

Summary:
New approaches are required for addressing the important and unmet need to expand the benefits of checkpoint blockade to patients with mismatch repair proficient (MMRp) colorectal cancer. Systematic profiling of the tumor immune microenvironment provides insights into potential mechanisms of resistance, predictive biomarkers, and novel combinatorial strategies for overcoming resistance.
Keywords: colorectal cancer, immunotherapy, tumor immune microenvironment
In this issue of Clinical Cancer Research, Llosa and colleagues describe a gene expression-based approach to characterize the tumor immune microenvironment (TiME) of patients with advanced mismatch repair proficient (MMRp) colorectal cancer (CRC) (1). This approach uncovers evidence that upregulation of intra-tumoral IL-17-mediated signaling may attenuate responses to checkpoint inhibition even in the context of an immunoreactive microenvironment within MMRp tumors. As such, the work by Llosa and colleagues provides a roadmap for significantly expanding the population of patients with advanced CRC eligible to receive therapy with checkpoint inhibitors
Currently, the role of immunotherapy in advanced CRC is limited to the mismatch repair deficient (MMRd) subtype, which represents only a minority (approximately 5%) of cases (2). As shown in multiple phase II studies, treatment of advanced MMRd CRC with anti-programmed cell death protein 1 (PD-1) antibody alone or in combination with anti-cytotoxic T lymphocyte associated antigen 4 (CTLA-4) antibody is associated with durable clinical benefit (2). The molecular genetic driver for this activity is clearly understood to be adaptive immune recognition of novel expressed peptides (neoantigens) produced from a high burden of somatic frameshift and point mutations in the tumor cell population.
While the benefit of checkpoint blockade in MMRd CRC is now well-established, there has been considerable effort to understand potential mechanisms of primary and secondary (acquired) resistance in this subtype. Primary resistance appears to be uncommon, although misdiagnosis of MMR status was recently implicated in a substantial proportion of cases (3). Shin and colleagues also identified inactivating JAK1/2 mutations as a potential cell-intrinsic mechanism of primary resistance (4). For secondary resistance, current evidence points towards the acquisition of defects in antigen processing and presentation as playing a major role (e.g. inactivation of β-2-microglobulin or mutation of human leukocyte antigen [HLA] class I). These insights will require further exploration and validation, but nonetheless will inform future therapeutic strategies for patients with advanced MMRd CRC.
In contrast, limited progress has been made to date with respect to the utility of checkpoint inhibitors for patients with MMRp CRC, which is the most common subtype where response rates to anti-PD-1 therapy are poor. In a landmark phase II study (KEYNOTE-016, clinicaltrials.gov ID ), Le and colleagues observed a disease control rate of only 11% (2 / 18) for patients with advanced MMRp CRC who received anti-PD-1 monotherapy in the second line or greater (5). Furthermore, unlike the case for other common cancers, PD-L1 expression does not appear to be a predictive biomarker for MMRp CRC. Consequently, two critical questions have emerged: 1) is there a molecularly-defined subset of MMRp CRC tumor that are immunosensitive; 2) can MMRp tumors convert from immunoresistant to immunosensitive?
These questions have been the focus of comprehensive efforts to define the tumor cell-intrinsic and TiME-mediated mechanisms of resistance to checkpoint blockade in MMRp CRC, which is a challenge shared across many tumor types (6). An important perspective emerging from this work is that the somatic landscape of MMRp CRC clearly plays a significant role in modulating the anti-tumor immune response. Elegant pre-clinical studies of murine CRC models have provided evidence that key oncogenic drivers, such as mitogen-activated protein kinase (MAPK) and transforming growth factor-β (TGFβ) activation, may promote the exclusion of CD8+ cytotoxic T cells from or recruitment of myeloid-derived suppressor cells to the TiME, respectively. By virtue of demonstrating enhanced efficacy with the combination of targeted small-molecule inhibitors and anti-PD-1 therapy in these models, such studies highlight not only the apparent plasticity of the MMRp CRC microenvironment, but also potential biomarkers as well. While these insights are promising, successful translation into therapeutic clinical trials has been proven difficult. For example, in a recent negative phase III study (IMblaze370, clinicaltrials.gov ID ) testing the efficacy of anti-PD-L1 with or without MEK inhibition in advanced CRC, RAS/RAF mutation status was not correlated with response rate (7).
In this context, the present study by Llosa and colleagues provides a significant step forward in our understanding of other clinically-meaningful TiME factors (1). The investigators adeptly leveraged a repository of primary tumor samples obtained from treatment-naïve patients with advanced MMRp CRC who had received anti-PD-1 monotherapy through the aforementioned KEYNOTE-016 trial. By systematically comparing the expression of 44 immune-related genes between MMRd tumors that responded (partial or complete) versus MMRp tumors that did not (stable or progressive disease), the investigators discovered a concise 10-gene signature that predicts immunosensitivity. Upon further analysis and validation, approximately 3 (21%) out of 14 unselected MMRp CRC tumors harbored the immunosensitivity signature, suggesting that such tumors may response to checkpoint inhibition (however this was not formally tested). In addition, CD8+ tumor infiltrating lymphocytes (TILs) from these predicted immunosensitive tumors displayed an exhaustion phenotype, characterized by upregulation of PD-1 / PD-L1 and CXCL13, and downregulation of interferon gamma (IFNγ). Most strikingly, 4 (36%) of the remaining 11 MMRp CRC tumors that were putatively classified as non-immunosensitive were found to be enriched for an IL-17+ TIL population co-existing in the background of an exhausted phenotype.
The gene expression-based signature presented by Llosa and colleagues holds interesting potential as a novel predictive biomarker. However, a major confounding factor of their approach is that the immunosensitive signature was derived by comparing MMRd responders to MMRp non-responders. A direct comparison of MMRp responders to non-responders might capture other important elements in the TiME that were not seen in the present study. Yet, due to the rarity of MMRp responders, such an analysis is not feasible without pooled outcomes data and gene expression profiles from multiple therapeutic trials involving checkpoint blockade and MMRp CRC.
Nonetheless, as reported, the prospect of identifying 10–20% of patients with MMRp CRC who might benefit from checkpoint inhibition would represent a major advancement in the standard-of-care. Furthermore, the observation that IL-17+ T cells are enriched in a subset of putatively immune-resistant tumors is especially interesting considering the multitude of roles that IL-17 signaling may play in maintaining gut and microbiota homeostasis (8), and the clinical availability of IL-17 inhibitors. This finding warrants further investigation, as it may point towards a novel combinatorial strategy (e.g., anti-IL17 and anti-PD) for overcoming resistance to checkpoint blockade in MMRp.
The work by Llosa and colleagues also raises key questions: (1) how does the immunosensitive TiME signature correlate with relevant patient demographics and other well-established biomarkers for MMRp CRC (e.g. RAS/RAF mutation status, tumor sidedness, consensus molecular subtype); (2) how does exposure to cytotoxic chemotherapy or targeted therapy change the TiME, if at all; and (3) does the signature capture biology that is unique to the microenvironment of the primary tumor or biology that is relevant across different metastatic sites (e.g. liver, lung, peritoneum) as well?
Moving forward, independent validation of the immunosensitive TiME signature as a predictive biomarker may be challenging, especially given the limited number of treatment-naïve MMRp CRC patients who have received anti-PD-1 monotherapy through clinical trial. Nonetheless, this work provides a useful framework for understanding the heterogenous nature of the MMRp CRC immune microenvironment (Figure 1). It also exemplifies the critical need to embed correlative multi-omic analyses within therapeutic trials for advanced MMRp CRC.
Figure 1.
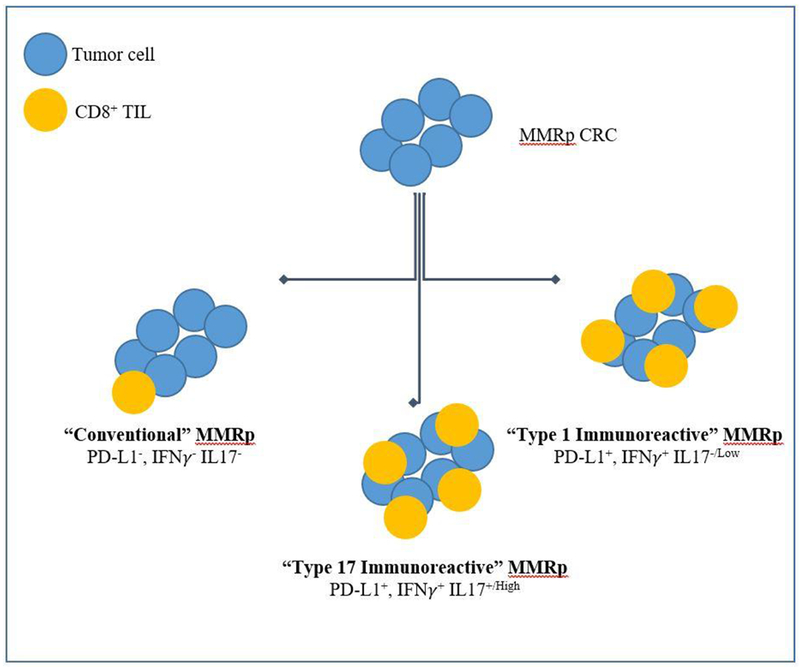
Subtypes of MMRp colorectal cancer as defined by comprehensive profiling of the tumor microenvironment.
Acknowledgements:
This work was supported by grant Research Training in Academic Medical Oncology T32 Award CA009666-23 (U.S. National Institutes of Health/National Cancer Institute) to J.W.; R01 CA219463 (US National Institutes of Health/National Cancer Institute) and a gift from the Feinberg Family to E.V.; and P30 CA016672 (US National Institutes of Health/National Cancer Institute) to the University of Texas Anderson Cancer Center Core Support Grant.
Abbreviations:
- CRC
colorectal cancer
- TiME
tumor immune microenvironment
- MSS
microsatellite stable
- MMR
mismatch repair
- MMRd
MMR deficient
- MMRp
MMR proficient
- PD-1
programmed cell death protein 1
- PD-L1
programmed death-ligand 1
- TIL
tumor-infiltrating lymphocytes
Footnotes
Conflict of Interest: EV: has a consulting role with Janssen Research and Development. MO: has consulting role with Bristol Myers Squibb, Novartis, Roche, Medimmune/Astra Zeneca and research funding from Bristol Myers Squibb, Merck, Roche, Medimmune/Astra Zeneca. No other disclosures are reported.
References:
- 1.Llosa NJ. Intratumoral adaptive immunosuppression and type 17 immunity in mismatch repair proficient colorectal tumors. Clinical Cancer Research 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nature Reviews Gastroenterology & Hepatology 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen R, Hain E, Buhard O, Guilloux A, Bardier A, Kaci R, et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer With Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncology 2019;5(4):551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK½ Mutations. Cancer Discovery 2017;7(2):188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine 2015;372(26):2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature Reviews Drug Discovery 2019;18(3):197–218. [DOI] [PubMed] [Google Scholar]
- 7.Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. The Lancet Oncology 2019 [DOI] [PubMed] [Google Scholar]
- 8.Hurtado CG, Wan F, Housseau F, Sears CL. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018;155(6):1706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]