

Abstract
Eya proteins (EYA1–4) are critical developmental transcriptional cofactors that contain an Eya domain (ED) harboring Tyr phosphatase activity. Eya proteins are largely downregulated post-embryogenesis but are re-expressed in cancers, and their Tyr phosphatase activity plays an important role in the DNA damage response and tumor progression. We previously identified a class of small molecule allosteric inhibitors that specifically inhibit the Tyr phosphatase activity of Eya2. Herein, we determined the crystal structure of the Eya2 ED in complex with NCGC00249987 (a representative compound in this class), revealing that it binds to an induced pocket distant from the active site. NCGC00249987 binding leads to a conformational change of the active site that is unfavorable for Mg2+ binding, thereby inhibiting Eya2’s Tyr phosphatase activity. We demonstrated, using genetic mutations, that migration, invadopodia formation, and invasion of lung adenocarcinoma cells are dependent on Eya2 Tyr phosphatase activity, whereas growth and survival are not. Further, we demonstrate that NCGC00249987 specifically targets migration, invadopodia formation, and invasion of lung cancer cells, but that it does not inhibit cell growth or survival. The compound has no effect on lung cancer cells carrying an Eya2 F290Y mutant that abolishes compound binding, indicating that NCGC00249987 is on target in lung cancer cells. These data suggest that the NCGC00249987 allosteric inhibitor can be used as a chemical probe to study the function of the Eya2 Tyr phosphatase activity in cells, and may have the potential to be developed into an anti-metastatic agent for cancers reliant on Eya2’s Tyr phosphatase activity.
Keywords: Eya2, Tyrosine phosphatase, phosphatase inhibitors, Six1, lung cancer
Introduction
The Eya proteins (Eya1–4) are evolutionarily conserved transcriptional co-activators that interact with the Six family of homeobox proteins to activate transcription (1–4). Eya and Six1 proteins together form bipartite transcription factors, in which Six1 determines DNA binding specificity through its homeodomain, and Eya family members mediate transactivation through their N-terminal domains (4). While the Six1/Eya complex is known to be critical in normal embryogenesis but down regulated in normal adult tissues (5,6), both Six1 and Eya proteins are overexpressed in many types of tumors, where their overexpression is thought to reinstate developmental properties out of context (6). Recently, we demonstrated that knockdown (KD) of Eya2 in Six1-overexpressing breast cancer cells (MCF7) inhibits the ability of Six1 to induce TGF-β signaling, epithelial-mesenchymal transition (EMT), and tumor initiating cell (TIC) characteristics, properties that are all associated with Six1-induced tumorigenesis and metastasis (7). Furthermore, we demonstrated that ectopic expression of a naturally occurring Six1 mutant (V17E), that cannot bind to Eya but retains DNA binding activity, fails to cause metastasis in a xenograft model of breast cancer (2). Intriguingly, Eya2 may play an especially critical role in lung cancer, as a SNP in its locus is significantly associated with lung cancer patient survival (8), and it contributes to lung adenocarcinoma proliferation, migration, invasion, and metastasis (9–11). Moreover, enhanced expression of both Eya2 and Six1 is associated with increased invasiveness and shortened overall survival of lung cancer patients (9,10,12,13).
In addition to acting as transcriptional co-activators, the Eya proteins are unique in that they contain two regions with associated phosphatase activity (1). The N-terminal region of Eya proteins contains not only a transcriptional activation domain, but also an associated Thr phosphatase activity mediated through an interaction with protein phosphatase 2A (14). The C-terminal Eya Domain (ED) of Eya has intrinsic Tyr phosphatase activity (15–17). This domain contains a conserved motif from the haloacid dehalogenase (HAD) superfamily, a diverse collection of hydrolases that are well studied in prokaryotes. Eya proteins and other members of the HAD family of phosphatases use an Asp as their active site residue instead of the Cys residue more commonly used by other cellular Tyr phosphatases (18). This enzymatic activity requires the presence of Mg2+, which may also be important for the folding of this domain (19). All other known HAD protein phosphatases are Ser/Thr phosphatases, while the Eya domain of Eya targets phosphorylated Tyr (20,21). Although the role of Eya phosphatase activity is not yet well defined, mouse Eya proteins have been shown to utilize their intrinsic phosphatase activity to switch the Six1 transcriptional complex from a repressor to an activator complex for Six1-induced genes (17). The Tyr phosphatase activity of Eyas also regulate multiple cellular phenotypes which are associated with tumor progression. For example, the Tyr phosphatase activity of Eya proteins has been shown to be critical for transformation, migration, invasion, and metastasis in breast cancer (22).
To date, there are three identified Tyr phosphorylated substrates of Eya proteins, H2AX (20,21,23), estrogen receptor β (ER-β) (24), and WDR1 (25). When Tyr-142 on H2AX is dephosphorylated after DNA damage, cells are directed to the repair pathway instead of the apoptotic pathway (20,21). Thus, inhibitors of Eya’s Tyr phosphatase activity may increase the efficiency of radiation therapy or DNA damaging related therapy in cancers that are known to overexpress Eya proteins. Examples of such cancers include breast cancer (7,22,26), ovarian carcinomas (27), Wilms’ tumor (28), lung cancers (29) and Ewing sarcomas (EWS) (30), the latter where Eya loss is shown to chemosensitize cells (30). Further, because Eya proteins such as Eya2 dephosphorylate ER-β to inhibit its tumor suppressive activities, targeting its phosphatase activity could re-activate the tumor suppressive effects of ER-β in breast cancer (24). Although the impact of Eya-mediated dephosphorylation of WDR1 in cancer is unknown, given the role of WDR1 in regulation of the cytoskeleton (25), it is possible that WDR1 will also be a target of interest for Eya in cancer.
These data suggest that the Eya Tyr phosphatase activity may play a critical role in tumor progression. However, there are no compounds in clinical development for this protein, as the active site of phosphatases is typically considered not druggable. Generating chemical probes that specifically inhibit the Eya Tyr phosphatase would be valuable to enable dissection of Eya Tyr phosphatase function in tumor progression, and may be of future interest as an anticancer therapeutic. We previously identified a novel class of highly selective Eya2 phosphatase inhibitors (which are inactive against Eya3) through a high-throughput screen (31,32). Biochemical analyses suggested that this inhibitor works through an allosteric mechanism. In this study, we determined the co-crystal structure of Eya2 ED in complex with a representative compound of this class (NCGC00249987, abbreviated as 9987). The structure reveals that binding of 9987 to an allosteric site changes the active site conformation of Eya2, consequently inhibiting Eya2’s Tyr phosphatase activity. We demonstrate that this allosteric inhibitor is on target in cells, where its effect on lung cancer migration and invasion mimic what is observed with the Eya2 D274N mutant that is Tyr phosphatase dead. The compound also has no effect on lung cancer cells carrying an Eya2 F290Y mutant that abolishes compound binding in vitro. Our study thus demonstrates the utility of 9987 as a chemical probe to understand the function of the Eya2 phosphatase, and serves as a starting compound for possible future development of anti-metastatic agents.
Materials and methods
Compound synthesis and characterization
(E)-3-fluoro-N’-((5-(pyridin-2-ylthio)furan-2-yl)methylene)benzohydrazide (MLS000544460)
A solution of 5-(pyridin-2-ylthio)furan-2-carbaldehyde (100 mg, 0.487 mmol., 1.0 equiv.) and 3-fluorobenzohydrazide (75.1 mg, 0.487 mmol., 1.0 equiv.) in anhydrous ethanol (4.8 mL, 0.1 M) was stirred for 18 h at 90°C under nitrogen atmosphere. Upon completion, excess solvent was removed under reduced pressure and partitioned between dichloromethane and water. The organic phase was separated, washed with aqueous sodium chloride, dried over sodium sulfate and concentrated to dryness. The residue was purified using reverse phase chromatography (C18, eluent 70 – 90% acetonitrile (0.1% formic acid)/water (0.1% formic acid) to give product (33.3 mg, 0.0980 mmol.) as a pale yellow solid. 1H NMR of the compound in DMSO-d6 was collected and the chemical shifts of protons were obtained: δ 11.98 (s, 1H), 8.42 (d, J = 4.40 Hz, 1H), 8.33 (s, 1H), 7.68 – 7.75 (m, 3H), 7.59 (q, J = 7.60 Hz, 1H), 7.46 (t, J = 8.40 Hz, 1H), 7.21 – 7.24 (m, 1H), 7.13 – 7.18 (m, 2H), 6.99 (d, J = 8.40 Hz, 2H). LCMS (ESI) showed a m/z of 342 (MH+) with a purity of 95.3%.
(E)-3-fluoro-N’-((5-(pyrimidin-2-ylthio)furan-2-yl)methylene)benzohydrazide (NCGC00249987/9987)
A solution of 5-(pyrimidin-2-ylthio)furan-2-carbaldehyde (150 mg, 0.727 mmol., 1.0 equiv.) and 3-fluorobenzohydrazide (112 mg, 0.727 mmol., 1.0 equiv.) in anhydrous ethanol (7.3 mL, 0.1 M) was stirred for 18 h at 90°C under nitrogen atmosphere. Upon completion, the suspension was filtered and the residue washed with methanol to give product (140 mg, 0.409 mmol.) as off-white solid. 1H NMR of the compound in DMSO-d6 was collected and the chemical shifts of protons were obtained: δ 11.96 (s, 1H), 8.66 (d, J = 4.80 Hz, 2H), 8.32 (s, 1H), 7.74 (d, J = 7.60 Hz, 1H), 7.69 (d, J = 9.60 Hz, 1H), 7.59 (q, J = 7.60 Hz, 1H), 7.44 – 7.48 (m, 1H), 7.33 (t, J = 4.80 Hz, 2H), 7.10 – 7.14 (m, 2H). LCMS (ESI) showed a m/z of 343 (MH+) with a purity of 99.5%.
Protein expression and purification for crystallization
Human Eya2 ED (residues 253–538) was cloned into pGEX6P-1 and purified as described previously (31). Briefly, recombinant GST-Eya2 ED protein was expressed in E. coli and purified on a gravity column filled with glutathione resin. Purified protein was cleaved with PreScision protease (GE Healthcare) and GST was removed by gel filtration chromatography. Protein was purified in a buffer containing 20 mM HEPES, 150 mM NaCl, 0.5 mM EDTA, and 1 mM DTT. Protein was concentrated and kept at −80 °C after freezing in liquid nitrogen for long-term storage.
Crystallization
Purified Eya2 ED protein was mixed with 9987 (1:2 molar ratio) and concentrated to 5 mg/ml for co-crystallization experiments. In addition, 5mM TCEP was added to prevent oxidation of the protein sample. Optimal crystals were grown in a sitting drop containing 2:1 (v/v) ratio of protein to reservoir solution (0.1M HEPES 7.5, 200mM NaCl, 20% PEG 3350) at 293K. The crystals were cryoprotected using 25% glycerol and flash frozen in liquid nitrogen.
The X-ray diffraction dataset was collected at the MX2 beamline at the Australian Synchrotron. The collected data was processed and scaled using XDS (38). The structure of the Eya2 ED-9987 complex was solved using the Eya2 structure (PDB: 3HBO) as an initial model for the molecular replacement method in PHASER in PHENIX (39). Further model building and iterative refinement was performed using COOT and PHENIX, respectively (40,41).
Protein expression and purification for biochemical analyses
Human Eya2 ED (residues 253–538) was sub-cloned into a pET15b vector. The Eya2 ED F290Y mutant was generated via Site Directed Mutagenesis using PCR with Q5 polymerase (New England BioLabs). Human Eya1 ED (residues 307–592) was codon optimized, synthesized, and cloned into vector pJ404 by DNA2.0 (Currently ATUM). Human Eya4 ED (residues 355–639) was codon optimized, synthesized, and cloned into vector pGEX-6P1 by GenScript.
Eya2 ED was expressed in E. coli strain BL21(DE3), and Eya1 ED was expressed in E. coli strain XA90. The WT Eya1 ED, Eya2 ED and mutant Eya2 ED proteins were first loaded on a Ni-NTA column in a buffer containing 50 mM HEPES (pH7.5), 600 mM NaCl, 10% glycerol, 20 mM imidazole and 5 mM β-mercaptoethanol (BME). The proteins were then washed with the above buffer containing 20 mM imidazole and buffer containing 40 mM imidazole, before being eluted in the above buffer containing 250 mM imidazole. All proteins were further purified on a Superose 6 Increase column (GE Healthcare) in FPLC buffer containing 50 mM HEPEs (pH7.5), 250 mM NaCl, 5 mM MgCl2, 5% glycerol and 1 mM TCEP.
Eya4 ED was expressed in E. coli strain XA-90 and purified as described previously for Eya2 ED (31). Briefly, GST-fusion protein was first purified on glutathione resin, cleaved using GST-tagged PreScission protease, and Eya4 ED protein was eluted with 50 mM HEPEs (pH7.5), 250 mM NaCl, 5% glycerol and 1 mM DTT.
Eya2 full length (FL) was sub-cloned into a pMALX(wt) vector (42), with an MBP-tag at the N-terminus. The full length Eya2 protein was expressed in E. coli strain XA-90. The protein was purified on Amylose resin (New England BioLabs) in 40 mM Tris (pH8.0), 450 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, and 5% glycerol, and eluted using the same buffer containing 10mM Maltose. FL Eya2 was further purified on a Superose 6 Increase column in the FPLC buffer.
Phosphatase assay
Eya phosphatase activity was measured in 50 μl reactions, using black, 96-well, half-volume microtiter plates (GreinerBio-one) with OMFP (3-O-methylfluoresceinphosphate, Sigma-Aldrich) as a substrate, which is converted to a fluorescent product OMF upon dephosphorylation. First, 25 μl of Eya2 ED, Eya2 ED F290Y, or FL Eya2 (final concentration 150 nM) in the reaction buffer (25 mM HEPES (pH6.5), 50 mM NaCl, 5 mM MgCl2, 0.33% BSA, and 5 mM DTT) was added into wells, then 0.5 μl 9987, that was serially diluted in DMSO at eight different concentrations, was added to the corresponding wells. The plate was incubated for 10 minutes at room temperature. Reactions were started with the addition of OMFP (final concentration 50 μM), incubated for 1 hour at room temperature in the dark, then terminated with the addition of EDTA (final concentration 75 mM). Phosphatase assays for different Eya family members were carried out using 150 nM Eya1 ED, Eya2 ED, or Eya4 ED, 50 μM OMFP, and 100 μM of 9987. Fluorescence intensity was measured at 485/515 nm excitation/emission on a Fluoromax-3 plate reader (Horiba Jobin Yvon). The data were analyzed and IC50s were obtained using the Prism software (GraphPad).
Cell culture
All lung cancer cell lines were cultured in RPMI-1640 medium (Hyclone SH30027.01) + 10% fetal bovine serum (Corning 35–010-CV) + 1% penicillin/streptomycin (Hyclone SV30010) in Forma Steri-Cycle CO2 incubator (ThermoFisher 370) at 37 °C with 5% CO2. Parental Calu-6 cells were retrovirally infected with empty, WT or D274N pMSCV-IRES-YFP vector (Addgene 52108). After serial passaging, cells were fluorescently sorted by YFP to select cells with permanent integration of vectors. Cell line identity was authenticated by STR analysis performed by University of Colorado Tissue Culture Core in Sept. and Oct. of 2018 after work was completed.
Immunoblotting
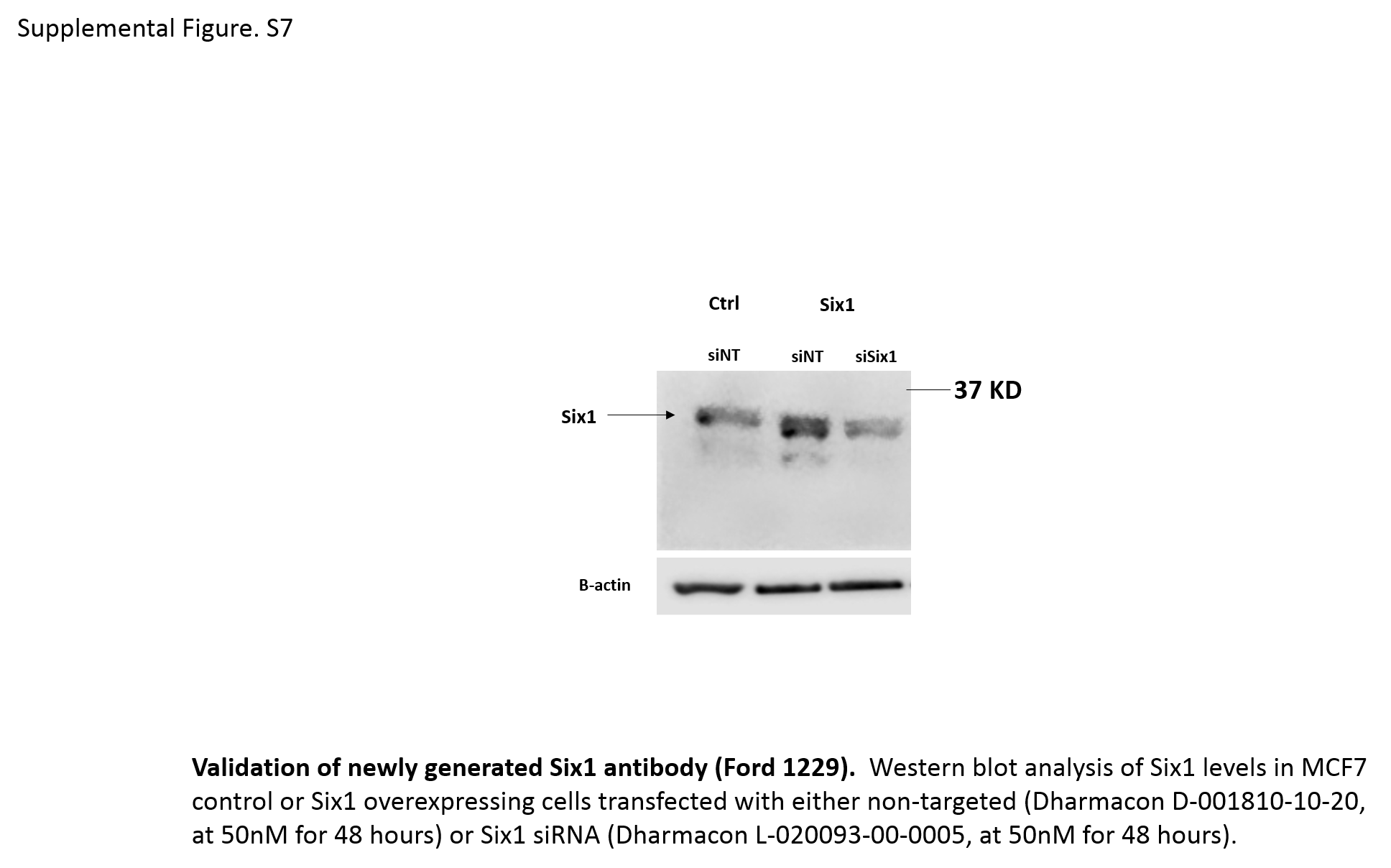
As described previously (14), protein lysates were collected using RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) containing protease inhibitors (Thermo Scientific, A32965), and were then electrophoresed on 10% SDS-Page gels. Proteins were then transferred from gels onto nitrocellulose or PVDF membranes, after which the membranes were blocked in 5% milk and incubated with primary antibody as follows: A new Anti-Six1 antibody (Ford 1229, 1:1000), made by MAbVista against GST-Cterm Six1 and MBP-cleaved Cterm Six1 fusion proteins (the latter as the boost) was used to detect Six1 levels (Supplementary Fig. S7). Other antibodies used include Anti-Eya2 (Sigma HPA027024, 1:1000), Anti-FN1 (BD bioscience 610077, 1:1000), Anti-E-cadherin (Cell signaling 3195S, 1:3000), Anti-Vimentin (Cell signaling D21H3, 1:1000), Anti-CK18 (Abcam ab82254, 1:1000), Anti-GAPDH (Cell signaling D16H11, 1:5000), Anti-β-Actin (Sigma A5316, 1:5000), Anti-HDAC1 (Santa Cruz sc-7872, 1:3000), and Anti-β-Tubulin (Sigma T4026, 1:5000). After washing the membranes in TBST (20mM Tris base, 136 mM NaCl and 0.1% Tween-20 in ddH2O, pH=7.6) appropriate secondary antibodies were applied to membranes (polyclonal goat anti-rabbit IgG-HRP (Sigma A9169, 1:5000) or rabbit anti-mouse IgG-HRP (Sigma A9044, 1:5000). Membranes were then washed in TBST 3 times, and chemiluminescence was used to measure the signal.
IncuCyte imaging for growth and Caspase 3/7 activation
As described previously (26), 1500 cells were seeded in each well of 96-well plates and live images of cells in culture were taken at various time points after seeding (at a 6-hour intervals) using IncuCyte (Essenbioscience). Cell growth was evaluated by quantitating images taken using bright-field (% confluence), while Caspase 3/7 activation was detected using IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (Essenbioscience 4440) and assessed by green fluorescent puncta intensity count.
2D Clonogenic Assay
500 cells were seeded in each well of 24-well plates and cells were allowed to grow for 7 days. Cells were then fixed by methanol at room temperature for 20 mins, stained using 0.1% crystal violet and photographed in grey scale using ImageQuant LAS4000 (GE Health Care Life Sciences 28955810). Colony intensity was quantified using ImageJ based on the purple intensity of the images.
Cell cycle analysis
Cells were plated at ~70% confluency and allowed to attach overnight, after which they were treated with 1μg/ml BrdU (Sigma B5002) for 45 min. After incubation, cells were collected and fixed using 70% ethanol for 1 hr, prior to sequential 2M HCl (with 0.5% Triton X-100) and 0.1M sodium borate treatment. Cells were then stained using anti-BrdU IgG (Santa Cruz sc-51514, 1:200) and Alexa 488 conjugated secondary antibody (Invitrogen A21202, 1:300), and propidium iodode (PI, BD Pharmingen 51–66211E) (with RNAse A) was added for a 30-min incubation in the dark. BrdU incorporation and cell cycle progression were analyzed by flow cytometry. Diploid cells were gated for analysis.
Migration assay
5×104 cells were seeded on each side of migration inserts (ibidi 81176). After cells attached (overnight), the inserts were removed and cells were allowed to migrate for 12/24 h (for Calu-6 and HCC4006, respectively). Cells were photographed at 0 h after removal of inserts and again at 12/24 h. Distance migrated was determined by measuring the remaining gap over 10 representative points between two migrating fronts, and an average was determined for each time point. The experiments were repeated at least five times.
Gelatin degradation assay
Lung cancer cell invadopodia formation was examined using the QCM™ Gelatin Invadopodia Assay kit (EMD Millipore ECM670). Gelatin-coated glass surfaces were prepared as follows: 8-well glass chamber slides were coated with poly-L-Lysine for 20 mins, followed by 15 mins. of glutaraldehyde crosslinking. Gelatin was then introduced to the surface of the slides for 10 mins, after which the slides were disinfected with 70% ethanol. After gelatin-coating, 1×104 cells were seeded in each well of chamber slides and allowed to attach for 24 hrs. Cells were then fixed and permeabilized for fluorescent staining of F-actin using TRITC-phalloidin. Finally, invadopodia (actin-rich and spike-like protrusions) were quantified via fluorescent microscopy, more than 6 fields and 180 cells were examined per condition.
Invasion assay
As previously described (26), 1×105 cells in serum free medium were added to the top of matrigel (Corning 354234)-coated 8μm-pore transwells (Corning CLS3422) that were submerged in serum containing medium. After 18/24 hrs (for HCC4006 and Calu-6, respectively), cells were fixed, and then stained using 0.1% crystal violet and photographed using bright field microscopy. At last, invasion was quantified using ImageJ based on the invaded percentage of the images. More than 6 fields were examined per condition.
AlphaScreen assay
The AlphaScreen assay was carried out using 6xHis-Eya2 ED and GST-Six1 (1–198) proteins following the manufacturer’s protocol (PerkinElmer, Waltham, MA, USA) unless otherwise specified. 6xHis-Eya2 ED and GST-Six1 (1–198) were purified as described in the protein purification section above or as previously reported (2) (protein purification section). 50 nM 6xHis- Eya2 ED and 50 nM GST-Six1 (1–198) were used in the assay in the presence of varying concentrations of 9987. The compound inhibition curve was analyzed using the GRAPHPAD PRISM software (GraphPad Software, San Diego, CA, USA).
Statistical Analyses
As previously described (26), comparison over a period of time (growth curves) was subjected to a mixed-effects model to fit the data, while significant differences across different conditions were examined via a linear regression test and the corresponding P value was reported. For comparisons within multiple conditions at a static time point, 1-way ANOVA was conducted followed by Tukey’s post-test to report the P value. All graphs were generated using GraphPad Prism, with **P<0.01, ***P<0.001.
Results
9987 inhibits full length Eya2 but not other Eya family members
After identifying initial inhibitors of the Tyr phosphatase activity of Eya2 ED (31), we were able to synthesize an analog, NCG00249987 (9987), that retains similar potency as the original hit [MLS000544460 (31)] but has a much higher solubility (>51 ug/ml versus 8.9 ug/ml in PBS) (Fig. 1A). Because the high-throughput screen was carried out using recombinant Eya2 ED, we performed an in vitro phosphatase assay, using MBP-fused full length Eya2 (it is difficult to obtain sufficient quantities of purified full length Eya2 on its own), and found that 9987 inhibits the full-length Eya2 similarly to the Eya2 ED (Fig. 1B). In addition, we have previously shown that 9987 specifically inhibits the Tyr phosphatase activity of Eya2 ED, but does not inhibit that of the Eya3 ED (31). Herein we expressed and purified Eya1 and Eya4 ED from E. coli and demonstrated that these proteins have phosphatase activity and their corresponding phosphatase-dead mutants (Eya1 ED D328N and Eya4 ED D375N) abolish the phosphatase activity (Fig. 1C). 9987 does not inhibit the Eya1 or Eya4 ED, while EDTA does (Fig. 1D). These data demonstrate that 9987 is highly specific for Eya2. It is worth noting that 9987 has no effect on the interaction between Eya2 and its transcriptional partner, Six1 (Supplementary Fig. S1).
Figure 1.

Compound NCGC00249987 (abbreviated as 9987) inhibits the phosphatase activity of Eya2 full length similarly to the Eya2 ED, but does not inhibit Eya1 and Eya4 ED. A, Chemical structures of the original hit and analog 9987. B, Phosphatase activity of Eya2 FL or ED in the presence of increasing concentrations of 9987, as measured in a malachite green based phosphatase assay using OMFP as a substrate. All activities are normalized to the activity of protein without any compound which is set at 100%. “ns” indicates statistically non-significant, as calculated using two-sample t test. C, Phosphatase activity of Eya1, Eya2, or Eya4 ED and their corresponding phosphatase-dead mutants, as measured in a malachite green based phosphatase assay using OMFP as a substrate. The activity of mutant protein is normalized to the activity of WT protein which is set at 100%. All proteins were expressed and purified from E. coli. D, Phosphatase activity of Eya1, Eya2, or Eya4 ED on their own (untreated), and in the presence of DMSO, EDTA, and 9987 (100 μM), as measured in a malachite green based phosphatase assay using OMFP as a substrate. All activities are normalized to the activity of untreated protein which is set at 100%.
Crystal structure reveals the mechanism of 9987 as an allosteric Eya2 inhibitor
Biochemical kinetics and mutagenesis analyses suggest that 9987 is an allosteric Eya2 inhibitor (31). To better understand how 9987 acts as an allosteric inhibitor, we set out to determine the crystal structure of the Eya2 ED in complex with 9987. We screened for crystallization conditions using the complex in the absence and presence of EDTA and only the sample with EDTA formed well-diffracting crystals (crystals from the sample without EDTA only diffracted to ~5 Å), consistent with our earlier observation that the original hit MLS000544460 binds to Eya2 ED more tightly in the presence of EDTA (31). The co-crystal structure was determined at 3.2 Å resolution and contains three Eya2 ED-9987 complex molecules in one asymmetric unit (Supplementary Fig. S2A). Data collection and refinement statistics are shown in Table 1. The overall structure of Eya2 ED adopts a similar fold as apo Eya2 ED (19) and Eya2 ED in complex with Six1 (2) (Fig. 2A).
Table 1.
Data collection and refinement statistics (PDB access code 5ZMA).
| Data Collection Statistics | Eya2 ED-9987 complex |
|---|---|
| Wavelength (Å) | 0.9538 |
| Resolution range(Å) | 47.8 – 3.2 (3.3 – 3.2) |
| Space group | I 1 2 1 |
| Unit cell | 143.9 49.4 144.9 90 101.2 90 |
| Total reflections | 33909 (3226) |
| Unique reflections | 17211 (1650) |
| Multiplicity | 2.0 (2.0) |
| Completeness (%) | 98.99 (95.65) |
| Mean I/sigma(I) | 10.20 (1.96) |
| Wilson B-factor | 85.48 |
| R-merge | 0.045 (0.350) |
| R-meas | 0.064 (0.495) |
| R-pim | 0.045 (0.350) |
| CC1/2 | 0.998 (0.781) |
| CC* | 1 (0.936) |
| Reflections used in refinement | 17202 (1649) |
| Reflections used for R-free | 1727 (161) |
| R-work | 0.228 (0.304) |
| R-free | 0.265 (0.374) |
| CC(work) | 0.953 (0.791) |
| CC(free) | 0.926 (0.702) |
| Number of non-hydrogen atoms | 5970 |
| macromolecules | 5898 |
| ligands | 72 |
| Protein residues | 758 |
| RMS(bonds) | 0.002 |
| RMS(angles) | 0.57 |
| Ramachandran favored (%) | 94.47 |
| Ramachandran allowed (%) | 5.39 |
| Ramachandran outliers (%) | 0.13 |
| Rotamer outliers (%) | 0.00 |
| Clashscore | 8.79 |
| Average B-factor | 85.45 |
| macromolecules | 85.75 |
| ligands | 61.34 |
Statistics for the highest-resolution shell are shown in parentheses.
Rmerge =ΣhΣi|Ihi-<Ih>|/Σh,i Ihi, where Ihi is the ith observation of the reflection h, while <Ih> is its mean intensity.
Rfactor =Σ ||Fobs| - |Fcalc|| / Σ |Fobs|.
Rfree was calculated with 5% of reflections excluded from the whole refinement procedure.
Figure 2.

Crystal structure of the Eya2 ED-9987 complex. A, Superposition of the inhibitor bound (blue) and apo (wheat (PDB: 4EGC)) crystal structures. Mg2+ bound at the active site of the apo structure is shown as a brown sphere. The major conformational changes in Eya2 ED are indicated by arrows. B, 9987 shown in green (stick representation) is bound at the interface between the catalytic domain (blue) and HBM (grey). The 2Fo-Fc map of 9987 (contoured at 1σ) is shown in blue. Consensus motif I-III of HAD family are colored in yellow and catalytic triad shown in stick representation. C, Surface representation of the Eya2 ED-9987 complex. The binding pocket for 9987 in Eya2 ED induced upon its binding is depicted. D, Surface representation of Eya2 ED apo structure shows the lack of the binding pocket. E, Hydrophobic residues lining the binding pocket of 9987 (<4Å) in Eya2 ED are shown in sticks. The 2Fo-Fc map of 9987 (contoured at 1σ) is shown in blue. F, Overlay of Eya2 ED-9987 (blue) and Eya2-Six1 (wheat) complex structures showing the rearrangements of hydrophobic residues of the binding pocket. Significant rearrangements of side chains upon compound binding are indicated by arrows. G, Hydrogen bond between T278, T421, L425 and H523 with the inhibitor is shown in dotted lines. The β1-α1 loop is displayed in yellow with residue T278 in stick representation. Residues T421, L425 and H523 from the allosteric site are displayed in blue sticks. H, D274, D276, T278 and D502 from apo and the complex structures are shown in wheat and yellow sticks, respectively. The compound induced conformational changes of the β1-α1 loop between the apo and the complex structure lead to changes in Mg2+ binding residues. In particular, T278 interacts with D274 and D502 in the absence of 9987, whereas in the compound-bound structure, T278 is pushed away to form hydrogen bond with the compound, which is indicated with an arrow.
Compound 9987 is bound at the interface between the catalytic domain and the Helical Bundle Motif (HBM) (Fig. 2B). An overlay of the structures of Eya2 ED in the presence and absence of 9987 show that the inhibitor binds away from the active site region (Fig. 2A), consistent with the allosteric nature predicted in previous biochemical and mutagenesis analyses (32). The active site of Eya2 ED is defined by three conserved sequence motifs (Motifs I, II and III) and a bound Mg2+, a feature shared by members of the HAD family (19). Mg2+ plays an important role in catalysis and it is coordinated by D274, D276, and D502 residues in the apo Eya2 ED (19) and Eya2 ED-Six1 structures (2). In the Eya2 ED-9987 complex structure, the β1- α1 loop (residues 274–282) containing Motif I (residues 274–278) undergoes significant conformational changes, and the catalytic and HBM domains are also slightly shifted away from each other at the interface to accommodate the inhibitor (Fig. 2A). These changes lead to a protein conformation around the active site that does not favor Mg2+ binding, as illustrated below through a detailed analysis of the interaction between 9987 and its binding pocket.
Unlike conventional compound binding pockets, 9987 binding creates a tunnel-like pocket that is not present in the absence of 9987 (Fig. 2C & 2D). 9987 interacts with Eya2 ED mainly through hydrophobic and hydrogen bond interactions. For example, the 9987-binding pocket is lined by many hydrophobic residues (I279, F282, F290, M308, L417, L420, W424, L425) and the aromatic rings of Y294 and Y461 (Fig. 2E). Of these residues, I279 and F282 on the β1- α1 loop moved significantly upon 9987 binding to accommodate the compound (Fig. 2F). Apart from these hydrophobic interactions, multiple hydrogen bond interactions are also observed between Eya2 ED and 9987. Residues T421, L425, and H523 from the allosteric binding site contribute to the majority of hydrogen bond interactions with 9987 (Fig. 2G). In addition, T278 of Motif I in the β1- α1 loop forms a hydrogen bond between its carbonyl oxygen and the hydrazine moiety of the inhibitor (Fig. 2G). T278 plays a crucial role in stabilizing the conformation of residues D274 and D502 which coordinate Mg2+ in the active site (19). In the presence of 9987, the conformational change of the β1- α1 loop causes residue T278 to move away from the active site to interact with the inhibitor (Fig. 2G) and T278 no longer interacts with D274 and D502 (Fig. 2H). This, together with the conformational changes in the catalytic triad residues D274 and D276 in Motif I, lead to an unfavorable conformation for Mg2+ binding. Therefore, the conformational change of the β1- α1 loop in the active site and the lack of a binding environment for Mg2+ provide the mechanism of how the binding of 9987 at the allosteric site inhibits the Eya2 phosphatase activity. It is worth noting that the 9987-binding pocket observed in our crystal structure overlaps with the proposed 9987-binding pocket based on mutagenesis analyses (31) (Supplementary Fig. S2B). Multiple residues (R414, L417, E418, L425) whose mutations significantly reduce compound binding are shared between the predicted and the actual 9987-binding pocket.
To elucidate the molecular mechanism of the selectivity of 9987 which is only active against Eya2, but not other Eya family members (31) (Fig. 1D), we mutated the only two residues in the 9987-binding pocket that are different between Eya2 and Eya3 (F290 and L417 in Eya2 whose counterparts are Y325 and I452 in Eya3, respectively). While L417I has a minimal effect on 9987 inhibition, the F290Y mutation abolishes compound inhibition (Fig. 3A). The Y290 residue can potentially form a hydrogen bond with T421, essentially closing the pocket to the compound (Fig. 3B). Alternatively, the extra hydroxyl group on Y290 (compared to F290) could create steric hindrance for compound binding. The equivalent residue of F290 in Eya2 is Y in all other Eya family members (Eya1, 3, 4) (Fig. 3C), which may explain why 9987 specifically inhibits Eya2, but does not inhibit any of the other Eya family members. However, mutating Eya3 Y325 (equivalent to F290 in Eya2) to F does not make Eya3 sensitive to 9987 inhibition (Supplementary Fig. S3). This data suggests that having an F residue in this position is necessary, but not sufficient, for compound inhibition. Thus, additional residues outside the binding pocket that are different between Eya2 and Eya3 must indirectly contribute to compound binding in Eya2.
Figure 3.

Eya2 ED F290Y mutation abolishes compound inhibition. A, Phosphatase activity of WT Eya2 ED or an F290Y or L417A mutant in the presence of increasing concentrations of 9987, as measured in a malachite green based phosphatase assay using OMFP as a substrate. All activities are normalized to the activity of protein without any compound which is set at 100%. “*” indicates p<0.05 using two-sample t test. B, Structure of the Eya2 ED + 9987 showing that F290, if changed to Y, can potentially form a hydrogen bond with T421 which will close the pocket for compound access or sterically interfere with compound binding through the extra hydroxyl group on Y. C, Corresponding residues around Eya2 F290 in Eya1–4.
The Tyr phosphatase activity of Eya2 enhances motility and invasiveness of lung cancer cells, but does not influence growth
To determine whether 9987’s cellular effects are on target, we first set out to identify a model system in which we could specifically assess the function of the Tyr phosphatase activity of Eya2. Although the Tyr phosphatase activity of Eyas has been shown to be tumor promotional in breast cancer (22), its role in lung cancer has never before been examined. Because Eya2 regulates numerous aspects of lung adenocarcinoma growth and progression, and because it has been linked to poor survival in lung cancer patients (10,11), we decided to use lung cancer as our model system to first determine the role of the Eya2 Tyr phosphatase in mediating the aggressive properties attributed to Eya2, and to then test whether our novel Eya2 allosteric Tyr phosphatase inhibitor is on target in these cells.
To this end, we first examined Eya2 and Six1 protein levels across several human lung cancer cell lines: Calu-6, H1299, HCC4006 and A549. We found that HCC4006 cells express high levels of endogenous Eya2 protein, while it is almost undetectable in A549 and Calu-6 cells. The main transcriptional partner of Eya2, Six1, is expressed in all of the lung cancer cell lines except for the A549 line (Supplementary Fig. S4A). We thus chose the Calu-6 cell line, which does not express high levels of Eya2, but does express Six1, as a system in which we could introduce either wildtype (WT) or Tyr phosphatase dead (D274N) Eya2 to the cells while maintaining its interaction with Six1 (which is endogenously present).
After introduction of either an empty vector (EV), wildtype (WT), or Tyr phosphatase dead mutant (D274N) of Eya2 into Calu-6 cells (Supplementary Fig. S4B), we first examined whether the D274N mutation alters cellular localization of Eya2 which is important for its proper function (33). We found that localization of the Eya2 D274N mutant was not altered compared to Eya2 WT (Supplementary Fig. S4C). We then used incucyte live-cell imaging to examine cell growth in response to overexpression of WT or D274N mutant Eya2, while colony formation capability was assessed using a 2D clonogenic assay. We found that both Eya2 WT and D274N promote Calu-6 growth (Fig. 4A) and colony forming ability (Fig. 4B). Using BrdU and caspase activity assays, we found that Eya2 mediates growth not through altering the proliferative capacity of the cells (Fig. 4C), but rather via inhibiting cell death (Fig. 4D). Importantly, these data demonstrate that Eya2 regulates growth and cell death in Calu-6 lung cancer cells independent of its Tyr phosphatase activity (Fig. 4A–D), as the D274N Eya2 mutant prevents cell death to the same extent as WT Eya2. We also examined whether Eya2 WT and D274N cells could alter characteristics associated with the epithelial to mesenchymal transition (EMT) in lung cancer cells. We found that similar to the effects on growth, both WT and D274N mutant Eya2 decreased expression of E-cadherin, while increasing expression of fibronectin. Thus, Eya2 also regulates the expression of markers associated with EMT in a Tyr phosphatase independent manner (Supplementary Fig. S4D). In contrast, we observed that only Eya2 WT, but not D274N, enhances Calu-6 cell migration (Fig. 4E), invadopodia formation (Fig. 4F), and invasion (Fig. 4G). Taken together, these data demonstrate that the Tyr phosphatase activity of Eya2 is critical for migratory and invasive phenotypes in lung cancer cells, but it does not influence lung cancer growth. Thus, the divergent phenotypic effects of Eya2 WT versus Eya2 D274N in the Calu-6 model could be used to distinguish whether our small molecule phosphatase inhibitor, 9987, acts on target in lung cancer cells.
Figure 4.

Function of the Eya2 Tyr phosphatase in Calu-6 lung cancer cells. A, Representative images of Calu-6 EV, Eya2 WT, or Eya2 D274N cells taken using IncuCyte Zoom 72 hours after seeding (left, scale bar=200μm). Overall growth curves of Calu-6 EV, Eya2 WT, or Eya2 D274N cells graphed based on fold change of confluence over time point 0 (right), error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001. B, Representative 2D clonogenic assay images of Calu-6 EV, Eya2 WT, or Eya2 D274N cells taken 7 days after seeding (left), as well as quantification of colony intensity (stained in purple). Error bars represent standard deviations of demonstrated individual experiment, n≥3 (right), *** p<0.001, * p<0.05. C, Representative cell cycle analysis of Calu-6 EV, Eya2 WT, or Eya2 D274N cells determined using BrdU incorporation/PI staining and flow cytometry is shown (left), and percentage of cells in G1, S and G2-M phase is graphed (right), error bars represent standard deviations of demonstrated individual experiment, n≥3. D, Representative images of activated Caspase 3/7 in Calu-6 EV, Eya2 WT, or Eya2 D274N cells taken using IncuCyte Zoom 72 hours after seeding (left, scale bar=200μm). Quantitation of green fluorescence intensity (representing activated Caspase 3/7), error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001 (right). E, Representative images of Calu-6 EV, Eya2 WT, or Eya2 D274N cells from a migration assay are shown (left), as well as quantification of distance migrated. Error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001 (right). F, Representative images of actin-rich and spike-like protrusions (invadopodia) formed by Calu-6 EV, Eya2 WT, or Eya2 D274N cells plated on a gelatin-coated surface (left). Quantification of percentage of invadopodia-possessing cells is plotted (right), error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001. G, Representative images of Calu-6 EV, Eya2 WT, or Eya2 D274N cells from a transwell invasion assay are shown (left), and quantification of the invaded area (%) (stained in purple) (right). Error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001.
Because the interaction of Eya2 with Six1 has been shown to be critical for phenotypes associated with Eya2 (34,35), we also as asked whether the Tyr phosphatase activities of Eya2 were dependent on Six1 function. Using a mutant of Eya2 that cannot bind Six1 (A532R), we repeated the above experiments. Intriguingly, we found that all phenotypes associated with Eya2 (whether Tyr phosphatase dependent or independent) require an interaction of Eya2 with Six1, demonstrating the critical role of Six1 in Eya2 function within lung cancer cells (Supplementary Fig. S5).
9987 inhibits Eya2 Tyr phosphatase-mediated phenotypes in lung cancer cells
We next evaluated the effect of 9987 in lung cancer cells to determine whether it mimics the effects of genetic disruption of the Eya2 Tyr phosphatase activity. Calu-6 cells were treated either with vehicle (DMSO) or with 9987 at 5μM and 10μM, and its effects on Eya2 localization, growth, colony formation ability, proliferation, cell death, EMT markers expression, migration, invadopodia formation and invasion were evaluated. Consistent with the effect of the Tyr phosphatase dead mutation (Fig. 4 & Supplementary Fig. S4C & S4D), we found that 9987 treatment does not affect Eya2 localization, overall growth, colony formation ability, proliferation, cell death, or EMT marker expression in EV or Eya2 WT expressing Calu-6 cells (Fig. 5A–F). In contrast, 9987 treatment effectively suppresses migration (Fig. 5G), invadopodia formation (Fig. 5H) and invasion (Fig. 5I) in Eya2 WT expressing Calu-6 cells, but not in EV Calu-6 cells. Moreover, growth, colony formation ability, motility and invasion of HCC4006 cells, which express high endogenous levels of Eya2, were also measured after 9987 treatment (Supplementary Fig. S6). We found that growth and colony formation of HCC4006 cells were both unaffected by 9987 treatment (Supplementary Fig. S6 A & B), whereas 9987 inhibited migration and invasion of these cells (Supplementary Fig. S6 C & D). Thus, the phenotypes inhibited by 9987 in HCC4006 cells are in line with those affected by the D274N mutation in Eya2, suggesting that 9987 is on target in lung cancer cells.
Figure 5.

Cellular activity of 9987. Calu-6 EV or Eya2 WT cells were treated with 9987 at 5 or 10 μM, and, similar to Figure 4, the impact of 9987 was tested on A, Eya2 localization, B, overall growth, C, colony formation, D, proliferation, E, Caspase 3/7 activation, F, EMT markers expression, G, motility, H, invadopodia formation and I. invasion. Error bars are standard deviations of demonstrated individual experiment, n≥3, ** p<0.01, *** p<0.001. A, Western blot analysis of cytosolic and nuclear Eya2 and Six1 levels in Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987. B, Representative images of Calu-6 EV or Eya2 WT cells treated with 9987, taken using IncuCyte Zoom 72 hours after seeding (scale bar=200μm). C, Representative 2D clonogenic assay images of Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987, taken 7 days after seeding. D, Representative cell cycle analysis of Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987 as determined using BrdU incorporation/PI staining and flow cytometry. E, Representative images of activated Caspase 3/7 in Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987, taken using IncuCyte Zoom 72 hours after seeding (scale bar=200μm). F, Western blot analysis FN1, E-cadherin, Vimentin and CK18 levels in Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987 for 2 days. G, Representative images of Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987 from a migration assay. H, Representative images of actin-rich and spike-like protrusions (invadopodia) formed by Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987 plated on a gelatin-coated surface. I, Representative images of Calu-6 EV or Eya2 WT cells treated without or with varying doses of 9987 from a transwell invasion assay.
9987 has no effect on lung cancer cells expressing an Eya2 F290Y mutant.
As mentioned, a single amino acid mutation (F290Y) in Eya2 confers resistance to 9987 inhibition in a biochemical assay). We took advantage of this mutant and generated Calu-6 cells overexpressing Eya2 F290Y to further evaluate whether 9987 is on target in cells (Fig. 6A). Eya2 F290Y enhances migration and invasion of Calu-6 cells to a similar level as Eya2 WT, but importantly, these phenotypes are not suppressed by 9987 (Fig. 6B & C). These data provide strong support that the effect of 9987 in lung cancer cells is due to its ability to specifically inhibit the Eya2-Tyr phosphatase activity.
Figure 6.

The F290Y mutation in Eya2 inhibits its targeting by 9987 in Calu-6 lung cancer cells. A, Western blot analysis of Eya2 levels in Calu-6 cells transfected with an EV, Eya2 WT or a Tyr phosphatase inhibition resistant version of Eya2 (Eya2 F290Y). B, Representative images of Calu-6 EV, Eya2 WT, or Eya2 F290Y cells from a migration assay are shown (left), as well as quantification of distance migrated. Error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001 (right). C, Representative images of Calu-6 EV, Eya2 WT, or Eya2 F290Y cells from a transwell invasion assay are shown (left), and the invaded area (%) (stained in purple) is quantified (right). Error bars represent standard deviations of demonstrated individual experiment, n≥3, *** p<0.001, * p<0.05.
Discussion
In this study, we sought to better understand the mechanism of action of a unique Eya2 Tyr phosphatase inhibitor, and to examine its effects in lung cancer, where Eya2 may play a particularly critical role. We chose lung cancer as our preferred model, given the previously described function of Eya2 in lung cancer growth, and the fact that it is overexpressed in this context, and that it correlates with poor prognosis (9,10,12,13). Similar to work done in ovarian cancer (27), we found that Eya2 stimulates growth of lung cancer cells in vitro largely through increasing cell survival instead of affecting proliferation. More importantly for this work, we find that Eya2-mediated growth and survival is not dependent on its Tyr phosphatase activity. We also show that Eya2 stimulates migration, invapodia formation, and invasion, all attributes associated with metastasis, and that these phenotypes are dependent on its Tyr phosphatase activity. It has been controversial as to whether the Tyr phosphatase activity of Eya is required for growth (22,36). Our findings are in line with those described by Hegde and colleagues in breast cancer (22), in that we did not observe a role for the Tyr phosphatase activity of Eya in mediating growth, via either effects on proliferation or survival. Similar to their observations in breast cancer, we demonstrate that Eya2 Tyr phosphatase does mediate migration and invasion in lung cancer cells. Taken together, these data suggest that Tyr phosphatase inhibitors of Eya2 may be used to limit tumor progression by affecting migration and invasion, but will likely not limit the growth of tumor cells, which may be more dependent on the interaction of Eya with Six family members (2) or on the role of Eyas in regulating the immune microenvironment (26).
Our structural data conclusively demonstrate that 9987 is an allosteric Eya2 Tyr phosphatase inhibitor. It is well known that developing phosphatase inhibitors is challenging due to low druggability of phosphatase active site (37). However, we demonstrate that allosteric inhibitors that are highly specific can be identified for the Eya phosphatases. The crystal structure of Eya2 bound to 9987 reveals how 9987 binds to an allosteric site in Eya2, and consequently inhibits its activity by inducing conformational changes in the catalytic core that creates an unfavorable conformation for Mg2+ binding (Fig. 2). The structure will be a valuable resource for future chemical modification of 9987 to improve its potency, which can potentially be achieved by filling in the empty spaces within the binding pocket observed, although challenges still remain. Residues 275–282 are relatively flexible and adopt variable conformations dependent on binding ligands. The high flexibility of these residues may form different pockets when the compound structure is altered. In addition, Mg2+ is an abundant ion under physiological conditions; a more potent compound that can potentially compete with this ion is needed for pre-clinical and clinical studies.
Nonetheless, our data demonstrate that 9987 can be used as a novel chemical probe that specifically targets the Tyr phosphatase activity of Eya2. Treatment of Calu-6 lung adenocarcinoma cells with the compound demonstrate that it inhibits migration, invadopodia formation, and invasion, all phenotypes associated with the Tyr phosphatase activity of Eya2, whereas it does not inhibit growth or survival mediated by Eya2, properties that are not dependent on its Tyr phosphatase activity. The compound also has no effect on lung cancer cells expressing an Eya2 F290Y mutant which abolishes compound binding in vitro. These data demonstrate that 9987 is on target in Calu-6 lung cancer cells, providing a powerful tool to investigate the mechanisms by which Eya2, as a Tyr phosphatase, functions both molecularly and cellularly.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Joshua Cabrera for all analysis on the Six1 specific antibody used in this work. We thank Dr. David Jones at the University of Colorado Denver Anschutz Medical Campus for help with NMR experiments in early stage of this project.
Financial Information
We thank the support from the Singapore Ministry of Health’s National Medical Research Council under its open fund individual research grant (OFIRG17may050, NMRC/OFIRG/0051/2017). We thank the support over the years from the NIH R03DA030559, NIH R41CA180347, Colorado Bioscience Discovery and Evaluation Grant, and Cancer League of Colorado Pilot Grant (to R.Z. and H.L.F.).
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Zhou H, Zhang L, Vartuli RL, Ford HL, Zhao R. The Eya phosphatase: Its unique role in cancer. The international journal of biochemistry & cell biology 2018;96:165–70 doi 10.1016/j.biocel.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patrick AN, Cabrera JH, Smith AL, Chen XS, Ford HL, Zhao R. Structure-function analyses of the human SIX1–EYA2 complex reveal insights into metastasis and BOR syndrome. Nat Struct Mol Biol 2013;20(4):447–53 doi 10.1038/nsmb.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cellular and molecular life sciences : CMLS 2009;66(4):565–83 doi 10.1007/s00018-008-8335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rebay I. Multiple Functions of the Eya Phosphotyrosine Phosphatase. Mol Cell Biol 2015;36(5):668–77 doi 10.1128/MCB.00976-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen KL, Patrick AN, McCoy EL, Ford HL. The six family of homeobox genes in development and cancer. Advances in cancer research 2008;101:93–126 doi 10.1016/S0065-230X(08)00405-3. [DOI] [PubMed] [Google Scholar]
- 6.Blevins MA, Towers CG, Patrick AN, Zhao R, Ford HL. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin Ther Targets 2015;19(2):213–25 doi 10.1517/14728222.2014.978860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-[beta] signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 2012;31(5):552–62 doi 10.1038/onc.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YT, Heist RS, Chirieac LR, Lin X, Skaug V, Zienolddiny S, et al. Genome-wide analysis of survival in early-stage non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009;27(16):2660–7 doi 10.1200/JCO.2008.18.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Y, Zheng S, Li Q, Xiang X, Gao T, Ran P, et al. Overexpression of miR-30a in lung adenocarcinoma A549 cell line inhibits migration and invasion via targeting EYA2. Acta biochimica et biophysica Sinica 2016;48(3):220–8 doi 10.1093/abbs/gmv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Qiu R, Qiu X, Tian T. EYA2 promotes lung cancer cell proliferation by downregulating the expression of PTEN. Oncotarget 2017;8(67):110837–48 doi 10.18632/oncotarget.22860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang Y, Xu X, Wang T, Li Y, You W, Fu J, et al. The EGFR/miR-338–3p/EYA2 axis controls breast tumor growth and lung metastasis. Cell death & disease 2017;8(7):e2928 doi 10.1038/cddis.2017.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mimae T, Okada M, Hagiyama M, Miyata Y, Tsutani Y, Inoue T, et al. Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(4):945–55 doi 10.1158/1078-0432.CCR-11-1946. [DOI] [PubMed] [Google Scholar]
- 13.Xia Y, Zhu Y, Ma T, Pan C, Wang J, He Z, et al. miR-204 functions as a tumor suppressor by regulating SIX1 in NSCLC. FEBS letters 2014;588(20):3703–12 doi 10.1016/j.febslet.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Zhou H, Li X, Vartuli RL, Rowse M, Xing Y, et al. Eya3 partners with PP2A to induce c-Myc stabilization and tumor progression. Nature communications 2018;9(1):1047 doi 10.1038/s41467-018-03327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003;426(6964):247–54 doi 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 16.Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, et al. Eyes absent represents a class of protein tyrosine phosphatases. Nature 2003;426(6964):295–8 doi 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 17.Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, et al. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 2003;426(6964):299–302 doi 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- 18.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 2006;7(11):833–46. [DOI] [PubMed] [Google Scholar]
- 19.Jung SK, Jeong DG, Chung SJ, Kim JH, Park BC, Tonks NK, et al. Crystal structure of ED-Eya2: insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2010;24(2):560–9 doi 10.1096/fj.09-143891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, et al. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem 2009;284(24):16066–70 doi 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009;458(7238):591–6 doi 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, et al. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene 2010;29(25):3715–22 doi 10.1038/onc.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Long YH, Wang SQ, Li YF, Zhang JH. Phosphorylation of H2A.X(T)(yr39) positively regulates DNA damage response and is linked to cancer progression. The FEBS journal 2016;283(24):4462–73 doi 10.1111/febs.13951. [DOI] [PubMed] [Google Scholar]
- 24.Yuan B, Cheng L, Chiang HC, Xu X, Han Y, Su H, et al. A phosphotyrosine switch determines the antitumor activity of ERbeta. The Journal of clinical investigation 2014;124(8):3378–90 doi 10.1172/JCI74085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mentel M, Ionescu AE, Puscalau-Girtu I, Helm MS, Badea RA, Rizzoli SO, et al. WDR1 is a novel EYA3 substrate and its dephosphorylation induces modifications of the cellular actin cytoskeleton. Sci Rep 2018;8(1):2910 doi 10.1038/s41598-018-21155-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vartuli RL, Zhou H, Zhang L, Powers RK, Klarquist J, Rudra P, et al. Eya3 promotes breast tumor-associated immune suppression via threonine phosphatase-mediated PD-L1 upregulation. The Journal of clinical investigation 2018;128(6):2535–50 doi 10.1172/JCI96784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer research 2005;65(3):925–32. [PubMed] [Google Scholar]
- 28.Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, et al. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol 2002;160(6):2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao T, Zheng S, Li Q, Ran P, Sun L, Yuan Y, et al. Aberrant hypomethylation and overexpression of the eyes absent homologue 2 suppresses tumor cell growth of human lung adenocarcinoma cells. Oncology reports 2015;34(5):2333–42 doi 10.3892/or.2015.4245. [DOI] [PubMed] [Google Scholar]
- 30.Robin TP, Smith A, McKinsey E, Reaves L, Jedlicka P, Ford HL. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Molecular cancer research : MCR 2012;10(8):1098–108 doi 10.1158/1541-7786.MCR-12-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krueger AB, Drasin DJ, Lea WA, Patrick AN, Patnaik S, Backos DS, et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J Biol Chem 2014;289(23):16349–61 doi 10.1074/jbc.M114.566729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krueger AB, Dehdashti SJ, Southall N, Marugan JJ, Ferrer M, Li X, et al. Identification of a selective small-molecule inhibitor series targeting the eyes absent 2 (Eya2) phosphatase activity. J Biomol Screen 2013;18(1):85–96 doi 10.1177/1087057112453936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohto H, Kamada S, Tago K, Tominaga SI, Ozaki H, Sato S, et al. Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol 1999;19(10):6815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 2012;31(5):552–62 doi 10.1038/onc.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patrick AN, Cabrera JH, Smith AL, Chen XS, Ford HL, Zhao R. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nat Struct Mol Biol 2013;20(4):447–53 doi 10.1038/nsmb.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, et al. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer research 2013;73(14):4488–99 doi 10.1158/0008-5472.CAN-12-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazo JS, McQueeney KE, Sharlow ER. New Approaches to Difficult Drug Targets: The Phosphatase Story. SLAS discovery : advancing life sciences R & D 2017:2472555217721142 doi 10.1177/2472555217721142. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W. Xds. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 2):125–32 doi 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007;40(Pt 4):658–74 doi 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 4):486–501 doi 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta crystallographica Section D, Biological crystallography 2012;68(Pt 4):352–67 doi 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon AF, Mueller GA, Zhong X, Pedersen LC. A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein science : a publication of the Protein Society 2010;19(5):901–13 doi 10.1002/pro.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.