous ligand 1:1 model, with KDs of 0.008 μM and 1.2 μM (Fig. 6C-D, Table 3). The KD of 0.008 μM for zfBGO-D2 is not likely meaningful, as the response attained upon injection of zfBGO-D2 over TGF-β2 (Fig. 6D) was significantly lower compared to that of zfBGO when injected at the same concentrations over the same TGF-β2 surface (Fig. 2B). Thus, both zfBGO subdomains bind TGF-β2 weakly, suggesting that both simultaneously engage TGF-β2 to enable high affinity binding (KD 27 nM) of TGF-β2 by full-length zfBGO (Fig. 2B, Table 1). To rule out the possibility that the weak binding of TGF-β2 by zfBGO-D1 or zfBGO-D2 was due to misfolding of the domains, NMR natural abundance 1H-13C shift correlation spectra of the methyl resonances were recorded. These spectra both revealed a well-dispersed pattern of peaks, both with several resonances upfield of 0.0 ppm 1H (Fig. 6E-F), indicating that both were natively folded.
Figure 6. Binding of zfBGO-D1 and zfBGO-D2 to TGF-β2.

A, B. SEC chromatograms of complexes between zfBGO and zfBGO-D1 (A) and zfBGO and zfBGO-D2 (B) with TGF-β2DM and TβRII are depicted in blue and red, respectively. Chromatograms of the TGF-β2DM:TβRII binary complex and zfBGO-D1/zfBGO-D2 alone are shown in purple and orange curves, respectively. C, D. SPR sensorgrams for binding of zfBGO-D1 (C) and zfBGO-D2 (D) to immobilized TGF-β2. Kinetic fit is shown in red over the experimental data shown in black. SPR sensor chip with immobilized TGF-β2 is the same as that used for the experiments shown in Figs 2 and S2. E-F. Methyl region of natural abundance 1H-13C HSQC spectra of BGO-D1 (E) and BGO-D2 (F).
Structural modeling of the complex of BGO with TGF-β2 and TβRII
The finding that BGO bound TGF-β dimers with 1:1 stoichiometry, together with results above that BGO binds more than just the finger region and does so by contacting the GF with both O-D1 and O-D2, suggest an entirely different manner of GF binding compared to ENGO. To determine, at low-resolution, how BGO might bind TGF-β, 1:2:1 TGF-β2TM:TβRII:BGO complexes formed with both zfBGO and rBGO were isolated by SEC and analyzed by SAXS over a range of concentrations (1, 2, and 4 mg mL−1 and 2 and 4 mg mL−1, respectively) (Fig. 7A-B). The overall shape of the scattering curves of the two complexes were similar, consistent with the SEC and SEC-MALS results (Fig. 2D-G, Fig. S2D-G) which showed that zfBGO and rBGO form similar complexes in solution. There was, however, a concentration-dependent increase in scattering in the low-q region for the zebrafish complex, indicative of aggregation (Fig. 7A). Thus, all subsequent modeling was performed with the scattering data for the rat protein, which was almost perfectly behaved (Fig. 7B).
Figure 7. Structural modeling of the complex of BGO with TGF-β2 and TβRII.

A-B. SAXS data for 1:2:1 TGF-β2TM:TβRII:zfBGO (A) or 1:2:1 TGF-β3:TβRII:rBGO (B) complexes at the concentrations shown (scattering curves are scaled by the expected dilution factor). Insets highlight concentration-dependent changes in the low-q region. C-E. Comparison of the experimental SAXS data for the 1:2:1 TGF-β3:TβRII:rBGO complex (red) with the scattering curve calculated (blue) (C) from a 1:2:2 TGF-β3:TβRII:rBGO model with rBGO positioned in an hENGO-like manner (D). Calculated electron density map for the 1:2:1 TGF-β3:TβRII:rBGO complex contoured at 1.5 sigma with the hENGO-like 1:2:2 TGF-β3:TβRII:rBGO model fitted into the density. F-I. Comparison of the experimental SAXS data for the 1:2:1 TGF-β3:TβRII:rBGO complex (red) with the calculated scattering curve (blue) (F) from the best scoring 1:2:1 TGF-β3:TβRII:rBGO model derived from positioning rBGO onto the 1:2 TGF-β3:TβRII binary complex using pyDockSAXS (G, H). Calculated electron density map for the 1:2:1 TGF-β3:TβRII:rBGO complex contoured at 1.5 sigma with the pyDockSAXS 1:2:1 TGF-β3:TβRII:rBGO model fitted into the density. Estimated resolution of the electron density map shown in panels E and I from Fourier shell correlation analysis is 30.9 Å. See also Figure S10.
To determine whether an alternative manner of binding was required, a model was constructed with rBGO placed onto the 1:2 TGF-β3:TβRII complex (Hart et al., 2002) in the same 2:1 manner that hENGO binds BMP-9 (structure of rBGO was obtained by building a zfBGO-based homology model) (Fig. 7D). The scattering curve was then calculated from this model and compared with the experimentally measured scattering curve for the rat complex. This revealed large systematic deviations over the entire scattering curve and a very high χ2 value (65.2) (Fig. 7C), indicating that this model poorly describes the structure of the TGF-β:TβRII:rBGO ternary complex in solution. This model was also poorly described by the electron density calculated from the experimental scattering data, with a significant portion of both molecules of rBGO protruding from the density (Fig. 7E).
To determine whether models with an altered stoichiometry (1:1 BGO:TGF-β) and manner of binding (direct binding by O-D1 and O-D2 at sites beyond the fingertip region) might be more consistent with the experimental data, the program pyDockSAXS (Jimenez-Garcia et al., 2015) was used to construct 1:2:1 TGF-β2:TβRII:rBGO models that optimized favorable van der Waals, H-bonding, and electrostatic contacts at the interface, and consistency with the experimental SAXS data. The complex that best met these criteria, shown in Fig. 7G-H, had the orphan domain straddling the TGF-β dimer, with O-D1 and O-D2 in contact with residues protruding from the C-terminal end of the TGF-β N-terminal helix, α1, and the heel helix, α3, within each monomer. This manner of binding, which had the pseudo-symmetry axis of rBGO near, but not coincident with the symmetry axis of TGFβ2, agreed well with the experimental scattering data (χ2 1.38) and resulted in no significant protrusions of the structure from the calculated electron density (Fig. 7F, I). The manner of binding described above was well-represented within the ensemble of 10 best scoring structures, with first 6 best scoring structures having an overall RMSD within 1.0 Å of that shown in Fig. 7G-H. The next two best scoring structures had a similar staggered positioning of the orphan domain, but were rotated by approximately 100° around an axis connecting the centroid of O-D1 and O-D2. The next two structures within the ensemble were bound closer to the fingertips in a manner that would enable two molecules of the orphan domain to bind, which based on the SEC-MALS data shown in Figs. 2F and S2F, is not feasible. The experimental SAXS data is therefore sufficient to rule out a 2:1 ENGO-like manner of binding, and while it suggests and is consistent with a pseudo-symmetric 1:1 manner of binding, it is not sufficient to unambiguously determine the structure of such a complex.
Discussion
The TGF-β pathway has expanded and diversified as organisms have diversified, with 3 family members in nematodes, 7 in flies, and more than 30 in vertebrates (Hinck, 2012). Though the type I and type II receptors have also similarly expanded, there are consistently fewer signaling receptors than GFs – flies, for example, have three type I and two type II receptors for 7 family GFs (Upadhyay et al., 2017), while humans have seven type I and five type II receptors for 33 family GFs (Hinck, 2012). The type I receptors of the family additionally couple to and activate just two classes of R-Smads: the GFs of the TGF-β/activin branch of the pathway bind type I receptors that activate R-Smads 2 and 3, while those of the BMP/GDF branch bind type I receptors that activate R-Smads 1, 5, and 8 (Chen et al., 1998; Hinck, 2012). The functional diversity that can be attained through intrinsic differences in signaling is therefore limited and cannot account for the functional diversity of the 33 TGF-β family GFs in humans.
The membrane-anchored co-receptors of the TGF-β family, BG and ENG, have essential roles potentiating the signaling activity of TGF-β2 and inhA and BMP-9 and BMP-10, respectively (Lewis et al., 2000; Lopez-Casillas et al., 1993; Nickel et al., 2018; Scharpfenecker et al., 2007). The selectivity with which BG and ENG recognize these GFs (Castonguay et al., 2011; Henen et al., 2018), together with the restricted pattern of their expression, is vital in enabling unique temporal-spatial patterns of signaling that underlie the unique functions of these GFs in vivo – for TGFβ2, morphogenetic transformation of endothelial progenitors in the developing heart and liver (Brown et al., 1999; Stenvers et al., 2003), for inhibin A antagonism of activin A and B in the anterior pituitary (Bernard et al., 2002; Lewis et al., 2000), and for endoglin activation of BMP-9 and BMP-10 signaling on endothelial cells required for normal development and maintenance of the vasculature (Roman and Hinck, 2017).
The previous studies aimed at uncovering how BG and ENG recognize their cognate GFs and potentiate assembly of their signaling complexes showed that they differ in terms of the overall manner with which they engage their respective GFs: ENG exists as a covalent dimer and uses only its orphan domain to engage the GF dimer with 2:1 stoichiometry (Alt et al., 2012; Castonguay et al., 2011), while BG, which exists as a monomer, uses both its orphan and ZP-C domains to engage the GF dimer with 1:1 stoichiometry (Esparza-Lopez et al., 2001; Mendoza et al., 2009; Villarreal et al., 2016). The two co-receptors also have different effects on the binding of the signaling receptors, with ENGO competing against type II receptors that BMP-9 and -10 bind, such as ActRII and ActRIIB, but not the type I receptor Alk1 (Alt et al., 2012; Castonguay et al., 2011), while BGO potentiates the binding of the TGF-β type II receptor, TβRII, but competes against the TGF-β type I receptor, TβRI (Villarreal et al., 2016).
The recent structural studies of the human ENG ZP domain alone, and human ENG orphan domain, both alone and bound to its cognate growth factor BMP-9, showed that ENG binds the GF dimer with 2:1 stoichiometry in an antibody-like manner, with the covalently dimerized ZP domain homologous to the Fc region, and the orphan domain, bound to the growth factor by formation of a super β-sheet with the extended finger region, homologous to the Fab region (Figs. 1C, 4D) (Saito et al., 2017). If BGo were to bind TGF-β2 in the same manner as the ENGO domain, it would on the one hand be consistent with the observed effects of ENG and BG on type II receptor binding as structural studies show that ActRII/ActRIIB and TβRII bind their cognate GFs differently: ActRII/ActRIIB bind the knuckle in a way that would directly overlap with the co-receptor (Greenwald et al., 2003; Thompson et al., 2003; Townson et al., 2012), while TβRII binds the fingertips in a way that would not directly overlap with the co-receptor (Fig. S9B) (Groppe et al., 2008; Hart et al., 2002; Radaev et al., 2010). On the other hand, this manner of binding, in addition to being inconsistent with the observed 1:1 binding of BGO to TGF-β dimers (Villarreal et al., 2016), would also be inconsistent with effects on type I receptor binding as prior structural studies showed Alk1 and TβRI both bind in similar, although not identical manner, on the underside of the fingers of one monomer and the heel helix of the other monomer in a way that would not directly overlap with that of the co-receptor (Groppe et al., 2008; Radaev et al., 2010; Townson et al., 2012).
The structure of zfBGO shows that while it shares the same tandem β-sandwich architecture as hENGO, it nonetheless differs in that there is an additional helix (α1) and β-strand (β8) following β7 within subdomain 1 (O-D1). The additional β-strand, β8, is shown to pair with β6. This would block BG O-D1 from pairing and forming a super β-sheet with finger 4 of TGF-β, as does β6 of ENG O-D1. Three observations consistent with the α1-β8 insertion fulfilling this “blocking” role are as follows: 1) The electron density for BGO α1 and β8, as well as most of the meandering loop that follows, is well-defined (Fig. 4G). 2) The backbone conformation, and even the conformation of many of the sidechains in the region spanning from β6-α2, are consistently determined in the orthorhombic and tetragonal zfBGO structures, indicating that this region is relatively rigid and would not likely be displaced by binding of the GF (Fig. 4F). 3) Residues involved in the formation of the additional α-helix (α1) and β-strand (β8) in BG O-D1 are either conserved or invariant in species ranging from fish to mammals (Fig. S3B), consistent with these fulfilling the same role in blocking β6 as they do in zfBGO.
The structural modification above, together with the absence of any additional edge β-strands that could possibly form a super β–sheet with the GF, suggest that BGO engages TGF-β2 in an entirely different manner than ENGO engages BMP-9. To test this, binding studies were performed with full-length zfBGO and rBGO and an engineered TGF-β monomer that had the finger region and base of the fingers with the cystine knot intact, but lacked the heel helix as well as all of the second monomer. To further test this, binding studies were also performed with zfBGO constructs that included only subdomain 1, O-D1, or subdomain 2 O-D2 and intact TGF-β2 dimers. The results demonstrated weak binding, both for intact zfBGO and rBGO to the engineered TGF-β monomer, and both of the orphan subdomains to intact TGF-β2. This suggested that the orphan domain recognizes and binds to features beyond those present in the engineered monomer and that the BG orphan domain gains its high affinity by utilizing both of its subdomains to contact TGF-β2 dimers. This sets BGO apart from ENGO as the latter recognizes only the finger region of the GF and does so by utilizing only subdomain 1, O-D1. The alternative explanation, that weak binding is a consequence of an altered conformation or misfolding of the domain-deleted constructs, is unlikely as the engineered monomer was shown to bind TβRII through the fingertips with same affinities as native TGF-β dimers and NMR spectra clearly showed that both zfBG O-D1 and O-D2 were natively folded. Thus, binding studies with domain deleted constructs are consistent with the structure-based hypothesis that BGO will be unable to bind to TGF-β by formation of a super β-sheet as does ENGO, and as a consequence, must bind in an alternative manner.
The observations above, together with the previous observations that the orphan domain binds TGF-β dimers with 1:1 stoichiometry and in a manner that partially blocks binding of TβRI, but not TβRII, suggested that the orphan domain binds across the TGF-β dimer interface with at least one of its subdomains protruding into the TβRI site, but neither subdomain protruding into the TβRII site. This is supported by the SAXS data for the 1:2:1 TGF-β2DM:TβRII:rBGO complex, which was most consistent with models of the complex in which the orphan domain straddled the dimer interface and subdomains 1 and 2 both contacted the N-terminal and heel helices (α1 and α3, respectively). This manner of binding, in addition to accounting for the lack of competition against TβRII binding and competition against TβRI binding, had the pseudo-symmetry axis of the orphan domain near the symmetry axis of TGF-β, but not exactly coincident. This accounts for the 1:1 stoichiometry with which BGO binds TGF-β dimers as positioning of a second symmetry-related molecule is impossible due to steric overlap. There were notably alternative models that were nearly as consistent with the SAXS data, but were rotated around an axis passing through the centroid of orphan subdomains 1 and 2. To determine if the predicted modes of binding might involve regions of BGO with a high density of conserved surface exposed residues, the product of an amino acid conservation index and the solvent accessible surface (SAS) area was calculated and smoothed over a 10-residue window (Fig. S10). This highlighted three regions with high conservation/SAS index, residues 199-211, 249-255, and 308-322, the first and second of which had close contact with TGF-β in the first and second class of models. This provides further, albeit indirect support for the SAX-derived models, however even with this additional information, there is insufficient information to uniquely determine the positioning of the orphan domain on TGF-β2. There is, however, ample evidence to rule out 2:1 models with an ENGO-like manner of binding, as these are inconsistent with the SEC and SEC-MALS data, the analysis of the domain deleted forms of TGF-β and BGO, and the experimental SAXS data. Thus, it is concluded that BGO binds its cognate GF, TGF-β2, in completely different manner compared to ENGO and that the general features of the complex are likely similar to that shown in Figure 7G-H. However, because the docking algorithm used does not allow for flexibility of the backbone and because no effort was made to validate the interfaces, the actual structure, which must await the direct determination of the TGF-β:BGO complex using crystallography, could certainly differ from that presented in Fig. 7G-H.
The alternative manner by which evolutionary latecomers to the family, such as the TGF-βs, bind their type II receptor, TβRII, compared to other more ancestral GFs of the family, such as BMPs, GDFs, and activins (Allendorph et al., 2006; Greenwald et al., 2003; Groppe et al., 2008; Hart et al., 2002; Hinck, 2012; Kirsch et al., 2000; Radaev et al., 2010; Weber et al., 2007) has already shown how structurally homologous proteins of the TGF-β family can diverge from one another in terms of the interfaces they use to assemble into functional complexes. The alternative interfaces expand the range of receptor binding specificity and provide an effective means of segregating the binding, and thus function, of different subclasses of TGF-β family GFs from one another. The different modes of GF binding for BG and ENG are therefore not unprecedented. The different modes likely engender BG and ENG with the ability to selectively bind their cognate GFs, imparting them with the ability to selectively target the heart, liver, and anterior pituitary and vasculature, respectively, without interfering with one another or TGF-β family GFs.
Star methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew P. Hinck (ahinck@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The BL21(DE3) strain of E. coli was purchased from EMD-Millipore (Cat# 69450-3) and plasmids of interest used for protein expression that encode β-lactamase for antibiotic resistance were directly transformed into the competent cells provided and plated onto LB plates with 50 μg/mL carbenicillin. The resistant cells were cultured at 37 °C, both before and after the induction of protein expression, on liquid medium containing 50 μg/mL carbenicillin with shaking at 250 rpm in baffled 2.8 L Fernbach flasks.
The expi293 strain of HEK-293F (female) cells (Cat# A14527) and the corresponding expi293 growth medium (Cat# A14351-01) were purchased from Invitrogen. Cells were grown in baffled flasks with shaking (125 rpm) at 37 with 8% CO2. Cells are passaged every 3-4 days when the cells reach 5 × 106 cells/mL by diluting the cells in fresh media to 5 × 105 cells mL−1. Low passage stocks of 1 × 107 cells mL−1 were frozen back with 10% DMSO. For protein expression, the day before transfection cells were diluted to 2.5 × 106 cells mL−1 and allowed to continue growing for 20 – 24 h. The transfection solution was prepared in 1/10th the volume of the total desired expression growth. For the transfection solution, CsCl purified DNA (1.5 μg per mL total culture volume) was diluted with media to 1/20th the culture volume. Linear polyethylenimine MW 2500 (Polysciences Cat# 23966-1) (4.5ug per mL total volume) was diluted with media to 1/20th the culture volume. These two components were then combined and gently agitated. The transfection solution was allowed to incubate at room temperature for 30 min before being added to enough of the overnight cell culture to make 2.5 × 106 cells mL−1 of the desired volume. Medium was then added to adjust the volume to the desired amount. Cells were grown in the presence of the transfection solution for 14-16 hrs before protein production was stimulated by the addition of 2.2 mM valproic acid (Alfa Aesar Cat# A12962-18). Cells were allowed to continue to grow for 4 days and the conditioned media containing the desired protein was harvested.
METHOD DETAILS
Protein Preparation
Recombinant human TbRII extracellular domain and TGF-βs, including TGF-β2, TGF-β2DM, TGF-β2TM, mmTGF-β2-7M, avi-mmTGF-β2-7M, and TGF-β3, were expressed in E. coli at 37 °C in the form of insoluble inclusion bodies, refolded, and purified as described (Hinck et al., 2000; Huang and Hinck, 2016; Kim et al., 2017). Zebrafish and rat betaglycan constructs were expressed in transiently transfected suspension cultured human embryonic kidney (HEK) 293 expi cells (Invitrogen, Carlsbad, CA) and purified by metal affinity chromatography (NiNTA) and size exclusion chromatography (SEC) as described (Qin et al., 2016). Iodoacetamide treated zfBGO for crystallization was prepared by dialyzing NiNTA-purified protein into 25 mM sodium carbonate, 50 mM NaCl, pH 8.0, followed by the addition of freshly prepared iodoacetamide to a concentration of 20 mM. Following a 1 hr incubation at 25 °C, the reaction was quenched with ethanolamine and the protein was purified by SEC. Sequences, along with database references and short descriptions, are provided in Supporting Information (Table S1, S2).
SPR measurements
Minimally biotinylated TGF-β2 for SPR was prepared by reaction of 5 nmol TGF-β2 in 40% DMSO with 60 nmol ethyl-3-[3- dimethylaminopropyl]carbodiimide hydrochloride (EDC, Pierce, Rockford, IL), 120 nmol sulfo-N-hydroxysulfosuccinimide (Sulfo-NHS, Pierce, Rockford, IL), and 480 nmol (+)- biotinyl-3,6,9,- trioxaundecanediamine (EZ-Link Amine-PEG3-Biotin, Pierce, Rockford, IL) for 1 h at 25 °C at pH 6.0. Electrospray ionization time of flight mass spectrometry showed that roughly 10% of the growth factor was singly modified, while the remaining 90% was unmodified. Biotinylated avi-tagged mmTGF-β2-7m for SPR was prepared by enzymatic biotinylation with BirA biotin ligase as previously described (Kim et al., 2017).
SPR binding studies were performed with a BIAcore X100 instrument (GE Lifesciences, Piscataway, NJ) with biotinylated GFs captured onto neutravidin-coated CM-5 sensor chips (GE Lifesciences, Piscataway, NJ) at a density of 50 – 150 RU. Kinetic binding assays were performed by duplicate injections of the analytes in 20 mM HEPES, pH 7.0, 150 mM NaCl, 0.05% surfactant P20 (Pierce, Rockford, IL) at 100 uL min−1. Regeneration of the surface was achieved by a 30 sec injection of 4 M guanidine hydrochloride. Baseline correction was performed by subtracting the response both from the reference surface with no immobilized ligand and 5 – 10 blank buffer injections. Kinetic analyses were performed by fitting the results to either a simple 1:1 model with a single set of kinetic constants (ka, kd) using the program Scrubber (BioLogic Software, Canberra, Australia) or a 1:1 model with high and low affinity classes of immobilized ligands and two sets of kinetic constants (ka1, kd1 for binding to one class of immobilized ligand and ka2, kd2 for binding to the other class of immobilized ligand) using the program BIAevaluation (GE Lifesciences, Piscataway, NJ). Importantly the latter model was used only if the data could not be satisfactorily fit to the simpler model and was needed only in three instances, binding of zfBGZP-C, rBGZP-C, or zfBGO-D2 to TGF-β2.
SEC and SEC-MALS
Protein complexes were prepared by combining 1:2.5 TGF-β2DM:TβRII binary complex with a concentrated stock of full-length betaglycan or its component subdomain at the desired ratio. Samples for SEC and SEC-MALS were prepared in 25 mM HEPES, 150 mM arginine, pH 7.4, concentrated to 100 μL, and loaded onto a Superdex 200 Increase 10/300 GL column (GE Lifesciences, Piscataway, NJ) equilibrated in the same buffer. SEC measurements were made using an Akta FPLC (GE Lifesciences, Piscataway, NJ), while SEC-MALS measurements were made using a Waters high-performance liquid chromatography system (Waters, Milford, MA) and a Wyatt DAWN HELEOS-II multiangle light scattering detector and Optilab T-rEX refractive index detector (Wyatt, Santa Barbara, CA). SEC-MALS instrument control and data analyses were performed with the ASTRA software package (Wyatt, Santa Barbara, CA).
Crystallization, structure determination, and refinement
Native orthorhombic crystals were grown using hanging-drop vapor diffusion at 25 °C with drops consisting of 2.0 μL 9.2% v/v PEG 5K mono methyl ether, 4.6% w/v PEG 20K, 0.1M MES pH 6.2, 1.6 μL 20 mg mL−1 iodoacetamide-treated zfBGO in 10 mM MES pH 6.0, and 0.4 μL crushed zfBGO crystals (formed from non-iodoacetamide treated protein) as micro-seeds. For data collection, harvested crystals were mounted in nylon loops, cryo-protected in well solution containing 10% ethylene glycol, and flash-cooled in liquid nitrogen. Data were acquired at the Advanced Photon Source (APS) SER-CAT 22-ID and integrated and scaled using XDS (Kabsch, 2010). For phasing, platinum and iodide derivatized crystals were prepared by soaking the native crystals in crystallization well buffer with 5 mM K2PtCl4 or 4% saturated KI for 20 min and 20 s, respectively. For the platinum-derivatized crystals, data were collected at the APS SER-CAT 22-ID with the X-ray wavelength optimized for platinum anomalous scattering. Iodide-derivatized crystal data were collected at the Cu K-α wavelength with a Rigaku FR-E generator and RAXIS HTC image plate detector at the University of Pittsburgh Department of Structural Biology X-ray facility. Data reduction and processing of the derivative data sets were performed using iMOSFLM (Battye et al., 2011) and Pointless, Aimless, and Ctruncate (Collaborative Computational Project, 1994; Winn et al., 2011). MIRAS phasing to 2.80 Å in autoSHARP (Bricogne et al., 2003) yielded an overall figure of merit of 0.52 for acentric reflections and 0.49 for centric reflections. Solvent flattening yielded interpretable electron density, which after autobuilding with phenix.autobuild (Adams et al., 2010) and manual adjustment with COOT (Emsley et al., 2010), could be traced to build nearly the complete model for Chain A except for four loop regions with weak density (Table S3). Chain B had weaker density and additional breaks, especially in domain 2 (Table S3).
Native tetragonal crystals were grown using hanging-drop vapor diffusion at 25 °C with drops consisting of 2 μL 8% v/v PEG 5K mono methyl ether, 4% w/v PEG 20K, 0.1M MES pH 6.5, 1.6 μL 20 mg mL−1 non-iodoacetamide treated protein, and 0.4 μL micro-seeds (formed from non-iodoacetamide treated protein). For data collection, harvested crystals were mounted in nylon loops, cryo-protected in well solution containing 10% ethylene glycol, and flash-cooled in liquid nitrogen. Data were acquired at the Advanced Photon Source (APS) SER-CAT 22-ID and integrated and scaled using XDS (Kabsch, 2010). For phasing, structure model A from the asymmetric unit of the orthorhombic crystal form was used as a search model for molecular replacement in Molrep (Vagin and Teplyakov, 2010).
Coordinates were refined using Buster (Bricogne G. and Roversi P, 2018) with TLS (Schomaker and Trueblood, 1968), alternated with manual rebuilding in COOT (Emsley et al., 2010). TLS groups were identified with the TLSMD server (Painter and Merritt, 2006) and corresponded to most of domain 1 (residues 233-360), all of domain 2 (residues 58-215), and the extended segments that exit (residues 29-48) and re-enter domain 1 (residues 216-232) from domain 2. The refinement procedure was supported/accompanied by analysis of composite omit maps (Terwilliger et al., 2008). X-ray sources, data collection and refinement statistics are provided in Table 2. Figures were prepared using UCSF Chimera (Pettersen et al., 2004), except for Figs. 4G and S4 which was prepared using PyMOL (DeLano).
NMR spectroscopy
Natural abundance 1H-13C HSQC spectra of the methyl bearing residues of BGO-D1 and BGO-D2 were collected by preparing samples at 3 - 4 mg mL−1 in 25 mM sodium phosphate, 50 mM NaCl, pH 7.0 and by recording spectra at 30°C using a Bruker AVI 700 or AV-II 900 MHz spectrometer equipped with a 5 mm 1H{13C,15N} TCI cryogenically cooled probe (Bruker Biospin, Billerica, MA).
Small angle X-ray scattering
Experimental SAXS data of zfBGO and rBGO, both alone and as SEC-purified complexes with either TβRII:TGF-β2TM or TβRII:TGF-β3, were collected at APS beamline 12-ID-B. Samples were prepared at 1.0, 2.0, and 4.0 mg mL−1 in 20 mM CHES, 100 mM NaCl, pH 9.5. Scattering data was recorded with sample oscillation as 30 2 sec exposures with a 1 sec delay between exposures. Raw scattering data were radially averaged, and after removing outliers, the data were averaged and subtracted from a buffer alone reference using the program PRIMUS (Konarev et al., 2003). Data corresponding to different concentrations were scaled, and if shown to not exhibit any systematic deviation over the 0 < q < 0.5 range, were merged using PRIMUS (Konarev et al., 2003). Solution scattering curves were calculated from PDB coordinate files and fit to the concentration merged curves using the program CRYSOL (Svergun et al., 1995). Electron density envelopes were calculated from the merged scattering curves using the program DENSS (Grant, 2018). Models of ratBGO bound to the 1:2 TGF-β3:TβRII complex were generated using the program pyDockSAXS (Jimenez-Garcia et al., 2015), with the merged scattering curve for the 1:2:1 TGF-β3:TβRII:rBGO complex, the structure of the 1:2 TGF-β3:TβRII complex (extracted from the structure of the TGF-β3:TβRII:TβRI complex, PDB 2PJY (Groppe et al., 2008)), and a homology model of rBGO (built based on the zfBGO structure) as input.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistics of the X-ray crystallographic data processing, phasing, refinement, and structure validation are summarized in Table 2. The quality of the fit of the experimental SAXS data to that calculated from the corresponding structures was determined using the program CRYSOL, and the molecular weights and corresponding errors estimated from the SEC-MALS measurements were determined using the ASTRA software package (Wyatt, Santa Barbara, CA). The quality of the fit of the experimental SPR data to the kinetic constants and Rmax was determined using either Scrubber (for data fit to a 1:1 binding model) or Biaeval (for data fit to a 1:1 heterogenous ligand model).
DATA AND CODEAVAILABILITY
The accession number for the structure factor and coordinate files of zfBGO reported in this paper are Protein Data Bank (PDB) 6MZP and 6MZN (orthorhombic and tetragonal forms, respectively).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | EMD-Millipore | Cat# 69450-3 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| sulfo-N-hydroxysulfosuccinimide | Thermo | Cat# 24510 |
| Ethyl-3-[3- dimethylaminopropyl]carbodiimide hydrochloride | Thermo | Cat# 22980 |
| (+)- biotinyl-3,6,9,- trioxaundecanediamine | Thermo | Cat# 21347 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Structure of TGF-β3:TβRII:TβRI | Groppe et al., 2008 | PDB:2PJY |
| Structure of mmTGF-β27M:TβRII | Kim et al., 2017 | PDB:5TX4 |
| Structure of ENGO | Saito et al., 2017 | PDB:5I04 |
| Structure of ENGO:BMP-9 | Saito et al., 2017 | PDB:5HZW |
| Experimental Models: Cell Lines | ||
| Human: Expi293F | Invitrogen | Cat# A14527 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA1 | ||
| pET32a-TGF-β2 | De Crescenzo et al., 2006 | Hincklab #225 |
| pET32a-TGF-β2DM | Baardsnes, et al., 2009 | Hincklab #253 |
| pET32a-TGF-β2TM | De Crescenzo et al., 2006 | Hincklab #225 |
| pET32a-mmTGF-β27M | Kim et al., 2017 | Hincklab #267 |
| pET32a-avi-mmTGF-β27M | Kim et al., 2017 | Hincklab #273 |
| pET32a-TGF-β3 | Groppe et al., 2008 | Hincklab #27 |
| pET32a-TβRII | Hinck et al., 2000 | Hincklab #249 |
| pcDNA3.1+− zfBGO-ZP | This study | Hincklab #305 |
| pcDNA3.1+− zfBGO | This study | Hincklab #309 |
| pcDNA3.1+− zfBGZP-C | This study | Hincklab #310 |
| pcDNA3.1+− zfBGO-D1 | This study | Hincklab #391 |
| pcDNA3.1+− zfBGO-D2 | This study | Hincklab #392 |
| pcDNA3.1+− rBGO-ZP | Villarreal et al., 2016 | Hincklab #276 |
| pcDNA3.1+− rBGO | Villarreal et al., 2016 | Hincklab #281 |
| pcDNA3.1+− rBGZP-C | Villarreal et al., 2016 | Hincklab #282 |
| Software and Algorithms | ||
| XDS | Kabsch, 2010 | https://strucbio.biologie.uni-konstanz.de/xdswiki/index.php/Main_Page |
| iMOSFLM | Battye et al., 2011 | https://www.mrc-lmb.cam.ac.uk/harry/imosflm/ver722/introduction.html |
| Pointless | Evans, 2006; Evans, 2011 | http://www.ccp4.ac.uk/dist/html/pointless.html |
| Aimless | Evans, 2006; Evans, 2011 | http://www.ccp4.ac.uk/dist/html/aimless.html |
| Ctruncate | Evans, 2006; Evans, 2011 | http://www.ccp4.ac.uk/html/ctruncate.html |
| MolRep | Vagin and Teplyakov, 2010 | http://www.ccp4.ac.uk/html/molrep.html |
| autoSHARP | Bricogne et al., 2003 | https://www.globalphasing.com/sharp/ |
| Phenix.autobuild | Adams et al., 2010 | https://www.phenix-online.org/documentation/reference/autobuild.html |
| COOT | Emsley, et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Buster | Bricogne G. and Roversi P, 2018 | https://www.globalphasing.com/buster/ |
| FFT | Ten Eyck, 1973 | http://www.ccp4.ac.uk/dist/html/fft.html |
| TLSMD | Painter and Merritt, 2006 | http://skuld.bmsc.washington.edu/~tlsmd/ |
| PDBePISA | Krissinel and Henrick, 2007 | https://www.ebi.ac.uk/pdbe/pisa/ |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Pymol | Schrodinger LLC | http://www.pymol.org |
| PRIMUS | Konarev et al., 2003 | https://www.embl-hamburg.de/biosaxs/primus.html |
| CRYSOL | Svergun et al., 1995 | https://www.embl-hamburg.de/biosaxs/crysol.html |
| DENSS | Grant, 2018 | https://denss.ccr.buffalo.edu |
| pyDockSAXS | Jimenez-Garcia et al., 2015 | https://life.bsc.es/pid/pydocksaxs |
| Scrubber | BioLogic Software | http://www.biologic.com.au/scrubber.html |
| BiaEval | Biacore | https://www.biacore.com/lifesciences/index.html |
| ASTRA 6.0 | Wyatt Technology | https://www.wyatt.com/products/software/astra.html |
| Jalview | Waterhouse et al., 2009 | http://www.jalview.org |
| Clustal Omega | Sievers et al., 2011 | http://www.clustal.org |
| Other | ||
| CM-5 SPR sensor chip | GE Lifesciences | Cat# 29149604 |
| Ni-NTA agarose | Qiagen | Cat# 30210 |
| Superdex 200 16/60 SEC Column | GE Lifesciences | Cat# 17-1069-01 |
| Superdex 200 Increase 10/300 GL SEC Column | GE Lifesciences | Cat# 28-9909-44 |
Highlights.
Structure of the betaglycan orphan domain, BGO, was determined by crystallography
Structure reveals an insertion compared to the endoglin orphan domain, ENGO
Insertion blocks the edge β-strand used in the ENGO to bind BMP-9 or BMP-10
BGO binds TGF-β in a different manner than ENGO binds BMP-9 or BMP-10
Acknowledgements
The authors would like to thank Doowon Lee for assistance with the X-ray instrumentation, Mike Delk for assistance with the NMR instrumentation, Drs. Lixin Fan and Xiaobing Zuo for assistance with SAXS data collection, Drs. Christopher Barnes and Guillermo Calero for their assistance with structure determination of rBGO, and Dr. Łukasz Wieteska for providing valuable comments on the manuscript. This research was supported by the NIH (GM58670 and CA172886), the U.S. Department of Defense (DoD W81XWH-17-1-0429), and the University of Pittsburgh Vascular Medicine Institute. S.K.K. was supported by training grants provided by CPRIT (RP1450105) and the AHA (15PRE25550015) and F.L-C. is supported by CONACYT (254046). X-ray diffraction data were collected at the SER-CAT 22-ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory (ANL). SER-CAT is supported by its member institutions and equipment grants (RR25528 and RR028976) from the NIH. SAXS data was collected through the SAXS core facility of Center for Cancer Research, National Cancer Institute (NCI) which utilized ANL APS beamline 12-ID-B under PUP-24152. Molecular graphics and analyses were performed with UCSF Chimera, which is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco and supported by NIGMS P41-GM103311. The X-ray Crystallography Core Laboratory at UT Health Science Center at San Antonio is supported by the Office of the Vice President for Research and the Mays Cancer Center, the center home to the UT Health San Antonio MD Anderson Cancer Center (NIH P30 CA054174).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorph GP, Vale WW, and Choe S (2006). Structure of the ternary signaling complex of a TGF-beta superfamily member. Proc Natl Acad Sci U S A 103, 7643–7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt A, Miguel-Romero L, Donderis J, Aristorena M, Blanco FJ, Round A, Rubio V, Bernabeu C, and Marina A (2012). Structural and functional insights into endoglin ligand recognition and binding. PLoS One 7, e29948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baardsnes J, Hinck CS, Hinck AP, and O'Connor-McCourt MD (2009). TbetaR-II discriminates the high- and low-affinity TGF-beta isoforms via two hydrogen-bonded ion pairs. Biochemistry 48, 2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard DJ, Chapman SC, and Woodruff TK (2002). Inhibin binding protein (InhBP/p120), betaglycan, and the continuing search for the inhibin receptor. Mol Endocrinol 16, 207–212. [DOI] [PubMed] [Google Scholar]
- Bier E, and De Robertis EM (2015). EMBRYO DEVELOPMENT. BMP gradients: A paradigm for morphogen-mediated developmental patterning. Science 348, aaa5838. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Church RH, Surae S, Godson C, and Martin F (2015). BMP signalling: agony and antagony in the family. Trends Cell Biol 25, 249–264. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Vonrhein C, Flensburg C, Schiltz M, and Paciorek W (2003). Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr 59, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Bricogne G, B.E., Brandl M, Flensburg C, Keller P, Paciorek W,, and Roversi P, S.A., Smart OS, Vonrhein C, Womack TO (2018). BUSTER version 2.10.2 (Cambridge, United Kingdom, Global Phasing Ltd.). [Google Scholar]
- Brown CB, Boyer AS, Runyan RB, and Barnett JV (1999). Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science 283, 2080–2082. [DOI] [PubMed] [Google Scholar]
- Brown MA, Zhao Q, Baker KA, Naik C, Chen C, Pukac L, Singh M, Tsareva T, Parice Y, Mahoney A, et al. (2005). Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J Biol Chem 280, 25111–25118. [DOI] [PubMed] [Google Scholar]
- Castonguay R, Werner ED, Matthews RG, Presman E, Mulivor AW, Solban N, Sako D, Pearsall RS, Underwood KW, Seehra J, et al. (2011). Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem 286, 30034–30046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S, Hernandez H, Laiho M, ten Dijke P, Iwata KK, and Massague J (1990). Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J Biol Chem 265, 20533–20538. [PubMed] [Google Scholar]
- Chen YG, Hata A, Lo RS, Wotton D, Shi Y, Pavletich N, and Massague J (1998). Determinants of specificity in TGF-beta signal transduction. Genes Dev 12, 2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cull MG, and Schatz PJ (2000). Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol 326, 430–440. [DOI] [PubMed] [Google Scholar]
- De Crescenzo G, Hinck CS, Shu Z, Zuniga J, Yang J, Tang Y, Baardsnes J, Mendoza V, Sun L, Lopez-Casillas F, et al. (2006). Three key residues underlie the differential affinity of the TGFbeta isoforms for the TGFbeta type II receptor. J Mol Biol 355, 47–62. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2019). The PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC.). [Google Scholar]
- Diestel U, Resch M, Meinhardt K, Weiler S, Hellmann TV, Mueller TD, Nickel J, Eichler J, and Muller YA (2013). Identification of a Novel TGF-beta-Binding Site in the Zona Pellucida C-terminal (ZP-C) Domain of TGF-beta-Receptor-3 (TGFR-3). PLoS One 8, e67214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza-Lopez J, Montiel JL, Vilchis-Landeros MM, Okadome T, Miyazono K, and Lopez-Casillas F (2001). Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. J Biol Chem 276, 14588–14596. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, and Ten Dijke P (2018). TGF-beta Signaling in Control of Cardiovascular Function. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Zwijsen A, Ten Dijke P, and Bailly S (2018). Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant TD (2018). Ab initio electron density determination directly from solution scattering data. Nat Methods 15, 191–193. [DOI] [PubMed] [Google Scholar]
- Greenwald J, Groppe J, Gray P, Wiater E, Kwiatkowski W, Vale W, and Choe S (2003). The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell 11, 605–617. [DOI] [PubMed] [Google Scholar]
- Groppe J, Hinck CS, Samavarchi-Tehrani P, Zubieta C, Schuermann JP, Taylor AB, Schwarz PM, Wrana JL, and Hinck AP (2008). Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell 29, 157–168. [DOI] [PubMed] [Google Scholar]
- Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, and Hinck AP (2002). Crystal structure of the human TbetaR2 ectodomain--TGF-beta3 complex. Nat Struct Biol 9, 203–208. [DOI] [PubMed] [Google Scholar]
- Hata A, and Chen YG (2016). TGF-beta Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henen MA, Mahlawat P, Zwieb C, Kodali RB, Hinck CS, Hanna RD, Krzysiak TC, Ilangovan U, Cano KE, Hinck G, et al. (2018). TGF-beta2 uses the concave surface of its extended finger region to bind betaglycan's ZP domain via three residues specific to TGF-beta and Inhibin-alpha. J Biol Chem in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck AP (2012). Structural studies of the TGF-betas and their receptors - insights into evolution of the TGF-beta superfamily. FEBS Lett 586, 1860–1870. [DOI] [PubMed] [Google Scholar]
- Hinck AP, Mueller TD, and Springer TA (2016). Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck AP, Walker KP 3rd, Martin NR, Deep S, Hinck CS, and Freedberg DI (2000). Sequential resonance assignments of the extracellular ligand binding domain of the human TGF-beta type II receptor. J Biomol NMR 18, 369–370. [DOI] [PubMed] [Google Scholar]
- Huang T, and Hinck AP (2016). Production, Isolation, and Structural Analysis of Ligands and Receptors of the TGF-beta Superfamily. Methods Mol Biol 1344, 63–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Garcia B, Pons C, Svergun DI, Bernado P, and Fernandez-Recio J (2015). pyDockSAXS: protein-protein complex structure by SAXS and computational docking. Nucleic Acids Res 43, W356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamaid A, Molina-Villa T, Mendoza V, Pujades C, Maldonado E, Ispizua Belmonte JC, and Lopez-Casillas F (2015). Betaglycan knock-down causes embryonic angiogenesis defects in zebrafish. Genesis 53, 583–603. [DOI] [PubMed] [Google Scholar]
- Kim SK, Barron L, Hinck CS, Petrunak EM, Cano KE, Thangirala A, Iskra B, Brothers M, Vonberg M, Leal B, et al. (2017). An engineered transforming growth factor beta (TGF-beta) monomer that functions as a dominant negative to block TGF-beta signaling. J Biol Chem 292, 7173–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T, Sebald W, and Dreyer MK (2000). Crystal structure of the BMP-2-BRIA ectodomain complex. Nat Struct Biol 7, 492–496. [DOI] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, and Svergun DI (2003). PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36, 1277–1282. [Google Scholar]
- Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, and Vale W (2000). Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404, 411–414. [DOI] [PubMed] [Google Scholar]
- Li Y, Fortin J, Ongaro L, Zhou X, Boehm U, Schneyer A, Bernard DJ, and Lin HY (2018). Betaglycan (TGFBR3) Functions as an Inhibin A, but Not Inhibin B, Coreceptor in Pituitary Gonadotrope Cells in Mice. Endocrinology 159, 4077–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Hu Y, Zhu J, Woodruff TK, and Jardetzky TS (2011). Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-beta binding. Proceedings of the National Academy of Sciences of the United States of America 108, 5232–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F, Wrana JL, and Massague J (1993). Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73, 1435–1444. [DOI] [PubMed] [Google Scholar]
- Mendoza V, Vilchis-Landeros MM, Mendoza-Hernandez G, Huang T, Villarreal MM, Hinck AP, Lopez-Casillas F, and Montiel JL (2009). Betaglycan has two independent domains required for high affinity TGF-beta binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 48, 11755–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, and Wrana JL (2017). TGF-beta Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel J, Ten Dijke P, and Mueller TD (2018). TGF-beta family co-receptor function and signaling. Acta Biochim Biophys Sin (Shanghai) 50, 12–36. [DOI] [PubMed] [Google Scholar]
- Nolan K, and Thompson TB (2014). The DAN family: modulators of TGF-beta signaling and beyond. Protein Sci 23, 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J, and Merritt EA (2006). Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62, 439–450. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qin T, Barron L, Xia L, Huang H, Villarreal MM, Zwaagstra J, Collins C, Yang J, Zwieb C, Kodali R, et al. (2016). A novel highly potent trivalent TGF-beta receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget 7, 86087–86102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaev S, Zou Z, Huang T, Lafer EM, Hinck AP, and Sun PD (2010). Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J Biol Chem 285, 14806–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson IB, and Rifkin DB (2013). Unchaining the beast; insights from structural and evolutionary studies on TGFbeta secretion, sequestration, and activation. Cytokine Growth Factor Rev 24, 355–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman BL, and Hinck AP (2017). ALK1 signaling in development and disease: new paradigms. Cell Mol Life Sci 74, 4539–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Bokhove M, Croci R, Zamora-Caballero S, Han L, Letarte M, de Sanctis D, and Jovine L (2017). Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep 19, 1917–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, and Doetschman T (1997). TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, and ten Dijke P (2007). BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci 120, 964–972. [DOI] [PubMed] [Google Scholar]
- Schomaker V, and Trueblood KN (1968). On the rigid-body motion of molecules in crystals. Acta Crystallogr B24, 63–76. [Google Scholar]
- Stenvers KL, Tursky ML, Harder KW, Kountouri N, Amatayakul-Chantler S, Grail D, Small C, Weinberg RA, Sizeland AM, and Zhu HJ (2003). Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol Cell Biol 23, 4371–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, and Koch MHJ (1995). CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28, 768–773. [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, and Hung LW (2008). Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr D Biol Crystallogr 64, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TB, Woodruff TK, and Jardetzky TS (2003). Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-beta ligand:receptor interactions. EMBO J 22, 1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, et al. (2012). Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem 287, 27313–27325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay A, Moss-Taylor L, Kim MJ, Ghosh AC, and O'Connor MB (2017). TGF-beta Family Signaling in Drosophila. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, and Teplyakov A (2010). Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Villarreal MM, Kim SK, Barron L, Kodali R, Baardsnes J, Hinck CS, Krzysiak TC, Henen MA, Pakhomova O, Mendoza V, et al. (2016). Binding Properties of the Transforming Growth Factor-beta Coreceptor Betaglycan: Proposed Mechanism for Potentiation of Receptor Complex Assembly and Signaling. Biochemistry 55, 6880–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RG, Poggioli T, Katsimpardi L, Buchanan SM, Oh J, Wattrus S, Heidecker B, Fong YW, Rubin LL, Ganz P, et al. (2016). Biochemistry and Biology of GDF11 and Myostatin: Similarities, Differences, and Questions for Future Investigation. Circ Res 118, 1125–1141; discussion 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber D, Kotzsch A, Nickel J, Harth S, Seher A, Mueller U, Sebald W, and Mueller TD (2007). A silent H-bond can be mutationally activated for high-affinity interaction of BMP-2 and activin type IIB receptor. BMC Struct Biol 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiater E, Harrison CA, Lewis KA, Gray PC, and Vale WW (2006). Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actions. J Biol Chem 281, 17011–17022. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, and Massague J (1994). Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Zinski J, Tajer B, and Mullins MC (2018). TGF-beta Family Signaling in Early Vertebrate Development. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.