

SUMMARY
The unique membrane composition of cilia is maintained by a diffusion barrier at the transition zone that is breached when the BBSome escorts signaling receptors out of cilia. Understanding how the BBSome removes proteins from cilia has been hampered by a lack of structural information. Here, we present a nearly complete Cα model of BBSome purified from cow retina. The model is based on a single-particle cryo-electron microscopy density map at 4.9-Å resolution that was interpreted with the help of comprehensive Rosetta-based structural modeling constrained by cross-linking mass-spectrometry data. We find that BBSome subunits have a very high degree of interconnectivity, explaining the obligate nature of the complex. Furthermore, like other coat adaptors, the BBSome exists in auto-inhibited state in solution and must thus undergo a conformational change upon recruitment to membranes by the small GTPase ARL6/BBS3. Our model provides the first detailed view of the machinery enabling ciliary exit.
Graphical Abstract
eTOC BLURB
The BBSome transports activated signaling receptors across the transition zone, a diffusion barrier that separates the ciliary from the plasma membrane. Chou et al. combined cryo-electron microscopy with structural modeling to deduce an almost complete Ca model for the BBSome and show that it is autoinhibited in solution.
INTRODUCTION
Cilia function as hubs for phototransduction, olfaction, Hedgehog and other signaling pathways by dynamically concentrating signaling receptors and transducers into a compartment with a distinct composition of lipids and second messengers (Bangs and Anderson, 2017; Garcia et al., 2018; Nachury and Mick, 2019). To maintain their protein composition, cilia rely on the transition zone (TZ), a diffusion barrier that functionally separates ciliary and plasma membranes (Garcia-Gonzalo and Reiter, 2017). Unlike most other diffusion barriers, the TZ can be crossed by select membrane proteins that enter or exit cilia, often in a regulated manner. For example, Hedgehog signal transduction requires that upon pathway engagement the G protein-coupled receptor (GPCR) GPR161 exits cilia and that the GPCR Smoothened (SMO) accumulates within cilia (Bangs and Anderson 2017). A major unsolved question is how ciliary signaling receptors cross the TZ.
A first clue comes from studies of Bardet-Biedl Syndrome (BBS), a ciliopathy characterized by obesity, retinal degeneration, polydactyly and kidney cysts. The BBSome, an evolutionarily conserved complex of eight BBS proteins ferries GPR161, SMO and other ciliary membrane proteins across the TZ and out of cilia (Nachury, 2018; Wingfield et al., 2017). The BBSome acts in concert with another conserved BBS gene product, the ARF-like GTPase ARL6/BBS3, which recruits the BBSome to membranes (Jin et al., 2010) and enables TZ crossing of GPR161 (Ye et al., 2018). The BBSome also associates with intraflagellar transport (IFT) trains comprised of microtubule motors, IFT-A and IFT-B complexes. IFT trains undergo processive intraciliary transport and the BBSome functions as an adaptor complex between IFT complexes and cargoes (Nachury, 2018; Wingfield et al., 2018). First, the BBSome directly recognizes cytoplasmic determinants on signaling receptors such as SMO and GPR161 (Klink et al., 2017; Ye et al., 2018) and is required for the intraciliary movements of the cargo phospholipase D in Chlamydomonas (Liu and Lechtreck, 2018). Second, BBSome subunits consist mostly of domains characteristic of coat adaptors (α-solenoids, β-propellers and appendages, Fig. 1A). Finally, the polymerization of a membrane-apposed BBSome/ARL6 coat (Jin et al., 2010) is reminiscent of the clathrin coat adaptor AP-2, which polymerizes onto membranes (Elkhatib et al., 2017; Hinrichsen et al., 2006).
Figure 1. BBSome subunits and cryo-EM density map of the BBSome.

A. Domain organization of the eight BBSome subunits. The 29 domains making up the BBSome subunits are 4 β-propellers (BBS1/2/7/9), one 4-helix bundle inserted into a β-propeller (BBS1), 4 connector helices between the β-propellers and the GAE domains (some of which are predicted to form coiled coils) (BBS1/2/7/9), 4 GAE domains (BBS1/2/7/9), 3 platform domains (BBS2/7/9), 3 hairpins (BBS2/7/9), 3 helical bundles (BBS2/7/9), 3 α-solenoids (BBS4/8TPR1-2/8TPR3-13), 2 PH domains (BBS5), one 3-helix bundle (BBS5), and one helical micropeptide (BBS18). B. Silver-stained 4-12% SDS-PAGE gel of the BBSome purified from retinal extract. C. Density map of the BBSome at 4.9-Å resolution obtained by single-particle cryo-EM (Map 1), showing a prominent helical bundle that is located in between the base and the top hemisphere.
A major open question is how the BBSome in complex with GTP-bound ARL6 (ARL6GTP) enables cargo exit from cilia (Nachury and Mick, 2019). A better understanding of the molecular architecture of the BBSome may provide some answers to this question. While some progress has been made in determining the high-resolution structure of individual BBSome subunits (Knockenhauer and Schwartz, 2015; Mourão et al., 2016) and in analyzing the subunit organization of BBSome sub-complexes (Klink et al., 2017), structural information on the intact complex is currently lacking.
Here, we combine cryo-electron microscopy (cryo-EM) and single-particle reconstruction with comprehensive Rosetta-based modeling to arrive at a near-complete Cα model of the BBSome. Unexpectedly, the map of the BBSome shows that its predominant conformation does not permit binding of ARL6GTP. We conclude that the BBSome, like the coat adaptor complexes AP-1, AP-2 and COPI, exists mostly in an auto-inhibited, closed conformation in solution and becomes activated as it is recruited to membranes.
RESULTS AND DISCUSSION
BBSome purification and structure determination
Initial attempts at recombinant expression of the BBSome in insect cells produced an unstable complex with BBS2 and BBS7 readily dissociating from the remainder of the complex, consistent with a previous report (Klink et al., 2017). To study the fully assembled BBSome, we modified our previous purification of native BBSome from retinal extract (Jin et al., 2010) and recovered 50 μg of nearly pure BBSome from 100 g of bovine retina in two chromatographic steps (Fig. 1B, Fig. S1A-C). Purity was assessed by mass spectrometry, which detected the 8 BBSome subunits (Fig. S1D) and only trace amounts of common contaminants. We then vitrified the BBSome sample, imaged it by cryo-EM (Fig. S2A), selected ~600,000 particles and subjected them to image processing in Relion (Scheres, 2015) (Fig. S2B,C). 3D classification revealed that the BBSome adopts different conformations, and we refined the major class (Fig. S2C, Map 1) to a resolution of 4.9 Å (Fig. 1C and S2D,E).
The BBSome has a compact morphology with dimensions of 200 × 155 × 115 Å that comprises a conical base, a well-defined helical bundle at its meridian, and a multi-headed top hemisphere. At this resolution, individual domains (α-solenoids, β-propellers and appendages) were well-defined and showed clear densities for helices and sheets (Fig. 1C). Three β-propellers and a prominent coiled coil were readily identified in the top hemisphere. The 4-helix bundle at the center was composed of two long hairpins. The base featured one β-propeller surrounded by two superhelical α-solenoids and another distinct domain.
Assignment of BBSome domains to densities in the cryo-EM map
The recurrence of α-solenoids, β-propellers and appendage domains within BBSome subunits posed considerable challenges for subunit assignment (Fig. 1A). BBS2, 7 and 9 share an identical domain organization consisting of a β-propeller (²prop) followed by a coiled coil (cc), an appendage domain subdivided into a γ-adaptin ear (GAE) domain and a platform (pf) domain, and a C-terminal helical bundle (CtH); and BBS1 is very similar but truncated after the GAE domain. The crystal structures of BBS1βprop and BBS9βprop have revealed that these two β-propellers are more closely related to one another than to any other β-propeller in the Protein Data Bank (PDB) (Knockenhauer and Schwartz, 2015; Mourão et al., 2014). Meanwhile, both BBS4 and BBS8 consist of twelve tetratricopeptide (TPR) repeats. Individual TPR repeats are known to fold into a stacked pair of helices, and stacking of TPR repeats results in the formation of a right-handed superhelix. BBS5 is composed of two pleckstrin homology (PH) domains followed by a small C-terminal 3-helix bundle (Nachury et al., 2007), while the micropeptide BBS18 is predicted to be a mixture of α-helices and unstructured regions (Loktev et al., 2008).
Altogether, 29 domains distributed in 8 proteins spanning 4,375 aa had to be located (Fig. 1A). Since the resolution was not sufficient to identify residues, a first approach used differences in 3D classes, known binary interactions, and structural predictions to manually assign each domain to a specific region in the map. The entire process of subunit assignment is depicted in Movie S1 and detailed below. Binary interactions were collected from three published datasets: visual immunoprecipitation (VIP) (Katoh et al., 2015; Nozaki et al., 2018), co-immunoprecipitations (co-IP) (Zhang et al., 2013), and systematic yeast-two hybrid (Y2H) experiments (Woodsmith et al., 2017) (Figs. 2A and S3A,B, Table S1). In addition, we subjected the BBSome to chemical crosslinking/mass spectrometry (XLMS) and identified 42 inter-subunit and 34 intra-subunit crosslinks with high confidence (Fig. 2B and Table S2).
Figure 2. Manual assignment of BBSome subunit domains to densities in the cryo-EM map.

A. Diagram summarizing the binary interaction studies. Interactions were identified by yeast-two hybrid (YTH, Fig. S3B), visual immunoprecipitation (VIP), and co-immunoprecipitation (co-IP) experiments. Individual datasets are depicted in Fig. S3A. Domains are boxed when interactions were assigned to specific fragment of a given subunit. Each subunit has a different color, and domains within a subunit are shown in lighter color shades from N to C termini. The same color scheme is used in all panels. B. Inter-subunit crosslinks identified by mass spectrometry mapped onto the subunits. Each subunit is drawn to the scale of its length. The numbers identify the subunit and the superscripts denote the specific domain: βprop, β-propeller; cc, coiled coil; GAE, γ-adaptin ear; pf, platform; hp, hairpin; CtH, C-terminal helix bundle; ins, insert; link, linker; PH, pleckstrin homology. C. Upper panels: The same three views presented in Fig. 1C are shown after manual segmentation. Domains are labeled as in B. Lowerpanel:The BBSome is shown segregated into its three main structural components, the top lobe consisting of BBS2 and BBS7, the corkscrew consisting of BBS4, BBS18 and the β-propeller of BBS1, and the base, which is shown as BBS8 and the assembly of BBS5, BBS9 and the linker and GAE domains of BBS1. D. Binary interactions mapped onto flattened views of the segmented map that were used to manually assign domains of BBSome subunits to specific densities in the cryo-EM map. The symbols correspond to those used in A. F. Angular distribution for BBSome projections.
We first noticed that one 3D class (Fig. S2C, Map 2) lacked a region that made extensive contacts with the β-propeller in the base (Fig. S3C). Upon segmentation of Map 1 (Fig. 2C, upper panels), this region did not show features of α-solenoids, β-propellers or appendage domains but was instead a good fit for the two PH domains of BBS5. The identification of BBS5 made it possible to assign the closely interacting BBS9βprop based on the BBS9-BBS5 interaction (Figs. 2A). Similarly, the interaction between BBS9βprop and BBS8TPR1-6 (Figs. 2A) identified the α-solenoid ensconced within the base as BBS8 and instructed the polarity of BBS8. By exclusion, the more peripheral α-solenoid that connects the base to the top lobe is BBS4. Superhelical solenoids wrap around unstructured and helical ligands (Zeytuni and Zarivach, 2012), and a partially helical density snaking inside the BBS4 and BBS8 superhelices was thus assigned to the micropeptide BBS18, based on its ability to bridge BBS4TPR8-11 to BBS8 (Figs. 2A). We next queried the composition of the central helical bundle. Removing the densities of BBS4, BBS8 and BBS18 revealed that BBS9βprop connects to an appendage domain followed by a ~100 amino acid-long hairpin (hp) and a C-terminal 5-helix bundle. A nearly symmetrical structure was found on the other side of the bundle with a long hairpin connected to a 3-helix bundle and a characteristic platform region. Considering that BBS2 was the only remaining subunit with a straight hairpin over 90 aa long and that the C termini of BBS2 and BBS9 interact (Figs. 2A), we concluded that the central helical bundle is composed of BBS2hp and BBS9hp and we located BBS2pf and BBS2CtH on the opposite side from BBS9pf and BBS9CtH. While BBS2pf is connected to a well-defined density, that density is too large to consist solely of BBS2GAE. The association between BBS2 and BBS7 through their GAE and coiled-coil domains (Figs. 2A) suggested that the density in question contains both BBS2GAE and BBS7GAE. As segmentation of this density failed to yield two separate domains, we propose that BBS2GAE and BBS7GAE combine into a single structural domain. Next, BBS1Pβprop was identified because it is the sole β-propeller with a large insertion. BBS7βprop was assigned based on its interaction with BBS1βprop, and BBS2βprop was identified as the last remaining β-propeller. The β-propellers of BBS2 and BBS7 are each connected to a coiled-coil helix ending at the mixed GAE of BBS2/7. The last unassigned domain was identified as BBS1GAE, in agreement with its reported interaction with the C terminus of BBS9 (Figs. 2A). While the helix that connects BBS1GAE to BBS1βprop was resolved, the remainder of the linker was absent from the map. Consisting of 68 amino acids, this linker would span more than 200 nm if entirely unstructured and can readily span the observed gap between the C terminus of BBS1βprop and the N terminus of BBS1GAE. The only other domain missing from our map is the 3-helix bundle at the C terminus of BBS5.
As predicted by binary interaction studies (Katoh et al., 2015; Nozaki et al., 2018; Woodsmith et al., 2017; Zhang et al., 2012a), a striking feature of the BBSome is the high degree of interconnectivity between subunits, which accounts for the obligate nature of the complex and contrasts with other coat adaptors. In all other instances in which appendage domains have been described (COP-I, AP-1/2/3/4/5), they are connected to a trunk by flexible linkers. In contrast, appendages of BBS1/2/7/9 are tightly connected to other architectural elements inside the BBSome, thus illustrating the modularity in organization of sorting complexes.
The subunit domain assignment was consistent with nearly all known binary interactions (Fig. 2D, see also Klink et al., 2017) and allowed for a refined understanding of the BBSome organization (Fig. 2C, lower panel). The broad base consists of BBS5, BBS8, BBS9 and of the C-terminal region of BBS1, while the top lobe consists of BBS2 and BBS7. BBS4 and BBS1βprop form a corkscrew that also contains part of BBS18. The top lobe is connected to the base through the 4-helix bundle made by BBS2hp and BBS9hp while the corkscrew attaches to the base through BBS18 and the BBS1 linker.
In support of our model, in which BBS4 and BBS5 are the only strictly peripheral subunits, these two subunits are dispensable for BBSome assembly (Seo et al., 2011; Zhang et al., 2012b). Similarly, the position of BBS4 explains why the loss of BBS1 or BBS18 results in BBS4 detaching from the BBSome (Loktev et al., 2008; Zhang et al., 2012b), as BBS1 and BBS18 provide the two attachment points for BBS4 to the BBSome. The peripheral position of BBS4 is consistent with the prior proposal that BBS4 is the last subunit added to the BBSome (Zhang et al., 2012a), likely moving from centriolar satellites to the BBSome at the base of cilia before entry of the complete BBSome into the ciliary shaft (Loktev et al., 2008; Nachury et al., 2007). BBS4 is held at centriolar satellites via the PCM-1 protein which recognizes the unstructured N-terminus and TPR1 of BBS4 (Kim et al., 2004). Given that BBS1βprop also binds BBS4TPR1 in our structure, it will not be possible for BBS4 to associate with PCM-1 and BBS1βprop at the same time, thus rationalizing the release of BBS4 from centriolar satellites prior to incorporation in the BBSome.
Our model is further supported by the finding that the deletion of BBS2 or BBS7 dramatically affects BBSome assembly and rationalizes why loss of either protein results in decreased levels of BBS2, BBS7 and BBS9 (Zhang et al., 2012a, 2013). First, the interweaving of BBS2GAE and BBS7GAE predicts that BBS2 and BBS7 are co-dependent on one another for folding. Second, BBS9hp will likely become unstable in the absence of its partner hairpin from BBS2.
Peptide backbone model
To independently validate our subunit assignment and gain an understanding of the BBSome at amino-acid resolution, we derived a backbone model of the entire complex using Rosetta (Leaver-Fay et al., 2011) and constraints from XLMS (Fig. 2B). Building a Cα model of the assembly was challenging due to the similarity of many domains in the complex, the lack of homologous structural information for many of the domains present, and the limited resolution of the density map. Modeling was performed in three steps. In a first step, models were built for individual domains (Figs. 3A). Of the 29 domains found in the BBSome subunits, 25 could be modeled, 10 with Rosetta CM (guided by available high-resolution structures of homologs), and 15 with Rosetta ab initio (the models of BBS2GAE and BBS7GAE also made use of sequence coevolution data) (Fig. S4). Only three domains could not be modeled with confidence (BBS1link, BBS5CtH and BBS9cc). In a second step, the 25 individual domain models were assembled using Monte Carlo domain assembly based on our previous de novo model-building method (Wang et al., 2015). This process was guided by both density and crosslinking data and resulted in a structure in which 23 domains (except BBS2GAE and BBS7GAE, see below) were uniquely placed. In a last step, Rosetta ES was used to model the micropeptide BBS18 into the EM density in the context of the 23 already placed domain models. The hybrid modeling approach fitted 3,522 residues of the Cα backbone through 24 different domains (Fig. 3B and Movie S2). Of the 76 crosslink pairs mapped with high confidence, 61 pairs had both positions present in the model and 44 out of these 61 crosslinks were satisfied by the model (Fig. 3C). Although the modeling approach converged to a single model, it is conceivable that very similar domains (e.g., BBS2βprop and BBS7βprop) may be swapped in the actual structure. The future determination of a BBSome structure at near-atomic resolution will help solidify the domain assignment.
Figure 3. Rosetta-generated Cα model of the BBSome.

A. Cα models of the 24 domains from BBSome subunits that were obtained with three different Rosetta modeling protocols (CM, ab initio, and ES; see Materials and Methods for details) and could be assembled into the Cα model of the BBSome. Although the GAE domains of BBS2 and BBS7 could be modeled using co-evolutionary data (see Fig. S4), they are not shown because they could not be satisfactorily built into the cryo-EM density map. The colors and labels are as in Figure 2. B. Nearly complete Cα model of the BBSome obtained using Rosetta to assemble the 24 domains into a complex, guided by the cryo-EM density map and XLMS data (see Materials and Methods for details). The three views are the same as in Fig. 1C. The GAE domains of BBS2 and BBS7 are not included in the final Cα model but their general placement is indicated by coloring the density map. C. Magnified views of crosslink clusters in the final BBSome model. The yellow dotted lines indicate crosslinks that were satisfied by the final Rosetta molecular model. For clarity, only selected crosslinks of each cluster are shown. Depicted crosslinks are: Top Left: 9CtH[K789]- 4N[K116] and 9CtH[K810]-4N[K116]. Top Right: 5PH1[K87]-18[K90], 18[K90]-1GAE[K553], 18[K93]-1GAE[K553] and 1GAE[K553]-2hp[K638]. Bottom Left: 9βprop[K218]-8N[K181]. Bottom Middle: 7βprop[K56]-7cc[K352], 7βprop[K56]-2cc[K345], 2cc[K360]-7cc[K359], 2cc[K360]-7cc[K352], 7cc[K359]-7cc[K352], 2cc[K345]-7cc[K352] and 2cc[K345]-7cc[K338]. Bottom Right: 2 βprop[K9]-1βprop[K69], 2 βprop[K13]-7hp[K658], 4N[K20]-7hp[K659], 4N[K20]-7βprop[K222], 4N[K5]-1βprop[K143], 1βprop[K192]-4N[K25].
The models of BBS2GAE and BBS7GAE were the only ones that could not be satisfactorily placed into the cryo-EM density. While the application of evolutionary contacts led to well-converged models of the individual GAE domains, these could only be approximately placed into the density, and only their relative position and approximate orientation could be determined (Fig. 3B and Movie S2). Considering the lack of a clearly defined domain boundary in the density representing the GAE domains of BBS2 and BBS7 and the failure to incorporate BBS2 and BBS7 into a recombinant BBSome in insect cells (Klink et al., 2017), we surmise that strand exchange between BBS2GAE and BBS7GAE may preclude folding of the combined BBS2GAE/BBS7GAE domain in silico and in heterologous systems. In support of this interpretation, assembly of the BBSome requires a specialized machinery that comprises the chaperonin-like proteins BBS6, BBS10 and BBS12. The canonical group II chaperonin TCP-1 ring complex (TRiC) temporarily holds hydrophobic β-strands to fold complex structures such as actin, tubulin and some β-propellers (Yam et al., 2008). BBS6, 10 and 12 substitute for some of the TRiC subunits, forming an alternate TRiC/BBS complex that binds BBS2 and BBS7 (Seo et al., 2010) and incorporates BBS2/7 into the BBSome (Sinha et al., 2014). The intimate association of BBS2GAE and BBS7GAE may explain the requirement for a specialized folding machinery as hydrophobic β-edges in one GAE domain may need to be chaperoned until the partner polypeptide becomes available for co-folding. The binding of BBS2cc to BBS6 and the position of BBS2cc immediately prior to BBS2GAE in the polypeptide chain suggest that the alternate TRiC/BBS complex may become recruited to the nascent BBS2 polypeptide immediately before the GAE emerges from the ribosome.
Disease-causing variants
To date, 89 missense pathogenic variants in BBSome subunits have been found to cause monogenic disorders ranging from non-syndromic retinal degeneration or obesity to full-fledged Bardet-Biedl Syndrome (Figs. 4A and S5 and Table S3). The most common and best-studied missense variant is BBS1[c.1169T>G, p.Met390Arg], which affects a buried residue inside BBS1βProP and interferes with folding of the β-propeller (Mourão et al., 2014). Consequently, BBS1 levels are drastically reduced in the BBS1[p.Met390Arg] mouse model (Zhang et al., 2012a). Mapping each of the 78 BBS pathogenic variants onto the Cα model reveals that most of the affected positions are likely to be buried (Fig. 4B,C) and predicts that these variants affect folding of a specific domain, thereby resulting in BBSome assembly defects. We surmise that most BBS alleles converge on defective BBSome assembly.
Figure 4. Mapping of missense pathogenic variants onto the Cα model of the BBSome.

Missense variants causing Bardet-Biedl syndrome are shown in cyan and variants causing less severe disease phenotypes in magenta. A. Variants were placed on diagrams of each BBSome subunit. RP stands for Retinitis Pigmentosa, i.e., retinal degeneration. B. All variants were mapped onto the Cα model of the BBSome to show the spatial distribution of the variants. C. Close-up views of variants present in specific domains, with BBSome subunits colored as in Figure 3.
The distribution of pathogenic variants is non-even among BBSome domains, with BBS1Pβprop and BBS2βprop harboring nearly half of all missense variants (Fig. 4A and S5), suggesting considerable functional importance for these two domains. While BBS1βprop is known to bind ARL6GTP, no specific interactions have yet been described for BBS2βprop. Finally, despite being nearly entirely contained within the core of the BBSome, BBS8 is affected by only two pathogenic variations, both facing towards BBS1GAE. Similarly, missense variants on BBS4 are concentrated at the sites of interactions with BBS18 (BBS4TPR8-10) or BBS9 (BBS4TPR2-3).
Interestingly, visual inspection suggests that while BBS-associated pathogenic variants appear to affect residues predicted to be buried, pathogenic variants associated with non-syndromic retinal degeneration or obesity tend to be on residues that are predicted to be solvent-exposed (Fig. 4B and C). This finding suggests that the latter types of variants may affect specific interactions with cargoes or other partners, whereas BBS variants tend to disrupt the entire structure of the complex. A correlation between relatively mild variants and retinal degeneration is in agreement with the tremendous transport rate in photoreceptors that may heighten the sensitivity of this cell type to slight alterations in BBSome activity.
The BBSome is auto-inhibited in solution
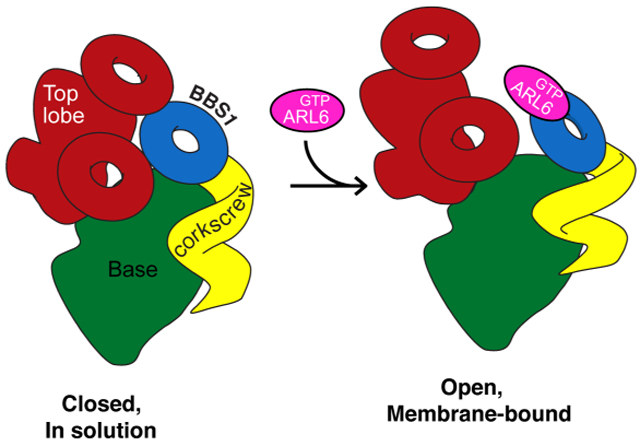
Because ARL6GTP recruits the BBSome to membranes (Jin et al., 2010) and enables TZ crossing (Ye et al., 2018), we sought to fit the crystal structure of the BBS1βprop/ARL6GTP complex (Mourão et al., 2014) into our cryo-EM map. Problematically, placing the BBS1βprop/ARL6GTP structure into our map resulted in extensive steric clashes between ARL6GTP and both BBS7Pβprop and BBS2cc (Fig. 5A). We considered and ruled out the possibilities that the subunit assignment, the orientation of BBS1βprop or the contacts between ARL6GTP and BBS1βprop in the crystal structure were erroneous. First, the convergence of the manual and the computational assignment approaches gives high confidence in the validity of the subunit assignment. Second, the presence of a helical insert between two successive blades of BBS1βprop provides a unique landmark that allows unambiguous fitting of the BBS1βprop crystal structure into one specific region of the map. Finally, the contacts between BBS1βprop and ARL6GTP identified in the crystal structure have been extensively validated through biochemical and cell biological assays (Mourão et al., 2014; Nozaki et al., 2018). We therefore conclude that BBS1βprop is inaccessible to ARL6GTP in the canonical structure and that a conformational change must take place for ARL6GTP to recruit the BBSome to membranes (Fig. 5B). The surprising observation that a BBSome purified by ARL6GTP affinity adopts a conformation that is refractory to ARL6GTP binding once released from the affinity column argues for a rapid interconversion between the closed form that we resolved and an open form.
Figure 5. BBSome recruitment to membranes is coupled to a conformational change.

A. Placement of the crystal structure of the BBS1βprop, which was determined in complex with ARL6GTP (PDB id: 4V0M; Stenson et al., 2017), into the corresponding density of the 4.9-Å cryo-EM map results in a major steric clash between ARL6GTP and BBS7 βprop. The crystal structures are shown in ribbon representation and the cryo-EM map as a colored segmented map. Left, overview. Right, close-up view. B. Diagram of the predicted conformational change in the BBSome induced by ARL6GTP binding. Left panel: In solution, the BBSome exists predominantly in a closed conformation, in which BBS1βprop is too close to the top lobe to allow binding of ARL6GTP. Right panel: Binding of ARL6GTP locks the membrane-bound BBSome in an open, active conformation. This conformation would allow the BBSome to interact with cargoes and/or IFT and to cross the TZ.
Autoinhibition is a recurring theme in coat adaptors, and the BBSome joins the ranks of COPI, AP-1 and AP-2 as an adaptor that undergoes a closed-to-open conformational change coincident with recruitment to membranes (Cherfils, 2014; Faini et al., 2013). The docking of the BBSlβpro/ARL6GTP structure (Mourão et al., 2014) into our map identifies the top lobe, consisting of BBS2 and BBS7, as the major autoinhibitory module of the BBSome. Since Drosophilae genomes lack BBS2 and BBS7, it appears that the small number of ciliated cells and the limited functions of cilia in Drosophiiaehave relaxed the demands on controlling BBSome activation in this organism. Congruent with the loss of BBS2/7 from the Drosophila BBSome, the C terminus of DrosophiJaBBS9, which provide the main attachment point for the BBS2/7 complex to the BBSome in our structure, has undergone extensive remodeling in Drosophila (shorter hairpin and C-terminal helix bundle as well as insertion before the hairpin).
For BBSomes from other organisms, the top lobe will have to move for ARL6GTP to bind (Fig. 5B). The conformation stabilized by ARL6GTP binding may then promote TZ crossing by increasing BBSome interactions with cargoes. Activating conformational changes coordinately induced by membrane and cargo binding are common features in coat adaptors (Cherfils, 2014; Faini et al., 2013). In the clathrin adaptor AP-2, four coplanar PI(4,5)P2-binding sites and two cargo-binding sites are cooperatively and jointly unmasked (Jackson et al., 2010). AP-1 and exomer undergo similar structural transitions upon binding to ARF1GTP (Paczkowski and Fromme, 2014; Ren et al., 2013) and COPI adopts an open conformation upon recruitment to membranes to bind cargoes (Langer et al., 2008; Dodonova et al., 2015, 2017). Alternatively, the BBSome conformation induced by ARL6GTP binding may allow TZ crossing by coupling to IFT trains. IFT38/CLUAP1 is the only IFT-B subunit to consistently interact with the BBSome in systematic affinity purification studies (Beyer et al., 2018; Boldt et al., 2016). Since the N terminus of IFT38 is involved in interactions with the rest of IFT-B (Taschner et al., 2016), the C terminus of IFT38 is the most likely region to mediate interactions with the BBSome. Future biochemical assays and a high-resolution structure of the open conformation will be necessary to understand which interactions are modulated by conformational opening of the BBSome and how it allows TZ crossing.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thomas Walz (twalz@rockefeller.edu).
METHOD DETAILS
Recombinant protein expression
The N-terminal amphipathic helix of human ARL6 was removed (ΔN16) to maximize solubility and the catalytic glutamine was mutated (Q73L) to limit hydrolysis of the bound GTP. N-terminally GST-tagged ARL6ΔN16[Q73L] was expressed from pGEX6P in Rosetta2(DE3)-pLysS cells grown in 2YT medium at 37°C. Once OD600 reached 0.3, the culture was transferred to 24°C. Isopropyl β-D-l-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM once OD600 reached 0.6, and protein expression was allowed to proceed for 16 h at 24°C. Bacteria were harvested, and pellets frozen in liquid nitrogen were ground to a fine powder with a porcelain pestle and mortar cooled with liquid nitrogen. 10 mL of bacteria powder (equivalent to 1 liter of bacterial culture) was resuspended in 30 mL of TBS* (50 mM Tris, pH 7.4, 150 mM NaCl, 5 μM GDP, 5 mM MgCl2, 1 mM DTT) and the resuspended homogenate was lysed by sonication. The lysate was clarified by spinning for 1 h at 30,000 × g at 4°C.
To purify ARL6ΔN16[Q73L], 35 mL of clarified extract was incubated with 8 mL Glutathione Sepharose 4B (GE Healthcare). The beads were washed three times with 40 mL TBS*, and ARL6ΔN16[Q73L] was eluted into 15 mL TBS* by cleavage with 2,000 units of PreScission protease overnight at 4°C. Eluates were concentrated by ammonium sulfate precipitation and ARL6ΔN16[Q73L] was further purified on Superdex 75 equilibrated in CB (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM DTT, 10% glycerol, 100 μM GDP, 1 mM MgClz).
Purification of native BBSome
The BBSome was purified from bovine retina (W.L. Lawson) by ARL6GTP-affinity chromatography followed by cation-exchange chromatography. 80 g of frozen retina were transferred to 240 mL of breaking buffer (25 mM Tris-HCl, pH 8.0, 500 mM KCl, 250 mM sucrose, 5 mM MgCl2, 1 mM DTT) supplemented with protease inhibitors (1 mM AEBSF, 0.8 μM Aprotinin, 15 μM E-64, 10 μg/mL Bestatin, 10 μg/mL Pepstatin A and 10 μg/mL Leupeptin), and the mixture was stirred in the cold room for 15 min to allow for tissue dissociation. All following steps were conducted at 4°C. Homogenization was completed by 10 strokes of Dounce. The retinal homogenate was cleared by ultracentrifugation in a Type 45Ti rotor at 45,000 rpm for 2 h.
To prepare an ARL6GTP- affinity chromatography column, a 1-mL GSTrap HP column was loaded with 12 mL of extract from bacteria expressing GST-tagged ARL6ΔN16[Q73L] and the nucleotide was exchanged by washing the column at 1 mL/min with a succession of 10 column volumes (CV) of NS100 (25 mM Tris-HCl, pH 8.0, 100 mM KCl, 5 mM MgCl2, 1 mM DTT) with 100 μM GTP, 10 CV NE (25 mM Tris-HCl, pH 8.0, 100 mM KCl, 10 mM EDTA, 5 mM MgCl2, 1 mM DTT), 10 CV NS500 (25 mM Tris-HCl, pH 8.0, 500 mM KCl, 5 mM MgCl2, 1 mM DTT) with 1 mM GTP, and 10 CV NS500 with 100 μM GTP. The ARL6GTP column was connected downstream of a 1-mL GSTrap 4B column in order to catch the GST from the retinal extract and to prevent displacement of the GST-ARL6GTP from the GSTrap HP column.
The clarified retinal extract was supplemented with 100 μM GTP, split into two equal halves and each half was applied onto a GSTrap 4B/GSTrap HP column at 0.5 mL/min. The columns were washed with 5 mL NS500 with 100 μM GTP at 0.5 mL/min, and the GSTrap HP column was washed with 20 mL NS500 with 100 μM GTP. The GSTrap HP column was brought to room temperature for 10 min, and protein was eluted with Elution Buffer II (25 mM Tris-HCl, pH 8.0, 2.5 M NaCl, 10 mM EDTA, 5 mM MgCl2, 1 mM DTT) containing protease inhibitors (1 mM AEBSF, 15 μM E-64, 10 μg/mL Bestatin, 10 μg/mL Pepstatin A and 10 μg/mL Leupeptin) and warmed to 30°C. Eight 0.5-mL fractions were collected, and the peak was identified by SDS-PAGE and silver staining. The peak typically straddled four fractions, and the peaks from each column were pooled.
The pooled peak fractions were dialyzed for 45 min each against 500 mL Dialysis Buffer 1 (25 mM Tris, pH 8.0, 1 M NaCl, 5 mM MgCl2, 1 mM DTT), 500 mL Dialysis Buffer 2 (25 mM HEPES, pH 7.0, 300 mM NaCl, 5 mM MgCl2, 1 mM DTT), 500 mL Dialysis Buffer 3 (25 mM HEPES, pH 7.0, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT),) and 500 mL H10 (40 mM HEPES, pH 7.0, 100 mM NaCl, 1 mM DTT). The dialysate was spun in a TLA100.3 at 80,000 rpm for 10 min at 4°C to clear any precipitate and loaded onto a MonoS PC1.6/5 column equilibrated in buffer H10 using a SMART system (Pharmacia). After washing the column with 20 CV H10, a gradient of 8 CV from H10 to H50 (40 mM HEPES, pH 7.0, 500 mM NaCl, 1 mM DTT) was applied. The BBSome eluted around 220 mM NaCl. Concentration was measured using a Nanodrop spectrophotometer at 280 nm (calculated extinction coefficient: 398,500 cm−1M−1 and MW: 488,000 g/mol) and the sample was processed for cryo-EM the next day.
Disease-causing variants
Missense variants in BBSome subunits were collected either from the Human Gene Mutation Database (HGMD; Stenson et al., 2017) or from our unpublished database of variants obtained from a previously described cohort of 350 French families (Redin et al., 2012). Variants present at high frequencies in 1000Genomes and gnomAD (http://www.internationalgenome.org: Li et al., 2015) were removed. 89 variants in 7 BBSome genes fulfilled the criteria of pathogenic variants.
Cryo-EM data collection and image processing
2.5 μL of the peak fraction from a BBSome purification (0.3-0.6 mg/mL) was applied to glow-discharged R1.2/1.3 holey carbon copper grids (Quantifoil). The grids were blotted for 2.5 s at 85% humidity and plunged into liquid ethane using a Gatan CryoPlunge 3. Grids were imaged with an FEI Polara electron microscope operated at 300 kV. Movie stacks were recorded with the UCSFImage4 data acquisition software (Li et al., 2015) on a Gatan K2 Summit direct electron detector in super-resolution counting mode. The defocus was varied from 1.0 and 3.0 μm, and the nominal magnification was set to 31,000x, corresponding to a calibrated pixel size of 0.65 Å. Two datasets were collected using an exposure of 300 ms per frame at a dose rate of 8 counts/pixel/sec, corresponding to a dose of 1.95 electrons/Å2/frame: 870 movie stacks were recorded with 24 frames and 1630 movie stacks with 46 frames, corresponding to total electron doses of 46.8 and 89.7 electrons/Å2, respectively.
Movie stacks were binned over 2 × 2 pixels, resulting in a final pixel size of 1.3 Å. The frames of each movie were aligned, dose-weighted and summed into a single image using MotionCor2 (Zheng et al., 2017). These summed images were used for further image processing, except for contrast transfer function (CTF) estimation using CTFFIND4 (Rohou and Grigorieff, 2015), for which images summed without dose weighting were used. About 10,000 particles were manually picked and subjected to reference-free 2D classification in Relion 1.3 (Scheres, 2015). 13 representative class averages were used as references for semi-automated particle picking with Relion, resulting in 153,332 and 435,492 particles for the two datasets. Particles picked from protein aggregates and carbon film were removed manually, and the remaining particles were used for 2D classification. After removing classes that produced poor 2D averages, the remaining 141,113 and 371,708 particles were combined and subjected to a first 3D classification into 10 classes. The reference map used for 3D classification was calculated using the validation of individual parameter reproducibility (VIPER) command in the SPARX software package (Hohn et al., 2007). Three classes appearing to show the complete BBSome complex were combined (total of 214,309 particles) and subjected to a second 3D classification into 6 classes. Five of the classes, which showed the BBSome in the same conformation, were combined (total of 179,280 particles) and refined, producing a density map at 4.9-Å resolution (Map 1). One class resulting from the first 3D classification (including 52,826 particles) showed a partial BBSome complex (later found to lack BBS5). This map was refined to 7.0-Å resolution (Map 2). The resolution of the two density maps was determined by Fourier shell correlation (FSC) of two independently refined half-maps using the 0.143 cut-off criterion (Rosenthal and Henderson, 2003). Local resolution was estimated from the two half-maps using the ResMap algorithm implemented in Relion. UCSF Chimera was used to visualize and segment density maps (Pettersen et al., 2004).
Prediction of structural domains
The boundaries of each BBSome subunit domain defined in (Jin et al., 2010) were refined using the consensus of 20 secondary structure prediction algorithms returned by the genesilico metaserver (Kurowski and Bujnicki, 2003).
Chemical crosslinking and cross-link mapping by LC-MS/MS
Buffer components for cross-linking were BioUltra grade (Sigma Aldrich), and LC-MS/MS was carried out with Thermo Scientific LC-MS grade reagents and solvents. Purified BBSome (0.3 mg/mL in 40 mM HEPES, pH 7.0, 300 mM NaCl, 1 mM DTT) was cross-linked with 200 μM of either BS3 (Thermo Scientific, 3.5 mM stock in water) or DSSeb (Proteochem, 4 mM stock in DMSO) for 30 min at room temperature. The reaction was quenched by addition of 100-fold molar excess of 1 M Tris-HCl, pH 7.5. Samples were reduced with 50 mM Tris(2-carboxyethyl)phosphine (TCEP) for 10 min at 60°C and alkylated with 50 mM iodoacetamide in the dark for 60 min at room temperature. In-gel digestion was carried out overnight at 37°C with 0.5 μg sequencing-grade modified trypsin (Thermo Scientific) in 100 mM ammonium bicarbonate. The resulting peptides were washed through C18 Spin Tips (Thermo Scientific) before elution with 40 μL of 0.1% trifluoroacetic acid in 80% acetonitrile. Eluted peptides were dehydrated in vacuum and resuspended in 20 μL 0.1% formic acid for MS analysis.
Peptides were analyzed in the Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an EASY-nLC (Thermo Scientific) liquid chromatography system. The peptides were eluted with a flow rate of 250 nL/min over a 120-min linear gradient from 96% buffer A (water) to 40% buffer B (acetonitrile) followed by a 20-min linear gradient to 98% buffer B. Each full MS scan (R = 60,000) was followed by 20 data-dependent MS2 scans (R = 15,000) with higher-energy collisional dissociation and an isolation window of 2.0 m/z. The normalized collision energy was set to 35. Precursors of charge state 4-6 were collected for MS2 scans; monoisotopic precursor selection was enabled and the dynamic exclusion window was set to 30.0 s.
Raw LC-MS/MS data files were converted into mgfformat and searched using pLink (Fan et al., 2015; Yang et al., 2012) with default FDR <5%, e-value set at <1, trypsin digest with up to 3 missed cleavages, constant modification at 1=carbamidomethyl[C], variable modification at 1=oxydation[M]. Cross-linker was set to BS3 ([K [K 138.068 138.068 156.079 156.079) or DSSeb ([K [K 166.099 166.099 181.11 181.11). Mass tolerances for fragments and precursors were left unaltered (default of 20 ppm). mgffiles were searched against a database comprising all fasta sequences of bovine BBSome subunits. Only peptide pairs detected at least once with an e-value under 0.001 were used to constrain the model.
Model building and refinement with Rosetta
In a first stage, we attempted to model the structure of the 29 domains that make up the BBSome:4 for BBS1 (BBS1βprop, BBS1ins, BBS1link and BBS1GAE), 6 for BBS2 (BBS2βprop,BBS2cc, BBS2GAE, BBS2pf, BBS2hp and BBS2CtH), 1 for BBS4, 3 for BBS5 (BBS5PH1, BBS5PH2 and BBS5CtH), 6 for BBS7 (BBS7βprop, BBS7cc, BBS7GAE, BBS7pf, BBS7hp and BBS7CtH), 2 for BBS8 (BBS8TPR1-2 and BBS8TPR3-12), 6 for BBS9 (BBS9βprop, BBS9cc, BBS9GAE, BBS9pf, BBS9hp and BBS9CtH), and 1 for BBS18.
The models of 10 domains could be built using high-resolution structures of homologous proteins as guides (and using alignments from hhpred (Söding, 2005)). These 10 domains are BBS1βprop, BBS1GAE, βprop, BBS4, BBS5PH1, BBS5PH2, BBS7βprop, BBS8TPR3-12, BBS9βprop and BBS9GAE (Fig. 3A). For each of these domains, 100 to 2000 models were built using RosettaCM (Song et al., 2013) (the number of models generated was based on convergence of low-energy structures). While models for all other domains were built without the use of the density map, models for BBS4 and BBS8TPR3-12 were built guided by the density of the two possible regions representing them in the map (half the models were generated from each of the two regions) to ensure that the curvature of the two subunits was consistent with the density map. For each of the 10 domains, the top 20 models obtained with Rosetta all-atom energy (Park et al., 2016) were saved for the next step. Of the remaining 19 domains, modeling with Rosetta ab initio (Bradley et al., 2005) produced converged models for 15 domains. These domains are BBS1ins, BBS2cc, BBS2GAE, BBS2pf, BBS2hp, BBS2CtH, BBS7cc, BBS7GAE, BBS7Ppf, BBS7hp BBS7CtH, BBS8TPR1-2, BBS9pf, BBS9hp, BBS9CtH (Figs. 3A and S4). 100,000 models were generated for these domains, which all are no more than 150 residues and consist mostly of helical bundles or coiled coils (except for the three platform domains). Overall convergence for BBS7hp, BBS7CtH and BBS9CtH was poor, but the models contained a consistent core that could be placed into the map, allowing the missing regions to be re-modeled based on their density in the map. The models for two other domains, BBS2GAE and BBS7GAE, only converged when metagenomic data were used to predict contacts from co-evolution (Ovchinnikov et al., 2017). The convergence of these 15 domains suggests that they represent near-native configurations of the domains (Ovchinnikov et al., 2017). As we did for the domains modeled with RosettaCM, the top 20 models of these 15 domains were saved for the next step. Of the remaining four domains, three domains, BBS1link, BBS5CtH and BBS9cc, could not be modeled with any confidence, and BBS18 was modeled at the very end (see below).
In a second stage, we assembled the complete complex using the domain models built as described above and guided by the cryo-EM density map and the crosslinking data. We used a modified version of a protocol used for de novo model building guided by cryo-EM data (Wang et al., 2015). This modified protocol was previously employed to determine the domain architecture of the Pex1/Pex6 ATPase complex (Blok et al., 2015). Briefly, the protocol first matches each domain individually to the density. Then, Monte Carlo assembly of the entire complex is carried out with a simple score function favoring: a) placement of as many domains as possible, b) maximizing agreement of the placed models with density, c) placement of domains within the same chain such that their termini are close enough to connect, and d) placements that maximize agreement with the crosslinking data. Since the protocol does not allow backbone movement, the crosslink distance was extended by 10 Å from ideal to account for possible backbone flexibility (note that changing this distance in between 5 Å and 20 Å did not change the final domain assignment). Following 10,000 independent trajectories, our Monte Carlo assembly yielded two low-energy solutions: one that was consistent with the manual domain assignment, and one in which the positions of BBS2βprop and BBS7βprop and their respective coiled-coil linker domains were reversed. A final flexible-backbone refinement of both configurations clearly identified the manual domain assignment as the correct one once the maximum crosslink distance between Cp atoms (27 Å for BS3 and 30 Å for DSSeb) was taken into account, with 44 crosslinks satisfied in the manual assignment compared to 41 for the flipped configuration. Flexible-backbone refinement was performed using RosettaCM guided by the cryo-EM density map and experimental crosslinks.
In the final step, BBS18, was visible as a mostly extended peptide snaking through the core of the complex. Since the configuration of this subunit appeared to be defined by contacts with the other subunits, we modeled this peptide in the context of the complete assembly. A fourteen-residue poly-alanine helix was initially docked into the clearest helical density region. Every possible fourteen-residue stretch of amino acids were threaded, and each was extended with RosettaES (Frenz et al., 2017). All resulting models were refined, and the lowest-energy configuration was accepted. For this lowest-energy configuration, the RosettaES ensemble was quite well converged. In addition to the 44 crosslinks that were fully compatible with our final model, 15 corresponded to amino acids that were not present in the final model and 17 were in conflict. Given the conformational flexibility of the BBSome, it is likely that crosslinks that are in conflict with the model built into Map 1 are satisfied in other conformations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed within published software as described in sections “Cryo-EM data collection and image processing” and “Model building and refinement with Rosetta”.
DATA AND CODE AVAILABILITY
Data Deposition
The maps have been deposited in wwPDB under ID codes EMD-7839 and EMD-7841 and the model has been deposited in PDB-Dev under ID code PDBDEV_00000018.
Code Availability
This study did not produce additional software. The software used here (summarized in Key Resources Table) has been published and is publicly available.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli Rosetta2(DE3)-pLysS | Millipore Sigma | 70956 |
| Biological Samples | ||
| Bovine retina | W.L. Lawson | Custom order |
| Experimental Models | ||
| E. coli Rosetta2(DE3)-pLysS | Millipore Sigma | 70956 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BS3 | Thermo Scientific | A39266 |
| DSSeb | Proteochem | c1136 |
| Sequencing-grade modified trypsin | Thermo Scientific | PI90305 |
| Deposited Data | ||
| Cryo-EM map of intact BBSome | This paper | EMD-7839 |
| Cryo-EM map of BBSome missing BBS5 | This paper | EMD-7841 |
| Cα model of BBSome | This paper | PDBDEV_00000018 |
| Coordinates of the BBS1βprop–ARL6GTP complex | (Mourão et al., 2014) | PDB: 4V0M |
| Recombinant DNA | ||
| pGEX6P-ARL6ΔN16[Q73L] | (Jin et al., 2010) | N/A |
| Software and Algorithms | ||
| UCSFImage4 | (Li et al., 2015) | https://cryoem.ucsf.edu/software/UCSFImage.html |
| MotionCor2 | (Zheng et al., 2017) | https://msg.ucsf.edu/software |
| CTFFIND4 | (Rohou and Grigorieff, 2015) | http://grigoriefflab.janelia.org/ctffind4 |
| Relion 1.3 | (Scheres, 2015) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| SPARX | (Hohn et al., 2007) | http://sparx-em.org/sparxwiki/SparxWiki |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| genesilico | (Kurowski and Bujnicki, 2003) | https://www.genesilico.pl/meta2/ |
| pLink | (Fan et al., 2015; Yang et al., 2012) | http://pfind.ict.ac.cn/software/pLink/index.html |
| hhpred | (Söding, 2005) | https://toolkit.tuebingen.mpg.de/#/hhpred |
| RosettaCM | (Song et al., 2013) | https://www.rosettacommons.org/software |
| Rosetta ab initio | (Bradley et al., 2005) | https://www.rosettacommons.org/software |
| RosettaES | (Frenz et al., 2017) | https://www.rosettacommons.org/software |
| Other | ||
| Quantifoil R1.2/1.3 holey carbon film on copper 200 mesh grids | Electron Microscopy Sciences | Cat #Q250CR-06 |
Supplementary Material
Movie S1, related to Figure 2: Assignment of BBSome domains to densities in the cryo-EM map using alternative 3D classes, binary interactions and subunit tracing. The color scheme is as in Figure 2C.
Movie S2, related to Figure 3: 360° view of the peptide backbone model. The density map is shown as transparent surface with the first rotation. The color scheme is as in Figure 3B. The placement of BBS2GAE and BBS7GAE is indicated by local coloring of the density map in red (BBS2GAE) and purple (BBS7GAE).
HIGHLIGHTS.
4.9-Å density map of the intact native BBSome obtained by single-particle cryo-EM
Cα model generated using Rosetta establishes the subunit organization
BBSome subunits have a very high degree of interconnectivity
In solution, the BBSome exists predominantly in an auto-inhibited state
ACKNOWLEDGMENTS
This work was funded by NIGMS (R01-GM089933, M.V.N.; R01-GM123089, F.D.), the Austrian Science Fund (FWF P30162, U.S.), a Damon Runyon Fellowship to A.R.N. (DRG 2160-13), and an NIGMS fellowship to S.R.W (F32-GM089218). This work was made possible, in part, by NEI EY002162 - Core Grant for Vision Research and by the Research to Prevent Blindness Unrestricted Grant (M.V.N.), and by the Howard Hughes Medical Institute and the Blavatnik Family Foundation (E.N.). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the RBVI at UCSF (supported by NIGMS P41-GM103311).
We thank the Nachury lab for comments, Jacqueline Cherfils and Soichi Wakatsuki for advice on ARL6 placement, Val Sheffield for help with BBS variant database, Anne-Sophie Leuvrey and Manuela Antin for technical assistance and Suling Wang for help with diagram preparation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bangs F, and Anderson KV (2017). Primary cilia and mammalian Hedgehog signaling. Cold Spring Harb. Perspect. Biol. 9, a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer T, Bolz S, Junger K, Horn N, Moniruzzaman M, Wissinger Y, Ueffing M, and Boldt K (2018). CRISPR/Cas9-mediated genomic editing of Cluap1/IFT38 reveals a new role in actin arrangement. Mol. Cell. Proteomics MCP 17, 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok NB, Tan D, Wang RY-R, Penczek PA, Baker D, DiMaio F, Rapoport TA, and Walz T (2015). Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 112, E4017–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen T-MT, Texier Y, van Beersum SEC, Horn N, Willer JR, Mans DA, et al. (2016). An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 7, 11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley P, Malmström L, Qian B, Schonbrun J, Chivian D, Kim DE, Meiler J, Misura KMS, and Baker D (2005). Free modeling with Rosetta in CASP6. Proteins Struct. Funct. Bioinforma. 61, 128–134. [DOI] [PubMed] [Google Scholar]
- Cherfils J (2014). Arf GTPases and their effectors: assembling multivalent membrane-binding platforms. Curr. Opin. Struct. Biol. 29, 67–76. [DOI] [PubMed] [Google Scholar]
- Dodonova SO, Chen J, Kovacs JM, Diestelkoetter-Bachert P, von Appen A, Peng H, Hagen WJH, Rits-Volloch S, Beck R, Lu J, et al. (2015). A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science 349, 195–198. [DOI] [PubMed] [Google Scholar]
- Dodonova SO, Aderhold P, Kopp J, Ganeva I, Röhling S, Hagen WJH, Sinning I, Wieland F, and Briggs JAG (2017). 9 Å structure of the COPI coat reveals that the Arf1 GTPase occupies two contrasting molecular environments. eLife 6, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkhatib N, Bresteau E, Baschieri F, Rioja AL, van Niel G, Vassilopoulos S, and Montagnac G (2017). Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science 356, eaal4713. [DOI] [PubMed] [Google Scholar]
- Faini M, Beck R, Wieland FT, and Briggs JAG (2013). Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol. 23, 279–288. [DOI] [PubMed] [Google Scholar]
- Fan S-B, Meng J-M, Lu S, Zhang K, Yang H, Chi H, Sun R-X, Dong M-Q, and He S-M (2015). Using pLink to analyze cross-linked peptides. Curr. Protoc. Bioinforma. 49, 8.21.1–19. [DOI] [PubMed] [Google Scholar]
- Frenz B, Walls AC, Egelman EH, Veesler D, and DiMaio F (2017). RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, Raleigh DR, and Reiter JF (2018). How the ciliary membrane Is organized inside-out to communicate outside-in. Curr. Biol. 28, R421–R434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, and Reiter JF (2017). Open Sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb. Perspect. Biol. 9, a028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichsen L, Meyerholz A, Groos S, and Ungewickell EJ (2006). Bending a membrane: How clathrin affects budding. Proc. Natl. Acad. Sci. U. S. A. 103, 8715–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, and Ludtke SJ (2007). SPARX, a new environment for Cryo-EM image processing. J. Struct. Biol. 157, 47–55. [DOI] [PubMed] [Google Scholar]
- Jackson LP, Owen DJ, Kelly BT, Mccoy AJ, Gaffry T, James LC, Collins BM, Höning S, and Evans PR (2010). A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, and Nachury MV (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Nozaki S, Hartanto D, Miyano R, and Nakayama K (2015). Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 128, 2351–2362. [DOI] [PubMed] [Google Scholar]
- Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, et al. (2004). The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat. Genet. 36, 462–470. [DOI] [PubMed] [Google Scholar]
- Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, and Wittinghofer A (2017). A recombinant BBSome core complex and how it interacts with ciliary cargo. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer KE, and Schwartz TU (2015). Structural characterization of Bardet-Biedl Syndrome 9 protein (BBS9). J. Biol. Chem. 290, 19569–19583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowski MA, and Bujnicki JM (2003). GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer JD, Roth CM, Béthune J, Stoops EH, Brügger B, Herten D-P, and Wieland FT (2008). A conformational change in the α-subunit of coatomer induced by ligand binding to γ-COP revealed by single-pair FRET. Traffic 9, 597–607. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman KW, Renfrew PD, Smith CA, Sheffler W, et al. (2011). Rosetta3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zheng S, Agard DA, and Cheng Y (2015). Asynchronous data acquisition and on-the-fly analysis of dose fractionated cryoEM images by UCSFImage. J. Struct. Biol. 192, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, and Lechtreck KF (2018). The Bardet-Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc. Natl. Acad. Sci. U. S. A. 115, E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, Slusarski DC, Sheffield VC, Jackson PK, and Nachury MV (2008). A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 15, 854–865. [DOI] [PubMed] [Google Scholar]
- Mourão A, Nager AR, Nachury MV, and Lorentzen E (2014). Structural basis for membrane targeting of the BBSome by ARL6. Nat. Struct. Mol. Biol. 21, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourão A, Christensen ST, and Lorentzen E (2016). The intraflagellar transport machinery in ciliary signaling. Curr. Opin. Struct. Biol. 41, 98–108. [DOI] [PubMed] [Google Scholar]
- Nachury MV (2018). The molecular machines that traffic signaling receptors into and out of cilia. Curr. Opin. Cell Biol. 51, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, and Mick DU (2019). Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. [DOI] [PubMed] [Google Scholar]
- Nozaki S, Katoh Y, Kobayashi T, and Nakayama K (2018). BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS ONE 13, e0195005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S, Park H, Varghese N, Huang P-S, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, and Baker D (2017). Protein structure determination using metagenome sequence data. Science 355, 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczkowski JE, and Fromme JC (2014). Structural basis for membrane binding and remodeling by the exomer secretory vesicle cargo adaptor. Dev. Cell 30, 610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Bradley P, Greisen P, Liu Y, Mulligan VK, Kim DE, Baker D, and DiMaio F (2016). Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 12, 6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Redin C, Le Gras S, Mhamdi O, Geoffroy V, Stoetzel C, Vincent M-C, Chiurazzi P, Lacombe D, Ouertani I, Petit F, et al. (2012). Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alstrom syndromes. J. Med. Genet. 49, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Farías GG, Canagarajah BJ, Bonifacino JS, and Hurley JH (2013). Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, and Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Scheres SHW (2015). Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Baye LM, Schulz NP, Beck JS, Slusarski DC, and Sheffield VC (2010). BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. U. S. A. 107, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Zhang Q, Bugge K, Breslow D, Searby CC, Nachury MV, and Sheffield VC (2011). A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 7, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Belcastro M, Datta P, Seo S, and Sokolov M (2014). Essential role of the chaperonin CCT in rod outer segment biogenesis. Invest. Ophthalmol. Vis. Sci. 55, 3775–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J (2005). Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960. [DOI] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY-R, Kim D, Miles C, Brunette T, Thompson J, and Baker D (2013). High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, and Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Weber K, Mourao A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, and Lorentzen E (2016). Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. EMBO J. 35, 773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY-R, Kudryashev M, Li X, Egelman EH, Basler M, Cheng Y, Baker D, and DiMaio F (2015). De novo protein structure determination from near-atomic resolution cryo-EM maps. Nat. Methods 12, 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Lechtreck K-F, and Lorentzen E (2018). Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery, Essays Biochem. 62, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsmith J, Apelt L, Casado-Medrano V, özkan Z, Timmermann B, and Stelzl U (2017). Protein interaction perturbation profiling at amino-acid resolution. Nat. Methods 14, 1213–1221. [DOI] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin H-TJ, Burlingame A, Gerstein M, and Frydman J (2008). Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 15, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Wu Y-J, Zhu M, Fan S-B, Lin J, Zhang K, Li S, Chi H, Li Y-X, Chen H-F, et al. (2012). Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906. [DOI] [PubMed] [Google Scholar]
- Ye F, Nager AR, and Nachury MV (2018). BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 217, 1847–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeytuni N, and Zarivach R (2012). Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20, 397–405. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Yu D, Seo S, Stone EM, and Sheffield VC (2012a). Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable Bardet-Biedl Syndrome protein complex, the BBSome. J. Biol. Chem. 287, 20625–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Seo S, Bugge K, Stone EM, and Sheffield VC (2012b). BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum. Mol. Genet. 21, 1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Nishimura D, Vogel T, Shao J, Swiderski R, Yin T, Searby C, Carter CS, Kim G, Bugge K, et al. (2013). BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci. 126, 2372–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, and Washburn MP (2006). Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1, related to Figure 2: Assignment of BBSome domains to densities in the cryo-EM map using alternative 3D classes, binary interactions and subunit tracing. The color scheme is as in Figure 2C.
Movie S2, related to Figure 3: 360° view of the peptide backbone model. The density map is shown as transparent surface with the first rotation. The color scheme is as in Figure 3B. The placement of BBS2GAE and BBS7GAE is indicated by local coloring of the density map in red (BBS2GAE) and purple (BBS7GAE).
Data Availability Statement
Data Deposition
The maps have been deposited in wwPDB under ID codes EMD-7839 and EMD-7841 and the model has been deposited in PDB-Dev under ID code PDBDEV_00000018.
Code Availability
This study did not produce additional software. The software used here (summarized in Key Resources Table) has been published and is publicly available.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli Rosetta2(DE3)-pLysS | Millipore Sigma | 70956 |
| Biological Samples | ||
| Bovine retina | W.L. Lawson | Custom order |
| Experimental Models | ||
| E. coli Rosetta2(DE3)-pLysS | Millipore Sigma | 70956 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BS3 | Thermo Scientific | A39266 |
| DSSeb | Proteochem | c1136 |
| Sequencing-grade modified trypsin | Thermo Scientific | PI90305 |
| Deposited Data | ||
| Cryo-EM map of intact BBSome | This paper | EMD-7839 |
| Cryo-EM map of BBSome missing BBS5 | This paper | EMD-7841 |
| Cα model of BBSome | This paper | PDBDEV_00000018 |
| Coordinates of the BBS1βprop–ARL6GTP complex | (Mourão et al., 2014) | PDB: 4V0M |
| Recombinant DNA | ||
| pGEX6P-ARL6ΔN16[Q73L] | (Jin et al., 2010) | N/A |
| Software and Algorithms | ||
| UCSFImage4 | (Li et al., 2015) | https://cryoem.ucsf.edu/software/UCSFImage.html |
| MotionCor2 | (Zheng et al., 2017) | https://msg.ucsf.edu/software |
| CTFFIND4 | (Rohou and Grigorieff, 2015) | http://grigoriefflab.janelia.org/ctffind4 |
| Relion 1.3 | (Scheres, 2015) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| SPARX | (Hohn et al., 2007) | http://sparx-em.org/sparxwiki/SparxWiki |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| genesilico | (Kurowski and Bujnicki, 2003) | https://www.genesilico.pl/meta2/ |
| pLink | (Fan et al., 2015; Yang et al., 2012) | http://pfind.ict.ac.cn/software/pLink/index.html |
| hhpred | (Söding, 2005) | https://toolkit.tuebingen.mpg.de/#/hhpred |
| RosettaCM | (Song et al., 2013) | https://www.rosettacommons.org/software |
| Rosetta ab initio | (Bradley et al., 2005) | https://www.rosettacommons.org/software |
| RosettaES | (Frenz et al., 2017) | https://www.rosettacommons.org/software |
| Other | ||
| Quantifoil R1.2/1.3 holey carbon film on copper 200 mesh grids | Electron Microscopy Sciences | Cat #Q250CR-06 |